Back to Archived Journals » Virus Adaptation and Treatment » Volume 6
Heterogeneous delivery is a barrier to the translational advancement of oncolytic virotherapy for treating solid tumors
Received 9 April 2014
Accepted for publication 29 April 2014
Published 29 July 2014 Volume 2014:6 Pages 11—31
DOI https://doi.org/10.2147/VAAT.S50110
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Amber C Miller,1,2 Stephen J Russell2,3
1Mayo Graduate School, Mayo Clinic, Rochester, Minnesota, USA; 2Department of Molecular Medicine, Mayo Clinic, Rochester, Minnesota, USA; 3Division of Hematology, Department of Medicine, Mayo Clinic, Rochester, Minnesota, USA
Abstract: Oncolytic viruses are a promising experimental anticancer therapy currently undergoing clinical translation. The development of oncolytic virotherapy offers a potential solution to the elusive “one-shot”cancer cure by providing targeted therapy to selectively infect and kill cancer cells while provoking adaptive anticancer immune responses as protection against distant metastasis and recurrent tumor challenge. While this technology has overcome barriers to safety and efficacy through cancer-specific targeting techniques, in order to reach full therapeutic potential, oncolytic therapies must still overcome barriers to intratumoral delivery and spread that result in heterogeneous intratumoral delivery and nonuniform response. This review will discuss barriers to oncolytic virotherapy translation related to mechanisms of delivering virus via tumor vasculature and distributing virus throughout the tumor microenvironment. Barriers include extravasation into the tumor that is dependent on adequate blood flow, tissue perfusion, and tumoral enhanced permeability and retention for transvascular transport. Subsequently, virions must undergo interstitial transport against dense stromal barriers and high interstitial fluid pressure to initiate infection. In order to achieve massive tumor regression, infection must spread to cover large volumes of tumor mass. Furthermore, virus bioavailability is quickly dampened upon systemic administration due to neutralization and sequestration. These barriers to delivery limit the amount of virus that effectively reaches and spreads within the tumor, forcing dose increases that impinge upon limits of production and increase possibility of adverse events. Techniques to overcome these barriers are discussed but largely remain to be translated into clinical use.
Keywords: oncolytic virotherapy, barriers, delivery, systemic therapy, translation
Introduction
The need for new cancer therapeutics is ever-present. Oncolytic virotherapy is an experimental cancer therapy that commandeers the ability of a virus to cause cellular death and incite adaptive immune responses to fight cancer. The use of oncolytic virotherapy as a treatment for malignant tumors has been studied both preclinically and in clinical trials. The history of oncolytic virotherapy development and clinical trials has been extensively reviewed.1,2 Briefly, the field was initially sparked by case reports of individuals with solid cancers who upon coincidental contraction of infectious disease (later identified as viral infection) showed clinical remission.3 The field lost momentum around the 1970s when development was met with limited efficacy and noncancerous tissue toxicity but has seen resurgence over the last quarter-century due to advances in understanding of genetic engineering and biological systems. Currently, viruses of naturally evolved or engineered cancer tropism are being used to specifically infect, replicate in, and kill cancer cells while leaving neighboring healthy cells unharmed due to cancer-specific abnormalities such as defective cellular signaling or receptor expression.4 Subsequent activation of the immune response during viral infection leads to cross-priming of the T-cells against tumor antigens leading to an antitumor adaptive immune response.5,6
There are many different types of oncolytic viruses currently undergoing clinical trial to treat a multitude of different late-stage refractory or recurrent cancers through various approaches of administration and combinations with other therapies (Table S1). Oncolytic viruses currently being translated to clinical practice include DNA viruses: adenovirus, herpes simplex virus, parvovirus, and vaccinia virus, and RNA viruses: coxsackie virus, Newcastle disease virus, Measles virus, poliovirus, reovirus, Seneca Valley virus, and vesicular stomatitis virus. Table S1 presents a list of clinical trials currently registered with the National Institutes of Health (clinicaltrials.gov), summarizing the translational status of oncolytic viruses and demonstrating the breadth and potential of this growing field. As the first oncolytic viruses near completion in Phase III testing, we recognize that the unique duality of the oncolytic paradigm embodies the potential for a one-shot cancer cure by both destroying existing tumor cells through induction of massive tumor cell lysis and maintaining an anticancer immune state to wipe out the total cancer cell population and protect against recurrence.7 Third-generation oncolytic viruses now in clinical trials have improved safety and efficacy, as they are better able to avoid off-target effects by targeting viral infection through the use of molecular techniques that allow for specific transduction, transcription, and replication.2,8,9 Oncolytic virotherapy also offers the ability to target the heterogeneous tumor-cell population, including quiescent cancer stem cells that can be a cause of tumor resistance and recurrence with standard therapies.10–14 Although many early-phase clinical trials have been completed using oncolytic virotherapies, only three have made it to Phase III testing due to variability in efficacy (adenovirus; NCT01869088, herpes simplex virus; NCT00169704/01368276, reovirus; NCT01166542; Table S1). This variability in efficacy is matched with preclinical results that show nonuniformity of intratumoral response to oncolytic therapies and even long-term viral persistence within the tumor in the absence of complete tumor response.15–17 This identifies a clear barrier in oncolytic virotherapy.
The benefits of these targeted therapies as single-agent cancer therapies are only as good as the ability to adequately deliver the virus to tumor cells. It is becoming increasingly apparent that intratumoral virus delivery and spread is a limiting factor in the translational success of oncolytic virotherapies. The barrier of delivery is of special importance in the use of systemic administration, which allows for targeted viruses to reach disseminated or numerous tumor deposits. Currently, limited success has been seen with systemic oncolytic virotherapy. Since the earliest reports of intravenously administered oncolytic viruses in humans, only five reasonably well documented complete responses to single agent virotherapy have been achieved (Table 1). Of these studies, detection of virus delivery to the tumor is variable. This highlights the need to optimize delivery of systemically administered therapy to tumor sites. Furthermore, our mathematical model derived to optimize oncolytic virotherapy parameters predicts that improvements in delivery can have large impacts on oncolytic efficacy.18 To achieve a one-shot cure with oncolytic virotherapy, access to all tumor cells must be maximized, requiring effective and uniform delivery of virus to the tumor. Therefore, this review will discuss issues pertaining to viral delivery, specifically focusing on barriers to intratumoral dissemination that are a major impediment to translational success.
