Back to Journals » International Journal of Nanomedicine » Volume 15
HeLa Cell-Derived Paclitaxel-Loaded Microparticles Efficiently Inhibit the Growth of Cervical Carcinoma
Authors Peng J, Zhao J, Zhao Y, Wu P ![]() , Gou L, Fu S
, Gou L, Fu S ![]() , Chen P
, Chen P ![]() , Lu Y, Yang L
, Lu Y, Yang L
Received 21 January 2020
Accepted for publication 30 July 2020
Published 25 August 2020 Volume 2020:15 Pages 6409—6420
DOI https://doi.org/10.2147/IJN.S246659
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yan Shen
Jinxia Peng,1,* Ju Zhao,2,* Yumei Zhao,2 Peng Wu,2 Lantu Gou,3 Shaozhi Fu,2 Ping Chen,2 Yun Lu,2 Linglin Yang2
1Department of Oncology, People’s Hospital of Xindu District, Chengdu, Sichuan 610500, People’s Republic of China; 2Department of Oncology, The Affiliated Hospital of Southwest Medical University Luzhou, Sichuan 646000, People’s Republic of China; 3West China Hospital, Sichuan University, Chengdu, Sichuan 610041, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Linglin Yang Email [email protected]
Aim: Tumor cell-derived microparticles (MP) can function as a targeted delivery carrier for anti-tumor drugs. Here, we aimed to generate paclitaxel-loaded microparticles (MP-PTX) from HeLa cells and examined its therapeutic potential on human cervical carcinoma.
Methods: MP-PTX was generated from HeLa cells by ultraviolet radiation and subsequent centrifugation. The particle size, drug loading rate, and stability of MP-PTX were examined in vitro. Flow cytometry and the MTT assay were performed to test the inhibitory effect of MP-PTX using different cell lines. Immunodeficient mice bearing HeLa cervical carcinoma were treated with 0.9% normal saline, MP, paclitaxel (PTX) (2.5 mg/kg), or MP-PTX (PTX content identical to PTX group) every day for 6 consecutive days. Tumor volume and animal survival were observed. Micro 18F-FDG PET/CT was performed to monitor the therapeutic efficacy. The proliferation activity of cells and microvessel density in tumor tissues were determined by immunohistochemical staining using Ki-67 and CD31, respectively.
Results: Dynamic laser scattering measurements showed that the particle size of MP-PTX was 285.58 ± 2.95 nm and the polydispersity index was 0.104 ± 0.106. And the particle size of MP-PTX was not change at 4°C for at least one week. More than 1% of PTX in the medium could be successfully encapsulated into HeLa cell-derived MP. When compared with PTX, MP-PTX treatment significantly increased apoptosis of tumor cells and reduced their proliferation. In addition, MP-PTX showed lower toxicity to normal human umbilical vein endothelial cells (HUVEC) than PTX. In vivo studies further demonstrated that MP-PTX treatment significantly inhibited the growth of cervical carcinoma, prolonged the survival of tumor-bearing mice, and reduced the toxicity of PTX. Immunohistochemical staining revealed that MP-PTX treatment led to decreased Ki-67 positive tumor cells and decreased microvessel density in tumor tissues.
Conclusion: Our results demonstrated that HeLa-derived MP-PTX significantly enhanced the anti-cancer effects of PTX with reduced toxicity, which may provide a novel strategy for the treatment of cervical carcinoma.
Keywords: cervical carcinoma, HeLa cells, microparticles, paclitaxel, vector
Introduction
Cervical cancer is the second most common type of tumor in women worldwide and is only surpassed by breast cancer.1 According to GLOBOCAN in 2018, the number and deaths of new cases due to cervical cancer are approximately to be 570,000 and 311,000, respectively.1,2 Currently, chemoradiotherapy is the most effective treatment in patients with locally advanced cervical cancer, which significantly improves both overall survival (OS) and disease-free survival (DFS).3,4 However, over 50% of patients with stage III to IV cervical cancer are resistant to therapy, and will finally develop recurrent disease or distant metastasis.5 In addition, severe toxicity, such as bone marrow suppression and impairment of liver and kidney function reduce the compliance of patients to treatments. Therefore, it is of utmost importance to improve the therapeutic effects of chemotherapeutic drugs and reduce the toxic side effects in the treatment of cervical carcinoma.
Paclitaxel is an antineoplastic agent, which has been used for the treatment of various types of cancers. In previous studies, it was demonstrated that paclitaxel can directly kill tumors by inhibiting the formation of microtubules. Recently, increasing evidence has suggested that paclitaxel can also significantly decrease the density of tumor microvessels through reducing the proliferation and migration of endothelial cells, thereby resulting in tumor growth inhibition.6 However, due to its poor water solubility, short half-life in circulation and poor selectivity to tumor cells, the concentration of paclitaxel that reaches the tumor site frequently fails to achieve the effect of killing tumor cells.7 Thus, improving the tumor-targeting ability of paclitaxel not only enhances its anti-tumor effects but also greatly reduces its toxic side effects.
