Back to Journals » Hepatic Medicine: Evidence and Research » Volume 10
Hepatic encephalopathy: current challenges and future prospects
Authors Swaminathan M, Ellul MA, Cross TJS
Received 6 April 2017
Accepted for publication 31 August 2017
Published 22 March 2018 Volume 2018:10 Pages 1—11
DOI https://doi.org/10.2147/HMER.S118964
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Gerry Lake-Bakaar
Mirashini Swaminathan,1 Mark Alexander Ellul,2 Timothy JS Cross1
1Department of Gastroenterology, Royal Liverpool University Hospital, 2Faculty of Health and Life Sciences, Institute of Infection and Global Health, University of Liverpool, Liverpool, UK
Abstract: Hepatic encephalopathy (HE) is a common complication of liver dysfunction, including acute liver failure and liver cirrhosis. HE presents as a spectrum of neuropsychiatric symptoms ranging from subtle fluctuating cognitive impairment to coma. It is a significant contributor of morbidity in patients with liver disease. HE is observed in acute liver failure, liver bypass procedures, for example, shunt surgery and transjugular intrahepatic portosystemic shunt, and cirrhosis. These are classified as Type A, B and C HE, respectively. HE can also be classified according to whether its presence is overt or covert. The pathogenesis is linked with ammonia and glutamine production, and treatment is based on mechanisms to reduce the formation and/or removal of these compounds. There is no specific diagnostic test for HE, and diagnosis is based on clinical suspicion, excluding other causes and use of clinical tests that may support its diagnosis. Many tests are used in trials and experimentally, but have not yet gained universal acceptance. This review focuses on the definitions, pathogenesis and treatment of HE. Consideration will be given to existing treatment, including avoidance of precipitating factors and novel therapies such as prebiotics, probiotics, antibiotics, laxatives, branched-chain amino acids, shunt embolization and the importance of considering liver transplant in appropriate cases.
Keywords: hepatic encephalopathy, pathogenesis, treatment, lactulose, rifaximin, probiotics, covert hepatic encephalopathy
Introduction
Hepatic encephalopathy (HE) is a hallmark of liver failure and affects up to 40% of patients with liver cirrhosis.1 It is defined as a multifactorial neuropsychiatric disorder presenting with a broad spectrum of cognitive impairment and neuromuscular dysfunction.1 HE is a significant contributor to repeated hospitalizations for patients with liver cirrhosis and severely impacts on the quality of life of both patients and caregivers.2 It is a marker of poor prognosis in cirrhotic patients, with reported rates of survival of only 36% at 1 year from its first presentation.3
Chronic liver disease is the fifth most common cause of death in the UK, with the mortality expected to rise due to increase in cirrhosis caused by alcohol-related liver disease, chronic hepatitis C and nonalcoholic fatty liver disease.4,5 Patients commonly present to primary and secondary care services with complications such as HE with or without a prior diagnosis of chronic liver disease. A milder form of the disorder, covert hepatic encephalopathy (CHE) or covert encephalopathy with subtle alterations of cognitive function, also exists.6 Although less severe, patients with CHE are at significant risk of interference with their quality of life, including increased falls, hospitalizations and progression to overt HE.6–8
The aims of this review are to provide a comprehensive, “state of the art”, account of the pathophysiology, clinical manifestations (classification, symptoms, signs and investigations), current treatments and future targets for the management of patients with HE. The review is targeted to physicians in primary or secondary care and also to health care professionals who are likely to encounter patients with liver disease in their professional roles.
Pathogenesis of HE
HE can be classified as three separate clinical entities. Type A HE is due to acute liver failure, Type B due to portosystemic shunting (e.g., transjugular intrahepatic portosystemic shunting procedures) and Type C results as a complication of liver cirrhosis.9
Type A HE is associated with an increased intracranial pressure that progresses rapidly and may lead to brain herniation. The pathophysiology of Type B and C HE is complex and remains under investigation. The main hypothesis involves the limited ability of the liver to effectively remove nitrogenous waste products, resulting in their accumulation and the deleterious effects on the brain due to portosystemic shunting.10,11 The key substrates implicated are ammonia and glutamine. Several studies have demonstrated that cirrhotic patients who had or were experiencing significant neuropsychiatric symptoms had elevated blood ammonia concentration.12,13 However, the levels were not predictive or consistent with the severity of HE.14,15 The exception is in type A HE, where Bernal et al have shown that a cut-off level of ammonia >200 µmol/L16 is predictive of raised intracranial pressure and death.16,17
Glutamine is a key amino acid that plays an important signaling role for processes including gene expression, cytokine production and cell proliferation. Enhanced activity of glutamine is primarily noted in skeletal muscle, brain, heart and hepatocytes. Glutamine is metabolized in the gut and kidney to ammonia and glutamate. Ammonia is then detoxified in the kidneys and liver and excreted as urea. In liver failure, ammonia escapes the urea cycle and is detoxified to glutamine in various tissues. A vicious cycle is formed, whereby increased ammonia concentration due to impaired detoxification in the liver further activates glutamine synthesis, leading to increased glutamine catabolism resulting in hyperammonemia. The enhanced activity of glutamine has been shown to exert adverse effects such as swelling of astrocytes in the brain and increases the catabolism of branched-chain amino acids (BCAAs) in skeletal muscles.18