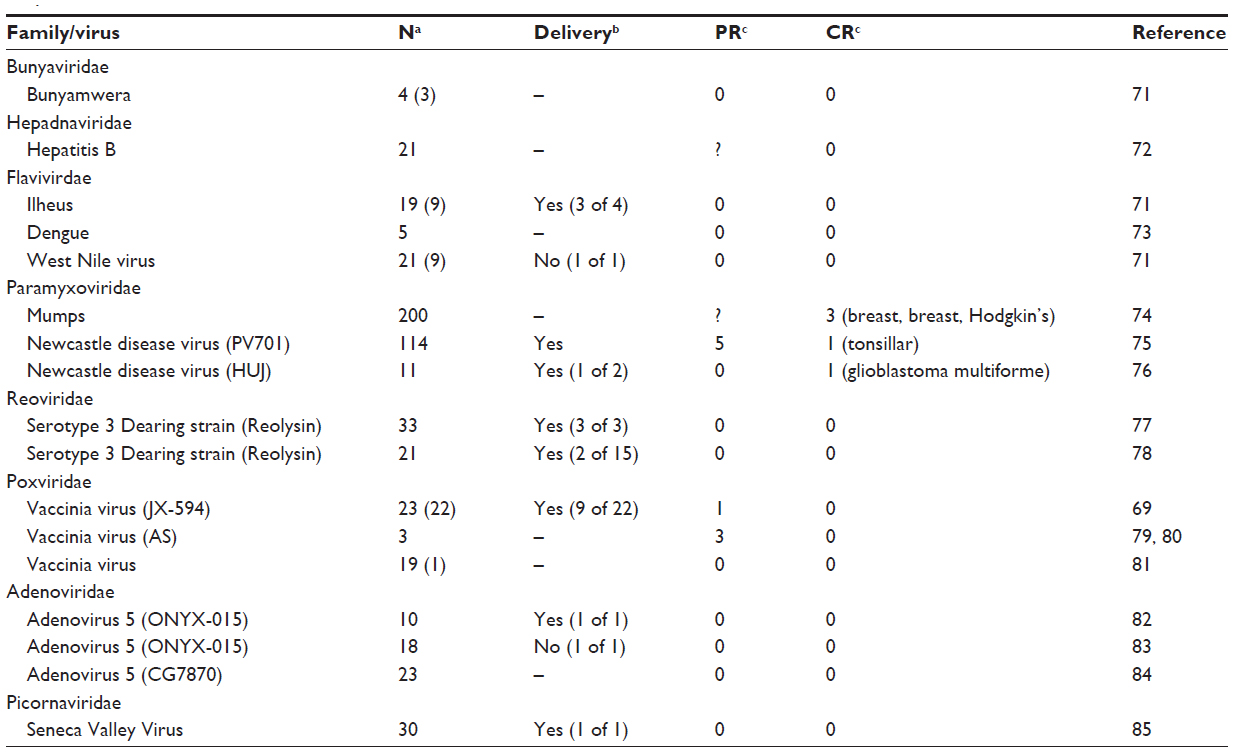 | Table 1 Clinical trials with intravenously administered oncolytic virotherapy monotherapy show variable delivery and limited response |
Barriers to success
While mechanisms to efficiently target viruses to cancer cells have been well established, the ability to effectively and uniformly deliver the virotherapy to the site of the tumor remains a barrier. Uniform coverage of the tumor is hindered by infection voids, locations where virus fails to initiate infection. Infection voids can result from inefficient delivery or extravasation from the blood vessel or the inability to achieve the viremic threshold necessary to seed infection. These voids correlate with regions of tumor-cell viability that remains after single-agent oncolytic virotherapy and limit efficacy (Figure 1). By focusing on the path of an individual virion en route to intratumoral infection, barriers to delivery can be easily identified (Figure 2). Virus arriving at the tumor site must first specifically extravasate into the tumor, bearing dependence on adequate blood flow, tissue perfusion, and tumoral enhanced permeability and retention (EPR) for transvascular transport. Subsequently, virions must undergo interstitial transport against dense stromal barriers and high interstitial fluid pressure to initiate infection. In order to achieve massive tumor regression, infection must spread to cover large volumes of tumor mass. Furthermore, virus bioavailability is quickly dampened upon systemic administration due to neutralization by circulating antibodies, association with factors in the blood, and sequestration by the mononuclear phagocytic system to the liver and spleen. These barriers to delivery limit the amount of virus that effectively reaches and spreads within the tumor, forcing dose increases that impinge upon limits of good manufacturing practice production and increase possibility of adverse events.
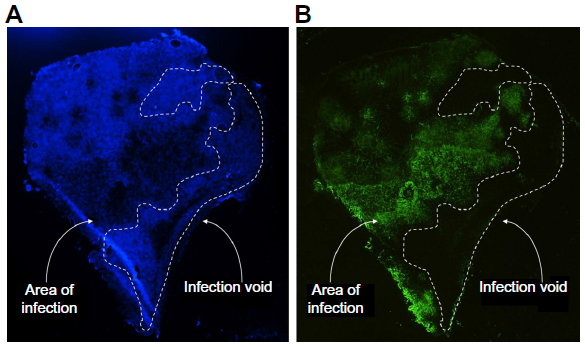 | Figure 1 Systemically administered oncolytic virotherapy success is limited by barriers to delivery that result in clinically relevant infection voids. |
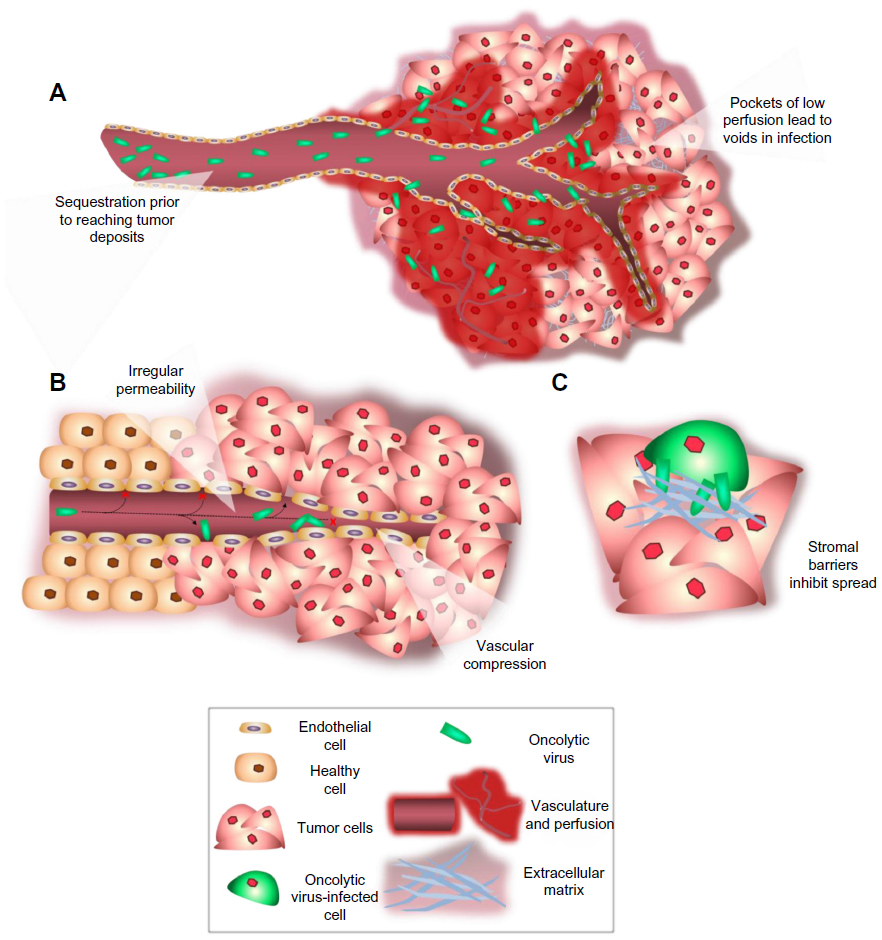 | Figure 2 Barriers to intratumoral delivery and distribution encountered by oncolytic viruses. |
Barriers to transvascular transport
When oncolytic virotherapy is systemically administered, access to the tumor is achieved through the blood supply. Delivery throughout the tumor is therefore dependent on tumor vasculature. Initial tumoral delivery is met by barriers to adequate tumor perfusion and vascular permeability necessary to get the virus into the tumor, including irregular blood flow, vascular occlusion, vascular organization, and permeability.
Tumor perfusion
A cause of heterogeneous deposition is the irregularity of blood flow due to inefficient and disorganized tumor vasculature, given that the rate and distribution of blood flow determines the amount of systemically administered virus that can reach specific regions of the tumor. Tumor vasculature is chaotic in terms of microvessel length, diameter, spatial distribution, and blood flow velocity and direction. Tumor vasculature can be further impacted by growing tumor cells and high interstitial pressure that can occlude flow and force vessels apart, thus decreasing microvascular density.19–21 The result is both spatially and temporally heterogeneous blood perfusion throughout the tumor. Poor perfusion causes pockets of low oxygenation due to low blood flow that result in transiently or chronically hypoxic tumor regions.22–24 As hypoxia is representative of perfusion state, it may also be indicative of therapeutic delivery; therefore, understanding of hypoxia is relevant to virotherapy delivery. It is known that areas of transient or chronic hypoxic tissue occur as microregions heterogeneously distributed within the tumor mass and may be located next to areas of normal perfusion.25,26 Transient hypoxia has been well documented in tumors in which markers of blood flow and perfusion show mismatched delivery after administration at different time points.23,27,28 Tracking changes in intratumoral partial oxygen pressure also shows cyclical turnover or fluctuations in blood flow that occur over time. Hypoxia is linked to tumor stem cell niches, treatment resistance, and metastasizing disease, as hypoxic tissue is a characteristically difficult target. Intriguingly, new technologies are available to monitor and target hypoxic regions.29 Therefore, hypoxia is a clear barrier and target for improving virus delivery.