Microparticles (MPs) are small vesicular structures with diameters that range from 100 to 1000 nm, which are released from cells upon stimulation with various physical and chemical factors.8 MPs play an important role in intercellular communication and transduction by transporting various types of molecules. Recent studies have shown that MP can be used as an ideal antitumor drug delivery carrier.8,9 Under UV irradiation or other types of stimulation, the cellular membrane becomes permeable to external molecules, including antitumor drugs. By protruding the cellular membrane, drugs are encapsulated in the MP, then they are released in a budding manner.10–13 The antitumor effects of MP-encapsulated drugs have been evaluated in several types of cancer, including ovarian cancer, liver cancer and breast cancers.14,15 These studies have demonstrated that drug-loaded MPs are efficiently targeted to tumor tissues, increasing the absorption of anticancer drugs by cancer cells, arsing a process known as “cell-eating phenomenon”.16 Moreover, drug-loaded MPs induce the formation of new drug-loaded MPs when they entering into cancer cells, which will kill the tumor cells in a form similar to the domino effect.10
The therapeutic and clinical potential of MP-encapsulated paclitaxel is still poorly documented. In this study, we try to explore the possible of application of paclitaxel-loaded microparticles (MP-PTX) for the treatment of cervical carcinoma. The generated MP-PTX were characterized in terms of their size, surface morphology and stability. In vitro antitumor activity of MP-PTX was evaluated by MTT assay and flow cytometry. We further evaluated the therapeutic effect of MP-PTX on the immunodeficient mice bearing human cervical carcinoma. Our results show that MP-PTX can efficiently induce tumor cell death, reduce tumor growth, and improve the survival of the animals, which suggest their potential application in the clinical treatment of cervical carcinoma.
Materials and Methods
Chemical Reagents, Cells and Animals
Paclitaxel (≥98%) was purchased from Shanghai Huicheng Biological Co, Ltd. (Shanghai, China). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (methyl thiazolyltetrazolium, MTT), NP-40 cracking solution and 0.25% trypsin were purchased from Biyuntian Biological Co, Ltd. (Shanghai, China). Fetal Bovine Serum (FBS) and Penicillin/Streptomycin were supplied by GIBCO (Grand Island, NY, USA). Dulbecco’s Modified Eagle’s Medium (DMEM) was purchased from HyClone (South Logan, UT, USA). Dimethyl sulfoxide (DMSO), methyl alcohol and acetonitrile (HPLC grade) were purchased from Kelong Co, Ltd. (Chengdu, China). Annexin V-FITC/Propidium iodide (PI) kit was purchased from BD Bioscience (San Jose, CA, USA). Phosphate-buffered solution (PBS) was supplied by Shanghai Genechem Co, Ltd. (Shanghai China). Pentobarbital sodium (Sigma-Aldrich, St. Louis, MO, USA) was supplied by Southwest Medical University. The anti-rabbit IgG was purchased from OriGene Biotechnology Co, Ltd. (Beijing China). The Ki-67 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, Dallas, TX, USA). The CD31 antibody and 3.3-diaminobenzidine tetrahydrochloride (DAB) kit were supplied by Abcam (Cambridge, MA, USA).
Human cervical carcinoma cells (HeLa cells) were donated by Sun Yat-sen University (Guangzhou, China) and preserved in the Experimental Medicine Center, at the Affiliated Hospital of Southwest Medical University (Luzhou, China). The use of HeLa cells was approved by the ethics committee of the Affiliated Hospital of Southwest Medical University. Human cervical carcinoma cells (SiHa cells) and human umbilical vein endothelial cells (HUVEC) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in an incubator at 37°C with 5% CO2 humidified atmosphere. Cells were kept in 25 cm2 culture flasks in 5 mL DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cells were passaged three times per week and used for the experiments when in the exponential growth phase.
Female nude mice (BALB/c, 17–21 g, 4–5 weeks of age) were purchased from Chengdu Dashuo Experimental Animal Co, Ltd. (Chengdu, China). Mice were kept at a controlled temperature of 20–22°C with a 50–60% relative humidity, and fed with standard laboratory chow and tap water ad libitum. Guidelines of the institutional animal ethics committee were followed for all in vivo experiments. All animals were handled in strict accordance with good animal practice as defined by the relevant animal welfare bodies. The protocols for animal assays were approved by the Institutional Animal Care and Treatment Committee of Southwest Medical University (Luzhou, China).
Preparation of MP-PTX
A total of 4 × 107 HeLa cells were collected at a density of 1 × 107 cells/mL and irradiated with ultraviolet irradiation (UBV, 300 Jm−2) for 1 hour, followed by paclitaxel (6 mg/mL). Cells were collected after incubation at 37°C and 5% CO2 for 16 hours. MP-PTX were extracted by high-speed centrifugation. Briefly, cell suspensions were collected and centrifuged for 10 min at 600 g. Then, the supernatant was centrifuged for 2 min at 14,000 g to remove debris. Finally, the supernatant was further centrifuged for 60 min at 14,000 g, to pellet MP-PTX.
Morphology and Particle Size of MP-PTX
The morphology of MP-PTX was observed by transmission electron microscopy (TEM; Tecnai G2F30, USA). MP-PTX were thin cut after fixation with 4% paraformaldehyde, then examined by TEM. The particle size of MP-PTX was measured by dynamic laser scattering (DLS, NanoBrook 90 Plus Zeta; Brookhaven, USA) in DMEM. To study MP-PTX stability, MP-PTX were maintained in FBS or DMEM at 4°C and particle size was evaluated by DLS every 24 hours for a week.
Determination of the Paclitaxel Content in MP-PTX
The concentration of PTX in MPs was measured by high-performance liquid chromatography (HPLC, Agilent 1260, USA), using a reverse phase C18 column. The column temperature was maintained at 35.5°C, sample solution was injected at a volume of 20 µL every 5 min for three times. Methyl alcohol/acetonitrile/water (38.1/23.8/38.1, v/v/v) was used as the mobile phase with a flow rate of 1 mL/min. Standard solutions of paclitaxel with different concentrations were filtered through a 220-nm filter and the peak area at 227 nm was measured to establish the standard curve (correlation coefficient R2 = 0.9998). MP-PTX were treated with NP-40 detergent for 5 min and the peak area of paclitaxel was measured by HPLC. The content of paclitaxel in MP-PTX was determined according to the standard curve of paclitaxel.
