Back to Journals » Clinical and Experimental Gastroenterology » Volume 9
Hereditary pancreatitis: current perspectives
Authors Raphael K, Willingham F
Received 2 December 2015
Accepted for publication 8 April 2016
Published 26 July 2016 Volume 2016:9 Pages 197—207
DOI https://doi.org/10.2147/CEG.S84358
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Andreas M. Kaiser
Kara L Raphael, Field F Willingham
Division of Digestive Diseases, Department of Medicine, Emory University School of Medicine, Atlanta, GA, USA
Abstract: Hereditary pancreatitis (HP) is a rare cause of acute, recurrent acute, and chronic pancreatitis. It may present similarly to other causes of acute and chronic pancreatitis, and often there has been a protracted evaluation prior to the diagnosis of HP. Since it was first described in 1952, multiple genetic defects that affect the action of digestive enzymes in the pancreas have been implicated. The most common mutations involve the PRSS1, CFTR, SPINK1, and CTRC genes. New mutations in these genes and previously unrecognized mutations in other genes are being discovered due to the increasing use of next-generation genomic sequencing. While the inheritance pathways of these genetic mutations may be variable and complex, sometimes involving coinheritance of other mutations, the clinical presentation of patients tends to be similar. Interactions with environmental triggers often play a role. Patients tend to present at an early age (prior to the second decade of life) and have a significantly increased risk for the development of pancreatic adenocarcinoma. Patients with HP may develop sequelae of chronic pancreatitis such as strictures and fluid collections as well as exocrine and endocrine insufficiency. Management of patients with HP involves avoidance of environmental triggers, surveillance for pancreatic adenocarcinoma, medical therapy for endocrine and exocrine insufficiency, pain management, and endoscopic or surgical treatment for complications. Care for affected patients should be individualized, with an emphasis on early diagnosis and multidisciplinary involvement to develop a comprehensive treatment strategy.
Keywords: pancreatic cancer, chronic pancreatitis, idiopathic pancreatitis, pancreatitis, familial pancreatitis, genetic mutations
Introduction
Hereditary pancreatitis (HP) is a rare cause of acute, recurrent acute, and chronic pancreatitis. It may present similarly to other causes of pancreatitis, such as alcohol, gallstones, autoimmune causes, anatomic variants, such as pancreas divisum, or hypertriglyceridemia. The pathway to the diagnosis of HP is often protracted, and many patients have been characterized as idiopathic before the detection and diagnosis of HP. HP can now be characterized with increasing specificity due to the increasing availability of testing for inherited genetic mutations. In the proper clinical scenario, patients with these genetic mutations are considered to have HP. In the absence of a detected mutation, HP may also be considered if the family history is suggestive of an inherited condition. More specifically, it has been suggested that the diagnosis of HP can be made without genetic testing if the patient has both a personal history of pancreatitis and recurrent acute or chronic pancreatitis diagnosed in two first-degree relatives or in three or more second-degree relatives, spanning at least two generations.1 Compared to the other causes of pancreatitis, HP has several unique features, including early onset of disease, as well as a significantly increased risk for pancreatic cancer. An overview of HP, including the most recent perspectives and updates, is reviewed.
History
In 1952, the pedigree of a family with four members diagnosed with chronic relapsing pancreatitis and two additional members with likely disease was outlined.2 These six family members spanned three generations, had the early onset of pancreatitis at or before the third decade of life, and experienced similar symptoms and sequelae in an autosomal dominant pattern. This was the first description of HP in the literature. In 1971, HP was defined as
inflammation of the pancreas, usually recurrent from childhood, with unusual prevalence among blood-related groups of persons in accordance with Mendelian laws.3
Over the next several decades, 100 additional families with multiple generations of individuals diagnosed with HP were cited in the literature.4–10 In 1996, a genome segregation analysis of a large family with 47 individuals affected with HP was performed, identifying several chromosomal markers on the long arm of chromosome 711; this was the first step toward the discovery of an HP gene. This work was confirmed by two independent groups,12,13 and shortly thereafter, the first genetic defect leading to HP was identified. This defect involved an arginine to histidine substitution in codon 122 of the cationic trypsinogen serine protease 1 gene, or the R122H mutation of PRSS1.14 Subsequently, numerous genetic mutations both on the PRSS1 gene and on several other genes have been identified as etiologic factors in the development of HP.
Epidemiology
Chronic pancreatitis is a disease with both a high morbidity and cost burden. The United States National Institutes of Health Database Studies identified chronic pancreatitis as the seventh most common digestive disease diagnosis in hospitalized patients, with health care utilization costs upward of 3 billion dollars.15 Chronic pancreatitis is thought to occur in 5–14/100,000 people in the United States with an annual prevalence of 50/100,000 people.16 Recently, the North American Pancreatitis Trial 2 identified alcohol as the most common etiologic factor in chronic pancreatitis, noting that 44.5% of cases were caused by alcohol use; of the “nonalcohol” etiologies, genetic factors accounted for ∼8.7% of cases.17 The estimated prevalence of HP is 0.3/100,000 people in France, 0.125/100,000 people in Germany, and 0.57/100,000 people in Denmark.18,19 The worldwide prevalence has not been estimated in the literature. However, these numbers likely underrepresent the true prevalence of HP due to infrequent testing outside of specialty centers. In 2015, in children previously diagnosed with idiopathic acute, recurrent acute, and chronic pancreatitis, 33%, 45.4%, and 54.4%, respectively, were associated with a genetic etiology.20 HP is one of the main causes of pancreatitis in childhood. A population-based study identified the mean age at diagnosis of chronic pancreatitis due to nonalcohol etiologies to be 64 years.21 However, it has been determined that the median age at diagnosis of HP ranges from 5 years to 19 years, with progression to chronic pancreatitis usually within the first 2 decades of life.19,22
Clinical features