By normalizing blood flow, the causes of spatial and temporal hypoxia can be reduced, thereby increasing perfusion throughout the entire tumor. Tumor blood flow can be increased for the purpose of therapeutic delivery through manipulation of the systemic vasculature. Physiological changes include increasing blood flow or perfusion pressure to the tumor by increased systemic blood pressure (pharmacologically or through exercise). This approach is based on the principle that irregular, poorly differentiated tumor vasculature does not maintain blood flow volume homeostasis like systemic vasculature does. In this way, while systemic vasculature that is exposed to vasoconstrictors or shear stress experiences increases in peripheral resistance and arteriolar blood pressure, the tumor vasculature experiences increased perfusion pressure, resulting in increased blood flow into the tumor.30,31 Small changes in blood flow over short periods of time, as evidenced in multiple tumor types by measuring tumor oxygenation, result in changes in the hypoxic regions surrounding tumor tissue.22 If these small changes can be therapeutically induced, they can be used advantageously to change areas of perfusion and therefore particle deposition. The vasoconstrictor angiotensin-II has been used in preclinical and clinical studies to increase tumor-specific blood flow and intratumoral delivery of chemotherapies and macromolecular complexes.30–35 While this idea of improving nanoparticle delivery by influencing blood pressure during infusion was first suggested by Matsumara and Maeda in 1986, its use with oncolytic viruses has yet to be reported.36
Enhanced permeability and retention
Once the virus arrives at the vascular wall, it must exit the vasculature. Passive diffusion from the circulation into the tumor parenchyma is mediated by the enhanced permeability and retention (EPR) effect, a phenomenon that results from irregular, leaky tumor vasculature with enhanced permeability and the lack of lymphatic drainage that allows for particle retention.36,37 However, the EPR effect is not consistent, with differences in vascular pore size and abrupt changes in tumoral blood perfusion resulting in heterogeneous accumulation.35,38 While passive EPR-based delivery may be unpredictable, nanodrugs consistently accumulate within a tumor.35 This is because the enhanced permeability is selective for particles above 40 kDa, which is far surpassed by the average oncolytic therapy.35 Beyond molecular mass, the dimensions of current oncolytic viruses undergoing translation are important in understanding physical barriers to viral movement (Table 2). Tumor leakiness is related to the size of tumor vessel interendothelial pores. The size of pores in tumor endothelium is heterogeneous and tumor dependent, ranging from less than 1 nm to greater than 1 μm, while the endothelial gaps in healthy tissue are only ~2–6 nm in diameter.21,39–41 Endothelial gap size determines the pore cutoff size, a functional measure of size exclusion during transvascular transport of nanoparticles. This is important in determining if oncolytic virotherapy is even a viable option for the tumor, as the virus must be able to extravasate between endothelial cells to disseminate throughout the tumor. In some tumors, especially primary brain tumors where the pore cutoff size is as small as 1 nm, systemically administered oncolytic virotherapy will be unable to deliver to the tumor.42 Interestingly, in animal models it has been shown that this pore cutoff size can change due to location of tumor growth and to growth conditions such as hormone depletion.40 It should be noted that the primary means of tumor interendothelial junction measurements has been observation of subcutaneously grown murine tumors of human xenografts. Although these observations may differ compared to those seen in patients, the distinctive discrepancy between tumor endothelium and that of healthy tissue allows for tumor-specific pathways of virus extravasation.
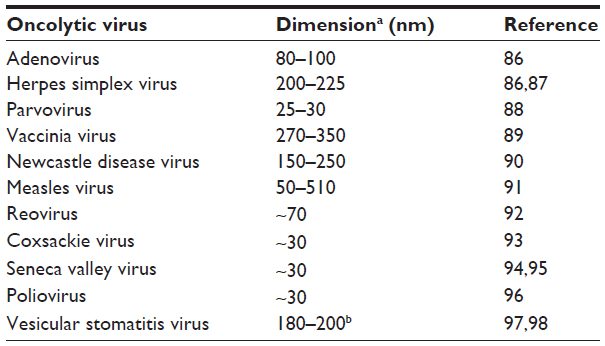 | Table 2 Approximate virion dimensions of oncolytic viruses currently undergoing clinical translation |
Learning how to use the EPR effect to our advantage will assist in overcoming obstacles related to diffusion and heterogeneous deposition. Permeability factors such as vascular endothelial growth factor (VEGF), bradykinin, nitric oxide, prostaglandins, tumor necrosis factor (TNF)-α, TNF-β, and interleukin-2 facilitate tumor-selective enhanced vascular permeability and improved delivery of macromolecular drugs in solid tumors.35 The use of permeability factors may be a promising combinatorial-therapy approach for oncolytic virotherapy. The use of VEGF has been shown to increase vascular leakiness and increased tumor transduction in combination with oncolytic Sindbis virus.43 Intravenous administration of TNF-α resulted in increased permeability and up to six-fold increase in adenovirus extravasation into tumor tissue.44 Many of these permeability factors remain to be explored for the purposes of selectively increasing tumor vasculature permeability and subsequent viral delivery. Another means of increasing permeability is the induction of transient hyperthermia. Mild hyperthermia (41°C–42°C) has been used to increase tumor-specific extravasation of nanoparticles to xenograft tumor models by increasing the pore cutoff size and increasing endothelial cell gaps up to 10 μm in some tumor models.45,46 Increased permeability was maintained up to 8 hours after heating.46 Hyperthermia presents another potential means of modifying tumor permeability for oncolytic virotherapy delivery that has yet to be explored.
Barriers to interstitial transport
Upon transvascular extravasation or release of progeny from an infected tumor cell, the virions must passively diffuse to uninfected tumor cells and establish infection. The EPR effect acts as a double-edged sword for nanoparticle distribution, since the poorly organized and leaky vasculature results in unregulated extravasation that contributes to increased interstitial fluid pressure (IFP). High IFP can cause intratumoral vessels to collapse, which in combination with dense tumoral extracellular matrix and lack of lymphatic flow results in loss of convective flow throughout the tumor, making macromolecular transport dependent on passive diffusion.17,47,48 Passive diffusion is limited by the composition and organization of the tumor microenvironment, including the dense extracellular matrix (ECM), and tight packing of tumor and normal stromal cells such as resident macrophages.38,49 Indeed, a mathematical model developed to provide insight into the causes of insufficient distribution predicted that an improvement in effective diffusion coefficient could result in a substantial increase in number of infected cells.50 One way to alter the diffusion coefficient is through degradation or manipulation of the ECM and surrounding interstitial space.
Interstitial fluid pressure
High IFP affects intravenous delivery by limiting movement of large molecules such as oncolytic viruses out of the vascular bed and limiting access to the tumor core where IFP is highest. The high pressures are comparable to that of the microvascular pressure, causing the loss of a pressure gradient necessary for convection-driven diffusion.51 Mechanical manipulation techniques have been developed to aid in spread against pressure gradients. Therapeutic ultrasound can be used to enhance delivery of therapeutic agents. Ultrasound induced inertial cavitation, which has been shown to successfully deliver macromolecules larger than several millimeters against pressure gradients, has been used to enhance distribution of oncolytic adenovirus in in vitro and in vivo models.52–55 For example, focused ultrasound increased systemically administered oncolytic adenovirus delivery to tumor up to 50-fold when coadministered with microbubbles for induction of inertial cavitation.53 Although this technique increases diffusion into solid tumor, this is not a viable option for disseminated or metastatic cancer where location is unknown or too numerous for ultrasound targeting. Solid stress alleviation via tumor debulking can also reduce IFP and reduce cellular infringement upon tumor vasculature, thereby improving virus delivery. Solid stress alleviation can be achieved by inducing cell death to reduce crowding in the interstitial space. The average distance between tumor cells is approximately 20 nm, smaller than the dimension of oncolytic viruses, therefore creating a physical barrier (Table 2).56 Not only do chemotherapies and oncolytic viruses work in synergy to enhance cancer survival, chemotherapies can actually benefit tumoral virus penetration. Channels formed by the spaces left behind by dead tumor cells independent of any ECM alterations will allow for increased tumor penetration by virus particles. Apoptosis-inducing chemotherapies in combination with oncolytic virotherapy have resulted in increased spread within tumors and synergistic antitumor effect. For example, induction of apoptosis using chemotherapies and TNF-related apoptosis-inducing ligand increased oncolytic herpes virus intratumoral spread and produced a more diffuse pattern of infection in mice with both collagen-rich and collagen-poor mammary tumors.56
Another approach to decrease IFP uses anti-VEGF or other antiangiogenic therapies to reduce irregular tumor vascularity and allow for tumor neovasculature normalization. This normalization reduces hypoxia and IFP, resulting in an intravascular–interstitial pressure gradient that allows increased delivery of drugs to the tumor and increased survival of patients given chemoradiotherapies.20,57–60 This technique has yet to be fully developed with oncolytic virotherapies, although work with oncolytic herpes simplex virus has shown that the order of treatment with anti-VEGF bevacizumab in combination with virus treatment matters. While Eshun et al61 saw an increase in overall tumor response when bevacizumab was given prior to virus compared to either therapy alone, a greater overall response was seen when bevacizumab was given after virus. This may be explained by the dual role VEGF plays in herpes simplex virus receptor expression. Perhaps the decrease in IFP after initiation of infection allowed for greater local spread and tumor control. However, vascular normalization also diminishes the vascular permeability necessary for extravasation. For this reason it will be necessary to balance any improvement in virus delivery gained by vascular normalization leading to increased tumor penetration against the parallel loss in extravasation of macromolecules due to decreased vascular permeability.62
Extracellular matrix
A major contributor to high IFP is the ECM, primarily made up of collagen, elastic fibers, glycosaminoglycans, and proteoglycans, which creates a gel-like medium difficult for viruses to traverse given that the size of viral vectors is comparable to or larger than the ~70–100 nm space between fibers17,21,63 (Table 2). It has been demonstrated that the greater the fibrillar collagen content, the lower the interstitial diffusion rate of macromolecules, and because of this higher collagen content, a greater infusion pressure is necessary to initiate flow into the tumor interstitium.49,64 The ECM composition, and therefore the IFP, can be altered by ECM-degrading enzymes. For instance, collagenases reduce IFP and increase transcapillary pressure gradients, improving uptake and distribution of immunoglobulin G in solid tumors.65 Overexpression of matrix metalloproteinases in tumors was found to enhance convection and viral distribution.66 Recombinant viruses expressing matrix-degrading proteins have been developed to successfully increase distribution and penetration in solid tumors. A multitude of oncolytic viruses in combination with or genetically engineered to express matrix-degrading proteins – including but not limited to bacterial collagenases, matrix metalloproteinases, relaxin, hyaluronidase, and heparanase to increase distribution, control tumor growth, and increase survival of tumor-bearing animals – have been reviewed previously.67 Table 3 summarizes the main matrix-degrading enzymes used in combination with oncolytic viruses to increase distribution of and tumor response to oncolytic viruses. As an example, increased levels of matrix metalloproteinases increased intratumoral virus delivery, distribution, and tumor response to adenovirus, herpes simplex virus, and vaccinia virus in multiple tumor models.62,66–68 However, the efficacy of these approaches above traditional oncolytic virotherapy paradigms is minimal, with the increase in delivery and distribution resulting in a nonsignificant survival benefit. Further, the associated risk of potentiating the spread of replicating viruses must be closely monitored as these techniques continue to be developed.
