Back to Journals » Clinical Ophthalmology » Volume 13
Hemoglobin level and macular thinning in sickle cell disease
Authors Hussnain SA, Coady PA, Slade MD, Carbonella J, Pashankar F ![]() , Adelman RA
, Adelman RA ![]() , Stoessel KM
, Stoessel KM
Received 19 November 2018
Accepted for publication 28 February 2019
Published 15 April 2019 Volume 2019:13 Pages 627—632
DOI https://doi.org/10.2147/OPTH.S195168
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
S Amal Hussnain,1–4 Patrick A Coady,1,5 Martin D Slade,6 Judith Carbonella,7 Farzana Pashankar,7 Ron A Adelman,1 Kathleen M Stoessel1
1Department of Ophthalmology and Visual Science, Yale University School of Medicine, New Haven, CT, USA; 2Department of Ophthalmology, Columbia University Vagelos College of Physicians and Surgeons, New York, NY, USA; 3Vitreous Retina Macula Consultants of New York, New York, NY, USA; 4Department of Ophthalmology, New York University School of Medicine, New York, NY, USA; 5New England Retina Associates, Hamden, CT, USA; 6Department of Internal Medicine, Yale University School of Medicine and Yale School of Public Health, New Haven, CT, USA; 7Department of Pediatric Hematology and Oncology, Yale University School of Medicine, New Haven, CT, USA
Purpose: To study the relationship between complete blood count (CBC) indices over time, particularly serum hemoglobin (Hb) levels, and severity of macular thinning on spectral domain optical coherence tomography (SD-OCT) in patients with sickle cell disease (SCD).
Methods: This is a single-center, retrospective analysis of 141 consecutive SCD patients over a 10-year period, of which 40 patients (79 eyes) had SD-OCT imaging of the macula and 29 (58 eyes, mean age 17.5 years) were eligible for the study. Investigators reviewed electronic medical records for documentation of retinopathy stage, disease genotype, CBC values, and SD-OCT imaging. SD-OCT parameters and CBC values were compared between different retinopathy stages and disease genotypes. Regression analyses were performed on SD-OCT parameters and CBC values.
Results: Of the 58 eligible eyes (34HbSS, 18HbSC, 4HbSβ +thal, 2HbS βthal), 18 had PSR (proliferative sickle retinopathy), 14 had NPSR (nonproliferative sickle retinopathy), and 26 had NSR (no sickle retinopathy). Hb values were higher in SC group compared to SS group. Macular thickness in the temporal inner (Δ=26±33 um, p=0.01) and outer (Δ=21±30 um, p=0.02) subfields was higher in SC compared to SS group. Patients with SD-OCT thinning below the 5th percentile in the temporal outer subfields had lower recorded Hb nadirs (6.0±0.9) compared to those with thickness within the top 95th percentile (9.1±2.3). Regression analysis showed temporal macular thickness to be positively correlated with Hb values in the SS group.
Conclusion: Macular thinning observed on SD-OCT in SCD patients with SS genotype may be related to the level of anemia in this population.
Keywords: sickle cell retinopathy, macular thinning, spectraldomain optical coherence tomography, hemoglobin
Introduction
Sickle cell disease (SCD) is one of the most common genetic disorders.1 In 1949, Linus Pauling et al localized the defect to a single amino acid substitution (glutamic acid to valine) at position 6 in the oxygen-carrying β-globin subunit of hemoglobin (Hb) in red blood cells (RBCs).2 This mutation leads to abnormal hemoglobin HbS which can combine with itself to cause HbSS, with normal Hb to cause sickle cell trait, or with another β-globin variant HbC to cause HbSC. Thalassemia, a blood disorder characterized by defective production of either the α or β-globin chain, can also coexist with HbS. Under conditions of hypoxia and acidosis, the mutated β-globin molecules polymerize resulting in sickling of RBCs. Sickled RBCs are prone to hemolysis and cause vascular occlusion given their fragile membranes and rigid structure. SCD affects many organs as well as the orbit, anterior segment, and posterior segment of the eye. Sickle cell retinopathy (SCR) is the most common ocular manifestation of SCD and can result in significant vision loss.3
Though peripheral retinopathy is considered the primary clinical manifestation of SCR, the advent of spectral domain optical coherence tomography (SD-OCT) and angiography (OCTA) has shifted the focus to detecting early changes in the macula.4 Subclinical thinning in the macula, especially in the temporal macula, can be detected with the help of SD-OCT.5,6 Focal macular thinning on SD-OCT has been shown to correlate with decreased retinal sensitivity in patients with SCD.7 In addition, changes at the level of the choroid have also been shown with the aid of SD-OCT in patients with SCD.8 Though it is unclear if these subclinical signs seen on SD-OCT predict progression in SCR, they may be important early indicators of disease activity in patients with SCD before SCR becomes clinically evident.