HP can present initially as recurrent acute pancreatitis with pancreatic inflammation, manifested by the sudden onset of abdominal pain, nausea, and vomiting. The pain may be postprandial and located in the epigastric region, with radiation to the back. There is no published data on the frequency with which bouts of pancreatitis occur in HP. Over time, as the number of recurrences increase, the pancreas is thought to sustain more parenchymal damage and duct distortion (Figure 1). Manifestations of chronic pancreatitis such as fibrosis, parenchymal calcification, ductal stricture, and peripancreatic fluid collections may develop. These morphologic changes can lead to clinical complications such as biliary obstruction, pancreatic duct stone formation, malabsorption, and steatorrhea due to pancreatic exocrine insufficiency, as well as diabetes mellitus due to pancreatic endocrine insufficiency. Weight loss, chronic pain, frequent hospitalizations, and contact with the medical system due to these complications are common.23 There have been two major studies that have attempted to characterize the clinical features of HP that may present during the natural history of the disease. In 2004, 418 patients with HP across multiple countries were characterized, and in 2009, 200 French patients with HP were evaluated. Epigastric pain was found in 83% of the patients, pseudocysts in 23%, calcifications in 61%, pancreatic exocrine insufficiency in 35%–37%, and diabetes mellitus in 26%–32%.1,19 Of the patients who developed diabetes mellitus, 60% were insulin dependent.19
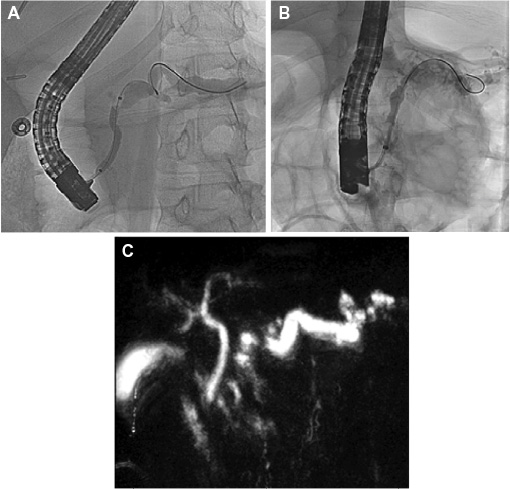 | Figure 1 Radiographic findings in hereditary pancreatitis. Notes: (A) Endoscopic retrograde cholangiopancreatography in a 15-year-old patient with hereditary pancreatitis due to a SPINK1 gene mutation demonstrating a tortuous and ectatic pancreatic duct. Balloon dilation is being performed on a severe stricture at the insertion. (B) Endoscopic retrograde cholangiopancreatography in a 7-year-old patient with hereditary pancreatitis due to a PRSS1 gene mutation demonstrating a markedly ectatic, irregular pancreatic duct with side branch ductal dilatation. (C) Magnetic resonance cholangiopancreatography in a 43-year-old patient with a CFTR gene mutation demonstrating an enlarged and tortuous pancreatic duct with filling defect at the tail. |
While these symptoms of HP are similar to those caused by other types of acute and chronic pancreatitis, there are several distinguishing features. As noted previously, HP tends to have an earlier age of onset, often in the first 2 decades of life. Patients with a maternal inheritance pattern tend to have an earlier age of onset of disease than those with a paternal inheritance pattern (9 years of age vs 14 years of age),19 as do patients with the R122H mutation of the PRSS1 gene as compared to patients with HP caused by other genetic mutations.1 The early age of onset of this disease may lead to unique challenges for the patients and their families. The protracted course of the disease starting in childhood also may carry extended psychological and financial burdens as compared to adult-onset disease. Additionally, while the time to develop exocrine and endocrine failure did not differ between patients with HP and patients with “mutation-negative” pancreatitis, the cumulative risk of exocrine insufficiency and diabetes is higher in HP than in other forms of pancreatitis (60.2% and 68.6%, respectively).1 Patients with HP also have a significantly increased risk of developing pancreatic cancer compared to the average population. The EUROPAC trial found a standardized incidence ratio (SIR) of 67,1 whereas the French cohort calculated the SIR of pancreatic cancer in HP to be 87.19
Genetic features
In the case of HP, the symptoms described earlier are thought to be the results of genetic mutations that cause an imbalance of naturally secreted proteases and their inhibitors, leading to autodigestion of the pancreatic parenchyma. Since the first mutation identified in 1996,14 numerous genetic defects have been discovered and the inheritance patterns of HP have been described. These mutations have a variable but typically high penetrance. The most common inheritance pattern of HP is the autosomal dominant pattern, where acquisition of a single abnormal parental gene leads to the phenotype of HP. This pattern is most often associated with the PRSS1 gene mutations. Rarely, other mutations leading to HP are inherited in an autosomal dominant pattern. In 2009, a pedigree analysis of a Polish family with 14 family members over four generations diagnosed with HP found that these family members had a new and unidentified mutation with 80% penetrance, for which genetic locus analysis is underway.24 A whole exome sequencing technique in individuals with HP identified a new one-base deletion in exon 1 of the serine protease inhibitor Kazal type 1 gene (SPINK1 c.27delC) that is also transmitted in an autosomal dominant fashion.25 Autosomal recessive HP, in which two mutated parental genes are needed for the development of symptomatic HP, is a less common inheritance pattern. HP due to mutations in the cystic fibrosis gene, as well as some of the mutations in SPINK1, is inherited in this manner. The final inheritance pattern that has been described in the literature is a complex type, in which environmental factors in addition to a genetic abnormality, or a combination of genetic mutations, are necessary for the patient to develop the clinical presentation of HP. For instance, coinheritance of a rare variant of the cystic fibrosis gene (CFTR p.R75Q) and SPINK1 genetic variants were described in 2010 leading to an increased incidence of HP. If the cystic fibrosis variant was inherited alone, however, a normal phenotype resulted without evidence of pancreatitis.26
PRSS1
Up to 80% of patients with HP have been found to have a mutation in the PRSS1 gene.19,23,27–29 PRSS1 encodes cationic trypsin, the most abundant isoform of trypsin secreted by the pancreas. The role of cationic trypsin is to convert inactive pancreatic zymogens secreted by the pancreas into active digestive enzymes in the duodenum when stimulated by food exposure. Trypsinogen is the inactive form of trypsin. Premature conversion of trypsinogen to trypsin leads to premature activation of these digestive enzymes before excretion from the pancreas, which in turn leads to autodigestion of the parenchyma, leading to the inflammation and damage that manifest clinically as pancreatitis. There are multiple physiologic defense mechanisms that guard against the premature conversion of trypsinogen to trypsin. Genetic mutations in the PRSS1 gene may interfere with these defense mechanisms, ultimately leading to clinical pancreatitis.