Manganese has also been implicated in the pathogenesis of HE, with elevated plasma levels due to inability of excretion by the liver causing deposition in the basal ganglia.19 This has been shown to correlate with pallidal signal hyperintensity observed on magnetic resonance imaging of cirrhotic patients.19,20 Furthermore, an animal study by Rivera-Mancia et al showed that manganese favored ammonia and glutamine accumulation in the brain.21
Additional factors involved in the pathogenesis of HE include gut microbiota dysbiosis and small intestinal bacterial overgrowth (SIBO). This pathway has gained interest for novel therapies. SIBO in patients with cirrhosis is a consequence of reduced gut motility, reduced gastric acid secretion, luminal IgA deficiency and malnutrition.22 SIBO results in impairment of intestinal barrier integrity, therefore increasing bacterial translocation and release of ammonia and endotoxins into the circulation.22 Zhang et al24 and Bajaj et al,23 in several well-designed studies, have found a distinct variation in the population of microbiota between cirrhotics and noncirrhotics. Cirrhotics were shown to have an increased Bacteriodes/Firmicutes ratio at the expense of commensal bacteria.25 Dominant species in cirrhotic patients included those of Streptococcaceae, Veillonellaceae, Alcaligenaceae and Porphyromonadaceae. These correlated significantly with ammonia levels and decreased cognitive function. Also, several other bacterial species demonstrated an association with increased inflammatory responses in patients with CHE.26 Tsai et al have recently shown that cirrhotic patients using proton-pump inhibitors (PPIs) were more likely to develop HE. It is hypothesized that PPIs cause gut dysbiosis leading to HE, from the previous findings of the association between PPI use and SIBO.27
Sarcopenia (muscle volume depletion) has been shown to predict the development of encephalopathy in cirrhotic patients.28 Skeletal muscle represents an alternative site of ammonia detoxification; therefore, reduced muscle volume results in hyperammonemia. The consumption of BCAAs in the detoxification of ammonia to glutamine in skeletal muscle results in low levels of BCAAs in blood plasma. Hanai et al demonstrated that patients with sarcopenia and HE due to cirrhosis have low levels of BCAA.28–31 The theory is that therapeutic supplementation with BCAAs can, therefore, reduce malnutrition and revert the loss of muscle cell mass and breakdown of protein driving hyperammonemia.31 An increase in muscle mass will also help drive increased extrahepatic ammonia detoxification. Another main driver of sarcopenia is thought to be myostatin, a negative regulator of satellite cell differentiation and proliferation. There are higher levels of serum and muscle myostatin in cirrhotic patients, and ammonia has been shown to stimulate myostatin expression.32
There are additional factors that can precipitate HE and act synergistically with ammonia in its pathogenesis (Table 1). For example, hyponatremia can exacerbate HE due to its osmotic effects on astrocytes,33,34 and patients with systemic inflammatory response syndrome are predisposed to HE. In patients with HE, there is an alteration in cerebral blood flow35 and enhanced sensitization of the brain to inflammatory cytokines by ammonia.36 Moreover, there is an increased oxidative stress caused by augmented permeability of the blood–brain barrier contributing to altered mental status.37–39
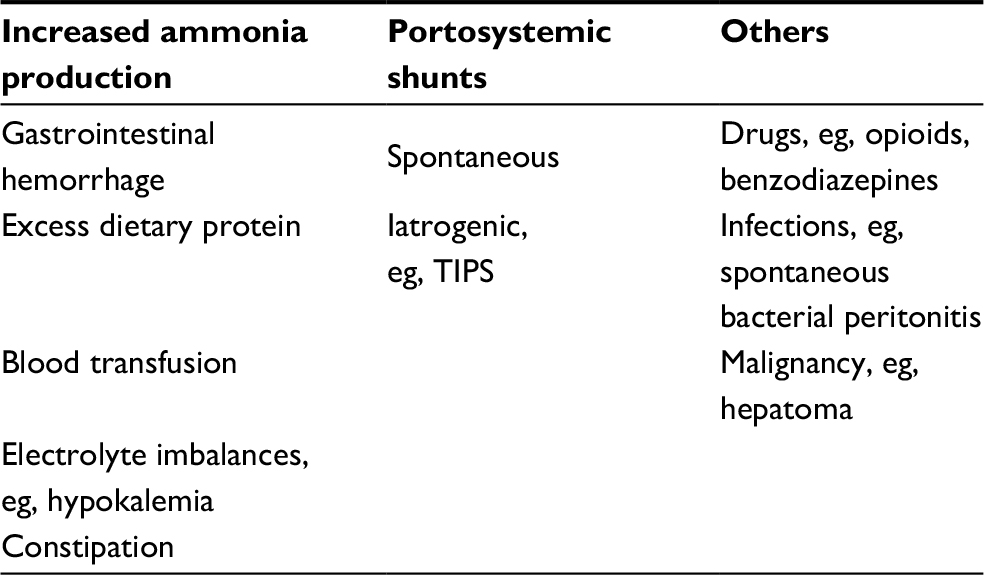 | Table 1 Precipitating factors to HE |
It is becoming increasingly recognized that no single entity is responsible for HE, but rather it is a synergistic effect of multiple mechanisms (Figure 1).
 | Figure 1 Contributing factors toward pathophysiology of HE. Notes: Ammonia is produced from nitrogenous products by bacterial metabolism of urea and proteins in the gut and from deamination of glutamine in the small intestine. Normally, ammonia is cleared by liver and kidneys and metabolized in skeletal muscle. However, as a result of liver dysfunction and portosystemic shunting, ammonia cannot be cleared adequately. **Increased ammonia levels in the plasma increases metabolism to glutamine (via glutamine synthetase) in astrocytes, which subsequently causes intracellular swelling and edema.109 Abbreviation: HE, hepatic encephalopathy. |
Diagnosis of HE
Clinical features
HE presents with a wide spectrum of neuropsychiatric symptoms which typically begin with subtle psychomotor changes.40 This progresses to confusion with the presence of asterixis, somnolence and, finally, its most severe form, coma. The most common clinical classification used to describe this continuum is the West Haven Criteria (Table 2).9
 | Table 2 West Haven Criteria for hepatic encephalopathy and symptoms Note: Data from a previous study.110 Abbreviation: WHC, West Haven Criteria. |
CHE or covert encephalopathy represents the initial stages of the disorder that can only be recognized by psychometric testing.41 The first feature to emerge is psychomotor slowing and difficulties in performing activities of daily living.42,43 Up to 50% of cirrhotic patients are affected by CHE.42 As HE progresses, symptoms become more apparent and can be detected clinically. There is alteration in conscious levels and disorientation, and disturbance of the sleep–wake cycle is often a feature.43 Personality changes may emerge, and there can also be motor system abnormalities including hypertonia, hyper-reflexia, dystonia, dyskinesia, upgoing plantars and asterixis. Asterixis or “flapping tremor” is a negative myoclonus with a loss of postural tone frequently seen in the hands, but can affect other parts of the body.44 The onset of disorientation and asterixis is described as overt encephalopathy.
The American and European Associations for the Study of the Liver 2014 practice guidelines recommend that HE be classified according to four factors:1
- the underlying etiology as described previously – Type A, B or C;
- severity – using grading system such as West Haven Criteria;
- time course – episodic, recurrent (>1 episode in 6 months) or persistent (symptoms always present and can have episodes of acute exacerbations); and
- nonprecipitated or precipitated by factors such as infections, medications or electrolyte disorders.
