Back to Journals » Journal of Blood Medicine » Volume 12
Hematopoietic Stem Cell Therapy for Wiskott–Aldrich Syndrome: Improved Outcome and Quality of Life
Authors Mallhi KK, Petrovic A, Ochs HD
Received 24 March 2021
Accepted for publication 13 May 2021
Published 11 June 2021 Volume 2021:12 Pages 435—447
DOI https://doi.org/10.2147/JBM.S232650
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Kanwaldeep K Mallhi,1– 3 Aleksandra Petrovic,1,2,4,* Hans D Ochs2,5,*
1Fred Hutchinson Cancer Research Center, Seattle, WA, USA; 2Department of Pediatrics, University of Washington School of Medicine, Seattle, WA, USA; 3Division of Hematology and Oncology, Seattle Children’s Hospital, Seattle, WA, USA; 4Division of Immunology and Division of Hematology and Oncology, Seattle Children’s Hospital, Seattle, WA, USA; 5Seattle Children’s Research Institute, Seattle, WA, USA
*These authors contributed equally to this work
Correspondence: Hans D Ochs
Department of Pediatrics, University of Washington School of Medicine, Seattle Children’s Research Institute, 1900 Ninth Avenue, Seattle, WA, 98101-1304, USA
Email [email protected]
Abstract: The Wiskott–Aldrich syndrome (WAS) is an X-linked disorder caused by mutations in the WAS gene resulting in congenital thrombocytopenia, eczema, recurrent infections and an increased incidence of autoimmune diseases and malignancies. Without curative therapies, affected patients have diminished life expectancy and reduced quality of life. Since WAS protein (WASP) is constitutively expressed only in hematopoietic stem cell-derived lineages, hematopoietic stem cell transplantation (HSCT) and gene therapy (GT) are well suited to correct the hematologic and immunologic defects. Advances in high-resolution HLA typing, new techniques to prevent GvHD allowing the use of haploidentical donors, and the introduction of reduced intensity conditioning regimens with myeloablative features have increased overall survival (OS) to over 90%. The development of GT for WAS has provided basic knowledge into vector selection and random integration of various viral vectors into the genome, with the possibility of inducing leukemogenesis. After trials and errors, inactivating lentiviral vectors carrying the WAS gene were successfully evaluated in clinical trials, demonstrating cure of the disease except for insufficient resolution of the platelet defect. Thus, 50 years of clinical evaluation, genetic exploration and extensive clinical trials, a lethal syndrome has turned into a curable disorder.
Keywords: Wiskott–Aldrich syndrome, WAS, X-linked thrombocytopenia, XLT, X-linked neutropenia, XLN, hematopoietic stem cell transplantation, HSCT, reduced intensity conditioning, gene therapy, GT, lentiviral vectors
Introduction
In 1937, Wiskott reported 3 brothers who died in early infancy with a history of early onset thrombocytopenia, frequent infections and severe eczema.1 Wiskott, examining blood smears of his patients, noticed that the platelets were tiny, in contrast to the large platelets characteristic for “Morbus Werlhofii”, the term used at that time for idiopathic thrombocytopenia (ITP). Seventeen years later, Aldrich observed a family with multiple generations where boys died during infancy of similar complications, demonstrating X-linked inheritance.2 Subsequently, the Wiskott–Aldrich Syndrome (WAS) was expanded to include adaptive and innate immune deficiency, autoimmunity and lymphoid malignancies.3–9 The identification of the molecular defect, mutations in the WAS gene,10 has broadened the clinical spectrum of the syndrome.11–15
Pathogenesis
The incidence of WAS has been estimated to be 1:100,000 live birth.16 With awareness of the different clinical phenotypes and the availability of reliable diagnostic tools, the incidence of pathologic entities caused by mutations in the WAS gene may be higher. The WAS protein (WASP) is constitutively expressed in all hematopoietic stem cell-derived lineages and is located predominantly in the cytoplasm with the highest protein expression toward the cell membrane. WASP is a member of a distinct family of proteins that link signaling pathways to actin cytoskeleton reorganization by inducing actin polymerization mediated by actin-related protein (Arp) 2/3. WASP is organized into several functional domains.17 The pleckstrin homology domain, located at the N-terminal region of WASP, interacts with lipids and may play a role in the localization of WASP in the immunologic synapse, the site of the interaction between various leukocyte subpopulations. A functional synapse facilitates the physical contact between antigen presenting cells, T and B cells required for a strong and lasting adaptive immune response, including class switch recombination, somatic hypermutation and cytokine production.3–6,18,19 While circulating NK cells are normal in number, the cytotoxicity of WAS-deficient NK cells is impaired as a result of defective synapse formation.20 For similar reasons, WASP-deficient patients as well as was-/- mice have regulatory T cells (Tregs) that fail to suppress effector cells in vitro and are incapable of controlling autoimmunity when studied in mouse models.21 While not required for the generation of Tregs in the thymus, WASP plays a crucial role in the homeostasis and function of these cells.22 The increased incidence of autoimmunity in WAS has been associated with deficient Treg cell function,21,22 B cell-intrinsic loss of tolerance by positive selection of self-reactive B cells23 and the production of a broad spectrum of autoantibodies.24 WASP-deficient neutrophils and monocytes exhibit impaired phagocytosis and chemotaxis,4,8,17,25,26 and monocytes, macrophages and dendritic cells from WAS patients lack the formation of podosomes and exhibit abnormal polarization and aberrant lamellipodia formation.9 Thrombocytopenia has been explained by ineffective thrombocytopoiesis,4 and reduced survival due to intrinsic platelet abnormalities or immune-mediated consumption.27,28