Over 20 mutations in PRSS1 that are associated with HP have been described,14,30–38 and more continue to be discovered. Almost all these mutations are located in areas of the PRSS1 gene that encode the two main regulatory regions of trypsin, one of which controls activation of trypsinogen into trypsin and the other that regulates destruction of activated trypsin. The three most common mutations on PRSS1 include the R122H mutation (78% of mutations, with a penetrance of 80%), the N29I mutation (12% of mutations, with a penetrance of 93%),19,39 and the A16V mutation (the third most common mutation, with a penetrance of 43%).35 The R122H mutation is a gain-of-function mutation that affects the destruction regulatory site; this mutation prevents autolysis of trypsin, leading to increased trypsin stability and high trypsin levels in the pancreas, allowing more digestive enzyme activation and autodigestion of the pancreas.34 The N29I mutation works at the other regulatory site, leading to increased and inappropriate autoactivation of trypsin, also leading to high trypsin levels and increased autodigestion of the pancreas.33 Thus, HP caused by either R122H or N29I PRSS1 mutations may have a similar clinical presentation. There were no differences in the clinical presentation of 27 patients with HP caused by the R122H or N291 mutations. Additionally, most patients with these mutations had mild disease.40 The mechanism of action of the A16V mutation is not as well understood. It is known that this mutation leads to an abnormal amino acid in the cleavage site of the mature trypsinogen protein31; however, despite this mutation, the trypsinogen protein is secreted in its normal amount and form.32 There is evidence that the A16V mutation increases the secretion of the chymotrypsin C (CTRC) protein, which leads to a fourfold increase in trypsin activation.41 This suggests that the A16V mutation is inherited in a complex pattern and may require coinheritance of other mutations to cause clinical pancreatitis, as compared to the autosomal dominant R122H and N291 mutations.
SPINK1
Another gene that has been found to cause HP is SPINK1. This gene encodes an acute-phase protein that is a trypsin inhibitor expressed in pancreatic acinar cells whose role is to inhibit activation of digestive enzymes by prematurely converted trypsin and to prevent autodigestion of the pancreatic parenchyma. Thus, loss-of-function mutations in SPINK1 lead to decreased levels of the inhibitor protein and cause an increased susceptibility to pancreatitis.27,42 The most common SPINK1 mutations associated with HP are the N34S mutation, the P55S mutation (more common in Europe and the US), and the IVS3 + 2TC mutation (more common in Asia).43 While these SPINK1 mutations may be directly associated with the development of HP in a minority of cases,44 their presence may increase the risk of pancreatitis by up to 23%.45–47 It is thought that few SPINK1 mutations directly associated with the development of HP are inherited in an autosomal recessive pattern,46 though at least one SPINK1 mutation (SPINK1 c.27delC) may be inherited in an autosomal dominant pattern.25 When they are inherited in a heterozygous form, as is the case the majority of the time, the phenotypic expression of pancreatitis may require interactions with other inherited genetic mutations and/or environmental factors to cause HP.44,46,48–50 Therefore, SPINK1 mutations are often considered disease-modifying mutations.
CFTR
Mutations in the cystic fibrosis transmembrane conductance regulator gene (CFTR) are also associated with the development of HP. The CFTR protein is involved in sodium, chloride, and bicarbonate transport across epithelial surfaces in the respiratory system, sweat glands, gallbladder, and pancreas. Cystic fibrosis is a multisystem disease caused by the CFTR mutations, manifested by thickened and excessive secretions, causing poor mucociliary clearance and subsequent lung disease and infection. It can also cause decreased insulin and digestive enzyme secretion due to low pancreatic fluid volumes, as well as increased acidity of the pancreatic lumen causing precipitation of proteins that obstruct the pancreatic ducts. This combination of pancreatic abnormalities ultimately results in diabetes mellitus, found in >50% of patients with cystic fibrosis over the age of 30 years,51 as well as pancreatic exocrine insufficiency, which is found in ∼85%–95% of patients with cystic fibrosis. In the remaining cystic fibrosis patients with enough intact pancreatic parenchyma, it has been estimated that 10%–20% may experience pancreatitis. Thus, ∼1.5% of all patients with cystic fibrosis may experience pancreatitis at some point in their lives.52,53 Pancreatitis caused by CFTR mutations can also occur independently of the cystic fibrosis syndrome.52,54
Over 2,000 CFTR mutations have been identified. An autosomal recessive inheritance of severe homozygous mutations, such as F508-delta/F508-delta, results in the typical multisystem cystic fibrosis phenotype that most often includes early-onset pancreatic insufficiency. Patients with this type of inheritance rarely develop recurrent acute pancreatitis in the form of HP.55 Conversely, inheritance of one of the mild variants of CFTR, such as the R75Q mutation, is associated with a more limited cystic fibrosis syndrome and a greater incidence of pancreatitis. In 2005, the presence of the R75Q mutation was associated with a 40- to 80-fold increased risk of developing pancreatitis as compared to the general population.56 Additionally, nine CFTR mutations associated with a deficiency in bicarbonate conductance (known as CFTR–BD mutations) were identified carrying a risk of acute and chronic pancreatitis in the setting of an otherwise mild cystic fibrosis syndrome.57 Finally, some CFTR mutations can be inherited in a complex-type pattern. When a patient is heterozygous with, for example, one CFTR mutation and an additional genetic mutation, such as SPINK1 or CTRC, the risk of pancreatitis is increased.26,58
CTRC
CTRC is a protease secreted by pancreatic acinar cells that has also been found to degrade trypsin and trypsinogen.41 This action is an example of another protective mechanism to prevent trypsin’s activation of digestive enzymes and autodigestion of the pancreas leading to pancreatitis. Mutations of CTRC cause loss of function by several mechanisms, including reduced CTRC secretion and increased CTRC degradation by trypsin, as well as a catalytic defect within the CTRC protein.59–61 Numerous CTRC mutations and four pathogenic variants that act by the above-mentioned mechanisms, including A73T, V235I, R253W, and K247_R254del, have been characterized. Ten other CTRC mutations were identified that were potentially pathogenic.60 Just as with SPINK1 mutations, CTRC mutations may not be causative of HP but may confer an increased risk via interactions with SPINK1 or CFTR gene mutations or with environmental factors.60 There have been several studies that investigated the frequency and effect size of CTRC mutations in patients with chronic pancreatitis in Europe, India, and Asia. It appears that these mutations are not very common in patients with HP (1%–3%) but have a significant effect size, conferring a four- to eightfold increased risk of developing HP.58–63
Others genetic mutations and copy number variants