In vivo acute ischemic events leading to macular thinning can be observed with SD-OCT. Paracentral acute middle maculopathy (PAMM) has been reported in SCD and shown to lead to macular thinning.9,10 It is unclear whether areas of macular thinning are only the result of acute ischemic events or could also be from chronic ischemia. Regardless of the mechanism of macular thinning, it seems plausible that systemic disease control in SCD may predict the degree of macular atrophy present. Lim et al recently showed that macular thinning in SCD is associated with age, retinopathy, and HbSS genotype.11 Furthermore, another recent study suggested that high HbF levels and chelation therapy are possible protective factors for the presence of sickle cell maculopathy.12
The purpose of this study is to investigate if there is a correlation in SCD between Hb levels, platelets (Plts), and mean corpuscular volume (MCV) over time and macular thinning as measured by SD-OCT.
Methods
The study complied with the Health Insurance Portability and Accountability Act of 1996 and followed the tenets of the Declaration of Helsinki. Informed consent was not required as this was a retrospective review where all personal health information was kept secure and confidential. This was a single-institution, IRB-approved (Yale University Institutional Review Board), retrospective medical record review of 141 consecutive SCD patients who underwent retinal evaluation at Yale Eye Center over a 10-year period. Of these, 40 patients had undergone SD-OCT imaging of the macula and 29 (58 eyes) were found to be eligible for the study.
All participants underwent slit-lamp biomicroscopy and dilated fundus ophthalmoscopy by a single retinal specialist (KMS). Presence of no sickle retinopathy (NSR), nonproliferative sickle retinopathy (NPSR), and proliferative sickle retinopathy (PSR) was documented based on clinical examination and fluorescein angiography.
Hb values, Plt levels, and MCVs were extracted by reviewing electronic medical records for patients included in this study. Patients on monthly blood transfusions were excluded. Only those patients who had complete blood count (CBC) drawn within 3 months of the OCT date and at least one CBC available more than a year prior to the OCT date were included. Most recent values within 3 months of OCT for Hb, MCV, and Plts were recorded. In addition, the highest, the lowest, and the average for these hematological markers over the 2 years preceding OCT imaging were noted as well. For patients with several CBC measurements within the last two years, the 15 most recent blood draws were used for analysis. Where available, hydroxyurea use, fetal Hb values (HbF), and other clinical data related to SCD activity were recorded.
All SD-OCT scans were performed on Cirrus HD-OCT 4,000 (Carl Zeiss Meditec, Inc., Dublin, CA, USA). The average macular thickness, macular volumes, and 9 subfield thicknesses according to Early Treatment Diabetic Retinopathy Study (ETDRS) were included for analysis with SD-OCT scans centered on the fovea.
Statistical analyses were performed using SAS 9.4 (SAS Institute Inc., Cary, NC, USA). Student’s t-test was utilized to compare continuous variables across two categories. One-way ANOVA was used to compare continuous variables across more than two categories. Mixed (fixed and random)-effects models were utilized for regression analyses in order to account for the nonindependence of left and right eye within a person. The mixed-effects statistical modeling incorporated person as a random effect that accounts for covariance between the two eyes of an individual. A backward elimination strategy was utilized to determine the parsimonious models. A p-value <0.05 was considered significant. Employing conservative Bonferroni correction to address multiplicity did not alter statistical findings. Different ETDRS subfields within the same eye were considered independent for statistical analyses.