In vitro Cytotoxicity Test
To study the cytotoxicity of MP-PTX in vitro and compare with paclitaxel alone, the MTT assay was used to evaluate the proliferation activity. In addition, the cytotoxicity of MP-PTX against other different cell lines in vitro was evaluated by an apoptosis study. In brief, HeLa cells were seeded in 96-well plates with 100 µL cell culture medium per well at a density of 1 × 105 cells/mL. Cells were treated with MP (extracted from 4 × 107 HeLa cells), PTX (0.1 µg) and MP-PTX (MP loaded with 0.1 µg paclitaxel). Then, cells were cultured at 37°C with 5% CO2, and viability was measured by MTT at 24 h, 48 h, and 72 h. The absorbance (OD value) was measured at 570 nm in a microplate reader (Bio-RAD, USA), and the survival rate of the cells was calculated as follows: Cell viability = (experimental group OD-blank group OD)/(control group OD-blank group OD) × 100%. In addition, apoptosis was analyzed by a double-staining method using Annexin V FITC/PI labeling according to the manufacturer’s instructions. Briefly, cells were seeded in six-well plates at a density of 1 × 106 cells per well. After adherence, cells were treated with MP (extracted from 4 × 107 HeLa cells), PTX (0.1 µg), and MP-PTX (MP loaded with 0.1 µg paclitaxel) for 48 h. After centrifugation at 2000 g for 5 min, cells were suspended, washed three times with cold PBS, and stained with propidium iodide (PI) and FITC-labeled Annexin in the dark for 15 min at room temperature. Cells were analyzed by flow cytometry (BD FACSVerse, Piscataway, NJ, USA).
Toxicity Stability of MP-PTX in vitro
MP-PTX were maintained in FBS or DMEM at 4°C for 1 week, then, the cytotoxic activity of MP-PTX was measured by flow cytometry. In brief, 1 × 106 HeLa cells were seeded in 6-well plates, and cultured in 1) DMEM+FBS, 2) DMEM+FBS+fresh MP-PTX (just prepared MP-PTX), 3) DMEM+FBS+MP-PTX in DMEM (MP-PTX maintained in DMEM after 1 week), 4) DMEM+FBS+MP-PTX in FBS (MP-PTX maintained in FBS after 1 week). After 48 hours, HeLa cells were collected to determine the apoptotic rate by flow cytometry (Becton Dickinson, CA, USA).
Anti-Tumor Efficacy of MP-PTX in vivo
In this study, BALB/c nude mice (17–21 g, 4–5 weeks of age) were used to establish cervical cancer models. Mice were subcutaneously injected with 100 µL of DMEM containing about 1 × 106 HeLa cells into the right flank. Tumors were allowed to grow until they reached a volume of 200 mm3 before initiating the treatment. The tumor volume was calculated according to the following formula: tumor volume (V) = 0.5 × length × width2. Nude mice were randomly divided into four groups (n = 6) and subjected to tail vein intravenous injection with 0.9% saline (control), MP, PTX (0.5 mg/mL, 2.5 mg/kg), and MP-PTX (PTX content identical to PTX group) once a day for six consecutive days. The tumor volume of each mouse was calculated every 2 days. On the seventh day, half of the mice per group were euthanized for the detection of creatinine and aminotransferase levels in peripheral blood. The remaining mice were used to record the changes in tumor volume change and survival time.
Tumor metabolic studies in the control, MP, PTX, and MP-PTX groups, were performed by micro PET/CT scans and image analysis using Inveon micro PET/CT (Siemens, Munich, Germany) animal scanner, one day after the end of treatment. An irregular region of interest (ROI), covering the entire tumor, was drawn by two experienced nuclear medicine physicians. Contralateral paraspinal muscles were also examined to draw the ROIs. The 18F-FDG maximum standard uptake values (SUV) of each lesion were obtained from the selected ROI and compared to the SUVs of the contralateral paraspinal to calculate the tumor/muscle (T/M) ratio. Glucose metabolism was measured by the T/M ratio.
Ki-67 and CD31 Immunohistochemistry
Immunohistochemistry was used to evaluate tumor proliferation and microangiogenesis, by Ki-67 and CD31, respectively. Tumor tissues were fixed in 4% formaldehyde and embedded in paraffin, and 4-µm thick sections were cut for immunohistochemical analysis using Ki-67 and CD31. Rabbit monoclonal anti-Ki67 or anti-CD31 antibody was used as a primary antibody and incubated in the appropriate secondary antibodies, goat anti-rabbit IgG, conjugated with horseradish peroxidase. After incubation, sections were exposed to DAB and then counterstained in hematoxylin. Sections were analyzed by a microscope at 400× (Leica TE2000-S Microscope, Tokyo, Japan) after being counter-stained with hematoxylin. The expression of Ki-67 and CD31 was evaluated by three experienced pathologists. In each section, Ki-67 positive cells were counted in five randomly selected regions and the ratio between Ki-67-positive cells and total cells was determined. Besides, by calculating the area percentage of positive expression of CD31, five high power visual fields (×400) were randomly selected for each tumor sample.
Statistical Analysis
Statistical analysis was performed with SPSS 17.0 software (Chicago, IL). The Kaplan–Meier method was used to plot the survival curve, and the results were presented as the survival time and a 95% confidence interval compared by the Log rank test. We used the Student’s t-test for two groups or one-way ANOVA for multiple groups. Data are presented as the mean ± SD. Statistical significance was considered at p < 0.05.