There are several less well-studied genes that when mutated are believed to cause HP.64 One of these genes is cathepsin B (CTSB), which is thought to prematurely activate trypsinogen when mutated. In 2006, one study suggested that CTSB mutations, assisted by SPINK1 mutations, led to the development of pancreatitis65; however, another study a year later failed to confirm the association.66 CASR, or the calcium-sensing receptor gene, plays a critical role in maintaining calcium homeostasis and trypsinogen and trypsin stability. Several studies indicate that mutations in this gene predispose a patient to pancreatitis by a complex inheritance pattern.67,68 Additionally, a high-risk mutation in the claudin-2 (CLDN-2) locus on the X chromosome was found to be highly associated with an increased risk for recurrent acute pancreatitis in a genome-wide susceptibility study.69 Due to its location on the X-chromosome, mutations in CLDN-2 appear to be inherited in an autosomal dominant fashion in men and in an autosomal recessive pattern in women.69–71 Another high-risk genetic defect has been found in the carboxypeptidase A1 (CPA1), which may confer increased risk of developing pancreatitis due to misfolding of the trypsin protein.72 Finally, recent studies have also investigated the relationship of copy number variants of known genes and their association with the development of HP. Several exons on the PRSS1 gene, as well as two exons on PRSS2 (which codes for anionic trypsinogen, a protein with functions similar to cationic trypsinogen), were found to be duplicated in patients with HP, causing gain-of-function mutations that increase the amount of trypsinogen produced.73 Two copy number gene mutations in SPINK1 associated with loss of function in a trypsin inhibitor were also described.74
As more genetic mutations associated with HP are discovered, the Chronic Pancreatitis Genetics Risk Factors Database is updated with these new variants. It includes an updated and comprehensive list of all known genetic mutations, both pathologic and nonpathologic, in the most common genes associated with HP.75
Pancreatic cancer
Regardless of the genetic mutation, HP of any etiology confers a greatly increased risk of pancreatic cancer. Overall, however, HP accounts for a small percentage of cases of pancreatic cancer. In 1997, the observed and expected rates of pancreatic cancer in 246 patients with HP from multiple countries were reviewed. The SIR of pancreatic cancer in patients with HP was 53. The cumulative risk of pancreatic cancer until the age of 70 years was 40% but was relatively low prior to the age of 50 years. The risk increases several decades after the initial presentation and appears to be equal in both men and women.76 These findings were confirmed in subsequent studies; however, the SIR of pancreatic cancer in other study populations was higher, at 87.1,19
Environmental factors also increase the risk of pancreatic cancer in patients with HP. Smoking tobacco and a history of diabetes mellitus conferred the greatest additional risk of developing pancreatic cancer.19 While smoking increases the risk of pancreatic cancer in patients with HP by the same proportion as it does in the general population (approximately doubling the risk), it appears that patients with HP who smoke develop pancreatic cancer ∼20 years earlier than their equivalents in the general population.77
The significantly increased risk of pancreatic cancer with an HP diagnosis has led to recommendations for pancreatic cancer screening, despite a lack of data regarding screening or prospective trials. In 2001, the first set of guidelines was published based on expert consensus from the Third International Symposium on Inherited Diseases of the Pancreas78; in 2007, the updated recommendations from the Fourth International Symposium were published. This panel recommended screening for pancreatic cancer in patients with HP on a 1- to 3-year basis, starting at age 40–45 years, or 10–15 years prior to the earliest age of onset of pancreatic cancer in the family.79 In February 2015, the American College of Gastroenterology published clinical guidelines on genetic testing and management of patients with hereditary gastrointestinal cancer syndromes. These guidelines included recommendations for patients who are at risk for developing hereditary pancreatic adenocarcinoma, including patients with HP. The recommendations were: 1) surveillance for pancreatic adenocarcinoma should be of individuals who are known mutation carriers of hereditary syndromes associated with increased risk of pancreatic cancer and should be performed in experienced centers using a multidisciplinary approach and under research conditions; 2) surveillance should be performed with endoscopic ultrasound and/or pancreatic magnetic resonance imaging, starting at age 50 years, or 10 years younger than the earliest age of pancreatic cancer in the family; 3) cystic lesions of the pancreas identified during surveillance require evaluation by centers experienced in the care of these high-risk individuals, and the decision for surgical intervention should be individualized and made by a multidisciplinary team assessment.80 Surgical resection must be carefully evaluated in this cohort, as patients may be more prone to complications such as pancreatic insufficiency (Table 1).81
 | Table 1 American College of Gastroenterology criteria for surveillance of hereditary pancreatic cancer Note: Adapted by permission from Macmillan Publishers Ltd: Am J Gastroenterol,80 copyright (2015). Abbreviations: HP, hereditary pancreatitis; EUS, endoscopic ultrasound; MRI, magnetic resonance imaging. |
Diagnostic evaluation
The workup for HP typically involves a detailed personal history to look for the clinical symptoms, an extended family history to identify possible inheritance patterns, and radiographic imaging to evaluate for signs of acute or chronic pancreatitis. Diagnostic endoscopic procedures with endoscopic ultrasound and endoscopic retrograde cholangiopancreatography (ERCP) may be helpful in understanding the patient’s pancreatobiliary anatomy as well as in evaluating or treating complications of chronic pancreatitis.23 HP patients have often been initially misdiagnosed with idiopathic pancreatitis. For those patients who are symptomatic and in whom HP is suspected, genetic testing for the most common etiologic mutations, including on the PRSS1, CFTR, SPINK1, and CTRC genes, is available. Table 2 lists the criteria that should be considered in the decision to offer genetic testing.23,82,83 It is recommended that genetic testing be performed with the assistance of a genetic counselor or an experienced provider who can assist with the interpretation of the results and provide family and social support.84 For predictive testing of asymptomatic patients, genetic testing can be helpful in the setting of suspected inheritance of autosomal dominant or recessive mutations, such as PRSS1 or CFTR. A negative test in a patient with a family history of HP with a known mutation essentially rules out the diagnosis. Alternatively, a positive test in that setting is thought to confer an 80% risk of developing HP in the patient’s lifetime.85 The decision to test asymptomatic patients under the age of 16 years should be individualized.82,84
 | Table 2 Criteria for genetic testing for HP Note: Reproduced from Patel MR, Eppolito AL, Willingham FF. Hereditary pancreatitis for the endoscopist. Therap Adv Gastroenterol. 2013;6(2):169–179.23 Abbreviation: HP, hereditary pancreatitis. |
Treatment
Patients with HP should be managed in the same way as patients who present with pancreatitis of other etiologies. Preventative measures, medical management, and endoscopic and surgical interventions may all be appropriate for a patient with HP.
Prevention
In the setting of a genetic mutation associated with HP, there are no clearly established methods of preventing development or progression of disease. Surveillance for pancreatic adenocarcinoma, as discussed previously, is required given the increased risk. An equally important preventative intervention involves advising the patients and their families to avoid environmental triggers that are known to exacerbate and worsen pancreatitis and are thought to be involved in the complex-type inheritance of HP. Smoking tobacco should be avoided, as it is known to increase the risk for pancreatic cancer by twofold.77 Alcohol use is another known cofactor for the development of pancreatitis and should be avoided, as no safe amount of alcohol use has been documented. Though harder to control, dietary fat and emotional stress are considered triggers and should also be avoided.19 Finally, the risk and benefit of the use of medications that have been associated with drug-induced pancreatitis (eg, ACE inhibitors, HMG co-A reductase inhibitors, and selective serotonin reuptake inhibitors)86 should be individualized to the patient.