Investigations
Diagnosis of HE should be made on a clinical basis after exclusion of various conditions that can mimic HE. These are outlined in Table 3. Arterial or venous ammonia levels can be helpful, but should not be used alone in diagnosis as they are often inconsistent, as outlined previously. Electroencephalography is valuable to investigate for the presence of subclinical seizures. Characteristic triphasic wave changes can be seen in HE, alongside subtle signs in CHE.45,46
 | Table 3 Differential diagnosis of HE |
Imaging modalities such as computed tomography and magnetic resonance imaging should be performed to exclude differential diagnosis.1,47 In patients with normal neurologic examinations, brain imaging is unlikely to be beneficial; also, there is probably an overuse of computed tomography scans in patients with cirrhosis and altered mental status alone.48 The main concern in patients with cirrhosis is intracranial bleeds due to coagulopathy; yet, this is rare in the absence of focal neurologic deficits.49 Other imaging abnormalities on magnetic resonance imaging include basal ganglia hyperintensity, and this may be a possible marker for HE and severity of liver failure.50–52 In the literature, there is a case describing a patient with recurrent HE associated with dynamic changes in the basal ganglia hyperintensity pre- and posttreatment.53 Similar changes have been seen using positron emission tomography, but this is currently used as an experimental tool to elucidate underlying pathophysiology.54
Diagnosing CHE remains a challenge, and clinicians should suspect presence of CHE from inquiring about the presence of signs and symptoms. It should be considered in patients complaining of difficulties with activities of daily living, loss of concentration, lack of ability to function at work or with relatives having noted change in cognition.47 Assessment is done by using validated psychometric tests, the gold standard being a combination of the psychometric hepatic encephalopathy score,46 but a combination of tests may be more accurate.55 A summary of the most established tests is highlighted in Table 4.
 | Table 4 Psychometric tests used in the evaluation of MHE Note: Data from a previous study.94 Abbreviations: CFF, critical flicker frequency; CRT, continuous reaction time; ICT, inhibitory control test; MHE, minimal hepatic encephalopathy; PHES, psychometric hepatic encephalopathy score. |
In an era of technologic advance, development of smartphone applications, such as the EncephalApp56 and Stroop App,57 are making these tests more accessible and user-friendly for clinicians and patients.
There are multiple other tools in development for a more accurate diagnosis of HE that are not yet validated, but are promising. The challenge is to keep the test simple for patients, but, at the same time, maintain its accuracy in diagnosing HE. Examples of these include the animal naming test (maximum number of animals listed in 1 minute), one study showed that scores obtained in the animal naming test directly correlated to grade of encephalopathy.58
Recently, a pilot study from Arasaradnam et al demonstrated the potential use of exhaled volatile organic compounds to detect HE.59
Management of HE
Patients presenting with overt HE typically will have an underlying precipitant such as infection, medications, gastrointestinal bleeding or other precipitating factors. The acute management relies on detailed history and examination to identify and treat these as appropriate. Ninety percent of patients can be treated by correcting the precipitating factor.47 It is important to recognize that a proportion of patients will have no underlying precipitant of HE.
Patients with CHE do not usually require treatment unless the condition is thought to be adversely affecting their quality of life. Current American Association for the Study of Liver Diseases (AASLD) guidelines focus on management strategies for overt HE. Patients should be managed empirically for HE, while the investigations for precipitating factors or mimics of HE are ongoing.47
Prebiotics, probiotics and symbiotics
Lactulose, a prebiotic, is the most widely used nonabsorbable disaccharide (NAD) in clinical practice and is recommended by AASLD/European Association for the Study of the Liver (EASL) guidelines as the first-line treatment for episodes of HE. Treatment should continue on resolution of symptoms to prevent further episodes. In the colon, lactulose is converted to lactic and acetic acid, reducing intraluminal pH and promoting the utilization of ammonia in the metabolism of gut bacteria (Figure 2). A 2016 Cochrane meta-analysis of 38 randomized clinical trials showed that when compared with placebo or no intervention, NAD may be associated with a beneficial effect on HE. In addition, it showed that NAD treatment can reduce serious adverse events associated with liver disease, including liver failure, hepatorenal syndrome and variceal bleeding.60 The analyses included data for treatment and prevention of HE. Lacitiol is a second-generation NAD that has been suggested to have similar effectiveness and better tolerance than lactulose,61 although a Cochrane review showed that it had no beneficial effects when compared to lactulose. The quality of evidence for lacitiol is poor and further research is needed.60
 | Figure 2 Mechanism of action of nonabsorbable disaccharides. Notes: Lactulose and lacitiol are not absorbed in the small intestine and enter the colon unchanged, where they are metabolized to hydrogen and VFA. Bacteria use these as preferred substrate, thereby reducing the production of ammonia and promoting its incorporation into stool for excretion. Abbreviation: VFA, volatile fatty acids. |
Probiotics are live microorganisms that are believed to confer health benefits in a variety of clinical settings.62 In HE, probiotics have been shown to act by modulating gut microbiota to reduce ammonia levels by several mechanisms, including decreased bacterial urease activity, decreased ammonia absorption by decreasing the pH and improved nutritional status of gut epithelium.63 The most commonly used probiotic currently is VSL#3. At present, all evidence relating to use of probiotics in the treatment of HE is of poor quality, as highlighted by the 2017 Cochrane review of 21 trials.64 Probiotics had no effect on all-cause mortality, when compared to placebo or no treatment. Probiotics may lead to improvements in the development of overt HE, quality of life and plasma ammonia concentrations, with minimal adverse effects. Yet, the review was unable to conclude if probiotics were superior to lactulose in the treatment of HE. The optimal dose, delivery and species of probiotics to use have not been determined.65
Symbiotics are a combination of prebiotics and probiotics and their clinical significance in HE remains uncertain. There are several randomized controlled trials that demonstrate the possible beneficial effects.25,66,67
Polyethylene glycol is a purgative laxative agent which has been shown in a randomized controlled trial by Rahimi et al to be superior to lactulose in improving HE with a quicker time for resolution.68 But, more data is required before it can be routinely recommended in preference to lactulose.