Results
MP-PTX Physicochemical Characterization
The specific steps for extracting MP-PTX are shown in Figure 1A. In this study, the morphological characteristics and vesicular structure of MP-PTX (Figure 1B) and MP (Figure 1C) were observed by TEM. The average diameter of MP-PTX was 285.58 ± 2.95 nm, as determined by DLS. The polydispersity index was 0.104 ± 0.106 (Figure 1D). The particle size of MP-PTX did not significantly alter when kept in PBS or DMEM solution for 7 days (Figure 1E). Moreover, we did not observe any aggregates, precipitates or turbidity were not observed by the naked eye, suggesting high physical stability of MP-PTX.
 |
Figure 1 Preparation and morphology of MP-PTX. (A) Protocol for extracting MP-PTX from HeLa cells. (B) Morphology of MP-PTX observed by TEM (magnification ×12,000). (C) Morphology of MP observed by TEM (magnification ×12,000). (D) The particle size of MP-PTX in DMEM was measured by DLS. (E) Particle size change of MP-PTX in FBS and DMEM. The particle size of the MP-PTX did not significantly change in 7 days. |
Determination of the Paclitaxel Content in MP-PTX
We used HPLC to determine the content of paclitaxel in MP-PTX. The chromatogram of paclitaxel standard samples is shown in Figure 2A. The retention time is ~2.7 min. The standard curve of paclitaxel is shown in Figure 2B: Y (peak area) = 41.464X (paclitaxel concentration) + 195.21, R2 = 0.9998. Quantitative HPLC of paclitaxel in MP-PTX is shown in Figure 2C. The retention time was the same as that of paclitaxel standard samples. Figure 2D indicates the paclitaxel content in MP-PTX extracted from 4 × 107 HeLa cells treated with different concentrations of paclitaxel. UV-irradiated HeLa cells were incubated with different concentrations of paclitaxel. Our results showed that for a content of paclitaxel within 250 µg, over 1% of paclitaxel was loaded into the microparticles.
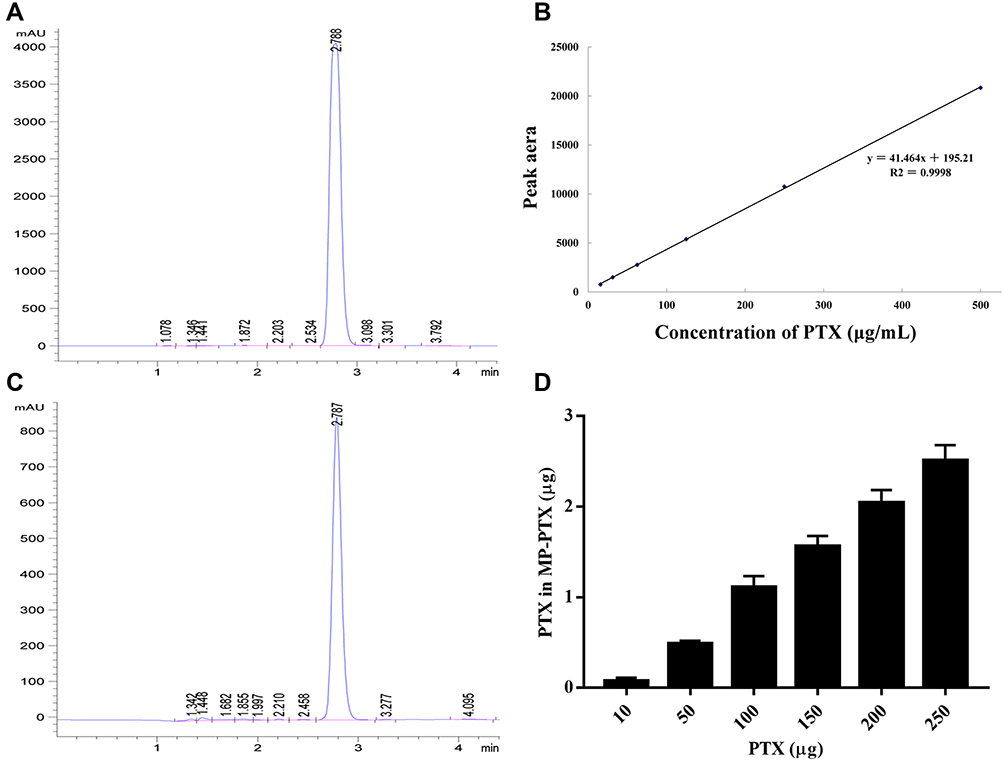 |
Figure 2 Determination of the paclitaxel content in MP-PTX. (A) Quantitative HPLC of PTX standard sample, the retention time is ~2.7 min. (B) The standard curve of PTX: Y = 41.464X + 195.21 and R2 = 0.9998. (C) Quantitative HPLC of PTX in MP-PTX, the retention time of MP-PTX is same as PTX. (D) Actual PTX content in MP-PTX extracted from 4 × 107 HeLa cells incubated with different concentration of PTX. More than 1% of PTX was loaded into the microparticles. |
Antitumor Activity and Toxicity Stability of MP-PTX in vitro
The results from cell viability experiments with MTT are shown in Figure 3A. The percentage of live cells of the MP-PTX group at 4872 h (58.59 ± 2.11%, 27.54 ± 3.26%, respectively) was significantly lower compared to that in the PTX group (73.93 ± 2.42% at 48 h, 47.73 ± 2.57% at 72 h, P < 0.05), the control group (93.63 ± 1.82% at 48 h, 95.08 ± 1.75% at 72 h, P < 0.05) and in the MP group (93.13 ± 1.61% at 48 h, 94.25 ± 1.32% at 72 h, P < 0.05) which indicated that MP-PTX was more efficient than PTX or MP alone. Our results suggested that the antitumor activity of MP-PTX is time-dependent. Furthermore, the percentage of live cells in the MP group was not significantly different from the control group at 24, 48, and 72 h, thereby suggesting that MP shows no cytotoxicity alone, but could enhance the uptake of paclitaxel by tumor cells, thus promoting tumor cell death.