Medical management
It is thought that medical management of pancreatitis should be delivered in a step-up approach, first with avoidance of triggers and dietary modification, then with exogenous pancreatic enzyme replacement, and finally with pain control.23 Pancreatitis-related pain in some patients has been shown to respond to pancreatic enzyme replacement87 and possibly with supplementation of antioxidants.88 The central nervous system and pain processing system have been implicated as a mechanism of pancreatitis-related pain,89 so the use of tricyclic antidepressants or gabapentin may be helpful. Additionally, while nonsteroidal anti-inflammatory agents are preferred over opioid analgesics for pain relief, they may be contraindicated due to the patient’s comorbidities or not tolerated due to side effects. If opioid analgesics are to be prescribed, long-acting formulations are preferred over short or intermediate acting forms.23 Amlodipine, octreotide, and somatostatin have shown no benefit in pain relief in patients with pancreatitis, and they are therefore not recommended for use in HP.90–92
Endoscopic therapy
As with other forms of pancreatitis, endoscopy can be extremely useful in managing sequelae of HP as well as pain. It has been shown that endoscopic decompression of obstructed pancreatic ducts (due to strictures or stones) with ERCP has been associated with long-term pain relief in patients with chronic pancreatitis, with the majority of patients (in the 2002 study, 76%) successfully avoiding surgical intervention.93,94 This finding was replicated in a group of 42 children who underwent therapeutic ERCP, in which 64% had complete resolution of pain and 81% reported decreased abdominal pain. Notably, 17% of these children did experience complications of ERCP, including mild and moderate post-ERCP pancreatitis or cholangitis.95 In 2010, the efficacy of therapeutic ERCP in a group of patients with HP was evaluated. These patients were followed for up to 39 years and were found to have significantly decreased pain, narcotic use, and hospitalizations after ERCP. It was also noted that 12 of the patients required surgical resection at some point; however, after surgery 92% of these patients still required subsequent endoscopic intervention.96 There has been debate over whether endoscopic management is preferred over surgical therapy in HP. A 2013 review aimed to compare endoscopic and surgical therapies in 87 patients with HP. Ceppa et al found that pain-free intervals between procedures were longer with surgical therapy than with endoscopic therapy. However, the decision to choose one intervention over the other was felt to be complex, with age, comorbidities, focality of the pancreatic lesion, and safety profiles all requiring consideration.97 A 2015 Cochrane Review of three randomized controlled trials concluded that surgical intervention was superior to endoscopic intervention in long-term pain relief.98 Another group proposed interventional recommendations for children with HP recommending a step-up strategy, starting with early ERCP and progressing to surgery if ERCP was unsuccessful.99 In our experience, therapeutic endoscopy should be considered as an important initial treatment option for patients with HP but must be individualized to the patient and performed by expert endoscopists.
Surgical therapy
Beyond decompression of obstructed pancreatic ducts, operative management of pancreatic necrosis or for drainage of pancreatic cysts may be indicated. Surgical options for the management of these conditions, as well as for prophylaxis against pancreatic cancer, include partial pancreatectomy, total pancreatectomy alone, or total pancreatectomy with islet cell autotransplantation. Reduction in pain and narcotic dependence after pancreatectomy, as well as maintenance of islet cell function after autotransplantation, have only been shown to be moderately successful, as well as age dependent, being more successful in younger patients.100,101 Pancreatectomy with islet cell autotransplantation is an irreversible procedure with rare but serious risks and commits the patient to a lifetime of exogenous pancreatic enzyme replacement as well as a high likelihood of insulin dependence over time. Thus, extensive risk–benefit analysis by a multidisciplinary team, which is individualized to the patient, needs to be undertaken prior to the decision to pursue surgical resection. However, the decision to evaluate auto-islet cell transplant should be considered earlier while there is still significant islet cell mass to enable a successful graft. Clearer guidelines would help providers determine which patients, in which settings, would benefit most from pancreatectomy with or without islet cell autotransplantation.
Prognosis
Patients with HP without pancreatic adenocarcinoma do not have an increased mortality compared to the general population. However, if a patient with HP develops pancreatic adenocarcinoma, it usually presents 20 years earlier than in the general population and thus results in an earlier and increased mortality.77 However, patients often have a significantly reduced quality of life due to their symptoms and the frequent need for interventions and hospitalizations.
Conclusion
HP is a rare cause of acute, recurrent acute, and chronic pancreatitis. The knowledge surrounding the disease has increased dramatically since it was first described in 1952. Multiple genetic defects in genes involved with inappropriate activation of digestive enzymes in the pancreas, such as PRSS1, CFTR, SPINK1, and CTRC, have been associated with the development of HP and can now be identified with genetic screening. Mutations continue to be discovered since the advent and increasing use of next-generation sequencing. The mechanism of disease induction by these mutations is complex and is only partially understood; however, it is often the interaction between mutations and environmental factors that leads to clinical pancreatitis. Thus, advising patients with HP to avoid medications and environmental triggers known to exacerbate pancreatitis, such as tobacco, alcohol, and a high-fat diet, is essential in management. Patients with HP experience similar complications as patients with other forms of chronic pancreatitis, such as pain, obstruction due to strictures and stones, cysts, and exocrine and endocrine insufficiency. HP differs from other forms of pancreatitis in the early onset as well as the significantly increased risk of pancreatic adenocarcinoma. As the specific forms of HP are more precisely identified and our understanding of their particular manifestations grows, it is hoped that we can offer more timely triage to the best care for each patient.
Disclosure
The authors report no conflicts of interest in this work.