Results
Men and women comprised 59% and 41% of the study participants, respectively. Forty-four percent, 24%, and 31% of the eyes showed NSR, NPSR, and PSR, respectively. This breakdown was similar between SS and SC genotype groups. Average age was 17 years for NSR and NPSR and 19 years for PSR (Table 1). Eighty-three percent of the patients had a history of sickle cell crisis, 34% had a history of acute chest syndrome, and none had a history of cerebrovascular accident. Fifty-two percent of the patients in our sample were on hydroxyurea treatment for their SCD.
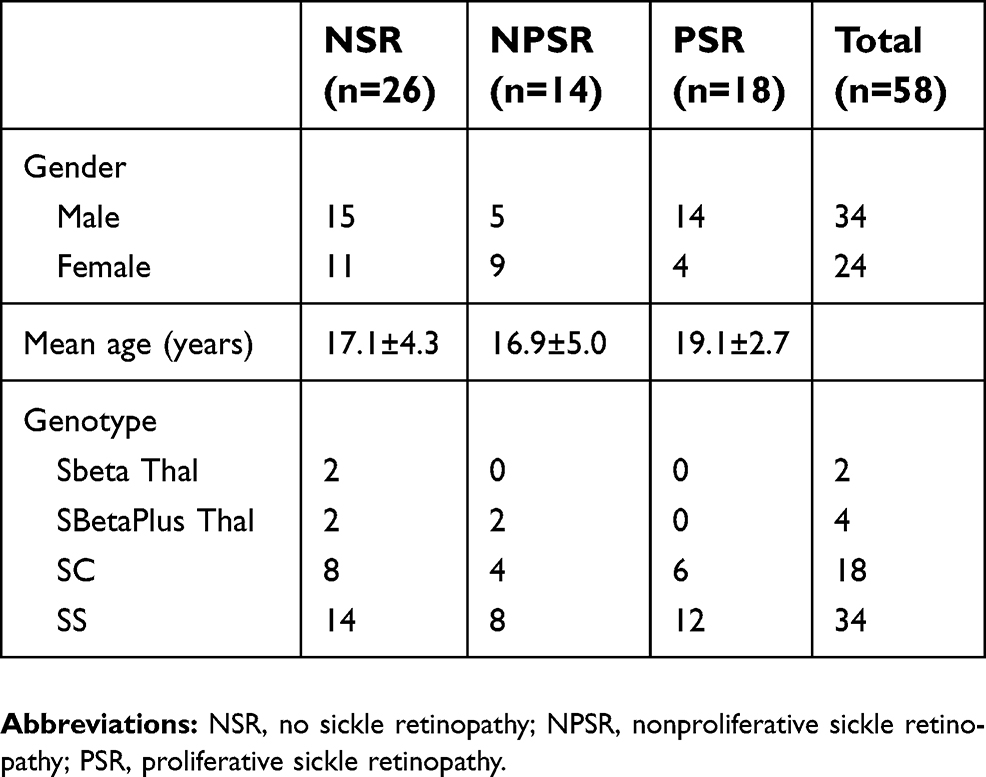 | Table 1 Patient characteristics and disease classification of study eyes |
Table 2 summarizes comparison of CBC indices, ie, Hb, Plt, and MCV between SS and SC genotypes. For each CBC index, four values were recorded: most recent value within 3 months of SD-OCT date, lowest as well as highest value over the 2 years preceding the SD-OCT date, and the average of all values that were obtained over the 2 years preceding the SD-OCT date. For all four categories, Hb was higher in patients with SC genotype compared to SS (p<0.001), whereas Plt and MCV values were higher in patients with SS genotype (p<0.01, except for Plt 2 years low).
 | Table 2 Comparison of CBC indices between SS and SC genotypes |
Macular thinning was more prevalent in SS genotype. Macular thickness values were generally higher for the SC group, with the difference being statistically significant for the temporal subfields (p=0.002 for inner and 0.005 for outer), as well as the central (0.022) and nasal (p=0.019) inner subfields (Table 3). No statistically significant difference was found in CBC indices based on retinopathy stage, ie, NSR, NPSR, and PSR. There was a trend (p=0.24) toward higher average macular thickness in PSR (287±21 μm) compared to NSR (276±26 μm) and NPSR (275±18 μm), but it was statistically significant only for inferior outer subfield.
