Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
Guidelines for acute management of hyperammonemia in the Middle East region
Authors Alfadhel M ![]() , Al Mutairi F, Makhseed N, Al Jasmi F, Al-Thihli K, Al-Jishi E, Al-Sayed M, Al-Hassnan ZN, Al Murshedi F, Häberle J, Ben-Omran T
, Al Mutairi F, Makhseed N, Al Jasmi F, Al-Thihli K, Al-Jishi E, Al-Sayed M, Al-Hassnan ZN, Al Murshedi F, Häberle J, Ben-Omran T
Received 26 July 2015
Accepted for publication 23 December 2015
Published 31 March 2016 Volume 2016:12 Pages 479—487
DOI https://doi.org/10.2147/TCRM.S93144
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Majid Alfadhel,1,2 Fuad Al Mutairi,1,2 Nawal Makhseed,3 Fatma Al Jasmi,4 Khalid Al-Thihli,5 Emtithal Al-Jishi,6 Moeenaldeen AlSayed,7 Zuhair N Al-Hassnan,7,8 Fathiya Al-Murshedi,5 Johannes Häberle,9 Tawfeg Ben-Omran10
Middle East Hyperammonemia and Urea Cycle Disorders Scientific Group (MHUSG)
1Department of Pediatrics, Division of Genetics, 2King Saud bin Abdulaziz University for Health Sciences, King Abdulaziz Medical City, Riyadh, Saudi Arabia; 3Department of Pediatrics, Jahra Hospital, Ministry of Health, Jahra City, Kuwait; 4Department of Pediatric, College of Medicine and Health Sciences, United Arab Emirates University, Al-Ain, United Arab Emirates; 5Genetic and Developmental Medicine Clinic, Sultan Qaboos University Hospital, Muscat, Sultanate of Oman; 6Salmaniya Medical Complex, Arabian Gulf University, Manama, Bahrain; 7Department of Medical Genetics, King Faisal Specialist Hospital & Research Center, 8The National Newborn Screening Program, Ministry of Health, Riyadh, Saudi Arabia; 9Department of Pediatrics, Division of Metabolism and Children’s Research Center, University Children’s Hospital Zurich, Zurich, Switzerland; 10Division of Clinical and Metabolic Genetics, Department of Pediatrics, Hamad Medical Corporation, Doha, Qatar
Background: Hyperammonemia is a life-threatening event that can occur at any age. If treated, the early symptoms in all age groups could be reversible. If untreated, hyperammonemia could be toxic and cause irreversible brain damage to the developing brain.
Objective: There are major challenges that worsen the outcome of hyperammonemic individuals in the Middle East. These include: lack of awareness among emergency department physicians about proper management of hyperammonemia, strained communication between physicians at primary, secondary, and tertiary hospitals, and shortage of the medications used in the acute management of hyperammonemia. Therefore, the urge to develop regional guidelines is extremely obvious.
Method: We searched PubMed and Embase databases to include published materials from 2011 to 2014 that were not covered by the European guidelines, which was published in 2012. We followed the process of a Delphi conference and involved one preliminary meeting and two follow-up meetings with email exchanges between the Middle East Hyperammonemia and Urea Cycle Disorders Scientific Group regarding each draft of the manuscript.
Results and discussion: We have developed consensus guidelines based on the highest available level of evidence. The aim of these guidelines is to homogenize and harmonize the treatment protocols used for patients with acute hyperammonemia, and to provide a resource to not only metabolic physicians, but also physicians who may come in contact with individuals with acute hyperammonemia.
Conclusion: These suggested guidelines aim to ease the challenges faced by physicians dealing with acute hyperammonemia in the region. In addition, guidelines have demonstrated useful collaboration between experts in the region, and provides information that will hopefully improve the outcomes of patients with acute hyperammonemia.
Keywords: hyperammonemia, inborn errors of metabolism, urea cycle, acute management, ammonia, Middle East
Introduction
Acute hyperammonemia is defined as elevated plasma ammonia levels associated with muscular hypotonia, seizures, vomiting, and impaired consciousness.1 The clinical features are heterogeneous depending on the age of the patient and on the type and severity of the underlying cause.2 The early symptoms in all age groups are loss of appetite and vomiting, which could be reversible if recognized and treated early. In newborns, common early symptoms are poor feeding, vomiting, lethargy, hyperventilation resulting often in respiratory alkalosis, and irritability that rapidly progresses to seizures, deep coma, and even death if not urgently treated. In infants, vomiting mimics pyloric stenosis, cow milk intolerance, or gastroenteritis. In older children and adults, vomiting, protein aversion, ataxia, confusion, disorientation, hallucinations, or abnormal behavior point to central nervous system or psychiatric disorders.2 If untreated, severe hyperammonemia will be neurotoxic and will cause irreversible brain damage both to the developing as well as to the mature brain.1–5 Hyperammonemic encephalopathy is associated with high mortality rates.6,7 The total duration of a hyperammonemic coma and the extent of hyperammonemia are the most relevant prognostic factors and are negatively correlated with the patient’s neurological outcome.6–8 Therefore, prompt identification and treatment of hyperammonemia are vital to optimize the outcome.