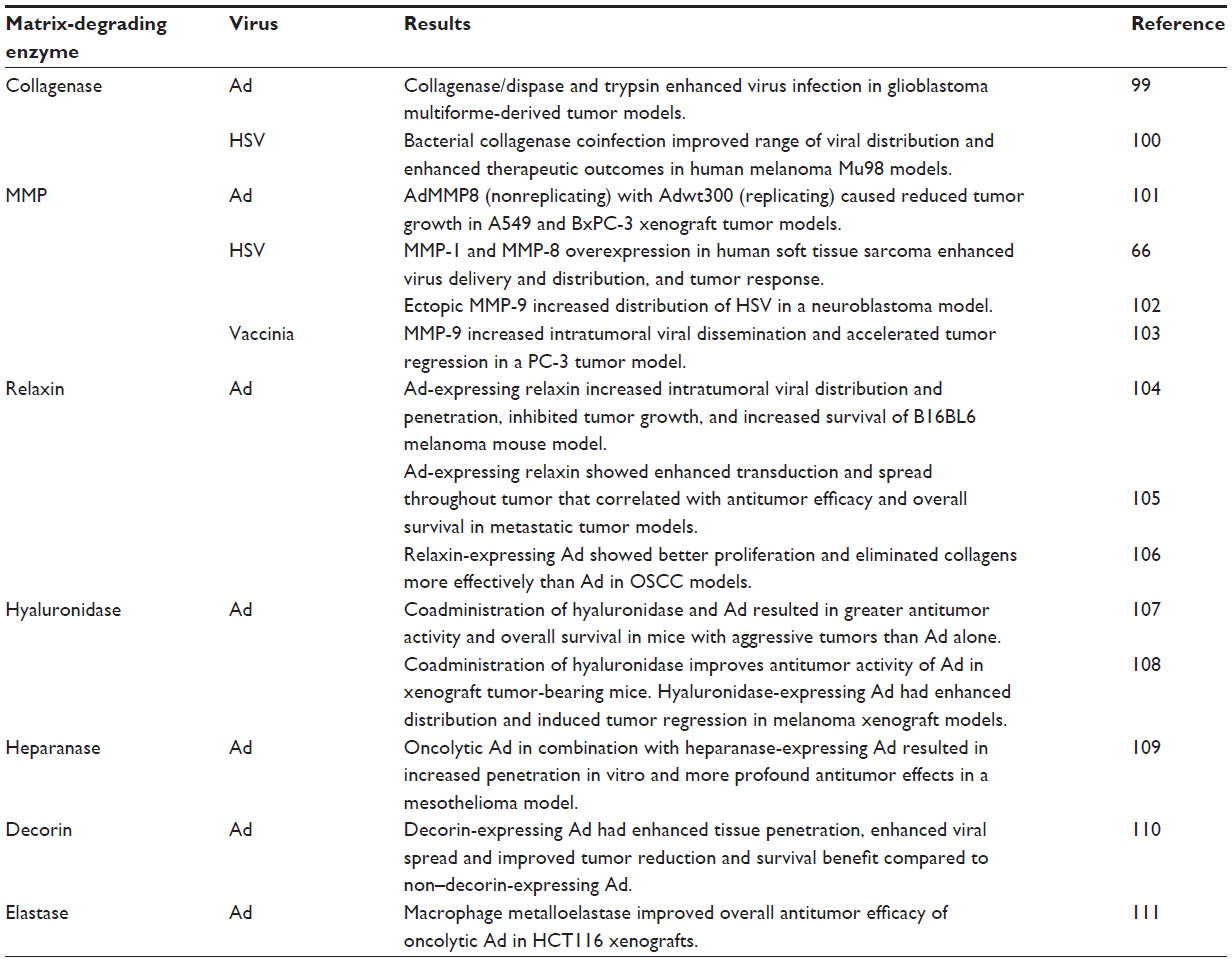 | Table 3 Examples of extracellular matrix degradation techniques used in combination with oncolytic viruses in preclinical work that increase intratumoral virus delivery and spread |
Other translational barriers
Administration, extravasation, and immune modulation
Routes of administration must also be addressed, as they can affect initial viral delivery. Current routes of virus administration include intravenous administration, intratumoral injection, and regional delivery including intracavitary, intrapleural, intraperitoneal, and regional perfusion. While intratumoral or regional administration offer increased delivery specificity, this type of delivery is limited to local spread in the absence of secondary viremia and requires tumors to be injectable, making disseminated disease or metastases difficult targets. Intravenous administration allows for systemic delivery of the virus and is therefore a necessary focus for optimization of the one-shot cure. Systemic administration depends on the ability of the virus to traverse the vascular barrier at sites of tumor growth while evading neutralization and sequestration. Whereas virion size plays a large role in the ability of the virus to traverse the physical barriers primarily focused on in this review, it should be noted that the physical size of the virion is not the determining factor in oncolytic virotherapy efficacy, which is also dependent on factors such as receptor availability, the ability of tumors to support virus infection, and immune status. Viruses are prone to neutralization by pre-existing antibodies, complement proteins, and coagulation and other serum factors that lead to opsonization. Recognition by the mononuclear phagocyte system, including splenic macrophages and hepatic Kupffer cells, results in phagocytosis and rapid clearance from circulation and can result in liver toxicity.4,68 Approaches to reduce neutralization and sequestration include serum depletion, saturation of the mononuclear phagocyte system and other virus recognition systems, shielding of viruses through engineered coats or surface modifications, and the use of cell carriers to protect and transport therapies to tumor sites. Many strategies to reduce neutralization and sequestration have been employed in preclinical testing and reviewed previously.4 Table 4 highlights successful strategies used in the oncolytic virotherapy field to eliminate binding, increase persistence in circulation, reduce sequestration, and improve tumor transduction. Barriers to transvascular transport must be remembered when any type of shielding of the virus is used, as shielding can increase particle dimensions and thereby limit extravasation. Similarly, immune depletion used prior to virotherapy to increase delivery could limit the efficacy of the subsequent immunotherapeutic phase of oncolytic virotherapy. It is therefore necessary to balance improvement in delivery with potential losses in efficacy as these techniques continue to be developed.
 | Table 4 Selected examples of strategies to reduce neutralization and sequestration of oncolytic virotherapy in preclinical work that show increased tumoral deposition and decreased sequestration |
Overcoming the barriers placed by the immune system and taking advantage of viral-initiated antitumor immunity will be critical to the success of systemic virotherapy and has been extensively reviewed.5 Because the role of the host antiviral and virus-initiated antitumor immune response in the therapeutic outcome of oncolytic virotherapies is another potential barrier to translation, it will be briefly discussed here. The host antiviral immune response decreases the amount of virus available to establish intratumoral infection. As discussed previously, soluble immune mediators, along with nonspecific blood-factor binding and nonspecific extravasation, limit viral efficacy following systemic delivery by reducing the bioavailability of the therapy. However, immunosuppression that accompanies cancer or the induction of an immunosuppressed state may both aid and prevent oncolytic efficacy. Thus, immunosuppression leads to enhanced virus delivery and more extensive intratumoral virus spread, but at the same time it limits cross-priming of the immune system, thereby reducing any immunotherapeutic benefit gained from the therapy. However, cross-priming of the antitumor immune response after oncolytic virus administration may be more the exception than the rule, and the host immune response may remain completely focused on the virus, not seeing the tumor antigens at all. Therefore, viral immunodominance may be an important obstacle to effective oncolytic immunotherapy. Considerable additional research in this area is needed.