Nutrition
Nutritional therapy is relevant in HE as a modulator of nitrogen metabolism. Historically, dietary protein restriction had been advised to reduce intestinal ammonia production. But, this may contribute to sarcopenia and actually worsen HE.69 Thus, it was recommended by the International Society for Hepatic Encephalopathy and Nitrogen Metabolism that 1.2–1.5 g/kg of protein be given in small meals distributed throughout the day with a late night snack of complex carbohydrates.70 A nasogastric feeding tube should be considered if the patient is unable to achieve their dietary targets.
If patients are unable to maintain dietary protein intake, supplementation with BCAA is an alternative. Oral BCAA supplements consist of several essential amino acids thought to improve ammonia detoxification. A recent 2015 Cochrane review of 16 randomized clinical trials found high-quality evidence of clinical benefit, but no effect on mortality, quality of life or nutrition parameters.31
Deficiencies of vitamins and electrolytes should also be addressed, as they can be associated with a wide range of neuropsychiatric symptoms.71 Although not directly implicated in the pathophysiology of HE, they can compound or mimic symptoms. The use of additional zinc supplementation has been previously studied; a systematic review showed a potential improvement in psychometric tests, but its use did not affect the recurrence rates of HE.72 More recently, Mousa et al conducted a randomized trial which demonstrated that antioxidant and zinc supplementation led to a significant improvement in baseline neuropsychometric tests in patients with CHE, when compared to lactulose therapy.73
Antimicrobials
The use of oral antibiotics to modulate gut flora and reduce ammonia production has been researched as a tool to treat HE. Neomycin, an aminoglycoside antibiotic which is poorly absorbed and reaches high concentrations in the gut, acts as a glutaminase inhibitor, thereby reducing ammonia levels.74 It was the first antibiotic agent to be widely used in HE. However, the adverse effects associated with neomycin and the development of newer agents preclude its use in current clinical practice.
Rifaximin is a semi-synthetic nonabsorbable antibiotic derived from rifamycin. When compared with neomycin, rifaximin was found to be at least as effective in reducing blood ammonia levels, while having less adverse effects such as ototoxicity and nephrotoxicity.75–77 It exerts its effects by several mechanisms – modulating gut microbiota composition and metabolism and also exhibiting anti-inflammatory properties.78 EASL and AASLD guidance recommends the use of rifaximin for secondary prophylaxis of overt HE in patients who have had further episodes while on lactulose therapy. These recommendations are based on evidence from a large well-conducted randomized controlled trial.79 Combination therapy of rifaximin with lactulose has been shown to be more effective than the use of rifaximin alone.80
Sidhu et al demonstrated in a randomized open-label trial that there was no difference in improvement of cognitive function or quality of life in patients with CHE treated with rifaximin alone versus lactulose alone.81 An upcoming clinical trial, RiMINI, aims to assess the influence of rifaximin versus combination therapy of rifaximin and lactulose on the microbiota in patients with CHE.82 Currently, there is no evidence for primary or secondary prophylaxis for patients with CHE.
Other antimicrobials such as metronidazole and vancomycin have been investigated, but their significant adverse effect profile limits their use.83,84
Other therapies
Several other therapies are currently under investigation for use as treatment for HE, most of which aim to lower serum ammonia levels. l-Ornithine-l-aspartate is used as a supplement that acts by stimulating the urea cycle and glutamine synthesis, an important mechanism in the detoxification of ammonia.85 Good-quality data from meta-analyses have demonstrated that l-ornithine-l-aspartate is more effective in improvement of symptoms and reduction in serum ammonia levels in cirrhotic patients with CHE and HE, when compared to placebo or no intervention control.86
Intravenous albumin infusion is commonly used in patients with cirrhosis after reports showed that it improves outcomes in cirrhotic patients with spontaneous bacterial peritonitis or hepatorenal syndrome. The mechanism of action is thought to be improvement in circulatory dysfunction by plasma expansion and reduction of oxidative stress.87 Two randomized clinical trials have, however, demonstrated that albumin infusion does not have a significant impact on HE in cirrhotics.88,89
A more promising development is the use of ammonia scavengers, such as glycerol phenylbutyrate (GPB) and ornithine phenylacetate, which lowers ammonia levels by providing substrates as an alternative pathway to urea for nitrogen metabolism.90 The Phase IIb study (HALT-HE [NCT00999167]) on the potential treatment with GPB and ornithine phenylacetate is now complete, and results are awaited along with the Phase III study (not yet registered with clinicaltrials.gov).
See Table 5 for a summary of other potential therapies for HE.
 | Table 5 Summary of other potential therapies for HE Note: *Not available in the USA. Abbreviations: BCAA, branched-chain amino acid; GPB, glycerol phenylbutyrate; HAS, human albumin solution; HE, hepatic encephalopathy; LOLA, l-ornithine-l-aspartate; OP, ornithine phenylacetate. |
Treatment-resistant HE
HE resistant to optimal medical treatment should raise the suspicion of large portosystemic shunts, and these can be treated by embolization therapy.91 Evidence from studies suggest that the procedure decreases hospital admissions and improves survival, despite the risks of de novo gastroesophageal varices, worsening ascites and renal dysfunction due to contrast-induced nephropathy.92,93 In Type B HE caused by iatrogenic shunts created via transjugular intrahepatic portosystemic shunt (TIPS) procedure, a reduction of the stent, that is, reducing caliber of the TIPS, can lead to clinical improvement and should be considered in patients with severe post-TIPS HE.
Liver transplantation is the only definitive treatment option for resistant HE and should be considered in suitable candidates presenting with HE. Discussions with transplant centers should be initiated early. Strict criteria apply and liver transplantation is not without risk, but an assessment should be considered in all patients with HE, provided that investigations for potential neurodegenerative disorders that may worsen posttransplant have been undertaken.47
Unfortunately, there are some patients (e.g., with significant comorbidities such as heart failure and disease, renal disease and coexisting malignancy) with resistant HE who are not suitable candidates for liver transplantation. In these patients, the emphasis of care should then be switched to controlling distressing symptoms and providing holistic support for the patient and caregivers. Multidisciplinary team input is needed, and support should be provided for both patients and caregivers.69 Education about preventing constipation is paramount, and it may be necessary for administration of phosphate enemas in the community. Despite their relative contraindication in end-stage liver disease, benzodiazepines and opioids may be required, as priorities change to reduce distress rather than preserve lucency.94
Future prospects
The management of HE has evolved over the last 10 years with the addition on rifaximin into current treatment guidelines. There has been an increase in the number of randomized controlled trials with good-quality evidence describing the use of lactulose, rifaximin, GPB and others. More studies are needed to ascertain the pathophysiology that will lead to new treatment options of HE. Results of several randomized clinical trials, such as HALT-HE and STOP-HE, are awaited.