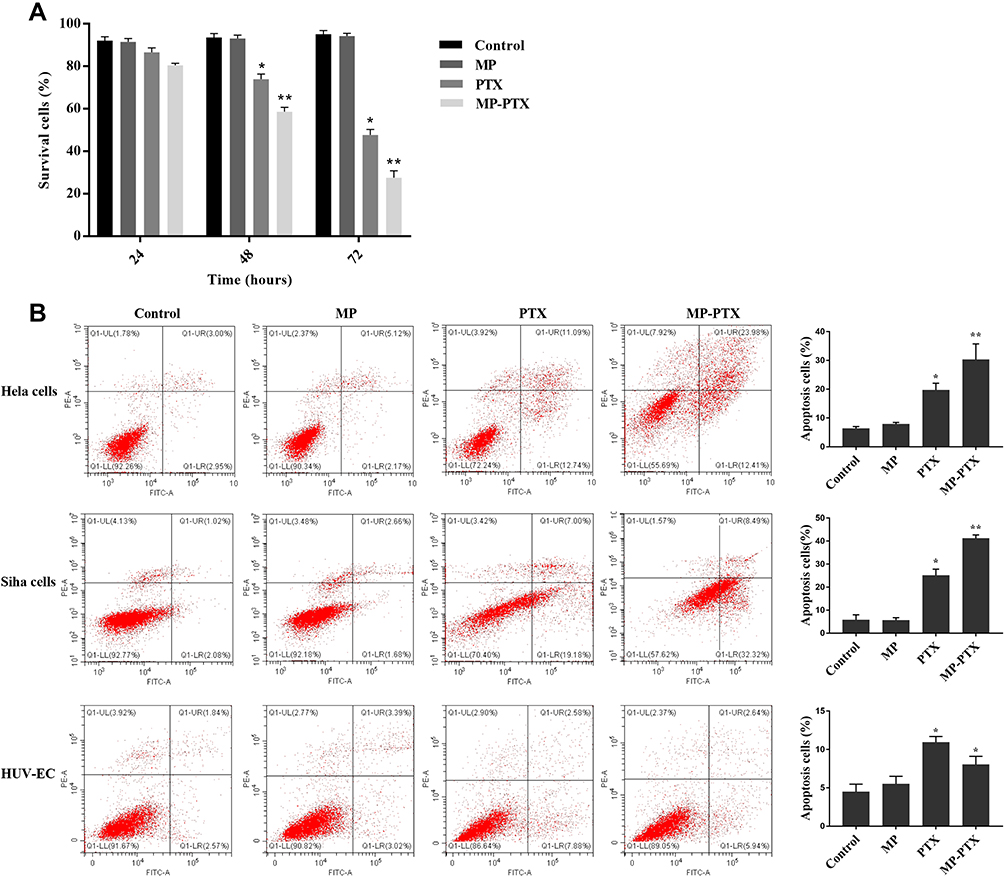 |
Figure 3 Antitumor activity of MP-PTX in vitro. (A) The MTT assay was used to determine the cytotoxic effects in control, MP, PTX, and MP-PTX group at 24h, 48h and 72h. PTX and MP-PTX groups showed cytotoxicity to Hela cells when compared to the control group, and the antitumor activity of MP-PTX was better than that of PTX alone. (B) The cytotoxicity of MP-PTX against other cervical cancer cells and non-cancer cells in vitro for 48 h was evaluated by the apoptosis study. PTX and MP-PTX groups showed cytotoxicity to Hela cells, SiHa cells, and HUV-EC when compared to the control group. The cytotoxicity of MP-PTX to SiHa cells was stronger than that of PTX, and the results were the same as that of Hela cells. The cytotoxicity of MP-PTX to HUVEC was weaker than that of PTX alone (*p < 0.05, **p < 0.01). |
The cytotoxicity of MP-PTX against other cervical cancer cells and non-cancer cells in vitro was evaluated by an apoptosis study (Figure 3B). Apoptosis in HeLa cells, the PTX group (19.70 ± 2.37%), and the MP-PTX group (30.25 ± 5.49%) was significantly higher compared to the control group (6.33 ± 0.70%, p < 0.05), and that of the MP-PTX group was higher compared to the PTX group (p < 0.05). The antitumor activity of MP-PTX was better than PTX alone in HeLa cells, similar as shown by the MTT assay. Regarding apoptosis in SiHa cells, no significant differences were observed between the control group (5.71 ± 2.28%) and the MP group (5.55 ± 1.16%, P > 0.05). As for HeLa cells, the cytotoxicity of the MP-PTX group (41.147 ± 1.55%) in SiHa cells was stronger than that of the PTX group (25.10 ± 2.71%, p < 0.05). These data showed that HeLa cell-derived MP-PTX remain valid for other cervical cancer cell lines. The apoptosis rate of the PTX group (10.93 ± 0.76%) and the MP-PTX group (8.01 ± 1.09%) was significantly higher than that of the control group (4.46 ± 1.02%) and the MP group (5.50 ± 1.01%) (p < 0.05) in HUVEC. However, the cytotoxicity of the MP-PTX group in HUV-EC was lower than that of the PTX group (p < 0.05), which indicated that the toxicity of paclitaxel decreased after encapsulation with microparticles.