Dose and manufacturing
As oncolytic therapies approach approval through late-phase clinical trials, focus must also be placed on issues of translation outside of safety and therapeutic efficacy. These include the production and cost bottleneck. In order to achieve a clinically relevant infection, viral delivery must exceed the threshold necessary to establish infection. As seen in the Phase I clinical trial of intravenously administered JX-594, virus was recoverable from tumor biopsies only at doses above 109 infectious units, demonstrating that tumoral deposition and most likely destruction is dose dependent.4,69 Since delivery is dose dependent, current delivery barriers severely drive dose up, impinging on the limits of production capability. Currently, production facilities that employ good manufacturing practice have the capability to produce high-titer virus preparations, but it is costly and time intensive. Manufacture requires high-grade purification techniques to remove cellular proteins and nucleic acid contaminants while maintaining gentle conditions for shear-sensitive viruses such as measles virus.70 With the idea that more virus delivered to the tumor will lead to more tumor infected, we can actively seek to maximize efficacy of current doses or even decrease the dose required by optimizing the current barriers that stand in the way of therapeutic efficacy, namely, deficiencies in delivery and distribution that result in nonuniform response.
Public acceptance
Perhaps unique to oncolytic viruses are other areas of safety that must be addressed, including pre-existing immunity, pathogenicity, and transmission. The idea of a replicating therapy is bound to bring controversy and apprehension to public acceptance. While certain genetic modifications provide built-in mechanisms of protection against infectious outbreaks by increasing tumor specificity and reducing the ability for the virus to spontaneously revert to a harmful strain, modifications such as matrix-degrading enzymes may have contradicting effects that increase likelihood of extratumoral dissemination of replicating viruses. It is difficult to tell what public perception of oncolytic virotherapies will be, as there are currently no therapies approved by the US Food and Drug Administration and little knowledge in the general public. It can be predicted that public acceptance of the use of replicating virotherapy will be met with hesitation, similar to that seen with the use of viruses for gene therapies and vaccines. However, patient acceptability and adherence may be improved compared to standard anticancer regimens as the field of oncolytics moves towards a one-shot cure that will reduce the need for repeat dosing and hospital stays.
Conclusion
Oncolytic viruses present a new paradigm in cancer treatment that allows for selective cancer-cell killing with built-in maintenance mechanisms to protect against tumor recurrence and to target distant metastases. As the technology advances along the path of translation, we are learning that although oncolytic virotherapy has shown efficacy in early-phase clinical trials, oncolytic virotherapies are still limited by deficiencies in tumor coverage. By looking at the specific path that is taken by a virion after systemic administration, we have elucidated key barriers to uniform delivery and distribution throughout the tumor, including sequestration and neutralization prior to reaching the tumor site, homogeneous distribution via the tumor circulation followed by transvascular transport, and interstitial transport to establish sites of infection, summarized in Table 5. Preclinical work has been done to address many of the barriers pertaining to stages of delivery and shows potential to overcome barriers to uniform delivery. Many of these techniques, however, have yet to be translated or used in current oncolytic virotherapy clinical trials (Table S1). This highlights a translational gap that will need to be addressed as this field seeks to optimize virotherapies. Taking the insight learned in preclinical and early-phase clinical trials regarding variable response rates matched with nonuniform therapeutic distribution, extensive work has been done to improve the delivery and spread of oncolytic viruses throughout the tumor. Knowledge of the delivery barriers and techniques to overcome them is the first step in translating a truly optimized oncolytic virotherapy.
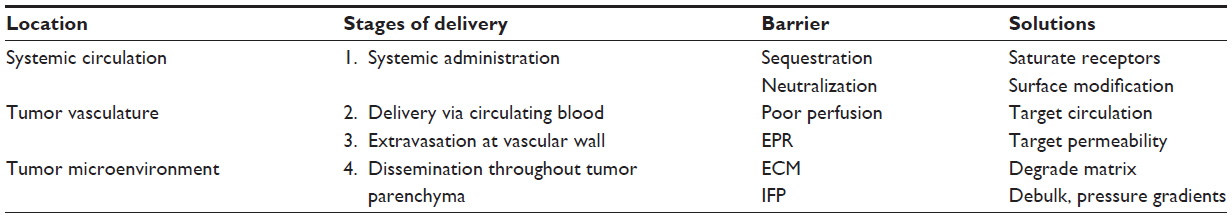 | Table 5 Stages of oncolytic virotherapy delivery with barriers that limit therapeutic efficacy |
Acknowledgments
Amber C Miller is the Mayo Clinic Gary and Anita Klesch Predoctoral fellow.
Disclosure
Stephen J Russell is Chief Manager of ImaNIS Life Sciences, LLC. The authors report no other conflicts of interest in this work.
References
Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15(4):651–659. | |
Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007;4(2):101–117. | |
Dock G. The influence of complicating diseases upon leukemia. Am J Med Sci. 1904;127(4):563–592. | |
Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30(7):658–670. | |
Prestwich RJ, Harrington KJ, Pandha HS, Vile RG, Melcher AA, Errington F. Oncolytic viruses: a novel form of immunotherapy. Expert Rev Anticancer Ther. 2008;8(10):1581–1588. | |
Tong AW, Senzer N, Cerullo V, Templeton NS, Hemminki A, Nemunaitis J. Oncolytic viruses for induction of anti-tumor immunity. Curr Pharm Biotechnol. 2012;13(9):1750–1760. | |
Naik S, Nace R, Federspiel MJ, Barber GN, Peng KW, Russell SJ. Curative one-shot systemic virotherapy in murine myeloma. Leukemia. 2012;26(8):1870–1878. | |
Cattaneo R, Miest T, Shashkova EV, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol. 2008;6(7):529–540. | |
Naik S, Russell SJ. Engineering oncolytic viruses to exploit tumor specific defects in innate immune signaling pathways. Expert Opin Biol Ther. 2009;9(9):1163–1176. | |
Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. | |
Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284. | |
Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. | |
Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY. Nanomedicine therapeutic approaches to overcome cancer drug resistance. Adv Drug Deliv Rev. 2013;65(13–14):1866–1879. | |
Park CH, Bergsagel DE, McCulloch EA. Mouse myeloma tumor stem cells: a primary cell culture assay. J Natl Cancer Inst. 1971;46(2):411–422. | |
Harrison D, Sauthoff H, Heitner S, Jagirdar J, Rom WN, Hay JG. Wild-type adenovirus decreases tumor xenograft growth, but despite viral persistence complete tumor responses are rarely achieved – deletion of the viral E1b-19-kD gene increases the viral oncolytic effect. Hum Gene Ther. 2001;12(10):1323–1332. | |
Sauthoff H, Hu J, Maca C, et al. Intratumoral spread of wild-type adenovirus is limited after local injection of human xenograft tumors: virus persists and spreads systemically at late time points. Hum Gene Ther. 2003;14(5):425–433. | |
Yun CO. Overcoming the extracellular matrix barrier to improve intratumoral spread and therapeutic potential of oncolytic virotherapy. Curr Opin Mol Ther. 2008;10(4):356–361. | |