References
Howes N, Lerch MM, Greenhalf W, et al; European Registry of Hereditary Pancreatitis and Pancreatic Cancer (EUROPAC). Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol. 2004;2(3):252–261. | ||
Comfort MW, Steinberg AG. Pedigree of a family with hereditary chronic relapsing pancreatitis. Gastroenterology. 1952;21(1):54–63. | ||
Gross JB, Jones JD. Hereditary pancreatitis: analysis of experience to May 1969. In: Beck I, Sinclair D, editors. The Exocrine Pancreas. London: J and A Churchill Ltd; 1971:247–270. | ||
Beall JH Jr, Bell JW, Jesseph JE, Nyhus LM. Fatal acute hemorrhagic pancreatitis occurring simultaneously in identical twins. Gastroenterology. 1960;39:215–218. | ||
Henderson J, Ingram D, House T. Acute pancreatitis in identical twins. Med J Aust. 1982;1(10):432–434. | ||
Mäkelä P, Aärimaa M. Pancreatography in a family with hereditary pancreatitis. Acta Radiol Diagn (Stockh). 1985;26(1):63–66. | ||
Kattwinkel J, Lapey A, Di Sant’Agnese PA, Edwards WA. Hereditary pancreatitis: three new kindreds and a critical review of the literature. Pediatrics. 1973;51(1):55–69. | ||
Gross JB, Comfort MW. Hereditary pancreatitis: report on two additional families. Gastroenterology. 1957;32(5):829–854. | ||
Gross JB, Gambill EE, Ulrich JA. Hereditary pancreatitis. Description of a fifth kindred and summary of clinical features. Am J Med. 1962;33:358–364. | ||
Le Bodic L, Schnee M, Georgelin T, et al. An exceptional genealogy for hereditary chronic pancreatitis. Dig Dis Sci. 1996;41(7):1504–1510. | ||
Le Bodic L, Bignon JD, Raguénés O, et al. The hereditary pancreatitis gene maps to long arm of chromosome 7. Hum Mol Genet. 1996;5(4):549–554. | ||
Pandya A, Blanton SH, Landa B, et al. Linkage studies in a large kindred with hereditary pancreatitis confirms mapping of the gene to a 16-cM region on 7q. Genomics. 1996;38(2):227–230. | ||
Whitcomb DC, Preston RA, Aston CE, et al. A gene for hereditary pancreatitis maps to chromosome 7q35. Gastroenterology. 1996;110(6):1975–1980. | ||
Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14(2):141–145. | ||
Go V, Everhart J. Pancreatitis. In: Everhart J, editor. Digestive Diseases in the United States: Epidemiology and Impact. Vol. 2009. Washington, DC: US Department of Health and Human Services, Public Health Service, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, US Government Printing Office; 1994:691–712. | ||
Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144(6):1252–1261. | ||
Cote GA, Yadav D, Slivka A, et al; North American Pancreatitis Study Group. Alcohol and smoking as risk factors in an epidemiology study of patients with chronic pancreatitis. Clin Gastroenterol Hepatol. 2011;9(3):266–273. quiz e27. | ||
Joergensen M, Brusgaard K, Crüger DG, Gerdes AM, de Muckadell OB. Incidence, prevalence, etiology, and prognosis of first-time chronic pancreatitis in young patients: a nationwide cohort study. Dig Dis Sci. 2010;55(10):2988–2998. | ||
Rebours V, Boutron-Ruault MC, Schnee M, et al. The natural history of hereditary pancreatitis: a national series. Gut. 2009;58(1):97–103. | ||
Poddar U, Yachha SK, Mathias A, Choudhuri G. Genetic predisposition and its impact on natural history of idiopathic acute and acute recurrent pancreatitis in children. Dig Liver Dis. 2015;47(8):709–714. | ||
Yadav D, Timmons L, Benson JT, Dierkhising RA, Chari ST. Incidence, prevalence, and survival of chronic pancreatitis: a population-based study. Am J Gastroenterol. 2011;106(12):2192–2199. | ||
Sultan M, Werlin S, Venkatasubramani N. Genetic prevalence and characteristics in children with recurrent pancreatitis. J Pediatr Gastroenterol Nutr. 2012;54(5):645–650. | ||
Patel MR, Eppolito AL, Willingham FF. Hereditary pancreatitis for the endoscopist. Therap Adv Gastroenterol. 2013;6(2):169–179. | ||
LaFemina J, Roberts PA, Hung YP, et al. Identification of a novel kindred with familial pancreatitis and pancreatic cancer. Pancreatology. 2009;9(3):273–279. | ||
LaRusch J, Barmada MM, Solomon S, Whitcomb DC. Whole exome sequencing identifies multiple, complex etiologies in an idiopathic hereditary pancreatitis kindred. JOP. 2012;13(3):258–262. | ||
Schneider A, Larusch J, Sun X, et al. Combined bicarbonate conductance-impairing variants in CFTR and SPINK1 variants are associated with chronic pancreatitis in patients without cystic fibrosis. Gastroenterology. 2011;140(1):162–171. | ||
DiMagno MJ, DiMagno EP. Chronic pancreatitis. Curr Opin Gastroenterol. 2005;21(5):544–554. | ||
Applebaum-Shapiro SE, Finch R, Pfützer RH, et al. Hereditary pancreatitis in North America: the Pittsburgh-Midwest Multi-Center Pancreatic Study Group Study. Pancreatology. 2001;1(5):439–443. | ||
Howes N, Greenhalf W, Stocken DD, Neoptolemos JP. Cationic trypsinogen mutations and pancreatitis. Gastroenterol Clin North Am. 2004;33(4):767–787. | ||
Teich N, Ockenga J, Hoffmeister A, Manns M, Mössner J, Keim V. Chronic pancreatitis associated with an activation peptide mutation that facilitates trypsin activation. Gastroenterology. 2000;119(2):461–465. | ||
Witt H, Luck W, Becker M. A signal peptide cleavage site mutation in the cationic trypsinogen gene is strongly associated with chronic pancreatitis. Gastroenterology. 1999;117(1):7–10. | ||
Kereszturi E, Szmola R, Kukor Z, et al. Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen: a novel disease mechanism. Hum Mutat. 2009;30(4):575–582. | ||
Sahin-Tóth M. Human cationic trypsinogen. Role of Asn-21 in zymogen activation and implications in hereditary pancreatitis. J Biol Chem. 2000;275(30):22750–22755. | ||
Sahin-Tóth M, Tóth M. Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem Biophys Res Commun. 2000;278(2):286–289. | ||
Grocock CJ, Rebours V, Delhaye MN, et al; European Registry of Hereditary Pancreatitis and Pancreatic Cancer. The variable phenotype of the p.A16V mutation of cationic trypsinogen (PRSS1) in pancreatitis families. Gut. 2010;59(3):357–363. | ||