Clinical Spectrum of Mutations in the WAS Gene
As originally described by Wiskott, the syndrome is characterized by the triad of frequent infections caused by immunodeficiency, eczema and hemorrhagic diathesis due to micro-thrombocytopenia.1 This “classic” WAS phenotype was subsequently expanded to include autoimmunity initiated by immune dysregulation, and an increased incidence of hematologic malignancies. The generation of a large data base of patients with WAS mutations revealed variable clinical phenotypes and allowed a reasonably accurate genotype/phenotype correlation.29–31 The introduction of a scoring system based on disease severity and clinical phenotype (Table 1) has facilitated the categorization of patients, prognostic predictions and individualized therapeutic decisions.30 A score of 1 or 2 defines patients with XLT, while a score of 3 and 4 is reserved for those with a classic WAS phenotype; a score of 5 identifies patients who have developed autoimmunity and/or malignancies. Patients with intermittent XLT are given a score of <1 and those with XLN a score of “0”. The WAS score merely reflects severity of the clinical phenotype, without considering the type of mutation.30 Of note, the most consistent genotype/phenotype correlation was obtained when the patients were divided into two categories: WASP positive if peripheral blood mononuclear cells (PBMC) express the mutated protein at normal size, and WASP negative if the protein is absent or truncated.29–31 The clinical phenotype may evolve over time and is often incomplete in children younger than 2 years of age. For this reason, scoring disease severity at a very young age is of little value.30
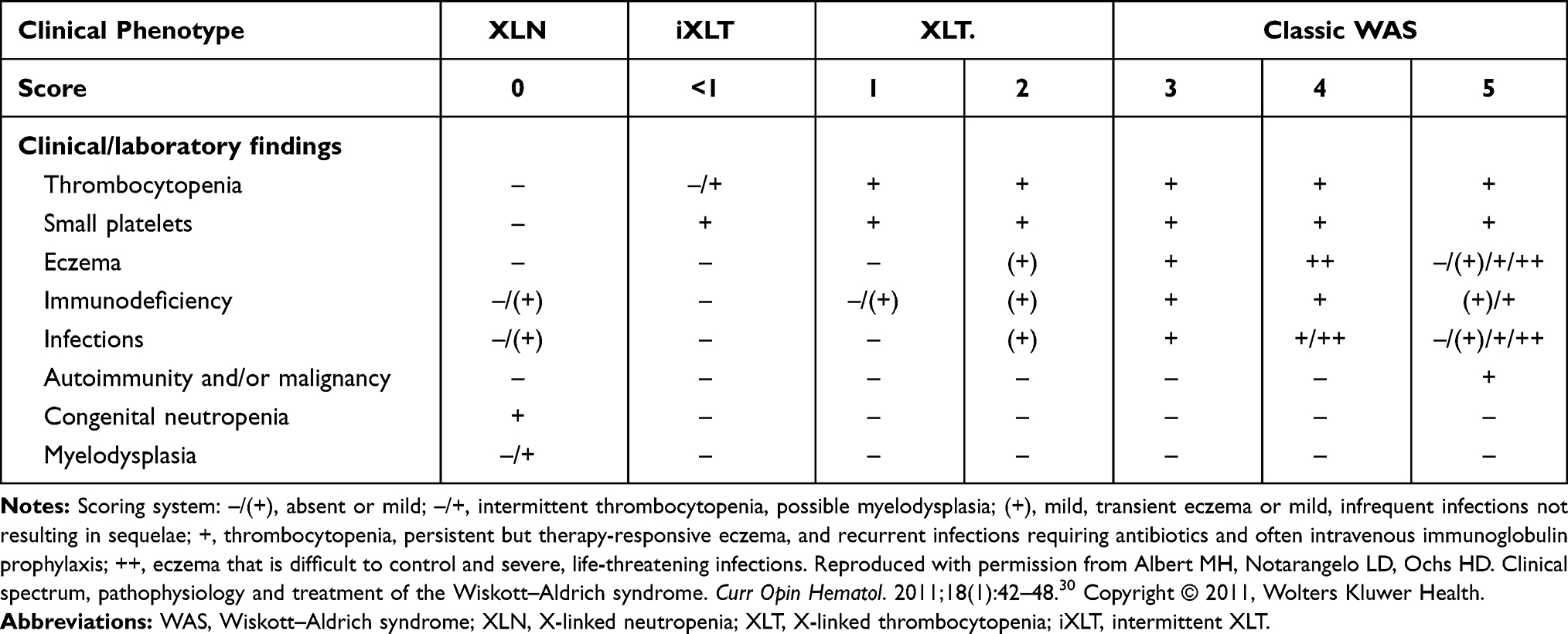 |
Table 1 Scoring System to Define Clinical Phenotypes Associated with Mutations in the WAS Gene |
WAS patients with the severe phenotype and a score of 3 or 4 present typically in early childhood with bloody diarrhea, petechiae, severe eczema and recurrent, often life-threatening bacterial, viral and fungal infections. They suffer from combined immune deficiency with abnormal antibody responses, declining T cell numbers and reduced lymphocyte proliferation to anti-CD3 and to specific antigens. IgA and IgE are elevated, while IgG and IgM are generally within normal range.17 Unless full hematologic and immunologic reconstitution by hematopoietic stem cell transplantation (HSCT) or gene therapy (GT) is achieved, quality of life is severely impacted and life expectancy reduced.3,6
The discovery of the WAS gene confirmed that “familial sex-linked thrombocytopenia”32 was in fact a mild variant of WAS11,12 with normal life expectancy, but severely reduced event-free survival probability due to life-threatening infections, cerebral hemorrhage, autoimmunity or cancer.14 Recognizing these risks in therapeutic decision-making is important, especially in view of the frequent misdiagnosis of XLT patients as ITP. The high success rate of HSCT in WAS patients makes this option more attractive for this group of patients.33,34
While loss of function mutations in the WAS gene cause WAS/XLT, unique gain of function (GOF) mutations in the GTPase-binding domain interfere with the autoinhibitory confirmation of the WAS protein, leading to increased and continuous actin polymerization resulting in congenital XLN. These patients have profound neutropenia, at times associated with lymphopenia, reduced in vitro lymphoproliferation in response to anti-CD3 and are at risk for myelodysplastic changes in the bone marrow.15,35 The neutropenia of XLN patients respond to G-CSF, but two patients were reported to have developed a myelodysplastic syndrome and acute myelogenous leukemia, respectively, both with somatic mutations in the G-CSF receptor gene and monosomy 7 in the leukemic cells.36
The diagnosis of WAS/XLT should be considered in any male patient presenting with petechiae, bruises and congenital or early-onset micro-thrombocytopenia. To confirm the diagnosis, a deleterious mutation in the WAS gene is required, often associated with absent or reduced WASP expression in PBMCs. Presence of mild or severe eczema, bloody diarrhea and a history of recurrent infections supports the diagnosis. The diagnosis of XLN has to be considered in any male patient presenting with severe congenital neutropenia, and is confirmed by sequencing the WAS gene with focus on the GTPase-binding site.15,35 WASP-interacting protein (WIP) stabilizes WASP. WIP deficiency, an autosomal recessive disorder, should be suspected in patients with features of WAS but normal sequence of the WAS gene and normal levels of mRNA but absent WAS protein. The diagnosis of WIP deficiency is confirmed by sequencing the WIPF1 gene; the treatment of choice is HSCT.37,38
Treatment of WAS/XLT is initiated with conventional measures including prophylactic antibiotics and antivirals for recurrent infections, and IgG replacement (IV or subcutaneous) for patients with demonstrated antibody deficiency. Platelet transfusions are used only to treat major bleeding episodes, such as acute CNS hemorrhage, gastrointestinal bleeding, or during major surgery. Eltrombopag, an oral thrombopoietin receptor agonist,39 or romiplostim given subcutaneously,40 have been used in WAS patients while waiting for HSCT but is less effective in raising platelet counts in WAS/XLT than in ITP. Immunosuppressive therapy, such as rituximab or other immunosuppressive therapies for autoimmune complications, may be required. Elective splenectomy has been advocated in selected patients with WAS or XLT to reverse the thrombocytopenia and prevent the bleeding tendency by increasing the number of circulating platelets.41 However, splenectomy markedly increases the risk of septicemia14,41 and is not recommended as routine procedure, especially not for those patients who are considered for future HSCT or gene therapy. Those who undergo splenectomy require lifelong antibiotic prophylaxis.