Bailey K, Kirk A, Naik S, et al. Mathematical model for radial expansion and conflation of intratumoral infectious centers predicts curative oncolytic virotherapy parameters. PLoS One. 2013;8(9):e73759. | |
Jain RK. Determinants of tumor blood flow: a review. Cancer Res. 1988;48(10):2641–2658. | |
Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31(17):2205–2218. | |
Wang Y, Yuan F. Delivery of viral vectors to tumor cells: extracellular transport, systemic distribution, and strategies for improvement. Ann Biomed Eng. 2006;34(1):114–127. | |
Cárdenas-Navia LI, Mace D, Richardson RA, Wilson DF, Shan S, Dewhirst MW. The pervasive presence of fluctuating oxygenation in tumors. Cancer Res. 2008;68(14):5812–5819. | |
Dewhirst MW. Relationships between cycling hypoxia, HIF-1, angiogenesis and oxidative stress. Radiat Res. 2009;172(6):653–665. | |
Dewhirst MW, Ong ET, Braun RD, et al. Quantification of longitudinal tissue pO2 gradients in window chamber tumours: impact on tumour hypoxia. Br J Cancer. 1999;79(11–12):1717–1722. | |
Höckel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93(4):266–276. | |
Sutherland RM. Cell and environment interactions in tumor microregions: the multicell spheroid model. Science. 1988;240(4849):177–184. | |
Brown JM. Evidence for acutely hypoxic cells in mouse tumours, and a possible mechanism of reoxygenation. Br J Radiol. 1979;52(620):650–656. | |
Chaplin DJ, Durand RE, Olive PL. Acute hypoxia in tumors: implications for modifiers of radiation effects. Int J Radiat Oncol Biol Phys. 1986;12(8):1279–1282. | |
Busk M, Horsman MR. Relevance of hypoxia in radiation oncology: pathophysiology, tumor biology and implications for treatment. Q J Nucl Med Mol Imaging. 2013;57(3):219–234. | |
Tozer GM, Shaffi KM. Modification of tumour blood flow using the hypertensive agent, angiotensin II. Br J Cancer. 1993;67(5):981–988. | |
Nagamitsu A, Greish K, Maeda H. Elevating blood pressure as a strategy to increase tumor-targeted delivery of macromolecular drug SMANCS: cases of advanced solid tumors. Jpn J Clin Oncol. 2009;39(11):756–766. | |
Hattori Y, Ubukata H, Kawano K, Maitani Y. Angiotensin II-induced hypertension enhanced therapeutic efficacy of liposomal doxorubicin in tumor-bearing mice. Int J Pharm. 2011;403(1–2):178–184. | |
Suzuki M, Hori K, Abe I, Saito S, Sato H. A new approach to cancer chemotherapy: selective enhancement of tumor blood flow with angiotensin II. J Natl Cancer Inst. 1981;67(3):663–669. | |
Li CJ, Miyamoto Y, Kojima Y, Maeda H. Augmentation of tumour delivery of macromolecular drugs with reduced bone marrow delivery by elevating blood pressure. Br J Cancer. 1993;67(5):975–980. | |
Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65(1):71–79. | |
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986; 46(12 Pt 1):6387–6392. | |
Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. | |
Prabhakar U, Maeda H, Jain RK, et al. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73(8):2412–2417. | |
Hashizume H, Baluk P, Morikawa S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156(4):1363–1380. | |
Hobbs SK, Monsky WL, Yuan F, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A. 1998;95(8):4607–4612. | |
Fox ME, Szoka FC, Fréchet JM. Soluble polymer carriers for the treatment of cancer: the importance of molecular architecture. Acc Chem Res. 2009;42(8):1141–1151. | |
Yuan F, Salehi HA, Boucher Y, Vasthare US, Tuma RF, Jain RK. Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res. 1994;54(17):4564–4568. | |
Tseng JC, Granot T, DiGiacomo V, Levin B, Meruelo D. Enhanced specific delivery and targeting of oncolytic Sindbis viral vectors by modulating vascular leakiness in tumor. Cancer Gene Ther. 2010;17(4):244–255. | |
Seki T, Carroll F, Illingworth S, et al. Tumour necrosis factor-alpha increases extravasation of virus particles into tumour tissue by activating the Rho A/Rho kinase pathway. J Control Release. 2011;156(3):381–389. | |
Kong G, Braun RD, Dewhirst MW. Hyperthermia enables tumor-specific nanoparticle delivery: effect of particle size. Cancer Res. 2000;60(16):4440–4445. | |
Li L, ten Hagen TL, Bolkestein M, et al. Improved intratumoral nanoparticle extravasation and penetration by mild hyperthermia. J Control Release. 2013;167(2):130–137. | |
Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Pathology: cancer cells compress intratumour vessels. Nature. 2004;427(6976):695. | |
Carlisle R, Coussios CC. Mechanical approaches to oncological drug delivery. Ther Deliv. 2013;4(10):1213–1215. | |
Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000;60(9):2497–2503. | |
Mok W, Stylianopoulos T, Boucher Y, Jain RK. Mathematical modeling of herpes simplex virus distribution in solid tumors: implications for cancer gene therapy. Clin Cancer Res. 2009;15(7):2352–2360. | |
Boucher Y, Baxter LT, Jain RK. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: implications for therapy. Cancer Res. 1990;50(15):4478–4484. | |
Bazan-Peregrino M, Arvanitis CD, Rifai B, Seymour LW, Coussios CC. Ultrasound-induced cavitation enhances the delivery and therapeutic efficacy of an oncolytic virus in an in vitro model. J Control Release. 2012;157(2):235–242. | |
Bazan-Peregrino M, Rifai B, Carlisle RC, et al. Cavitation-enhanced delivery of a replicating oncolytic adenovirus to tumors using focused ultrasound. J Control Release. 2013;169(1–2):40–47. | |
Rifai B, Arvanitis CD, Bazan-Peregrino M, Coussios CC. Cavitation-enhanced delivery of macromolecules into an obstructed vessel. J Acoust Soc Am. 2010;128(5):EL310–EL315. | |
Carlisle R, Choi J, Bazan-Peregrino M, et al. Enhanced tumor uptake and penetration of virotherapy using polymer stealthing and focused ultrasound. J Natl Cancer Inst. 2013;105(22):1701–1710. | |
Nagano S, Perentes JY, Jain RK, Boucher Y. Cancer cell death enhances the penetration and efficacy of oncolytic herpes simplex virus in tumors. Cancer Res. 2008;68(10):3795–3802. | |
Dickson PV, Hamner JB, Sims TL, et al. Bevacizumab-induced transient remodeling of the vasculature in neuroblastoma xenografts results in improved delivery and efficacy of systemically administered chemotherapy. Clin Cancer Res. 2007;13(13):3942–3950. | |
Gerstner ER, Duda DG, di Tomaso E, et al. VEGF inhibitors in the treatment of cerebral edema in patients with brain cancer. Nat Rev Clin Oncol. 2009;6(4):229–236. | |
Sorensen AG, Emblem KE, Polaskova P, et al. Increased survival of glioblastoma patients who respond to antiangiogenic therapy with elevated blood perfusion. Cancer Res. 2012;72(2):402–407. | |
Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64(11):3731–3736. | |
Eshun FK, Currier MA, Gillespie RA, Fitzpatrick JL, Baird WH, Cripe TP. VEGF blockade decreases the tumor uptake of systemic oncolytic herpes virus but enhances therapeutic efficacy when given after virotherapy. Gene Ther. 2010;17(7):922–929. | |
Zhang Q, Bindokas V, Shen J, Fan H, Hoffman RM, Xing HR. Time-course imaging of therapeutic functional tumor vascular normalization by antiangiogenic agents. Mol Cancer Ther. 2011;10(7):1173–1184. | |