The Toxicity Stability of MP-PTX in vitro
Figure 4A shows the cell apoptosis in control, fresh MP-PTX, MP-PTX in DMEM 7 days later, and MP-PTX in FBS 7 days later group. Cellular apoptosis was significantly increased in HeLa cells in the fresh MP-PTX group (24.86 ± 0.99%), MP-PTX maintained in FBS after 1 week group (23.70 ± 0.60%) and MP-PTX maintained in DMEM after 7 days group (24.06 ± 1.09%), compared to the control group (6.63 ± 0.69%) (p < 0.05) (Figure 4B). Furthermore, no significant differences were observed in apoptosis levels among the three MP-PTX groups, which indicated that the antitumor activity of MP-PTX did not decrease after 7 days of storage in FBS and in DMEM. MP-PTX has toxicity stability.
 |
Figure 4 The toxicity stability of MP-PTX in vitro. (A) Cell apoptosis in control, fresh MP-PTX, MP-PTX in DMEM 7 days later, and MP-PTX in FBS 7 days later. (B) Bar chart representing the percentage of apoptotic cells in each group. Three MP-PTX groups showed cytotoxicity to Hela cells when compared to control groups. The toxicity of MP-PTX in DMEM and FBS 7 days later did not change compared to fresh MP-PTX (**p < 0.01). |
In vivo Anti-Tumor Effects of MP-PTX
To evaluate the efficacy of MP-PTX on HeLa xenograft in mice, tumor volume and survival time were calculated and plotted every 2 days. After 20 days, the tumor volume of MP-PTX group was significantly lower than that of the PTX group (P < 0.05), MP group (P < 0.05) and control group (P < 0.05), and differences were observed at least until day 30 (Figure 5A). We did not observe statistically significant differences in the median survival time among MP-PTX, PTX, MP and control groups. However, albeit non-significant, mice treated with MP-PTX survived longer than in any other group (Figure 5B). This might be related to the combination of MP with PTX, which may enhance PTX activity, but is not so significant.
 |
Figure 5 Anti-tumor effect of MP-PTX in vivo. (A) Tumor volume curve in control, MP, PTX and MP-PTX-treated mice. Data are shown as the means ± SD (n = 6). (B) The Kaplan–Meier survival curve of control, MP, PTX and MP-PTX-treated mice. The anti-tumor effect upon MP-PTX treatment was better compared to that of other treatments. (C) 18F-FDG PET/CT imaging to explore the early therapeutic effect of MP-PTX, MP and PTX alone in nude mice. The white circle delineated the tumor tissue. (D) Bar chart representing the level of glucose metabolism in control, MP, PTX and MP-PTX. PTX group and MP-PTX group showed lower glucose metabolism compared to the control group. MP-PTX was more efficient than PTX in reducing glucose metabolism. (T) Tumor tissue; (M) Muscle; T/M = Tumor SUVmax/Muscle SUVmax (*P < 0.05, **p < 0.01). |
To study the early efficiency of MP-PTX treatment, we evaluated the glucose metabolism of mice in control, MP, PTX, and MP-PTX group (Figure 5C). T/M values (T/M = Tumor SUVmax/Muscle SUVmax) in each group were 2.81 ± 0.0628 (control), 2.71 ± 0.131 (MP), 2.12 ± 0.112 (PTX) and 1.10 ± 0.142 (MP-PTX). Mice from the MP-PTX group showed the lowest T/M (p < 0.05) (Figure 5D), showing that MP-PTX was more efficient than PTX in reducing glucose metabolism.
Effects of MP-PTX on Liver and Kidney
Our results confirmed that MP-PTX has potential as an antitumor agent. We investigated the side effects of MP-PTX in our xenograft mouse model, as well as in control mice. Our results showed that the levels of creatinine (48.72 ± 3.50 μmol/mL) (Figure 6A) and transaminase (35.60 ± 1.56 U/L) (Figure 6B) in the MP-PTX-treated group were not significantly increased when compared to the control group (49.73±5.17 μmol/mL, P > 0.05, respectively; 37.3 ± 1.12 U/L, P > 0.05, respectively). However, the levels of creatinine (81.31 ± 5.73 μmol/mL) and transaminase (70.43 ± 2.22 U/L) in the PTX group were higher than in the control group (P < 0.05). These findings indicated that MP-PTX have less impact on liver and renal function when compared to PTX alone and may be safely used in clinical.
 |
Figure 6 Side effects in liver and kidney by MP, PTX and MP-PTX. (A) Creatinine level in peripheral blood. (B) Glutamic-pyruvic aminotransferase level in peripheral blood. The toxicity of MP-PTX to liver and kidney was similar as control group, but the toxicity of PTX to liver and kidney was higher than control group. The toxicity of MP-PTX to liver and kidney was reduced compared to that PTX alone (*P < 0.05). |
Immunohistochemistry
Ki-67 is a marker of cell proliferation. As shown in Figure 7A, Ki-67 positive cells were observed in control, MP, PTX, and MP-PTX. A lower number of Ki-67 positive cells were observed in the MP-PTX group (24.40 ± 2.24%) compared to the PTX group (48.92 ± 4.32%, P < 0.05), control group (71.64 ± 1.28%, P < 0.05), or MP group (72.99 ± 1.78%, P < 0.05) (Figure 7B), indicating that MP-PTX could effectively inhibit tumor proliferation. CD31, also known as platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31), can be a marker of MVD. The microvessel percentage of the MVD area in the MP-PTX group (10.07 ± 2.47%) was significantly lower compared to control group (27.58 ± 4.15%, P < 0.05) and MP group (26.42 ± 2.56%, P < 0.05) (Figure 7C). However, no significant differences were observed between MP-PTX and PTX groups (12.52 ± 4.53%, P > 0.05) (Figure 7D). Our results demonstrated that MP-PTX significantly inhibited microvessel formation; however, no significant differences were observed between the MP-PTX group and the PTX group in inhibition of tumor micro vascularization. Thus, these findings indicated that the drug delivery form of paclitaxel loaded on microparticles did not reduce the ability of paclitaxel to inhibit tumor micro vascularization.