Hematopoietic Stem Cell Transplantation
Allogeneic hematopoietic stem cell transplantation (HSCT) is the primary curative approach in WAS patients providing long-term correction of the underlying immunodeficiency and thrombocytopenia. The first sibling donor bone marrow (BM) HSCT for WAS was performed in 1968. Initially, following minimal conditioning, the patient established donor-derived T-cell immunity while severe thrombocytopenia persisted.42 A second HSCT following myeloablative conditioning (MAC) regimen, from the same donor, was performed resulting in successful myeloid engraftment and normal platelet counts.42 Early experiences indicated reconstitution of lymphoid cells but not always platelet reconstitution, demonstrating the need for both immuno-ablation and myeloablation when transplanting WAS patients.42–45
Since these early attempts, HSCT outcomes have improved greatly (Table 2). Transplantation with HLA-identical sibling BM grafts has resulted in event-free survival (EFS) reaching 88% as early as 200146 and overall survival (OS) of 90–95% in subsequent years.33,47,48 Results of unrelated and alternative donor HSCT for treatment of WAS have also improved over time.49,50 Advances in high-resolution HLA allele typing and increasing availability of international donors and cord blood registries have facilitated optimal selection of unrelated donors for WAS patients. Several studies found comparable survival rates among recipients of HLA-matched sibling and HLA-matched unrelated donors. Earlier reports of long-term OS for recipients of unrelated bone marrow grafts ranged between 70%-78%,46,51,52 improving more recently to upwards of 90%.33,49 A collaborative study by Moratto et al, analyzing the outcomes of 194 WAS patients transplanted between 1980 and 2009, found improved transplant survival for all donor types, including matched unrelated donor grafts in those transplanted after 2000.47
 |
Table 2 Allogeneic HSCT for WAS: Evolution of Long-Term Outcomes |
Age at time of HSCT has been reported to consistently impact survival of WAS patients, likely related to the degree of comorbidities and higher WAS scores with increasing age. A 2001 study facilitated by the International Bone Marrow Transplant Registry (IBMTR) revealed that age greater than 5 years at time of transplant was associated with a strikingly increased risk of mortality46 and subsequent studies have endorsed this observation.47,48 In the cohort reported by Moratto et al, the 5-year OS was 91.9% for patients who received a matched unrelated donor transplant at younger than 2 years of age compared to 73.3% for those older than 5 years.47 Most recently, the Primary Immune Deficiency Treatment Consortium (PIDTC) reported the outcomes of 129 WAS patients (93 were <2 years old) who underwent HSCT between 2005 and 2015 in North America, and again confirmed that age was the only factor that had a significant impact on OS.33 Specifically, WAS patients < 5 years of age at time of HSCT had an OS of 94% compared to those ≥ 5 years of age who had a OS of 66%.33 Indications for early HSCT reflect a trend towards earlier and faster diagnosis of WAS, in addition to broader donor availability. While these factors support the strategy of early transplantation before the onset of disease-related complications, the potential late effects of intense conditioning at a young age remains to be determined.
While earlier studies demonstrated poor outcomes for WAS patients receiving alternative donor umbilical cord blood (UCB) and mismatched related/haploidentical HSCT,47,48 outcomes with the use of alternative donors have improved greatly over the past decades by overcoming barriers of graft rejection and graft-vs-host disease (GvHD). Shekhovtsova et al reported the use of UCB HSCT for 90 WAS patients following myeloablative conditioning between 1996 and 2013 and demonstrated an OS of 75% for the entire cohort, with an event-free survival (EFS) of 70% and graft failure rate of 11%.53 Multivariate analysis found age > 2 years was a risk factor for a worse outcome following UCB HSCT, and a trend towards improved overall survival in patients transplanted after 2007.53 Burroughs et al reported a cohort of 39 WAS patients receiving UCB HSCT between 2005 and 2015 with a 5-year OS of 90%.33 Overall, there was no significant difference in OS based on donor type comparing HLA-matched sibling, unrelated donor, or UCB grafts. This improvement over the past decade may be related to better UCB selection based on the degree of HLA-match, cell dose and improved supportive care during HSCT.
Historically, the selection of mismatched family donors required the use of ex-vivo T-cell depletion to overcome the risk of GvHD, which has been associated with inferior survival. The main challenge in using an HLA-mismatched related donors (MMRD)/haploidentical graft has been related to poor engraftment and incomplete immune reconstitution with high infectious complications. Earlier experiences with T-cell depleted MMRD HSCT for WAS patients resulted in OS between 37% and 55%.46,48,54 As has been the case with other graft sources, OS improved to 91.7% for recipients of MMRD-HSCT after 2000, compared to OS of 52.2% for those WAS patients transplanted between 1980 and 1999.47
Most recently, the introduction of post-transplant cyclophosphamide (PT-Cy)55,56 or the use of TCRαβ/CD19-depletion57–62 have yielded favorable results in alternative donor transplantation for WAS and other PIDs. Both methods reduce the incidence of acute and chronic GvHD and improve immune reconstitution. Laberko et al reported 98 PID patients (including 36 WAS patients) who underwent TCRαβ/CD19-depleted unrelated donors or MMRD HSCT59 and demonstrated an OS of 86% and 87%, respectively, with secondary graft failure occurring in 17% of patients. Late graft dysfunction noted in some WAS patients, was possibly related to the degree of donor chimerism, though the donor chimerism results were not presented in detail. A significantly lower OS of 52% was observed in patients who had poorly controlled infectious or autoimmune diseases at the time of HSCT. Encouragingly, the rates of grade II–IV acute GvHD and mild limited chronic GvHD were low at 17% and 9%, respectively, in the unrelated donor cohort, and 22% and 13%, respectively, in the MMRD group. Rate of CMV viremia was high with 17% of patients developing invasive disease. A similar rate of graft rejection was noted by Balashov et al in a cohort of 37 PID patients undergoing TCRαβ/CD19-depleted HSCT.60 To improve donor engraftment, they evaluated the addition of G-CSF and plerixafor to the conditioning regimen of 16 WAS patients undergoing TCRαβ/CD19-depleted HSCT.62 The OS reached 93.8% and no graft rejection was observed, with all participating patients achieving full donor chimerism in whole blood and in the CD3+ compartment.63
While the results are encouraging, TCRαβ/CD19-depletion is costly and resource restrictive. As such, the PT-Cy approach for GvHD prophylaxis with alternative donors is a viable option. Fernandes et al published the outcomes of haploidentical BM HSCT using PT-Cy in 73 patients with primary immunodeficiencies (PID), including 14 WAS patients, performed between 2012 and 2019.56 The 2-year OS for the WAS patients was 86%, while graft failure was experienced in 2 patients who received a reduced-intensity conditioning regimen. Using a MAC regimen reduced the incidence of graft failure and mixed chimerism. The incidence of grade II–IV aGVHD was slightly higher than the rates reported from studies with in vitro T cell-depletion techniques and TCRab/CD19 depletion, though comparable to those reported with other alternative sources, such as cord blood and MUDs. Yue et al reported good outcomes of 5 WAS patients receiving a busulfan-based MAC regimen with PT-Cy for GvHD prophylaxis following combined BM and peripheral blood (PB) stem cell haploidentical donor HSCT. All 5 patients engrafted, and are alive at last follow-up, with full donor chimerism and no post-HSCT autoimmune cytopenias.55
While the published number of patients is limited, there does not appear to be an increase incidence of post-transplant autoimmunity or immune dysregulation with PT-Cy, though larger studies are needed. Full intensity myeloablative conditioning is needed to overcome graft rejection with these methods, and further assessment of the degree and duration of lineage-specific donor chimerism achieved is being evaluated.