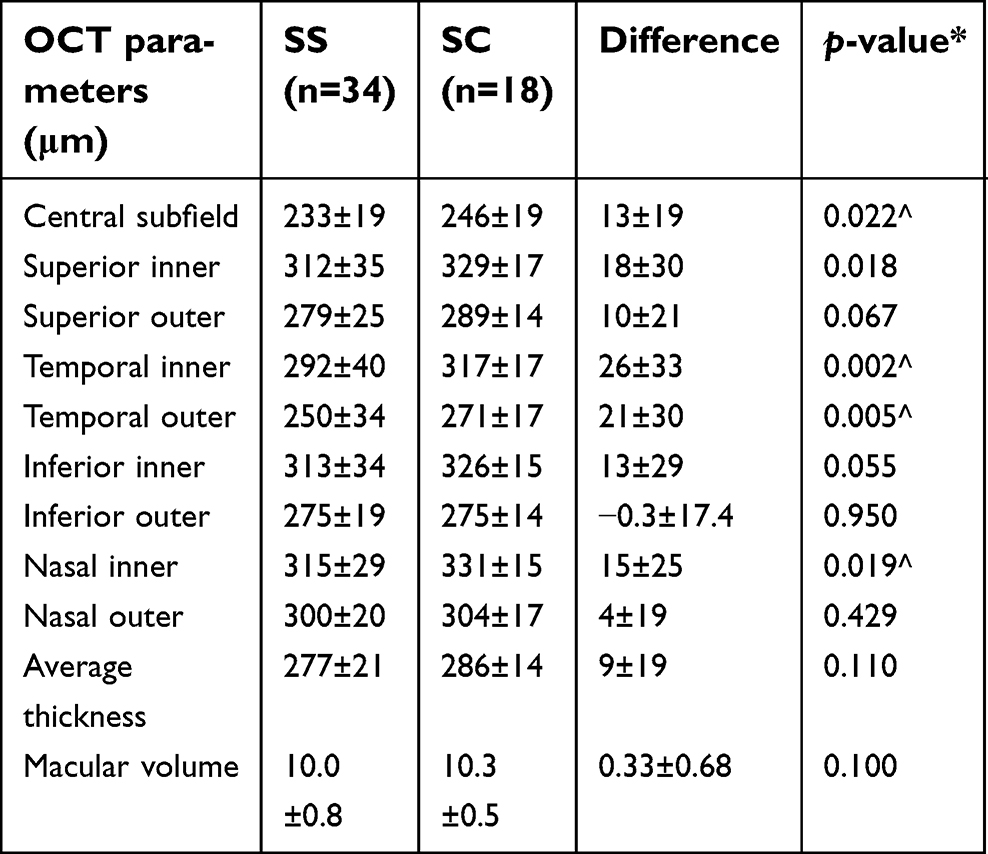 | Table 3 Comparison of OCT parameters between SS and SC genotype |
We used age-controlled normative database obtained from Zeiss for the Cirrus HD-OCT 4,000 macular subfields and looked at the average value for the 2-year low Hb values. The eyes with thinning below the 5th percentile for their age group had statistically lower Hb nadirs compared to those eyes in the top 95th percentile in the following categories: central subfield (p=0.016), superior inner (p=0.005), temporal inner (p<0.001), temporal outer (p<0.001), nasal inner (p=0.024), average thickness (p=0.043), and macular volume (p=0.043) (Table 4).