 |
Figure 7 Effects of MP-PTX in tumor proliferation and angiogenesis. (A) Ki-67 (200×) and CD31 (400×) immunohistochemistry in tumor tissue from control, MP, PTX and MP-PTX-treated mice. (B) Quantitative analysis of Ki-67 expression in tumors from control, MP, PTX and MP-PTX treated mice, PTX group and MP-PTX group showed lower number of Ki-67 positive cells compare to control group. MP-PTX effectively inhibit tumor proliferation compared to PTX alone. (C) Quantitative analysis of CD31 expression in tumors from control, MP, PTX and MP-PTX treated mice, PTX group and MP-PTX group showed smaller number of CD31 positive-neovascularized capillaries compare to control group. MP-PTX has a similar effect as PTX in inhibiting tumor microvessel formation (*P < 0.05, **p < 0.01). |
Discussion
Currently, combination chemoradiotherapy is the standard treatment in patients with locally advanced cervical carcinoma. Still, more than 50% of patients fail to reach effective treatment after combination chemoradiotherapy due to drug resistance, which results in disease recurrence, metastasis, and ultimately death.4 The related side effects such as liver and kidney insufficiency are also linked to the poor prognosis of patients. Therefore, the focus of this study was to explore novel effective anti-cancer agents that can improve patients’ treatment efficiency and reduce therapy-related side effects.
In this study, HeLa cells irradiated by ultraviolet radiation were incubated with paclitaxel, and paclitaxel-loaded microparticles (MP-PTX) were collected by high-speed centrifugation. The MP-PTX prepared in our study showed a single uniform spherical distribution under electron microscopy. The particle size in DMEM solution was 285.58 ± 2.95 nm, which was in accordance with other studies.14 Notably, we observed a sustained stability of the MP-PTX, with no significant changes in the particle size in FBS and DMEM solutions for up to one week, making it suitable for transportation and storage.
We performed in vitro evaluation of MP-PTX antitumor properties, using the MTT assay and a cell apoptosis assay. Our findings showed that MPs alone did not show toxicity to cancer cells and that MP-PTX could inhibit the growth of cervical cancer cells more effectively than PTX alone, in HeLa or SiHa cells. In addition, the results of HUV-EC apoptosis demonstrated that MP reduce PTX cytotoxicity to normal cells. These results suggested that encapsulation could enhance the toxic effect of chemotherapeutic drugs against cancer cells. The current hypothesis is that MP might have affinity for tumors (tumoritaxis), thereby improving chemotherapeutic drug delivery to the tumor site directionally, therefore increasing the concentration of drugs in the tumor cells.17,18 In addition, cancer cell-derived microparticles are highly compatible with cancer cells, and microparticles may be able to integrate into the membrane of cancer cells, thus inhibiting the efflux of chemotherapeutic drugs.19,20
To compare the antitumor effect of MP-PTX and PTX alone, we established a xenograft mouse model of cervical cancer. MP-PTX significantly delayed the growth of tumors when compared with the same dose of PTX alone. In previous studies, it was confirmed that 18F-FDG uptake in cancer tissues reflects the glucose metabolism and early drug therapeutic effects, which are closely related to disease progression.6 Our findings showed that MP-PTX could significantly reduce 18F-FDG uptake in cancer tissues when compared with PTX alone. However, in terms of survival time, the MP-PTX group and PTX group did not significantly prolong overall survival, which may be because combination chemoradiotherapy was the standard treatment of cervical cancer.2 Only paclitaxel, a chemotherapy drug, is far from enough to prolong survival.
Ki-67 protein is a nuclear protein, significantly associated with cell proliferation, with a role in mitosis and in maintaining DNA structure. Therefore, the expression of Ki-67 is a marker of cell proliferation.21,22 Our proliferation results, using the Ki-67 marker, support our observations of tumor growth retardation and reduced glucose metabolism in the MP-PTX treated mice. Our results showed that of the expression of Ki-67 in the MP-PTX group was significantly lower when compared to that in the other groups. These results indicate that the antitumor effect of MP-PTX on cervical cancer is associated with inhibition of tumor cell proliferation. CD31 participates in the formation of endothelial cells and is used to evaluate the formation of microvessels in tumors. Although MP-PTX and PTX alone could significantly reduce the ratio of CD31 positive microvessel area, no significant differences were observed between the two groups. It was previously reported that drug-loaded microparticles could inhibit angiogenesis of tumors.9,23 We did not observe this phenomenon in our study, which may be due to the different drug-loaded microparticles on the microvasculature in different types of tumors. We also found that MP-PTX did not affect liver and kidney function, which was in line with previous studies.14 We hypothesize that cancer cell-derived microparticles may display strong affinity for tumor cells. Compared with traditional nanomaterials, microparticles did not cause oxidative stress or allergic reactions.24,25 More importantly, the particle size of the microparticles was between 100 and 1000 nm, and the permeability of normal capillaries is limited to about 5~8 nm, while tumor-related capillaries with a diameter of 100~780 nm have high permeability.26–28 This facilitates drug transport through cancer cell-derived microparticles to the tumor site without causing damage to other tissues through capillaries.
Our results showed that MP-PTX could enhance the antitumor effects of PTX, but the microparticle composition was complex and the metabolic process of drug-loaded microparticles in vivo remains unclear. In addition, the drug loading rate of MP-PTX is relatively low, and it is necessary to solve the problem of preparation in large scale for clinical application. Therefore, clinical application of cancer-cell-derived microparticles still faces many problems. In summary, in this study, we developed new HeLa-derived MP-PTX. Our MP-PTX showed chemical and functional stability over time, and exhibited higher cytotoxic activity than PTX alone, both in vitro and in vivo. Moreover, MP-PTX treatment resulted in significantly reduced liver and kidney toxicity when compared to PTX alone, suggesting that our newly developed microparticles may enhance PTX function, with concomitant reduction of its associated side effects. Therefore, we suggest that MP-PTX is a promising anti-tumor agent candidate in the context of cervical carcinoma.