Despite an increase in the National Marrow Donor Program’s Registry size in recent years, racial disparities in access to matched unrelated donors have not improved, especially for African Americans.64 Thus, emphasizing the ongoing need to advance use of alternative graft sources such as cord blood transplant and haploidentical transplant in order to provide donor options in the ethnic groups not well represented in the donor databases is imperative. The results of alternative donor HSCT using UCB donors and TCRαβ/CD19-depletion or PT-Cy for mismatched/haploidentical donors is very promising, and provides new opportunities for successful curative therapy in WAS/XLT patients of minority ethnicities. Additionally, advances in gene therapy for WAS patients will increase treatment options for patients of minority ethnicity without suitable donors, though the limitations of high cost and restrictive resources will need to be addressed to make it widely accessible to all patients.
Regardless of the graft source, both myelo- and immuno-ablation are required to overcome graft rejection, establish myeloid and lymphoid engraftment and to correct the clinical manifestations of WAS. Myeloablative conditioning regimens have achieved excellent results in young WAS patients though short- and long-term toxicities remain a concern. While reduced intensity conditioning (RIC) regimens can significantly reduce early transplantation-related morbidity and mortality, especially in older patients, there is a greater risk for mixed chimerism, transplant associated complications and delayed graft failure.65,66 A more recent report found no difference in OS between patients receiving a MAC or RIC regimen, although significantly different donor T-cell, B-cell and myeloid engraftment was observed in patients receiving MAC vs RIC regimens.33 Stability or reversal of WAS symptoms post HSCT depends on the degree and long-term stability of donor cell engraftment in the hematopoietic lineage of interest. The degree of donor chimerism necessary for WAS symptom correction has been established.47,48 Lower levels of donor chimerism were observed more often in myeloid than in lymphoid lineages. Persistent thrombocytopenia after HSCT was strongly associated with mixed myeloid chimerism, suggesting that robust and stable engraftment of donor-derived myeloid cells is required to correct the defect.33,47 Burroughs et al noted that platelet count recovery post-HSCT was related to the degree of donor myeloid chimerism. At 1 year post-HSCT, patients with <50% donor myeloid engraftment were found to have a significantly lower median platelet count (40,000/mm3) compared to patients who attained ≥50% donor myeloid engraftment and achieved normal platelet counts,33 highlighting the need for ≥50% donor myeloid engraftment for correction of the thrombocytopenia. T and B-cell donor chimerism has also been shown to correlate with the intensity of the conditioning regimen.33
An association between mixed chimerism and post-transplant autoimmunity was reported in some WAS patients by several investigators. Specifically, in a multicenter European study of 96 patients who survived at least 2 years after HSCT, as many as 20% of the long-term survivors developed autoimmunity independent of chronic GvHD. The risk of autoimmunity was significantly higher for patients who developed mixed chimerism after receiving matched unrelated or MMRD HSCT.48 Cytopenias and endocrinopathies were predominant de-novo autoimmune disease following transplantation with time of onset and resolution being year and a half and four years respectively. Similarly, Moratto et al reported that autoimmune manifestations, predominantly cytopenias and endocrinopathies, occurred in 13.9% of patients following transplantation.47 These patients were noted to have a lower degree of chimerism in the T, B, and myeloid lineages compared with patients who did not develop autoimmunity, suggesting that mixed chimerism is a risk factor for developing autoimmune disease post-HSCT. However, subsequent studies did not confirm such an association.33,49,67 A retrospective analysis by the PIDTC evaluated if mixed donor chimerism (<95%) at day 100 or at 6 months following HSCT correlated with an increased risk for the development of de novo autoimmunity.33 Overall, 14% of patients developed 1 or more de novo autoimmune disease within the first year post-HSCT. However, unlike the results published earlier47,48 the analysis did not find a persistent association between mixed T-cell or B-cell donor chimerism and the development of de novo autoimmunity in long-term survivors. There was, however, an increased incidence of de novo autoimmunity in those with mixed myeloid donor chimerism at 6 months post-HSCT.33 Autoimmune cytopenia (AIHA being most prevalent) was the predominant de-novo autoimmunity with complete resolution occurring in most of the patients. Importantly, the presence of autoimmune disease prior to HSCT did not increase the risk of developing de novo autoimmune disorders after HSCT, nor did the WAS score and age at HSCT.33 Interestingly, none of the patients receiving a matched sibling donor HSCT developed de novo autoimmune disease post-HSCT, whereas 23% of patients receiving matched unrelated grafts and 9% receiving UCB grafts did.33 A smaller cohort of WAS patients followed prospectively at a single transplant center did not reveal an association between autoimmune cytopenia and mixed chimerism.67
Gene Therapy
HSCT outcomes have significantly improved over the last five decades since the first WAS patient was transplanted in 1968. Nevertheless, HSCT remains associated with significant short- and long-term problems, especially when a matched donor is not available. The need for intense conditioning to achieve successful myeloid and lymphoid engraftment, the risk of acute and chronic GvHD and the long-term sequelae associated with myeloablative conditioning have led to development of alternative treatment approaches.