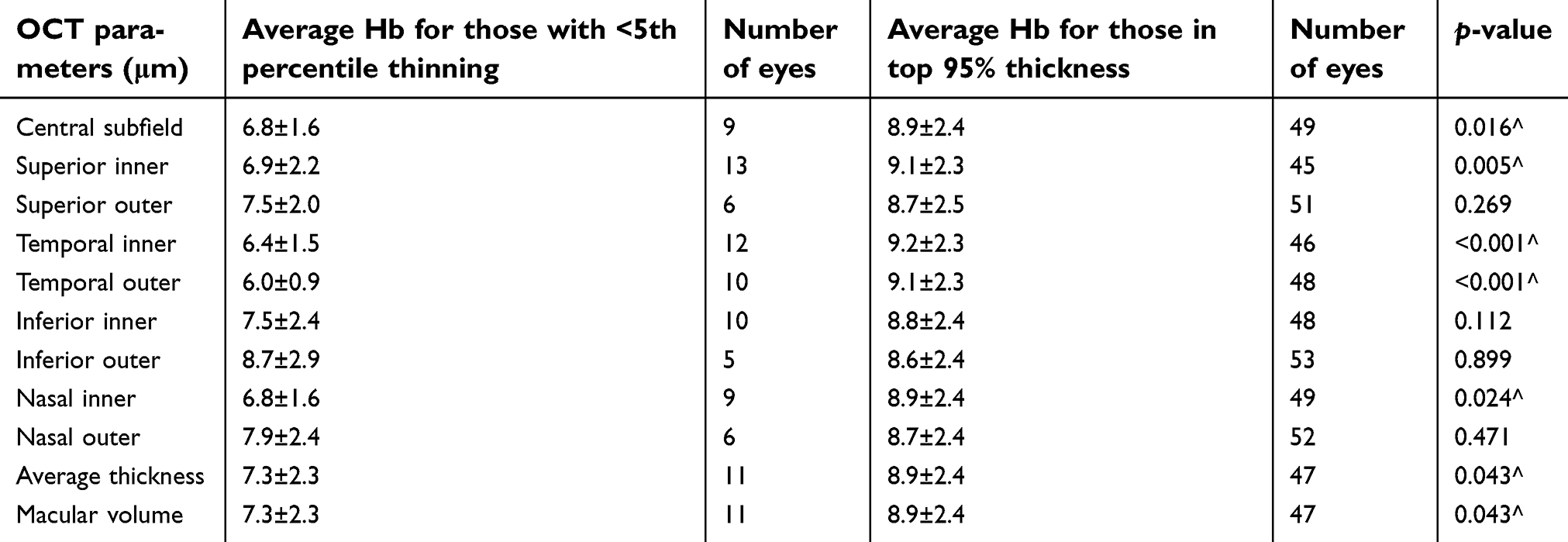 | Table 4 Comparison of average 2-year low hemoglobin (Hb) in eyes with thinning below the 5th percentile vs those with no thinning (top 95th percentile) |
A regression model adjusting for the effect of contributing variables that predicts change in temporal macular thickness as well as average macular volume and thickness based on CBC indices is shown in Table 5. This model predicts that a one-point drop in lowest Hb value was associated with a 7–10-micron drop in temporal macular thickness, 3-micron drop in average thickness, and a 0.12 mm3 drop in macular volume. When SS and SC genotype groups were analyzed separately, the correlation between lowest Hb level over the preceding 2 years (Hb 2 year low) and OCT thinning held for the SS population (p=0.01), but not for the SC population (p=0.25). A backward elimination strategy with a significance level to stay <0.05 showed that genotype dropped out of the model prior to Hb 2 year low.