In terms of pathophysiology, there should be an emphasis to discover how the different concepts act synergistically to lead to development of HE. The research into gut microbiota seems promising, and perhaps a look into the role of nutritional therapy in modulating gut microbiota would be of interest.
There is some evidence for the use of liver support systems such as the Molecular Adsorbent Recirculating System and Prometheus device. They act by removing circulating toxins that accumulate in the blood due to liver dysfunction.95 Both devices were well tolerated by patients in liver failure, and their effects on HE have been investigated in randomized trials.96 The MARS study showed that using the MARS system caused significant improvement in HE and responded significantly faster when compared to standard medical therapy.97 The Relief study also showed similar benefits, although survival benefits were not demonstrated.98 These devices may have a role in patients with incapacitating HE as a bridge to transplantation, but may not be appropriate in all causes, given the requirement for central venous access and the nonfinite timeline for treatment. More studies into the cost-effectiveness are needed, together with concerns over the development of sepsis.
Conclusion
HE is a significant contributor to morbidity in patients with cirrhosis associated with end-stage liver disease. The unpredictable nature of HE severely impacts on the quality of life for patients and relatives. Research into the complexities of HE has led to development of new and upcoming treatment options. Avoiding the precipitants of HE and combination treatment with lactulose and rifaximin remain the mainstay of treatment. Future studies should aim to further identify novel mechanisms and targets for future treatments with the hope of translating this into real benefit for patients with HE.
Disclosure
The authors report no conflicts of interest in this work.
References
American Association for the Study of Liver Diseases; European Association for the Study of the Liver. Hepatic Encephalopathy in Chronic Liver Disease: 2014 Practice Guideline by the European Association for the Study of the Liver and the American Association for the Study of Liver Diseases.– J Hepatol. 2014;61(3):642–659. | ||
Nabi E, Thacker LR, Wade JB, et al. Diagnosis of covert hepatic encephalopathy without specialized tests. Clin Gastroenterol Hepatol. 2014;12(8):1384–1389.e2. | ||
Jepsen P, Ott P, Andersen PK, Sorensen HT, Vilstrup H. Clinical course of alcoholic liver cirrhosis: a Danish population-based cohort study. Hepatology. 2010;51(5):1675–1682. | ||
Mokdad AA, Lopez AD, Shahraz S, et al. Liver cirrhosis mortality in 187 countries between 1980 and 2010: a systematic analysis. BMC Med. 2014;12(1):145. | ||
Public Health England. [webpage on the Internet]. Deaths from Liver Disease: Implications for end of life care in England. 2017. Accessed September 28, 2017. | ||
Patidar KR, Thacker LR, Wade JB, et al. Covert hepatic encephalopathy is independently associated with poor survival and increased risk of hospitalization. Am J Gastroenterol. 2014;109(11):1757–1763. | ||
Bajaj JS, Saeian K, Schubert CM, et al. Minimal hepatic encephalopathy is associated with motor vehicle crashes: the reality beyond the driving test. Hepatology. 2009;50(4):1175–1183. | ||
Roman E, Cordoba J, Torrens M, et al. Minimal hepatic encephalopathy is associated with falls. Am J Gastroenterol. 2011;106(3):476–482. | ||
Dharel N, Bajaj JS. Definition and nomenclature of hepatic encephalopathy. J Clin Exp Hepatol. 2015;5(Suppl 1):S37–S41. | ||
Shawcross D, Jalan R. The pathophysiologic basis of hepatic encephalopathy: central role for ammonia and inflammation. Cell Mol Life Sci. 2005;62(19–20):2295–2304. | ||
Desjardins P, Du T, Jiang W, Peng L, Butterworth RF. Pathogenesis of hepatic encephalopathy and brain edema in acute liver failure: role of glutamine redefined. Neurochem Int. 2012;60(7):690–696. | ||
STAHL J. Studies of the blood ammonia in liver disease. Its diagnostic, prognostic, and therapeutic significance. Ann Intern Med. 1963;58:1–24. | ||
Phear EA, Sherlock S, Summerskill WH. Blood-ammonium levels in liver disease and hepatic coma. Lancet. 1955;268(6869):836–840. | ||
Ong JP, Aggarwal A, Krieger D, et al. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med. 2003;114(3):188–193. | ||
Kundra A, Jain A, Banga A, Bajaj G, Kar P. Evaluation of plasma ammonia levels in patients with acute liver failure and chronic liver disease and its correlation with the severity of hepatic encephalopathy and clinical features of raised intracranial tension. Clin Biochem. 2005;38(8):696–699. | ||
Bernal W, Hall C, Karvellas CJ, Auzinger G, Sizer E, Wendon J. Arterial ammonia and clinical risk factors for encephalopathy and intracranial hypertension in acute liver failure. Hepatology. 2007;46(6):1844–1852. | ||
Kumar R, Shalimar, Sharma H, et al. Persistent hyperammonemia is associated with complications and poor outcomes in patients with acute liver failure. Clin Gastroenterol Hepatol. 2012;10(8):925–931. | ||
Holecek M. Evidence of a vicious cycle in glutamine synthesis and breakdown in pathogenesis of hepatic encephalopathy-therapeutic perspectives. Metab Brain Dis. 2014;29(1):9–17. | ||
Rose C, Butterworth RF, Zayed J, et al. Manganese deposition in basal ganglia structures results from both portal-systemic shunting and liver dysfunction. Gastroenterology. 1999;117(3):640–644. | ||