Over the last 25 years, the field of gene therapy (GT) has progressed from basic research to standard of care in patients with certain monogenic PID’s.68,69 WAS is an ideal candidate for gene therapy due to selective expression of WASP in cells of the hematopoietic lineage, which are expected to have proliferative advantage over the host WASP negative cells.70–72 The initial GT trial for WAS patients was started in 2006 and utilized a gibbon ape leukemia virus (GALV)- γ-retroviral vector.73 The first 2 patients, both 3 years of age, received autologous WASP-expressing retroviral vector transduced PB stem cells following Busulfan (8mg/kg) conditioning. Both patients showed evidence of successful correction of the myeloid and lymphoid defects, demonstrating a selective advantage of the vector transduced stem cells.73 Following these promising initial results, 8 additional patients were enrolled by 2009. In all 10 patients, autologous stem cells were isolated from peripheral blood mobilized with G-CSF (n=2) or G-CSF and Plerixafor (n=8). Participants were 2 to 14 years of age (2 patients were older than 5) and each received a robust stem cell product. Nine of ten patients demonstrated sustained engraftment with correction of WASP expression in myeloid and lymphoid lineages. GT resulted in partial or complete correction of infectious, autoimmune or bleeding manifestations of the disease. Eight patients had pre GT autoimmunity, with two patients having ongoing autoimmunity following treatment, however with milder manifestations. The participant with the lowest stem cell dose showed only partial lymphoid correction without myeloid or stem cell WASP expression noted and without improvement in disease manifestations. Unfortunately, despite early clinical success, 7 patients developed acute leukemia 16 to 60 months following therapy. Six patients developed T-ALL (2 of these patients subsequently developed AML during maintenance therapy for T-ALL) and 1 patient AML.74 Analysis of the retroviral integration demonstrated a polyclonal pattern during the early phase, followed by development of dominant clones with integration at the LMO2, MDS1 and MN1 loci. Unlike the insertional leukemogenesis observed in the X-SCID trials,75–77 expansion of clones in the WAS patients treated with GT was more rapid. Five patients have subsequently been successfully treated with HSCT.74 High vector copy number as well as strong enhancer/promoter elements, although associated with better efficacy, may lead to increased genotoxicity and leukemogenesis.
Additional 31 WAS patients were reported to have undergone GT since 2013 utilizing lentiviral vector mediated gene transfer. Transition to lentiviral vector use (based on human immunodeficiency virus) was driven by the ability of lentiviruses to easily transverse the nuclear membrane and transduce non-mitotic hematopoietic stem cells, as opposed to γ-retroviral vectors which require cell division for integration.78 To improve vector safety, a self-inactivating design has been adopted to decrease or eliminate activation of enhancer and promoter regions.79,80 Into these novel lentiviral constructs, a codon optimized human WAS cDNA was placed under the control of a fragment of the endogenous promoter.
The first three lentiviral treated patients were reported by the Milan group in 2013,81,82 followed by a publication in 2019 of their interim results.81,82 The 8 patients treated with GT had WAS scores of 3–5 and age range 1.1 to 12.4 years. Additionally, 9 patients were treated on an expanded access protocol (age 1.4–35.1 years) with a total of 14 patients having more than 1 year of follow-up.83 Five patients were older than 5 years of age at the time of GT. Bone marrow was the source of stem cells in the first 6 patients, with transition to mobilized PB stem cells in the remaining patients. Reduced intensity conditioning consisted of Rituximab 375mg/m2 given on day −22, with Busulfan (AUC 45–60 mg*hr/L) and Fludarabine 60mg/m2 given day −3 to day −1. Overall survival was 94% in 17 treated patients. Lentiviral transduced colonies were noted in the bone marrow at 3 months and persisted up to 8 years. High levels of multilineage engraftment and sustained WASP expression was noted in lymphocytes and platelets. All evaluable patients from the 2 cohorts were able to discontinue IVIG supplementation at a median time of 0.9 years (range 0.2–5 years) following GT. Median platelet counts improved from baseline of 19,500 to 39,800/mm3 (14,000–272,000/mm3) at 12 months following GT, with all patients attaining independence from platelet transfusions by 9 months following therapy. Pre GT autoimmunity resolved in all but 1 patient from the initial cohort who developed transient immune thrombocytopenia post GT. Reduction in severe infections, frequency and severity of bleeding episode as well as improvement or resolution of eczema were noted following GT. Stable polyclonal gene marking with high number of unique insertion sites were observed. No replication competent virus was noted in any subjects. Furthermore, significant clinical benefit was observed in patients over 5 years of age, which historically has been the cohort with poorest HSCT survival.
Hacein-Bey-Albina et al described 7 patients who underwent lentiviral GT following more intense myeloablative conditioning with Busulfan 12mg/kg given over 3 days, Fludarabine 120mg/m3 with Rituximab or Alemtuzumab serotherapy. All patients had a severe WAS phenotype (WAS scores of 3–5), lacked matched related or unrelated donors and ranged in age between 10 months and 15.5 years (4 patients were over 5 years of age).84 There was equal distribution of BM and mobilized PB stem cells with a robust median transduced CD34+ dose. One patient died 7 months following GT of refractory herpes infection. The remaining 6 patients demonstrated clinical improvement in eczema, bleeding severity and frequency, infections and autoimmunity. Only one patient continued to experience vasculitis following GT which was mild compared to severe limb vasculitis leading to impaired ambulation pre GT. Minimal improvement in platelet counts occurred in patients who were not splenectomized (either before or following gene therapy) with no patient reaching platelet counts greater than 50,000/mm3.
Five additional WAS patients were treated in the Boston based U.S trial.83 As in the other 2 trials, patients had severe WAS with age range of 1.4 to 8 years. Myeloablative conditioning was used with busulfan of 12–15mg/kg total (target AUC 70–80 mg*hr/L) and Fludarabine of 120mg/m2. Multilineage vector gene marking was sustained over time, however WASP expression even though increased from the baseline remained below normal. Two patients demonstrated an increase in platelet counts over 50,000/mm3. The clinical benefits observed were similar to those reported in the other trials, except for 2 patients whose autoimmunity failed to resolve with GT. The use of lentiviral vector was not associated with any events concerning safety, with highly polyclonal pattern of vector integration.
The first adult WAS patient treated with lentiviral GT was reported in 2017.85 He received the same conditioning described by Hacein-Bey-Albina et al GT led to resolution of autoimmunity and IVIG discontinuation. As this patient had prior splenectomy, GT-related platelet count recovery could not be evaluated.