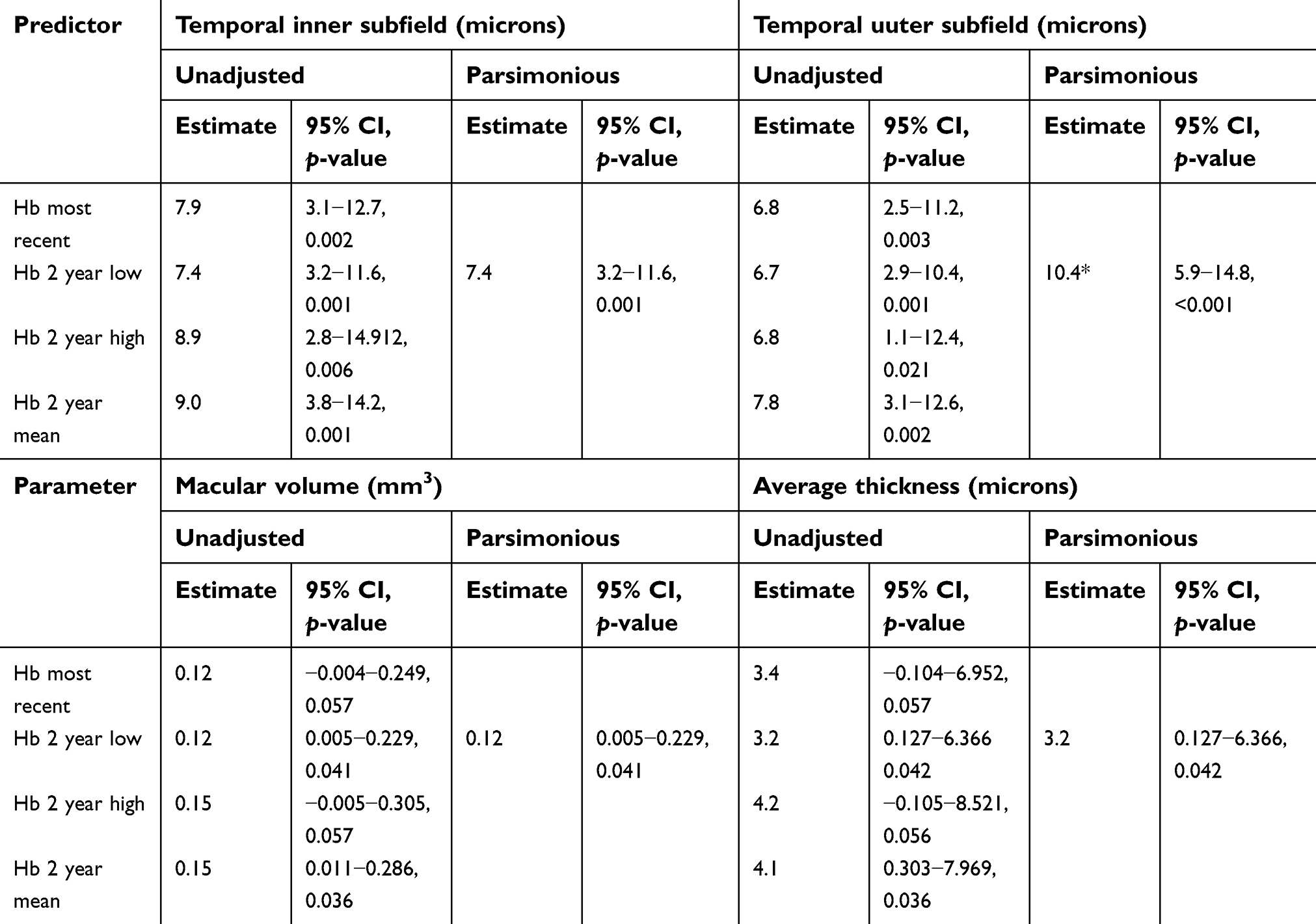 | Table 5 Regression model predicting macular thickness based on hemoglobin (Hb) values |
Discussion
In this study, we explore the relationship between recent Hb values over time and macular thinning on SD-OCT in SCD. Our results indicate that severe episodes of anemia (patients with the lowest Hb nadirs) in SCD due to HbSS are associated with macular thinning, most notably in the temporal area. We propose that macular thinning on SD-OCT should be used in screening and classification of SCR as well as a marker for systemic disease control in this population.