Conclusions
The data presented in our study showed that HeLa cell-derived MP-PTX has antitumor effect in vitro and in vivo, and its efficacy is stronger than that of PTX alone. The biological mechanism was related to targeted inhibition of tumor cell proliferation and reduction of the glucose metabolism of cancer cells. Microparticles are natural cellular products, whose main function is to carry molecules intra and intercellularly.9 We used microparticles to carry chemotherapeutic drugs, increase the anti-tumor effect, and reduce its side effects on liver and kidney. Therefore, microparticles may provide a novel strategy for the treatment of cervical carcinoma.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Freddie B, Jacques F, Isabelle S, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Marth C, Landoni F, Mahner S, et al. Cervical cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Supplement_4):iv262. doi:10.1093/annonc/mdy160
3. Mélanie D, Antonin L, Michele M, et al. The combination of the antiviral agent cidofovir and anti-EGFR antibody cetuximab exerts an antiproliferative effect on HPV-positive cervical cancer cell lines’ in-vitro and in-vivo xenografts. Anticancer Drugs. 2013;24(6):599–608. doi:10.1097/CAD.0b013e3283612a71
4. Yee Grace PC, de Souza P, Khachigian LM. Current and potential treatments for cervical cancer. Curr Cancer Drug Targets. 2013;13(2):205–220. doi:10.2174/1568009611313020009
5. Alina S, Richard P, Ulrik FL, et al. Image guided brachytherapy in locally advanced cervical cancer: improved pelvic control and survival in RetroEMBRACE, a multicenter cohort study. Radiother Oncol. 2016;120(3):428–433. doi:10.1016/j.radonc.2016.03.011
6. Yu YX, Xu S, You H, et al. In vivo synergistic anti-tumor effect of paclitaxel nanoparticles combined with radiotherapy on human cervical carcinoma. Drug Deliv. 2017;24(1):75–82. doi:10.1080/10717544.2016.1230902
7. Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25(18):2677–2681. doi:10.1091/mbc.e14-04-0916
8. Bence GR, Károly M, Eva P, et al. Detection and isolation of cell-derived microparticles are compromised by protein complexes resulting from shared biophysical parameters. Blood. 2011;117(4):e3948. doi:10.1182/blood-2010-09-307595
9. Ratajczak J, Wysoczynski M, Hayek F, et al. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia. 2006;20(9):1487–1495. doi:10.1038/sj.leu.2404296
10. Mause SF, Weber C. Microparticles: protagonists of a novel communication network for intercellular information exchange. Circ Res. 2010;107:1047–1057. doi:10.1161/CIRCRESAHA.110.226456
11. Tual-Chalot S, Leonetti D, Andriantsitohaina R, Martinez MC. Microvesicles: intercellular vectors of biological messages. Mol Interv. 2011;11:88–94. doi:10.1124/mi.11.2.5
12. The´ry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–593. doi:10.1038/nri2567
13. Boilard E, et al. Platelets amplify inflammation in arthritis via collagen- dependent microparticle production. Science. 2010;327:580–583. doi:10.1126/science.1181928
14. Tang K, Zhang Y, Zhang H, et al. Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat Commun. 2012;3:1282. doi:10.1038/ncomms2282
15. Ma J, Zhang Y, Tang K, et al. Reversing drug resistance of soft tumor-repopulating cells by tumor cell-derived chemotherapeutic microparticles. Cell Res. 2016;26(6):713–727. doi:10.1038/cr.2016.53
16. Abrahams VM, Straszewski-Chavez SL, Seth G, Gil M. First trimester trophoblast cells secrete Fas ligand which induces immune cell apoptosis. Mol Hum Reprod. 2004;10(1):55–63. doi:10.1093/molehr/gah006
17. Higgins Christopher F. Multiple molecular mechanisms for multidrug resistance transporters. Nature. 2007;446(7137):749–757. doi:10.1038/nature05630
18. Haber M, Henderson MJ, Norris MD, et al. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer. 2010;10(2):147–156. doi:10.1038/nrc2789
19. Keon J, Antoniw J, Carzaniga R, et al. Transcriptional adaptation of Mycosphaerella graminicola to programmed cell death (PCD) of its susceptible wheat host. Mol Plant Microbe Interact. 2007;20(2):178–193. doi:10.1094/MPMI-20-2-0178
20. Barnard AS. Nanohazards. Knowledge is our first defence. Nat Mater. 2006;5(4):245–248. doi:10.1038/nmat1615
21. Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133(4):1710–1715.
22. Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182(3):311–322. doi:10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9
23. Gong J, Jaiswal R, J-M M, Combes V, Grau GER, Bebawy M. Microparticles and their emerging role in cancer multidrug resistance. Cancer Treat Rev. 2012;38(3):226–234. doi:10.1016/j.ctrv.2011.06.005
24. Lewinski N, Colvin V, Drezek R. Cytotoxicity of nanoparticles. Small. 2008;4:26–49. doi:10.1002/smll.200700595
25. Zhiping Z, Si-Shen F. Nanoparticles of poly(lactide)/vitamin E TPGS copolymer for cancer chemotherapy: synthesis, formulation, characterization and in vitro drug release. Biomaterials. 2006;27(2):262–270. doi:10.1016/j.biomaterials.2005.05.104
26. Hu YL, Gao JQ. Potential neurotoxicity of nanoparticles. Int J Pharm. 2010;394(12):115–121. doi:10.1016/j.ijpharm.2010.04.026
27. Shvedova AA. Kagan VE and Fadeel B. Close encounters of the small kind: adverse effects of man-made materials interfacing with the nano-cosmos of biological systems. Annu Rev Pharmacol Toxicol. 2010;50:63–88. doi:10.1146/annurev.pharmtox.010909.105819
28. Karplus M, Kuriyan J. Molecular dynamics and protein function. Proc Natl Acad Sci U S A. 2005;102(19):6679–6685. doi:10.1073/pnas.0408930102
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.