Lentiviral GT trials utilizing the endogenous WAS promoter have demonstrated clinical efficacy, with improvement in infectious, autoimmune and bleeding complications in treated patients. Overall safety of the lentiviral vectors has been demonstrated, and so far, no adverse events associated with the use of transduced stem cells have occurred, with polyclonal pattern of vector integration and without evidence of replication competent lentivirus. However, reconstitution of the platelet count to normal range has not been achieved in the majority of patients. Platelet reconstitution was further evaluated by Sereni et al analyzing platelet phenotype, activation state, and overall function in WAS patients following GT.86 Post GT, platelets were found as having normal volume, granule content with greater than 90% WASP expressing platelets noted in patients followed longer than 2 years. De-novo autoimmunity following GT occurred at the comparable rate as seen with HSCT, with 3 patients reported.81–83 However, mean fluorescence intensity of WASP was low demonstrating suboptimal expression. Platelet activation and aggregation normalized following GT. However, while the dysregulated and activated state of platelets typical for WAS patients improved, it did not normalize, possibly contributing to the persistent thrombocytopenia frequently seen following gene therapy.
Conclusion
When originally recognized as a clinical entity, patients with classical WAS typically died during infancy/early childhood. Better understanding of the underlying hematologic and immunologic abnormalities has led to symptomatic therapies, including corticosteroids, antimicrobials and immunoglobulin replacement that improved modestly the quality of life and extended life expectancy into the early twenties. Since the initial success of HSCT in 1968, significant progress has been made in confirming the diagnosis earlier, establishing genotype phenotype correlation, and designing and improving curative therapies for WAS patients. As is the case in other PIDs, earlier intervention and disease correction has led to better outcomes and improved event-free survival. HSCT and GT have reached an OS above 90%, however some challenges remain. Potential late effects due to intense conditioning regimens, insufficient donor chimerism leading to only partial disease correction, difficulties achieving normal platelet recovery and function, and the potential risk of insertional mutagenesis following GT still need to be evaluated in the decades to come.
Disclosure
The authors reported no conflicts of interest for this work.
References
1. Wiskott A. Familiarer, angeborener Morbus Werlhofii? Mschr Kinderheilk. 1937;68:212.
2. Aldrich RA, Steinberg AG, Campbell DC. Pedigree demonstrating a sex-linked recessive condition characterized by draining ears, eczematoid dermatitis and bloody diarrhea. Pediatrics. 1954;13(2):133.
3. Cooper MD, Chae HP, Lowman JT, Krivit W, Good RA. Wiskott-Aldrich syndrome. An immunologic deficiency disease involving the afferent limb of immunity. Am J Med. 1968;44(4):499–513.
4. Ochs HD, Slichter SJ, Harker LA, Von Behrens WE, Clark RA, Wedgwood RJ. The Wiskott-Aldrich syndrome: studies of lymphocytes, granulocytes, and platelets. Blood. 1980;55(2):243–252.
5. Orange JS, Stone KD, Turvey SE, Krzewski K. The Wiskott-Aldrich syndrome. Cell Mol Life Sci. 2004;61(18):2361–2385.
6. Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr. 1994;125(6 Pt 1):876–885.
7. Rawlings SL, Crooks GM, Bockstoce D, Barsky LW, Parkman R, Weinberg KI. Spontaneous apoptosis in lymphocytes from patients with Wiskott-Aldrich syndrome: correlation of accelerated cell death and attenuated bcl-2 expression. Blood. 1999;94(11):3872–3882.
8. Rengan R, Ochs HD, Sweet LI, et al. Actin cytoskeletal function is spared, but apoptosis is increased, in WAS patient hematopoietic cells. Blood. 2000;95(4):1283–1292.
9. Burns S, Cory GO, Vainchenker W, Thrasher AJ. Mechanisms of WASp-mediated hematologic and immunologic disease. Blood. 2004;104(12):3454–3462.
10. Derry JM, Ochs HD, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. 1994;79(5):following 922.
11. Villa A, Notarangelo L, Macchi P, et al. X–linked thrombocytopenia and Wiskott–Aldrich syndrome are allelic diseases with mutations in the WASP gene. Nat Genet. 1995;9(4):414–417. doi:10.1038/ng0495-414
12. Zhu Q, Zhang M, Blaese RM, et al. The Wiskott-Aldrich syndrome and X-linked congenital thrombocytopenia are caused by mutations of the same gene. Blood. 1995;86(10):3797–3804.
13. Notarangelo LD, Mazza C, Giliani S, et al. Missense mutations of the WASP gene cause intermittent X-linked thrombocytopenia. Blood. 2002;99(6):2268–2269.
14. Albert MH, Bittner TC, Nonoyama S, et al. X-linked thrombocytopenia (XLT) due to WAS mutations: clinical characteristics, long-term outcome, and treatment options. Blood. 2010;115(16):3231–3238.
15. Ancliff PJ, Blundell MP, Cory GO, et al. Two novel activating mutations in the Wiskott-Aldrich syndrome protein result in congenital neutropenia. Blood. 2006;108(7):2182–2189.
16. Stray-Pedersen A, Abrahamsen TG, Froland SS. Primary immunodeficiency diseases in Norway. J Clin Immunol. 2000;20(6):477–485.
17. Ochs HD, Notarangelo LD. Wiskott-Aldrich syndrome. In: Ochs HD, Smith CIE, Puck JM, editors. Primary Immunodeficiency Diseases: A Molecular and Genetic Approach.
18. Dupre L, Aiuti A, Trifari S, et al. Wiskott-Aldrich syndrome protein regulates lipid raft dynamics during immunological synapse formation. Immunity. 2002;17(2):157–166.
19. Malinova D, Fritzsche M, Nowosad CR, et al. WASp-dependent actin cytoskeleton stability at the dendritic cell immunological synapse is required for extensive, functional T cell contacts. J Leukoc Biol. 2016;99(5):699–710.
20. Orange JS, Ramesh N, Remold-O’Donnell E, et al. Wiskott-Aldrich syndrome protein is required for NK cell cytotoxicity and colocalizes with actin to NK cell-activating immunologic synapses. Proc Natl Acad Sci U S A. 2002;99(17):11351–11356.
21. Marangoni F, Trifari S, Scaramuzza S, et al. WASP regulates suppressor activity of human and murine CD4(+)CD25(+)FOXP3(+) natural regulatory T cells. J Exp Med. 2007;204(2):369–380.
22. Humblet-Baron S, Sather B, Anover S, et al. Wiskott-Aldrich syndrome protein is required for regulatory T cell homeostasis. J Clin Invest. 2007;117(2):407–418.
23. Kolhatkar NS, Brahmandam A, Thouvenel CD, et al. Altered BCR and TLR signals promote enhanced positive selection of autoreactive transitional B cells in Wiskott-Aldrich syndrome. J Exp Med. 2015;212(10):1663–1677.