Spahr L, Butterworth RF, Fontaine S, et al. Increased blood manganese in cirrhotic patients: relationship to pallidal magnetic resonance signal hyperintensity and neurological symptoms. Hepatology. 1996;24(5):1116–1120. | ||
Rivera-Mancia S, Rios C, Montes S. Manganese and ammonia interactions in the brain of cirrhotic rats: effects on brain ammonia metabolism. Neurochem Res. 2012;37(5):1074–1084. | ||
Rai R, Saraswat VA, Dhiman RK. Gut microbiota: its role in hepatic encephalopathy. J Clin Exp Hepatol. 2015;5(Suppl 1):S29–S36. | ||
Bajaj JS, Hylemon PB, Ridlon JM, et al. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am J Physiol Gastrointest Liver Physiol. 2012;303(6):G675–G685. | ||
Zhang Z, Zhai H, Geng J, et al. Large-scale survey of gut microbiota associated with MHE Via 16S rRNA-based pyrosequencing. Am J Gastroenterol. 2013;108(10):1601–1611. | ||
Liu Q, Duan ZP, Ha DK, Bengmark S, Kurtovic J, Riordan SM. Synbiotic modulation of gut flora: effect on minimal hepatic encephalopathy in patients with cirrhosis. Hepatology. 2004;39(5):1441–1449. | ||
Bajaj JS, Heuman DM, Hylemon PB, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol. 2014;60(5):940–947. | ||
Tsai CF, Chen MH, Wang YP, et al. Proton pump inhibitors increase risk for hepatic encephalopathy in patients with cirrhosis in a population study. Gastroenterology. 2017;152(1):134–141. | ||
Hanai T, Shiraki M, Watanabe S, et al. Sarcopenia predicts minimal hepatic encephalopathy in patients with liver cirrhosis. Hepatol Res. Epub 2017 Feb 15. | ||
Davuluri G, Krokowski D, Guan BJ, et al. Metabolic adaptation of skeletal muscle to hyperammonemia drives the beneficial effects of l-leucine in cirrhosis. J Hepatol. 2016;65(5):929–937. | ||
Les I, Doval E, Garcia-Martinez R, et al. Effects of branched-chain amino acids supplementation in patients with cirrhosis and a previous episode of hepatic encephalopathy: a randomized study. Am J Gastroenterol. 2011;106(6):1081–1088. | ||
Gluud LL, Dam G, Les I, et al. Branched-chain amino acids for people with hepatic encephalopathy. Cochrane Database Syst Rev. 2015;(9):CD001939. | ||
Qiu J, Thapaliya S, Runkana A, et al. Hyperammonemia in cirrhosis induces transcriptional regulation of myostatin by an NF-kappaB-mediated mechanism. Proc Natl Acad Sci U S A. 2013;110(45):18162–18167. | ||
Guevara M, Baccaro ME, Torre A, et al. Hyponatremia is a risk factor of hepatic encephalopathy in patients with cirrhosis: a prospective study with time-dependent analysis. Am J Gastroenterol. 2009;104(6):1382–1389. | ||
Córdoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol. 1998;29(4):589–594. | ||
Jalan R, Olde Damink SW, Hayes PC, Deutz NE, Lee A. Pathogenesis of intracranial hypertension in acute liver failure: inflammation, ammonia and cerebral blood flow. J Hepatol. 2004;41(4):613–620. | ||
Aggarwal S, Kramer D, Yonas H, et al. Cerebral hemodynamic and metabolic changes in fulminant hepatic failure: a retrospective study. Hepatology. 1994;19(1):80–87. | ||
Marini JC, Broussard SR. Hyperammonemia increases sensitivity to LPS. Mol Genet Metab. 2006;88(2):131–137. | ||
Bai G, Rama Rao KV, Murthy CR, Panickar KS, Jayakumar AR, Norenberg MD. Ammonia induces the mitochondrial permeability transition in primary cultures of rat astrocytes. J Neurosci Res. 2001;66(5):981–991. | ||
Aldridge DR, Tranah EJ, Shawcross DL. Pathogenesis of hepatic encephalopathy: role of ammonia and systemic inflammation. J Clin Exp Hepatol. 2015;5(Suppl 1):S7–S20. | ||
Ferenci P, Lockwood A, Mullen K, Tarter R, Weissenborn K, Blei AT. Hepatic encephalopathy–definition, nomenclature, diagnosis, and quantification: final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology. 2002;35(3):716–721. | ||
Amodio P, Montagnese S, Gatta A, Morgan MY. Characteristics of minimal hepatic encephalopathy. Metab Brain Dis. 2004;19(3–4):253–267. | ||
Weissenborn K, Ennen JC, Schomerus H, Ruckert N, Hecker H. Neuropsychological characterization of hepatic encephalopathy. J Hepatol. 2001;34(5):768–773. | ||
Bajaj JS. Minimal hepatic encephalopathy matters in daily life. World J Gastroenterol. 2008;14(23):3609–3615. | ||
Bajaj JS, Wade JB, Sanyal AJ. Spectrum of neurocognitive impairment in cirrhosis: implications for the assessment of hepatic encephalopathy. Hepatology. 2009;50(6):2014–2021. | ||
Stewart J, Sarkela M, Koivusalo AM, et al. Frontal electroencephalogram variables are associated with the outcome and stage of hepatic encephalopathy in acute liver failure. Liver Transpl. 2014;20(10):1256–1265. | ||
Nardone R, Taylor AC, Höller Y, Brigo F, Lochner P, Trinka E. Minimal hepatic encephalopathy: A review. Neurosci Res. 2016;111:1–12. | ||
Vilstrup H, Amodio P, Bajaj J, et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Hepatology. 2014;60(2):715–735. | ||
Rahimi RS, Rockey DC. Overuse of Head Computed Tomography in Cirrhosis With Altered Mental Status. Am J Med Sci. 2016;351(5):459–466. | ||
Donovan LM, Kress WL, Strnad LC, et al. Low likelihood of intracranial hemorrhage in patients with cirrhosis and altered mental status. Clin Gastroenterol Hepatol. 2015;13(1):165–169. | ||
Inoue E, Hori S, Narumi Y, et al. Portal-systemic encephalopathy: presence of basal ganglia lesions with high signal intensity on MR images. Radiology. 1991;179(2):551–555. | ||
Pujol A, Pujol J, Graus F, et al. Hyperintense globus pallidus on T1-weighted MRI in cirrhotic patients is associated with severity of liver failure. Neurology. 1993;43(1):65–69. | ||