Schnúr A, Beer S, Witt H, Hegyi P, Sahin-Tóth M. Functional effects of 13 rare PRSS1 variants presumed to cause chronic pancreatitis. Gut. 2014;63(2):337–343. | ||
Dytz MG, Mendes de Melo J, de Castro Santos O, et al. Hereditary pancreatitis associated with the N29T mutation of the PRSS1 gene in a Brazilian family: a case-control study. Medicine (Baltimore). 2015;94(37):e1508. | ||
Rygiel AM, Beer S, Simon P, et al. Gene conversion between cationic trypsinogen (PRSS1) and the pseudogene trypsinogen 6 (PRSS3P2) in patients with chronic pancreatitis. Hum Mutat. 2015;36(3):350–356. | ||
Sossenheimer MJ, Aston CE, Preston RA, et al. Clinical characteristics of hereditary pancreatitis in a large family, based on high-risk haplotype. The Midwest Multicenter Pancreatic Study Group (MMPSG). Am J Gastroenterol. 1997;92(7):1113–1116. | ||
Keim V, Bauer N, Teich N, Simon P, Lerch MM, Mössner J. Clinical characterization of patients with hereditary pancreatitis and mutations in the cationic trypsinogen gene. Am J Med. 2001;111(8):622–626. | ||
Nemoda Z, Sahin-Tóth M. Chymotrypsin C (caldecrin) stimulates autoactivation of human cationic trypsinogen. J Biol Chem. 2006;281(17):11879–11886. | ||
Kume K, Masamune A, Ariga H, et al. Do genetic variants in the SPINK1 gene affect the level of serum PSTI? J Gastroenterol. 2012;47(11):1267–1274. | ||
Shimosegawa T, Kume K, Masamune A. SPINK1, ADH2, and ALDH2 gene variants and alcoholic chronic pancreatitis in Japan. J Gastroenterol Hepatol. 2008;23(suppl 1):S82–S86. | ||
Pfutzer RH, Barmada MM, Brunskill AP, et al. SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology. 2000;119(3):615–623. | ||
Teich N, Bauer N, Mössner J, Keim V. Mutational screening of patients with nonalcoholic chronic pancreatitis: identification of further trypsinogen variants. Am J Gastroenterol. 2002;97(2):341–346. | ||
Witt H, Luck W, Hennies HC, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000;25(2):213–216. | ||
Schneider A, Barmada MM, Slivka A, Martin JA, Whitcomb DC. Clinical characterization of patients with idiopathic chronic pancreatitis and SPINK1 mutations. Scand J Gastroenterol. 2004;39(9):903–904. | ||
Aoun E, Chang CC, Greer JB, Papachristou GI, Barmada MM, Whitcomb DC. Pathways to injury in chronic pancreatitis: decoding the role of the high-risk SPINK1 N34S haplotype using meta-analysis. PLoS One. 2008;3(4):e2003. | ||
Reddy DN, Prasad SS. Genetic basis of chronic pancreatitis in Asia Pacific region. J Gastroenterol Hepatol. 2011;26(suppl 2):2–5. | ||
Shimosegawa T, Kume K, Masamune A. SPINK1 gene mutations and pancreatitis in Japan. J Gastroenterol Hepatol. 2006;21(suppl 3):S47–S51. | ||
Hameed S, Jaffe A, Vergé CF. Cystic fibrosis related diabetes (CFRD) – the end stage of progressive insulin deficiency. Pediatr Pulmonol. 2011;46(8):747–760. | ||
Ooi CY, Durie PR. Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations in pancreatitis. J Cyst Fibros. 2012;11(5):355–362. | ||
De Boeck K, Weren M, Proesmans M, Kerem E. Pancreatitis among patients with cystic fibrosis: correlation with pancreatic status and genotype. Pediatrics. 2005;115(4):e463–e469. | ||
Rowntree RK, Harris A. The phenotypic consequences of CFTR mutations. Ann Hum Genet. 2003;67(pt 5):471–485. | ||
Modolell I, Alvarez A, Guarner L, De Gracia J, Malagelada JR. Gastrointestinal, liver, and pancreatic involvement in adult patients with cystic fibrosis. Pancreas. 2001;22(4):395–399. | ||
Cohn JA, Mitchell RM, Jowell PS. The impact of cystic fibrosis and PSTI/SPINK1 gene mutations on susceptibility to chronic pancreatitis. Clin Lab Med. 2005;25(1):79–100. | ||
LaRusch J, Jung J, General IJ, et al; North American Pancreatitis Study Group. Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLoS Genet. 2014;10(7):e1004376. | ||
Rosendahl J, Landt O, Bernadova J, et al. CFTR, SPINK1, CTRC and PRSS1 variants in chronic pancreatitis: is the role of mutated CFTR overestimated? Gut. 2013;62(4):582–592. | ||
Rosendahl J, Witt H, Szmola R, et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet. 2008;40(1):78–82. | ||
Beer S, Zhou J, Szabó A, et al. Comprehensive functional analysis of chymotrypsin C (CTRC) variants reveals distinct loss-of-function mechanisms associated with pancreatitis risk. Gut. 2013;62(11):1616–1624. | ||
Masson E, Chen JM, Scotet V, Le Maréchal C, Férec C. Association of rare chymotrypsinogen C (CTRC) gene variations in patients with idiopathic chronic pancreatitis. Hum Genet. 2008;123(1):83–91. | ||
Paliwal S, Bhaskar S, Mani KR, et al. Comprehensive screening of chymotrypsin C (CTRC) gene in tropical calcific pancreatitis identifies novel variants. Gut. 2013;62(11):1602–1606. | ||
Chang MC, Chang YT, Wei SC, et al. Association of novel chymotrypsin C gene variations and haplotypes in patients with chronic pancreatitis in Chinese in Taiwan. Pancreatology. 2009;9(3):287–292. | ||
Ravi Kanth V, Nageshwar Reddy D. Genetics of acute and chronic pancreatitis: an update. World J Gastrointest Pathophysiol. 2014;5(4):427–437. | ||
Mahurkar S, Idris MM, Reddy DN, et al. Association of cathepsin B gene polymorphisms with tropical calcific pancreatitis. Gut. 2006;55(9):1270–1275. | ||
Weiss FU, Behn CO, Simon P, Ruthenbürger M, Halangk W, Lerch MM. Cathepsin B gene polymorphism Val26 is not associated with idiopathic chronic pancreatitis in European patients. Gut. 2007;56(9):1322–1323. | ||
Felderbauer P, Hoffmann P, Einwächter H, et al. A novel mutation of the calcium sensing receptor gene is associated with chronic pancreatitis in a family with heterozygous SPINK1 mutations. BMC Gastroenterol. 2003;3:34. | ||
Murugaian EE, Premkumar RM, Radhakrishnan L, Vallath B. Novel mutations in the calcium sensing receptor gene in tropical chronic pancreatitis in India. Scand J Gastroenterol. 2008;43(1):117–121. | ||
Whitcomb DC, LaRusch J, Krasinskas AM, et al; Alzheimer’s Disease Genetics Consortium. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat Genet. 2012;44(12):1349–1354. | ||