24. Crestani E, Volpi S, Candotti F, et al. Broad spectrum of autoantibodies in patients with Wiskott-Aldrich syndrome and X-linked thrombocytopenia. J Allergy Clin Immunol. 2015;136(5):
25. Lorenzi R, Brickell PM, Katz DR, Kinnon C, Thrasher AJ. Wiskott-Aldrich syndrome protein is necessary for efficient IgG-mediated phagocytosis. Blood. 2000;95(9):2943–2946.
26. Badolato R, Sozzani S, Malacarne F, et al. Monocytes from Wiskott-Aldrich patients display reduced chemotaxis and lack of cell polarization in response to monocyte chemoattractant protein-1 and formyl-methionyl-leucyl-phenylalanine. J Immunol. 1998;161(2):1026–1033.
27. Shcherbina A, Rosen FS, Remold-O’Donnell E. Pathological events in platelets of Wiskott-Aldrich syndrome patients. Br J Haematol. 1999;106(4):875–883.
28. Marathe BM, Prislovsky A, Astrakhan A, Rawlings DJ, Wan JY, Strom TS. Antiplatelet antibodies in WASP(-) mice correlate with evidence of increased in vivo platelet consumption. Exp Hematol. 2009;37(11):1353–1363.
29. Jin Y, Mazza C, Christie JR, et al. Mutations of the Wiskott-Aldrich Syndrome Protein (WASP): hotspots, effect on transcription, and translation and phenotype/genotype correlation. Blood. 2004;104(13):4010–4019.
30. Albert MH, Notarangelo LD, Ochs HD. Clinical spectrum, pathophysiology and treatment of the Wiskott-Aldrich syndrome. Curr Opin Hematol. 2011;18(1):42–48.
31. Imai K, Morio T, Zhu Y, et al. Clinical course of patients with WASP gene mutations. Blood. 2004;103(2):456–464.
32. Vestermark B, Vestermark S. Familial sex-linked thrombocytopenia. Acta Paediatr. 1964;53:365–370.
33. Burroughs LM, Petrovic A, Brazauskas R, et al. Excellent outcomes following hematopoietic cell transplantation for Wiskott-Aldrich syndrome: a PIDTC report. Blood. 2020;135(23):2094–2105.
34. Oshima K, Imai K, Albert MH, et al. Hematopoietic stem cell transplantation for X-linked thrombocytopenia with mutations in the WAS gene. J Clin Immunol. 2015;35(1):15–21.
35. Beel K, Cotter MM, Blatny J, et al. A large kindred with X-linked neutropenia with an I294T mutation of the Wiskott-Aldrich syndrome gene. Br J Haematol. 2009;144(1):120–126.
36. Beel K, Vandenberghe P. G-CSF receptor (CSF3R) mutations in X-linked neutropenia evolving to acute myeloid leukemia or myelodysplasia. Haematologica. 2009;94(10):1449–1452.
37. Schwinger W, Urban C, Ulreich R, et al. The phenotype and treatment of WIP deficiency: literature synopsis and review of a patient with pre-transplant serial donor lymphocyte infusions to eliminate CMV. Front Immunol. 2018;9:2554.
38. Al-Mousa H, Hawwari A, Al-Ghonaium A, et al. Hematopoietic stem cell transplantation corrects WIP deficiency. J Allergy Clin Immunol. 2017;139(3):1039–1040e1034.
39. Gerrits AJ, Leven EA, Frelinger AL
40. Khoreva A, Abramova I, Deripapa E, et al. Efficacy of romiplostim in treatment of thrombocytopenia in children with Wiskott-Aldrich syndrome. Br J Haematol. 2021;192(2):366–374.
41. Lum LG, Tubergen DG, Corash L, Blaese RM. Splenectomy in the management of the thrombocytopenia of the Wiskott-Aldrich syndrome. N Engl J Med. 1980;302(16):892–896.
42. Bach FH, Albertini RJ, Joo P, Anderson JL, Bortin MM. Bone-marrow transplantation in a patient with the Wiskott-Aldrich syndrome. Lancet. 1968;2(7583):1364–1366.
43. Pai SY, Notarangelo LD. Hematopoietic cell transplantation for Wiskott-Aldrich syndrome: advances in biology and future directions for treatment. Immunol Allergy Clin North Am. 2010;30(2):179–194.
44. Kapoor N, Kirkpatrick D, Blaese RM, et al. Reconstitution of normal megakaryocytopoiesis and immunologic functions in Wiskott-Aldrich syndrome by marrow transplantation following myeloablation and immunosuppression with busulfan and cyclophosphamide. Blood. 1981;57(4):692–696.
45. Ochs HD, Lum LG, Johnson FL, Schiffman G, Wedgwood RJ, Storb R. Bone marrow transplantation in the Wiskott-Aldrich syndrome. Complete hematological and immunological reconstitution. Transplantation. 1982;34(5):284–288.
46. Filipovich AH, Stone JV, Tomany SC, et al. Impact of donor type on outcome of bone marrow transplantation for Wiskott-Aldrich syndrome: collaborative study of the International Bone Marrow Transplant Registry and the National Marrow Donor Program. Blood. 2001;97(6):1598–1603.
47. Moratto D, Giliani S, Bonfim C, et al. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980-2009: an international collaborative study. Blood. 2011;118(6):1675–1684.
48. Ozsahin H, Cavazzana-Calvo M, Notarangelo LD, et al. Long-term outcome following hematopoietic stem-cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation. Blood. 2008;111(1):439–445.
49. Elfeky RA, Furtado-Silva JM, Chiesa R, et al. One hundred percent survival after transplantation of 34 patients with Wiskott-Aldrich syndrome over 20 years. J Allergy Clin Immunol. 2018;142(5):1654–1656e1657.
50. Ngwube A, Hanson IC, Orange J, et al. Outcomes after allogeneic transplant in patients with Wiskott-Aldrich syndrome. Biol Blood Marrow Transplant. 2018;24(3):537–541.
51. Ozsahin H, Le Deist F, Benkerrou M, et al. Bone marrow transplantation in 26 patients with Wiskott-Aldrich syndrome from a single center. J Pediatr. 1996;129(2):238–244.
52. Pai SY, DeMartiis D, Forino C, et al. Stem cell transplantation for the Wiskott-Aldrich syndrome: a single-center experience confirms efficacy of matched unrelated donor transplantation. Bone Marrow Transplant. 2006;38(10):671–679.
53. Shekhovtsova Z, Bonfim C, Ruggeri A, et al. A risk factor analysis of outcomes after unrelated cord blood transplantation for children with Wiskott-Aldrich syndrome. Haematologica. 2017;102(6):1112–1119.