We found that mean Hb levels were lower, whereas mean Plts and MCV were higher in SS genotype compared to SC genotype. Our mean values for CBC indices for both SS and SC genotype groups were similar to those that Dr. Serjeant observed in a cohort of 740 SS and 441 SC patients.13 That study also showed that within SS and SC genotype groups, PSR was associated with higher Hb. However, in our study, there was no significant difference in Hb, Plt, and MCV indices between NSR, NPSR, and PSR groups.
Our study also demonstrated that macular thinning (defined as subthreshold values on ETDRS 9 subfield grid) is more prevalent in SS (35%) vs SC (11%) genotype. This finding is in agreement with Mathew et al who showed macular thinning on SD-OCT in 48% SS as opposed to 35% of SC eyes.8 A recent study by Lim et al also confirms that macular thinning is more prevalent in SS than SC.11 Although some previous studies have shown that macular thinning predicts PSR, in our study only 16% of patients with PSR displayed macular thinning on SD-OCT.
Patients with severe SD-OCT macular thinning (below 5th percentile) had worse anemia episodes (Hb 2 year low values) compared to eyes with normal thickness values. This was most striking and statistically significant in the temporal subfields. In addition, we found that a one-point drop in Hb during a severe episode of anemia was associated with a 7–10-micron decrease in temporal macular thickness and 3-micron decrease in average macular thickness in patients with SCD (Table 5). However, this correlation held only for SS genotype group, and not the SC group. It is possible that we were unable to detect a similar correlation between Hb nadirs and macular thinning for the SC population due to lower number of SC patients in our study. Alternatively, it might be due to the fact that most SC patients do not drop below the threshold Hb level needed for acute infarction and subsequent macular thinning. For statistical analysis, we focused on the temporal macula for two reasons: i) it is considered a watershed zone given vessels along the horizontal raphe are end branches and more prone to occlusion and ii) macular thinning is known to occur predominantly in the temporal macular region in SCD. Based on this relationship, we propose that SCD patients with frequent episodes of anemia should be closely monitored with OCT imaging.
Some of the observations made in our study may explain the differences in the pathophysiology of sickle retinopathy between SS and SC genotypes. A lower Hb as well as higher Plt count and MCV in SS genotype could contribute to acute occlusive episodes such as PAMM which might explain why macular thinning is more commonly seen in SS than SC genotype.8,10 This may also explain why PSR is less common in SS compared to SC, as complete vaso-occlusion in SS results in anoxia, as opposed to hypoxia in SC, and therefore no release of vasoproliferative factors. These findings are consistent with the pathophysiological model of sickle retinopathy put forth by Fox et al in 1990 that divides sickle cell patients into low, moderate and severe risk categories based on vaso-occlusive tendency.13 Based on this model, SC patients experience low-to-moderate ischemia resulting in hypoxia, release of vasoproliferative factors, and proliferative disease that is mainly seen in SC genotype. On the other hand, SS patients undergo severe vascular closure resulting in anoxia, and possibly occlusion of new proliferative vessels also, resulting in higher retinal thinning but lower PSR rates.14
Our study has several strengths. All study participants were referred for ophthalmic examination directly from our own department of hematology where these patients were closely followed with frequent blood draws. The diagnosis of SCD had therefore been confirmed with gel electrophoresis as opposed to being self-reported or derived from ICD codes in the charts. All of our patients were young without other co-existing ocular or systemic vascular pathology that might confound ocular findings. We were able to compare SD-OCT thickness values in our patients to an age-matched normative database provided by Zeiss for the Cirrus HD-OCT 4,000.
There are several limitations to our study and conclusions. This was a retrospective study with a small sample size and lacks modern imaging data such as OCT angiography and ultrawidefield imaging. As this is a pilot study, no adjustments were made for the multiple analyses undertaken. However, after adjusting for multiple comparisons through the use of the Bonferroni methodology, the association between Hb 2 year low and temporal inner and outer subfield thickness remained statistically significant. We could not include HbF values as a variable since a large number of our study subjects were missing this datapoint. However, HbF did not have a significant effect on our model fit when available HbF values were tested in a retrospective manner. We invite future studies that take into account these parameters as well as look at the correlation between macular thickness and other serious systemic manifestations of SCD such as stroke, pain crisis, and acute chest syndrome. We believe our study opens up a new avenue in the management of patients with SCD as it potentially establishes a relationship between severe episodes of anemia and temporal macular thinning that is seen in patients with SS SCD.