Derikx MH, Kovacs P, Scholz M, et al; PanEuropean Working group on Alcoholic Chronic Pancreatitis Members and Collaborators. Polymorphisms at PRSS1-PRSS2 and CLDN2-MORC4 loci associate with alcoholic and non-alcoholic chronic pancreatitis in a European replication study. Gut. 2015;64(9):1426–1433. | ||
Masamune A, Nakano E, Hamada S, Kakuta Y, Kume K, Shimosegawa T. Common variants at PRSS1-PRSS2 and CLDN2-MORC4 loci associate with chronic pancreatitis in Japan. Gut. 2015;64(8):1345–1346. | ||
Witt H, Beer S, Rosendahl J, et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat Genet. 2013;45(10):1216–1220. | ||
Chen JM, Masson E, Le Maréchal C, Férec C. Copy number variations in chronic pancreatitis. Cytogenet Genome Res. 2008;123(1–4):102–107. | ||
Chen JM, Mercier B, Audrezet MP, Ferec C. Mutational analysis of the human pancreatic secretory trypsin inhibitor (PSTI) gene in hereditary and sporadic chronic pancreatitis. J Med Genet. 2000;37(1):67–69. | ||
Genetic Risk Factors in Chronic Pancreatitis [homepage on the internet]. Available from: www.pancreasgenetics.org. Accessed June 13, 2016. | ||
Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1997;89(6):442–446. | ||
Lowenfels AB, Maisonneuve P, Whitcomb DC, Lerch MM, DiMagno EP. Cigarette smoking as a risk factor for pancreatic cancer in patients with hereditary pancreatitis. JAMA. 2001;286(2):169–170. | ||
Ulrich CD; Consensus Committees of the European Registry of Hereditary Pancreatic Diseases, Midwest Multi-Center Pancreatic Study Group, International Association of Pancreatology. Pancreatic cancer in hereditary pancreatitis: consensus guidelines for prevention, screening and treatment. Pancreatology. 2001;1(5):416–422. | ||
Brand RE, Lerch MM, Rubinstein WS, et al; Participants of the Fourth International Symposium of Inherited Diseases of the Pancreas. Advances in counselling and surveillance of patients at risk for pancreatic cancer. Gut. 2007;56(10):1460–1469. | ||
Syngal S, Brand RE, Church JM, et al; American College of Gastroenterology. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110(2):223–262. | ||
Whitcomb DC, Shelton CA, Brand RE. Genetics and genetic testing in pancreatic cancer. Gastroenterology. 2015;149(5):1252–1264.e4. | ||
Fink EN, Kant JA, Whitcomb DC. Genetic counseling for nonsyndromic pancreatitis. Gastroenterol Clin North Am. 2007;36(2):325–333, ix. | ||
Ellis I, Lerch MM, Whitcomb DC; Consensus Committees of the European Registry of Hereditary Pancreatic Diseases MM-CPSGIAoP. Genetic testing for hereditary pancreatitis: guidelines for indications, counselling, consent and privacy issues. Pancreatology. 2001;1(5):405–415. | ||
Solomon S, Whitcomb DC. Genetics of pancreatitis: an update for clinicians and genetic counselors. Curr Gastroenterol Rep. 2012;14(2):112–117. | ||
Ellis J, Greenhalf W, Howes N et al; Ireland EUROPAC Collaboration. Clinical and molecular findings of cationic trypsinogen mutations in 60 families with hereditary pancreatitis on the EUROPAC register (abstract). Digestion. 2000;61:266. | ||
Jones MR, Hall OM, Kaye AM, Kaye AD. Drug-induced acute pancreatitis: a review. Ochsner J. 2015;15(1):45–51. | ||
Halgreen H, Pedersen NT, Worning H. Symptomatic effect of pancreatic enzyme therapy in patients with chronic pancreatitis. Scand J Gastroenterol. 1986;21(1):104–108. | ||
Uomo G, Talamini G, Rabitti PG. Antioxidant treatment in hereditary pancreatitis. A pilot study on three young patients. Dig Liver Dis. 2001;33(1):58–62. | ||
Frøkjær JB, Olesen SS, Gram M, et al. Altered brain microstructure assessed by diffusion tensor imaging in patients with chronic pancreatitis. Gut. 2011;60(11):1554–1562. | ||
Morinville VD, Lowe ME, Elinoff BD, Whitcomb DC. Hereditary pancreatitis amlodipine trial: a pilot study of a calcium-channel blocker in hereditary pancreatitis. Pancreas. 2007;35(4):308–312. | ||
Malfertheiner P, Mayer D, Buchler M, Dominguez-Muñoz JE, Schiefer B, Ditschuneit H. Treatment of pain in chronic pancreatitis by inhibition of pancreatic secretion with octreotide. Gut. 1995;36(3):450–454. | ||
Uhl W, Anghelacopoulos SE, Friess H, Büchler MW. The role of octreotide and somatostatin in acute and chronic pancreatitis. Digestion. 1999;60(suppl 2):23–31. | ||
Okolo PI 3rd, Pasricha PJ, Kalloo AN. What are the long-term results of endoscopic pancreatic sphincterotomy? Gastrointest Endosc. 2000;52(1):15–19. | ||
Rosch T, Daniel S, Scholz M, et al; European Society of Gastrointestinal Endoscopy Research Group. Endoscopic treatment of chronic pancreatitis: a multicenter study of 1000 patients with long-term follow-up. Endoscopy. 2002;34(10):765–771. | ||
Li ZS, Wang W, Liao Z, et al. A long-term follow-up study on endoscopic management of children and adolescents with chronic pancreatitis. Am J Gastroenterol. 2010;105(8):1884–1892. | ||
Dever JB, Irani S, Brandabur J, Traverso LW, Kozarek R. Outcomes of interventional ERCP in hereditary pancreatitis. J Clin Gastroenterol. 2010;44(1):46–51. | ||
Ceppa EP, Pitt HA, Hunter JL, et al. Hereditary pancreatitis: endoscopic and surgical management. J Gastrointest Surg. 2013;17(5):847–856. discussion 856–857. | ||
Ahmed Ali U, Pahlplatz JM, Nealon WH, van Goor H, Gooszen HG, Boermeester MA. Endoscopic or surgical intervention for painful obstructive chronic pancreatitis. Cochrane Database Syst Rev. 2015;3:CD007884. | ||
Kargl S, Kienbauer M, Duba HC, Schöfl R, Pumberger W. Therapeutic step-up strategy for management of hereditary pancreatitis in children. J Pediatr Surg. 2015;50(4):511–514. | ||
Bellin MD, Gelrud A, Arreaza-Rubin G, et al. Total pancreatectomy with islet autotransplantation: summary of a National Institute of Diabetes and Digestive and Kidney diseases workshop. Pancreas. 2014;43(8):1163–1171. | ||
Bellin MD, Freeman ML, Gelrud A, et al. Total pancreatectomy and islet autotransplantation in chronic pancreatitis: recommendations from PancreasFest. Pancreatology. 2014;14(1):27–35. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.