54. Friedrich W, Schutz C, Schulz A, Benninghoff U, Honig M. Results and long-term outcome in 39 patients with Wiskott-Aldrich syndrome transplanted from HLA-matched and -mismatched donors. Immunol Res. 2009;44(1–3):18–24.
55. Yue Y, Shi X, Song Z, et al. Posttransplant cyclophosphamide for haploidentical stem cell transplantation in children with Wiskott-Aldrich syndrome. Pediatr Blood Cancer. 2018;65(8):e27092.
56. Fernandes JF, Nichele S, Arcuri LJ, et al. Outcomes after Haploidentical Stem Cell Transplantation with Post-Transplantation Cyclophosphamide in Patients with Primary Immunodeficiency Diseases. Biol Blood Marrow Transplant. 2020;26(10):1923–1929.
57. Shah RM, Elfeky R, Nademi Z, et al. T-cell receptor alphabeta(+) and CD19(+) cell-depleted haploidentical and mismatched hematopoietic stem cell transplantation in primary immune deficiency. J Allergy Clin Immunol. 2018;141(4):1417–1426 e1411.
58. Elfeky R, Shah RM, Unni MNM, et al. New graft manipulation strategies improve the outcome of mismatched stem cell transplantation in children with primary immunodeficiencies. J Allergy Clin Immunol. 2019;144(1):280–293.
59. Laberko A, Sultanova E, Gutovskaya E, et al. Mismatched related vs matched unrelated donors in TCRalphabeta/CD19-depleted HSCT for primary immunodeficiencies. Blood. 2019;134(20):1755–1763.
60. Balashov D, Shcherbina A, Maschan M, et al. Single-center experience of unrelated and haploidentical stem cell transplantation with TCRalphabeta and CD19 depletion in children with primary immunodeficiency syndromes. Biol Blood Marrow Transplant. 2015;21(11):1955–1962.
61. Kharya G, Nademi Z, Leahy TR, et al. Haploidentical T-cell alpha beta receptor and CD19-depleted stem cell transplant for Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2014;134(5):1199–1201.
62. Balashov D, Laberko A, Shcherbina A, et al. A conditioning regimen with plerixafor is safe and improves the outcome of TCRalphabeta(+) and CD19(+) cell-depleted stem cell transplantation in patients with wiskott-aldrich syndrome. Biol Blood Marrow Transplant. 2018;24(7):1432–1440.
63. Balashov D, Laberko A, Shcherbina A, et al. A conditioning regimen with plerixafor is safe and improves the outcome of TCRαβ. Biol Blood Marrow Transplant. 2018;24(7):1432–1440.
64. Barker JN, Boughan K, Dahi PB, et al. Racial disparities in access to HLA-matched unrelated donor transplants: a prospective 1312-patient analysis. Blood Adv. 2019;3(7):939–944.
65. Ochs HD, Filipovich AH, Veys P, Cowan MJ, Kapoor N. Wiskott-Aldrich syndrome: diagnosis, clinical and laboratory manifestations, and treatment. Biol Blood Marrow Transplant. 2009;15(1 Suppl):84–90.
66. Rao K, Amrolia PJ, Jones A, et al. Improved survival after unrelated donor bone marrow transplantation in children with primary immunodeficiency using a reduced-intensity conditioning regimen. Blood. 2005;105(2):879–885.
67. Shin CR, Kim MO, Li D, et al. Outcomes following hematopoietic cell transplantation for Wiskott-Aldrich syndrome. Bone Marrow Transplant. 2012;47(11):1428–1435.
68. Cicalese MP, Aiuti A. Clinical applications of gene therapy for primary immunodeficiencies. Hum Gene Ther. 2015;26(4):210–219.
69. Ferrua F, Twenty-Five AA. Years of gene therapy for ADA-SCID: from bubble babies to an approved drug. Hum Gene Ther. 2017;28(11):972–981.
70. Klein C, Nguyen D, Liu CH, et al. Gene therapy for Wiskott-Aldrich syndrome: rescue of T-cell signaling and amelioration of colitis upon transplantation of retrovirally transduced hematopoietic stem cells in mice. Blood. 2003;101(6):2159–2166.
71. Strom TS, Turner SJ, Andreansky S, et al. Defects in T-cell-mediated immunity to influenza virus in murine Wiskott-Aldrich syndrome are corrected by oncoretroviral vector-mediated gene transfer into repopulating hematopoietic cells. Blood. 2003;102(9):3108–3116.
72. Boztug K, Dewey RA, Klein C. Development of hematopoietic stem cell gene therapy for Wiskott-Aldrich syndrome. Curr Opin Mol Ther. 2006;8(5):390–395.
73. Boztug K, Schmidt M, Schwarzer A, et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med. 2010;363(20):1918–1927.
74. Braun CJ, Boztug K, Paruzynski A, et al. Gene therapy for Wiskott-Aldrich syndrome–long-term efficacy and genotoxicity. Sci Transl Med. 2014;6(227):227ra233.
75. Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302(5644):415–419.
76. Hacein-Bey-Abina S, Garrigue A, Wang GP, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118(9):3132–3142.
77. Gaspar HB, Cooray S, Gilmour KC, et al. Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med. 2011;3(97):97ra79.
78. Wiznerowicz M, Harnessing TD. HIV for therapy, basic research and biotechnology. Trends Biotechnol. 2005;23(1):42–47.
79. Qasim W, Gennery AR. Gene therapy for primary immunodeficiencies: current status and future prospects. Drugs. 2014;74(9):963–969.
80. Galy A, Roncarolo MG, Thrasher AJ. Development of lentiviral gene therapy for Wiskott Aldrich syndrome. Expert Opin Biol Ther. 2008;8(2):181–190.
81. Aiuti A, Biasco L, Scaramuzza S, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341(6148):1233151.
82. Ferrua F, Cicalese MP, Galimberti S, et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: interim results of a non-randomised, open-label, Phase 1/2 clinical study. Lancet Haematol. 2019;6(5):e239–e253.
83. Labrosse R, Chu J, Armant M, et al. Outcome of hematopoietic stem cell gene therapy for Wiskott-Aldrich syndrome. Blood. 2019;134(Supplement_1):4629.
84. Hacein-Bey Abina S, Gaspar HB, Blondeau J, et al. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA. 2015;313(15):1550–1563.
85. Morris EC, Fox T, Chakraverty R, et al. Gene therapy for Wiskott-Aldrich syndrome in a severely affected adult. Blood. 2017;130(11):1327–1335.
86. Sereni L, Castiello MC, Di Silvestre D, et al. Lentiviral gene therapy corrects platelet phenotype and function in patients with Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2019;144(3):825–838.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.