Xu X, Sabanayagam CR, Harrington DA, Farach-Carson MC, Jia X. A hydrogel-based tumor model for the evaluation of nanoparticle-based cancer therapeutics. Biomaterials. 2014;35(10):3319–3330. | |
McGuire S, Zaharoff D, Yuan F. Nonlinear dependence of hydraulic conductivity on tissue deformation during intratumoral infusion. Ann Biomed Eng. 2006;34(7):1173–1181. | |
Eikenes L, Bruland ØS, Brekken C, Davies Cde L. Collagenase increases the transcapillary pressure gradient and improves the uptake and distribution of monoclonal antibodies in human osteosarcoma xenografts. Cancer Res. 2004;64(14):4768–4773. | |
Mok W, Boucher Y, Jain RK. Matrix metalloproteinases-1 and -8 improve the distribution and efficacy of an oncolytic virus. Cancer Res. 2007;67(22):10664–10668. | |
Smith E, Breznik J, Lichty BD. Strategies to enhance viral penetration of solid tumors. Hum Gene Ther. 2011;22(9):1053–1060. | |
Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu Rev Immunol. 2002;20:825–852. | |
Breitbach CJ, Burke J, Jonker D, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477(7362):99–102. | |
Langfield KK, Walker HJ, Gregory LC, Federspiel MJ. Manufacture of measles viruses. Methods Mol Biol. 2011;737:345–366. | |
Southam CM, Moore AE. West Nile, Ilheus, and Bunyamwera virus infections in man. Am J Trop Med Hyg. 1951;31(6):724–741. | |
Hoster HA, Zanes RP Jr, Von Haam E. Studies in Hodgkin’s syndrome; the association of viral hepatitis and Hodgkin’s disease; a preliminary report. Cancer Res. 1949;9(8):473–480. | |
ERF LA. Human leukemia, with emphasis upon recent therapeutic experiences. Pa Med J. 1950;53(11):1172–1181. | |
Okuno Y, Asada T, Yamanishi K, et al. Studies on the use of mumps virus for treatment of human cancer. Biken J. 1978;21(2):37–49. | |
Lorence RM, Pecora AL, Major PP, et al. Overview of phase I studies of intravenous administration of PV701, an oncolytic virus. Curr Opin Mol Ther. 2003;5(6):618–624. | |
Freeman AI, Zakay-Rones Z, Gomori JM, et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther. 2006;13(1):221–228. | |
Vidal L, Pandha HS, Yap TA, et al. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res. 2008;14(21):7127–7137. | |
Galanis E, Markovic SN, Suman VJ, et al. Phase II trial of intravenous administration of Reolysin(®) (Reovirus Serotype-3-dearing Strain) in patients with metastatic melanoma. Mol Ther. 2012;20(10):1998–2003. | |
Kawa A, Arakawa S. The effect of attenuated vaccinia virus AS strain on multiple myeloma; a case report. Jpn J Exp Med. 1987;57(1):79–81. | |
Arakawa S Jr, Hamami G, Umezu K, Kamidono S, Ishigami J, Arakawa S. Clinical trial of attenuated vaccinia virus AS strain in the treatment of advanced adenocarcinoma. Report on two cases. J Cancer Res Clin Oncol. 1987;113(1):95–98. | |
Hunter-Craig I, Newton KA, Westbury G, Lacey BW. Use of vaccinia virus in the treatment of metastatic malignant melanoma. Br Med J. 1970;2(5708):512–515. | |
Nemunaitis J, Cunningham C, Buchanan A, et al. Intravenous infusion of a replication-selective adenovirus (ONYX-015) in cancer patients: safety, feasibility and biological activity. Gene Ther. 2001;8(10):746–759. | |
Hamid O, Varterasian ML, Wadler S, et al. Phase II trial of intravenous CI-1042 in patients with metastatic colorectal cancer. J Clin Oncol. 2003;21(8):1498–1504. | |
Small EJ, Carducci MA, Burke JM, et al. A phase I trial of intravenous CG7870, a replication-selective, prostate-specific antigen-targeted oncolytic adenovirus, for the treatment of hormone-refractory, metastatic prostate cancer. Mol Ther. 2006;14(1):107–117. | |
Rudin CM, Poirier JT, Senzer NN, et al. Phase I clinical study of Seneca Valley Virus (SVV-001), a replication-competent picornavirus, in advanced solid tumors with neuroendocrine features. Clin Cancer Res. 2011;17(4):888–895. | |
Hermens WT, Verhaagen J. Viral vectors, tools for gene transfer in the nervous system. Prog Neurobiol. 1998;55(4):399–432. | |
Grünewald K, Desai P, Winkler DC, et al. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science. 2003;302(5649):1396–1398. | |
Meng G, Zhang X, Plevka P, Yu Q, Tijssen P, Rossmann MG. The structure and host entry of an invertebrate parvovirus. J Virol. 2013;87(23):12523–12530. | |
Smith SA, Kotwal GJ. Immune response to poxvirus infections in various animals. Crit Rev Microbiol. 2002;28(3):149–185. | |
Sinkovics JG, Horvath JC. Newcastle disease virus (NDV): brief history of its oncolytic strains. J Clin Virol. 2000;16(1):1–15. | |
Liljeroos L, Huiskonen JT, Ora A, Susi P, Butcher SJ. Electron cryotomography of measles virus reveals how matrix protein coats the ribonucleocapsid within intact virions. Proc Natl Acad Sci U S A. 2011;108(44):18085–18090. | |
Kapikian AZ, Shope RE. Rotaviruses, reoviruses, coltiviruses, and orbiviruses. In: Baron S, editor. Medical Microbiology. 4th ed. Galveston, TX: University of Texas Medical Branch; 1996. | |
Xiao C, Bator-Kelly CM, Rieder E, et al. The crystal structure of coxsackievirus A21 and its interaction with ICAM-1. Structure. 2005;13(7):1019–1033. | |
Venkataraman S, Reddy SP, Loo J, Idamakanti N, Hallenbeck PL, Reddy VS. Structure of Seneca Valley Virus-001: an oncolytic picornavirus representing a new genus. Structure. 2008;16(10):1555–1561. | |
Venkataraman S, Reddy SP, Loo J, Idamakanti N, Hallenbeck PL, Reddy VS. Crystallization and preliminary X-ray diffraction studies of Seneca Valley virus-001, a new member of the Picornaviridae family. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008; 64(Pt 4):293–296. | |
Hogle JM, Chow M, Filman DJ. Three-dimensional structure of poliovirus at 2.9 A resolution. Science. 1985;229(4720):1358–1365. | |
Cureton DK, Massol RH, Whelan SP, Kirchhausen T. The length of vesicular stomatitis virus particles dictates a need for actin assembly during clathrin-dependent endocytosis. PLoS Pathog. 2010;6(9): e1001127. | |
Hodges J, Tang X, Landesman MB, et al. Asymmetric packaging of polymerases within vesicular stomatitis virus. Biochem Biophys Res Commun. 2013;440(2):271–276. | |
Kuriyama N, Kuriyama H, Julin CM, Lamborn K, Israel MA. Pretreatment with protease is a useful experimental strategy for enhancing adenovirus-mediated cancer gene therapy. Hum Gene Ther. 2000;11(16):2219–2230. | |
McKee TD, Grandi P, Mok W, et al. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006;66(5):2509–2513. | |
Cheng J, Sauthoff H, Huang Y, et al. Human matrix metalloproteinase-8 gene delivery increases the oncolytic activity of a replicating adenovirus. Mol Ther. 2007;15(11):1982–1990. | |
Hong CS, Fellows W, Niranjan A, et al. Ectopic matrix metalloproteinase-9 expression in human brain tumor cells enhances oncolytic HSV vector infection. Gene Ther. 2010;17(10):1200–1205. | |
Schäfer S, Weibel S, Donat U, et al. Vaccinia virus-mediated intra-tumoral expression of matrix metalloproteinase 9 enhances oncolysis of PC-3 xenograft tumors. BMC Cancer. 2012;12:366. | |
Kim JH, Lee YS, Kim H, Huang JH, Yoon AR, Yun CO. Relaxin expression from tumor-targeting adenoviruses and its intratumoral spread, apoptosis induction, and efficacy. J Natl Cancer Inst. 2006;98(20):1482–1493. | |