Acknowledgments
None of the authors have a proprietary interest related to this research. This study was supported in part by an unrestricted department grant from Research to Prevent Blindness. This study was completed at Yale University School of Medicine, Department of Ophthalmology and Visual Science, New Haven, CT.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rosenberg JB, Hutcheson KA. Pediatric sickle cell retinopathy: correlation with clinical factors. J AAPOS. 2011;15(1):49–53. doi:10.1016/j.jaapos.2010.11.014
2. Pauling, L, Itano, HA, Singer, SJ, Wells, IC. Sickle cell anemia, a molecular disease. Science. 1949;109(2835):443. doi:10.1126/science.109.2833.386-a
3. Elagouz M, Jyothi S, Gupta B, Sivaprasad S. Sickle cell disease and the eye: old and new concepts. Surv Ophthalmol. 2010;55:359–377. doi:10.1016/j.survophthal.2009.11.004
4. Wang M, Hussnain SA, Chen RWS. The role of retinal imaging in sickle cell retinopathy: a review. Int Ophthalmol Clin. 2019;59(1):71–82. doi:10.1097/IIO.0000000000000255
5. Hoang QV, Chau FY, Shahidi M, Lim JI. Central macular splaying and outer retinal thinning in asymptomatic sickle cell patients by spectral-domain optical coherence tomography. Am J Ophthalmol. 2011;151(6):990–994 e991. doi:10.1016/j.ajo.2010.12.010
6. Brasileiro F, Martins TT, Campos SB, et al. Macular and peripapillary spectral domain optical coherence tomography changes in sickle cell retinopathy. Retina. 2015;35(2):257–263. doi:10.1097/IAE.0000000000000309
7. Chow CC, Genead MA, Anastasakis A, Chau FY, Fishman GA, Lim JI. Structural and functional correlation in sickle cell retinopathy using spectral-domain optical coherence tomography and scanning laser ophthalmoscope microperimetry. Am J Ophthalmol. 2011;152(4):704–711 e702. doi:10.1016/j.ajo.2011.03.035
8. Mathew R, Bafiq R, Ramu J, et al. Spectral domain optical coherence tomography in patients with sickle cell disease. Br J Ophthalmol. 2015;99(7):967–972. doi:10.1136/bjophthalmol-2014-305532
9. Ilginis T, Keane PA, Tufail A. Paracentral acute middle maculopathy in sickle cell disease. JAMA Ophthalmol. 2015;133(5):614–616. doi:10.1001/jamaophthalmol.2014.6098
10. Hussnain SA, Coady PA, Stoessel KM. Paracentral acute middle maculopathy: precursor to macular thinning in sickle cell retinopathy. BMJ Case Rep. 2017;2017:1–2.
11. Lim JI, Cao D. Analysis of retinal thinning using spectral-domain optical coherence tomography imaging of sickle cell retinopathy eyes compared to age- and race-matched control eyes. Am J Ophthalmol. 2018;192:229–238. doi:10.1016/j.ajo.2018.03.013
12. Dell’Arti L, Barteselli G, Riva L, et al. Sickle cell maculopathy: identification of systemic risk factors, and microstructural analysis of individual retinal layers of the macula. PLoS One. 2018;13(3):e0193582. doi:10.1371/journal.pone.0193582
13. Fox PD, Dunn DT, Morris JS, Serjeant GR. Risk factors for proliferative sickle retinopathy. Br J Ophthalmol. 1990;74(3):172–176.
14. Hayes RJ, Condon PI, Serjeant GR. Haematological factors associated with proliferative retinopathy in homozygous sickle cell disease. Br J Ophthalmol. 1981;65(1):29–35.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.