Grover VP, Crossey MM, Fitzpatrick JA, et al. Quantitative magnetic resonance imaging in patients with cirrhosis: a cross-sectional study. Metab Brain Dis. 2016;31(6):1315–1325. | ||
Jiang M, Wang Z, Tsauo J, Li X. Basal ganglia hyperintensity may be a marker of hepatic encephalopathy secondary to portosystemic shunting. Clin Res Hepatol Gastroenterol. 2015;39(1):e5–e6. | ||
Stewart CA, Reivich M, Lucey MR, Gores GJ. Neuroimaging in hepatic encephalopathy. Clin Gastroenterol Hepatol. 2005;3(3):197–207. | ||
Giménez-Garzó C, Garcés JJ, Urios A, et al. The PHES battery does not detect all cirrhotic patients with early neurological deficits, which are different in different patients. PLoS One. 2017;12(2):e0171211. | ||
Allampati S, Duarte-Rojo A, Thacker LR, et al. Diagnosis of minimal hepatic encephalopathy using stroop encephalApp: a multicenter US-Based, Norm-Based Study. Am J Gastroenterol. 2016;111(1):78–86. | ||
Bajaj JS, Thacker LR, Heuman DM, et al. The Stroop smartphone application is a short and valid method to screen for minimal hepatic encephalopathy. Hepatology. 2013;58(3):1122–1132. | ||
Campagna F, Montagnese S, Ridola L, et al. The animal naming test: an easy tool for the assessment of hepatic encephalopathy. Hepatology. 2017;66(1):198–208. | ||
Arasaradnam RP, McFarlane M, Ling K, et al. Breathomics–exhaled volatile organic compound analysis to detect hepatic encephalopathy: a pilot study. J Breath Res. 2016;10(1):016s012. | ||
Gluud LL, Vilstrup H, Morgan MY. Non-absorbable disaccharides versus placebo/no intervention and lactulose versus lactitol for the prevention and treatment of hepatic encephalopathy in people with cirrhosis. Cochrane Database Syst Rev. 2016(5):CD003044. | ||
Camma C, Fiorello F, Tine F, Marchesini G, Fabbri A, Pagliaro L. Lactitol in treatment of chronic hepatic encephalopathy. A meta-analysis. Dig Dis Sci. 1993;38(5):916–922. | ||
Dhiman RK. Gut microbiota and hepatic encephalopathy. Metab Brain Dis. 2013;28(2):321–326. | ||
Poh Z, Chang PE. A current review of the diagnostic and treatment strategies of hepatic encephalopathy. Int J Hepatol. 2012;2012:480309. | ||
Dalal R, McGee RG, Riordan SM, Webster AC. Probiotics for people with hepatic encephalopathy. Cochrane Database Syst Rev. 2017;2:CD008716. | ||
Viramontes Hörner D, Avery A, Stow R. The effects of probiotics and symbiotics on risk factors for hepatic encephalopathy: a systematic review. J Clin Gastroenterol. 2017;51(4):312–323. | ||
Malaguarnera M, Greco F, Barone G, Gargante MP, Toscano MA. Bifidobacterium longum with fructo-oligosaccharide (FOS) treatment in minimal hepatic encephalopathy: a randomized, double-blind, placebo-controlled study. Dig Dis Sci. 2007;52(11):3259–3265. | ||
Pratap Mouli V, Benjamin J, Bhushan Singh M, et al. Effect of probiotic VSL#3 in the treatment of minimal hepatic encephalopathy: a non-inferiority randomized controlled trial. Hepatol Res. 2015;45(8):880–889. | ||
Rahimi RS, Singal AG, Cuthbert JA, Rockey DC. Lactulose versus polyethylene glycol 3350–electrolyte solution for treatment of overt hepatic encephalopathy: the HELP randomized clinical trial. JAMA Intern Med. 2014;174(11):1727–1733. | ||
Cross TJ. Liver disease in clinical practice. Springer International Publishing, UK; 2017. | ||
Atif Zaman M, MPH. Nutritional Management of Patients with Cirrhosis and Hepatic Encephalopathy. 2013. Available from http://www.jwatch.org/na31703/2013/07/19/nutritional-management-patients-with-cirrhosis-and-hepatic. Accessed September 28, 2017. | ||
Amodio P, Bemeur C, Butterworth R, et al. The nutritional management of hepatic encephalopathy in patients with cirrhosis: international society for hepatic encephalopathy and nitrogen metabolism consensus. Hepatology. 2017;58(1):325–336. | ||
Chavez-Tapia NC, Cesar-Arce A, Barrientos-Gutiérrez T, Villegas-López FA, Méndez-Sanchez N, Uribe M. A systematic review and meta-analysis of the use of oral zinc in the treatment of hepatic encephalopathy. Nutr J. 2013;12:74. | ||
Mousa N, Abdel-Razik A, Zaher A, et al. The role of antioxidants and zinc in minimal hepatic encephalopathy: a randomized trial. Therap Adv Gastroenterol. 2016;9(5):684–691. | ||
Hawkins RA, Jessy J, Mans AM, Chedid A, DeJoseph MR. Neomycin reduces the intestinal production of ammonia from glutamine. Adv Exp Med Biol. 1994;368:125–134. | ||
Mullen KD, Sanyal AJ, Bass NM, et al. Rifaximin is safe and well tolerated for long-term maintenance of remission from overt hepatic encephalopathy. Clin Gastroenterol Hepatol. 2014;12(8):1390–1397.e2. | ||
Pedretti G, Calzetti C, Missale G, Fiaccadori F. Rifaximin versus neomycin on hyperammoniemia in chronic portal systemic encephalopathy of cirrhotics. A double-blind, randomized trial. Ital J Gastroenterol. 1991;23(4):175–178. | ||
Sharma BC, Sharma P, Lunia MK, Srivastava S, Goyal R, Sarin SK. A randomized, double-blind, controlled trial comparing rifaximin plus lactulose with lactulose alone in treatment of overt hepatic encephalopathy. Am J Gastroenterol. 2013;108(9):1458–1463. | ||
Bajaj JS. Review article: potential mechanisms of action of rifaximin in the management of hepatic encephalopathy and other complications of cirrhosis. Aliment Pharmacol Ther. 2016;43(Suppl 1):11–26. | ||
Bass NM, Mullen KD, Sanyal A, et al. Rifaximin treatment in hepatic encephalopathy. N Engl J Med. 2010;362(12):1071–1081. | ||
Mohammad RA, Regal RE, Alaniz C. Combination therapy for the treatment and prevention of hepatic encephalopathy. Ann Pharmacother. 2012;46(11):1559–1563. | ||