Ganesh S, Gonzalez Edick M, Idamakanti N, et al. Relaxin-expressing, fiber chimeric oncolytic adenovirus prolongs survival of tumor-bearing mice. Cancer Res. 2007;67(9):4399–4407. | |
Lee SY, Park HR, Rhee J, Park YM, Kim SH. Therapeutic effect of oncolytic adenovirus expressing relaxin in radioresistant oral squamous cell carcinoma. Oncol Res. 2013;20(9):419–425. | |
Ganesh S, Gonzalez-Edick M, Gibbons D, Van Roey M, Jooss K. Intratumoral coadministration of hyaluronidase enzyme and oncolytic adenoviruses enhances virus potency in metastatic tumor models. Clin Cancer Res. 2008;14(12):3933–3941. | |
Guedan S, Rojas JJ, Gros A, Mercade E, Cascallo M, Alemany R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol Ther. 2010;18(7):1275–1283. | |
Watanabe Y, Kojima T, Kagawa S, et al. A novel translational approach for human malignant pleural mesothelioma: heparanase-assisted dual virotherapy. Oncogene. 2010;29(8):1145–1154. | |
Choi IK, Lee YS, Yoo JY, et al. Effect of decorin on overcoming the extracellular matrix barrier for oncolytic virotherapy. Gene Ther. 2010;17(2):190–201. | |
Lavilla-Alonso S, Bauer MM, Abo-Ramadan U, et al. Macrophage metalloelastase (MME) as adjuvant for intra-tumoral injection of oncolytic adenovirus and its influence on metastases development. Cancer Gene Ther. 2012;19(2):126–134. | |
Haisma HJ, Kamps JA, Kamps GK, Plantinga JA, Rots MG, Bellu AR. Polyinosinic acid enhances delivery of adenovirus vectors in vivo by preventing sequestration in liver macrophages. J Gen Virol. 2008; 89(Pt 5):1097–1105. | |
Liu YP, Tong C, Dispenzieri A, Federspiel MJ, Russell SJ, Peng KW. Polyinosinic acid decreases sequestration and improves systemic therapy of measles virus. Cancer Gene Ther. 2012;19(3):202–211. | |
Fulci G, Dmitrieva N, Gianni D, et al. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res. 2007;67(19):9398–9406. | |
Ciavarra RP, Taylor L, Greene AR, et al. Impact of macrophage and dendritic cell subset elimination on antiviral immunity, viral clearance and production of type 1 interferon. Virology. 2005;342(2):177–189. | |
Tao N, Gao GP, Parr M, et al. Sequestration of adenoviral vector by Kupffer cells leads to a nonlinear dose response of transduction in liver. Mol Ther. 2001;3(1):28–35. | |
Fisher KD, Seymour LW. HPMA copolymers for masking and retargeting of therapeutic viruses. Adv Drug Deliv Rev. 2010;62(2):240–245. | |
Prill JM, Subr V, Pasquarelli N, et al. Traceless bioresponsive shielding of adenovirus hexon with HPMA copolymers maintains transduction capacity in vitro and in vivo. PLoS One. 2014;9(1):e82716. | |
Tesfay MZ, Kirk AC, Hadac EM, et al. PEGylation of vesicular stomatitis virus extends virus persistence in blood circulation of passively immunized mice. J Virol. 2013;87(7):3752–3759. | |
De Geest B, Snoeys J, Van Linthout S, Lievens J, Collen D. Elimination of innate immune responses and liver inflammation by PEGylation of adenoviral vectors and methylprednisolone. Hum Gene Ther. 2005;16(12):1439–1451. | |
Gao JQ, Eto Y, Yoshioka Y, et al. Effective tumor targeted gene transfer using PEGylated adenovirus vector via systemic administration. J Control Release. 2007;122(1):102–110. | |
Doronin K, Shashkova EV, May SM, Hofherr SE, Barry MA. Chemical modification with high molecular weight polyethylene glycol reduces transduction of hepatocytes and increases efficacy of intravenously delivered oncolytic adenovirus. Hum Gene Ther. 2009;20(9):975–988. | |
Eto Y, Yoshioka Y, Ishida T, et al. Optimized PEGylated adenovirus vector reduces the anti-vector humoral immune response against adenovirus and induces a therapeutic effect against metastatic lung cancer. Biol Pharm Bull. 2010;33(9):1540–1544. | |
Shi HS, Yang LP, Wei W, et al. Systemically administered liposome-encapsulated Ad-PEDF potentiates the anti-cancer effects in mouse lung metastasis melanoma. J Transl Med. 2013;11:86. | |
Shikano T, Kasuya H, Sahin TT, et al. High therapeutic potential for systemic delivery of a liposome-conjugated herpes simplex virus. Curr Cancer Drug Targets. 2011;11(1):111–122. | |
Jiang ZK, Koh SB, Sato M, et al. Engineering polypeptide coatings to augment gene transduction and in vivo stability of adenoviruses. J Control Release. 2013;166(1):75–85. | |
Vetter A, Virdi KS, Espenlaub S, et al. Adenoviral vectors coated with PAMAM dendrimer conjugates allow CAR independent virus uptake and targeting to the EGF receptor. Mol Pharm. 2013;10(2):606–618. | |
Grünwald GK, Vetter A, Klutz K, et al. Systemic image-guided liver cancer radiovirotherapy using dendrimer-coated adenovirus encoding the sodium iodide symporter as theranostic gene. J Nucl Med. 2013;54(8):1450–1457. | |
Grünwald GK, Vetter A, Klutz K, et al. EGFR-targeted adenovirus dendrimer coating for improved systemic delivery of the theranostic NIS Gene. Mol Ther Nucleic Acids. 2013;2:e131. | |
Willmon C, Harrington K, Kottke T, Prestwich R, Melcher A, Vile R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol Ther. 2009;17(10):1667–1676. | |
Power AT, Bell JC. Taming the Trojan horse: optimizing dynamic carrier cell/oncolytic virus systems for cancer biotherapy. Gene Ther. 2008;15(10):772–779. | |
Russell SJ, Peng KW. The utility of cells as vehicles for oncolytic virus therapies. Curr Opin Mol Ther. 2008;10(4):380–386. | |
Mader EK, Maeyama Y, Lin Y, et al. Mesenchymal stem cell carriers protect oncolytic measles viruses from antibody neutralization in an orthotopic ovarian cancer therapy model. Clin Cancer Res. 2009;15(23):7246–7255. | |
Iankov ID, Msaouel P, Allen C, et al. Demonstration of anti-tumor activity of oncolytic measles virus strains in a malignant pleural effusion breast cancer model. Breast Cancer Res Treat. 2010;122(3):745–754. | |
Kottke T, Diaz RM, Kaluza K, et al. Use of biological therapy to enhance both virotherapy and adoptive T-cell therapy for cancer. Mol Ther. 2008;16(12):1910–1918. | |
Fulci G, Breymann L, Gianni D, et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci U S A. 2006;103(34):12873–12878. | |
Hasegawa N, Abei M, Yokoyama KK, et al. Cyclophosphamide enhances antitumor efficacy of oncolytic adenovirus expressing uracil phosphoribosyltransferase (UPRT) in immunocompetent Syrian hamsters. Int J Cancer. 2013;133(6):1479–1488. | |
Ungerechts G, Springfeld C, Frenzke ME, et al. An immunocompetent murine model for oncolysis with an armed and targeted measles virus. Mol Ther. 2007;15(11):1991–1997. | |
Ikeda K, Wakimoto H, Ichikawa T, et al. Complement depletion facilitates the infection of multiple brain tumors by an intravascular, replication-conditional herpes simplex virus mutant. J Virol. 2000;74(10):4765–4775. | |
Shashkova EV, Doronin K, Senac JS, Barry MA. Macrophage depletion combined with anticoagulant therapy increases therapeutic window of systemic treatment with oncolytic adenovirus. Cancer Res. 2008;68(14):5896–5904. |
Supplementary materials
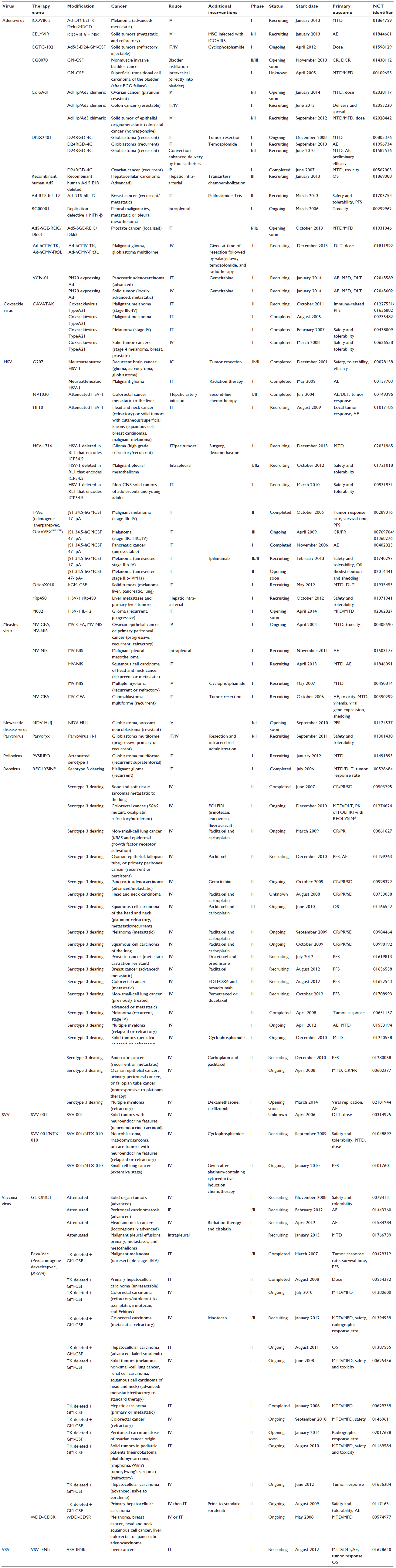 | Table S1 Current clinical trials registered with the US National Institutes of Health (clinicaltrials.gov) investigating the use of replicating oncolytic viruses as a cancer treatment gives an insight into the translational status of this therapy |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.