Sidhu SS, Goyal O, Parker RA, Kishore H, Sood A. Rifaximin versus. lactulose in treatment of minimal hepatic encephalopathy. Liver Int. 2016;36(3):378–385. | ||
Schulz C, Schütte K, Kropf S, et al. RiMINI – the influence of rifaximin on minimal hepatic encephalopathy (MHE) and on the intestinal microbiome in patients with liver cirrhosis: study protocol for a randomized controlled trial. Trials. 2016;17(1):111. | ||
Bajaj JS, O’Leary JG, Reddy KR, et al. Second infections independently increase mortality in hospitalized patients with cirrhosis: the North American consortium for the study of end-stage liver disease (NACSELD) experience. Hepatology. 2012;56(6):2328–2335. | ||
Loft S, Sonne J, Dossing M, Andreasen PB. Metronidazole pharmacokinetics in patients with hepatic encephalopathy. Scand J Gastroenterol. 1987;22(1):117–123. | ||
Ndraha S, Hasan I, Simadibrata M. The effect of L-ornithine L-aspartate and branch chain amino acids on encephalopathy and nutritional status in liver cirrhosis with malnutrition. Acta Med Indones. 2011;43(1):18–22. | ||
Bai M, Yang Z, Qi X, Fan D, Han G. l-ornithine-l-aspartate for hepatic encephalopathy in patients with cirrhosis: a meta-analysis of randomized controlled trials. J Gastroenterol Hepatol. 2013;28(5):783–792. | ||
Sort P, Navasa M, Arroyo V, et al. Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis. N Engl J Med. 1999;341(6):403–409. | ||
Simon-Talero M, Garcia-Martinez R, Torrens M, et al. Effects of intravenous albumin in patients with cirrhosis and episodic hepatic encephalopathy: a randomized double-blind study. J Hepatol. 2013;59(6):1184–1192. | ||
Riggio O, Nardelli S, Pasquale C, et al. No effect of albumin infusion on the prevention of hepatic encephalopathy after transjugular intrahepatic portosystemic shunt. Metab Brain Dis. 2016;31(6):1275–1281. | ||
Rockey DC, Vierling JM, Mantry P, et al. Randomized, double-blind, controlled study of glycerol phenylbutyrate in hepatic encephalopathy. Hepatology. 2014;59(3):1073–1083. | ||
Wijdicks EF. Hepatic encephalopathy. N Engl J Med. 2016;375(17):1660–1670. | ||
Laleman W, Simon-Talero M, Maleux G, et al. Embolization of large spontaneous portosystemic shunts for refractory hepatic encephalopathy: a multicenter survey on safety and efficacy. Hepatology. 2013;57(6):2448–2457. | ||
An J, Kim KW, Han S, Lee J, Lim YS. Improvement in survival associated with embolisation of spontaneous portosystemic shunt in patients with recurrent hepatic encephalopathy. Aliment Pharmacol Ther. 2014;39(12):1418–1426. | ||
Ellul MA, Gholkar SA, Cross TJ. Hepatic encephalopathy due to liver cirrhosis. BMJ. 2015;351:h4187. | ||
Hassanein TI, Schade RR, Hepburn IS. Acute-on-chronic liver failure: extracorporeal liver assist devices. Curr Opin Crit Care. 2011;17(2):195–203. | ||
Hassanein T. Current state of knowledge of hepatic encephalopathy (part IV): management of hepatic encephalopathy by liver support systems. Metab Brain Dis. 2017;32(2):303–306. | ||
Hassanein TI, Tofteng F, Brown RS Jr, et al. Randomized controlled study of extracorporeal albumin dialysis for hepatic encephalopathy in advanced cirrhosis. Hepatology. 2007;46(6):1853–1862. | ||
Banares R, Nevens F, Larsen FS, et al. Extracorporeal albumin dialysis with the molecular adsorbent recirculating system in acute-on-chronic liver failure: the RELIEF trial. Hepatology. 2013;57(3):1153–1162. | ||
Pantham G, Post A, Venkat D, Einstadter D, Mullen KD. A new look at precipitants of overt hepatic encephalopathy in cirrhosis. Dig Dis Sci. 2017;62(8):2166–2173. | ||
Romero-Gomez M, Cordoba J, Jover R, et al. Value of the critical flicker frequency in patients with minimal hepatic encephalopathy. Hepatology. 2007;45(4):879–885. | ||
Lauridsen MM, Thiele M, Kimer N, Vilstrup H. The continuous reaction times method for diagnosing, grading, and monitoring minimal/covert hepatic encephalopathy. Metab Brain Dis. 2013;28(2):231–234. | ||
Amodio P, Del Piccolo F, Marchetti P, et al. Clinical features and survivial of cirrhotic patients with subclinical cognitive alterations detected by the number connection test and computerized psychometric tests. Hepatology. 1999;29(6):1662–1667. | ||
Bajaj JS, Hafeezullah M, Franco J, et al. Inhibitory control test for the diagnosis of minimal hepatic encephalopathy. Gastroenterology. 2008;135(5):1591–1600.e1591. | ||
Zhu GQ, Shi KQ, Huang S, et al. Systematic review with network meta-analysis: the comparative effectiveness and safety of interventions in patients with overt hepatic encephalopathy. Aliment Pharmacol Ther. 2015;41(7):624–635. | ||
Ke Q, Yang RN, Ye F, et al. Impairment of liver regeneration by the histone deacetylase inhibitor valproic acid in mice. J Zhejiang Univ Sci B. 2012;13(9):695–706. | ||
Brunetti-Pierri N, Lanpher B, Erez A, et al. Phenylbutyrate therapy for maple syrup urine disease. Hum Mol Genet. 2011;20(4):631–640. | ||
Holecek M, Vodenicarovova M. Phenylbutyrate exerts adverse effects on liver regeneration and amino acid concentrations in partially hepatectomized rats. Int J Exp Pathol. 2016;97(3):278–284. | ||
Ventura-Cots M, Arranz JA, Simon-Talero M, et al. Safety of ornithine phenylacetate in cirrhotic decompensated patients: an open-label, dose-escalating, single-cohort study. J Clin Gastroenterol. 2013;47(10):881–887. | ||
Patidar KR, Bajaj JS. Covert and Overt Hepatic Encephalopathy: Diagnosis and Management. Clin Gastroenterol Hepatol. 2015;13(12):2048–2061. | ||
Ferenci P. Hepatic encephalopathy. Gastroenterology Report. 2017;5(2):138–147. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.