In the Middle East, the prevalence of urea cycle disorders and organic acidemias, which are the major causes of hyperammonemia, is probably higher than in other parts of the world.9–12 This is most likely due to the high rate of consanguinity leading to increased autosomal recessive disorders in the region.13–16 In addition to the high prevalence, there are several major challenges that must be considered as risk factors for the outcome of hyperammonemic individuals in the Middle East. These include lack of awareness of health care professionals about prompt diagnosis and adequate management of hyperammonemia, strained communication between physicians at primary, secondary, and tertiary hospitals, and lack of knowledge and availability of the diagnostic tools and medications required in the acute management of hyperammonemia. Finally, the recognition of late onset and milder cases diagnosed in adulthood would require the particular alertness among internists and adult intensive care units, a goal certainly not yet everywhere achieved. Based on these considerations, we regard the development of regional guidelines for the acute management of hyperammonemia as urgent and a chance to improve the patients’ outcome in the Middle East. In this article, therefore, we have developed consensus guidelines that in particular address the unique situation in the Middle East and have based them on the highest available level of evidence. The aim of these guidelines is to homogenize and harmonize the various regional treatment protocols currently used for patients with acute hyperammonemia, and to provide a resource for metabolic specialists and likewise for physicians who may come in contact with individuals with acute hyperammonemia.
Method
We have based our guidelines on the European guidelines,17 published in 2012, which followed a strict methodological protocol. To include all relevant recent information, we searched PubMed and Embase databases to include published materials from 2011 to 2014 that were not covered by the European guidelines.1,6,18–38 Development of the present guidelines followed the process of a Delphi conference and involved one preliminary meeting and two follow-up meetings with email exchanges between the Middle East Hyperammonemia and Urea Cycle Disorders Scientific Group regarding each draft of the manuscript. The Middle East Hyperammonemia and Urea Cycle Disorders Scientific Group include an expert panel of metabolic specialists from countries in the Middle East as well as renowned international advisors. The process of guideline development followed Grading of Recommendations Assessment, Development, and Evaluation methodology of classifying the evidence.39,40 Although these guidelines are developed with highest accuracy, such guidelines should be considered as recommendations that aim at guiding physicians, but should not be considered as a protocol that is blindly followed. In particular, as each patient is an individual and each situation may vary from what is described here, all medical decisions must be carefully made on an individual basis.
Evidence levels were classified in accordance with the Grading of Recommendations Assessment, Development, and Evaluation methodology.39–42
Results and discussion
The first part of the results and discussion will focus on identifying unknown patients at risk for hyperammonemia. We regard an earlier identification of affected patients with high index of suspicion as a key to overall improved prognosis. The second part describes recommendations for the acute management of hyperammonemia and is divided into several sections according to practical considerations. Finally, other issues related to management and prognosis of hyperammonemia will be discussed. Embedded in the discussion are some answers to valid questions that may rise during the management of hyperammonemia in an emergency setting.
Although these guidelines are built upon previously published guidelines, the novelty of these guidelines is the focus on answers to many practical questions that were not discussed in depth in the previously published materials.17,43 Examples for practical aspects are: the meaning and definition of a high caloric intake, the normal ammonia levels according to the age, the amount of potassium in intravenous (IV) fluids when hyperammonemia scavengers are used. Additionally, the responsible person for insertion of the line(s) for dialysis, and the dosage of insulin if it is used during initial management of hyperammonemia are discussed. The protocol and suggested guidelines are summarized in Tables 1 and 2, respectively.
 | Table 1 Protocol for acute management of primary hyperammonemia based on ammonia level |
 | Table 2 Summary of protocol of acute management of hyperammonemia |
Identifying unknown patients at risk for hyperammonemia
When to order ammonia analysis?
Patients from all age groups who present with encephalopathy of unknown etiology should undergo measurements of plasma ammonia (NH3).17 The signs and symptoms of hyperammonemia can vary according to age, but are nonspecific in all age groups. In neonates, lethargy, poor feeding, and vomiting are frequent; it may progress to central hyperventilation, seizures, coma, and death if left untreated. In older children, symptoms include unexplained change in consciousness, unexplained or unusual neurological or psychiatric illness, and acute liver failure. Chronically, hyperammonemia can present as recurrent vomiting, headache, ataxia, strange behavior, especially if it occurs episodically, developmental delay, and aversion to protein. It is strongly recommended to consider ammonia measurement in all encephalopathic patients, especially in newborns who require a septic screen and are sick looking at the same time.1,2,17,23,25 Hospitals may also decide to install an automatic red flag as warning for certain clinical circumstances as recently suggested.23
Statement #1: strong recommendation, low quality evidence (1C)
Any individual at any age who presents with any of the following symptoms should undergo prompt plasma ammonia determination:
- Acute unexplained encephalopathy
- Acute neurological illness
- Suspected sepsis in a newborn
- Developmental delay of unknown cause
- Recurrent vomiting
- Acute liver failure, elevated transaminases of unknown cause
- Ataxia
- Headache, especially if episodic in women
- Coma or stupor
- Psychiatric symptoms
- Aversion to protein.
Initial management when hyperammonemia is suspected
Once hyperammonemia is suspected, the patient should be managed in a hospital with access to basic metabolic tests, first-line hyperammonemia medications, dialysis facilities, and metabolic specialists. If any of these elements are not available, the patient should be transferred without delay to a specialist center after stabilization according to basic life support (circulation, airway, and breathing), addressing vital signs as with any critically ill patient and including blood glucose monitoring.44 In addition, the following should be done:
- Insert IV lines, if possible as a central venous access. If this cannot be achieved, an intraosseous access could be an alternative.
- Maintain airway: intubate and ventilate if necessary.
- Adequate rehydration using minimum of dextrose 10% glucose and high calorie intake, maintain normal blood pressure, and add vasopressors if necessary.
- Take blood and urine samples as outlined in Table 3.
 | Table 3 Laboratory testing that should be done in any patient suspected of having hyperammonemia |
What ammonia levels require immediate action?
The normal ammonia level varies based on the patient’s age. There is no scientifically sound consensus about the upper normal limit for each age group. However, in general hyperammonemia is considered if plasma ammonia is >50 μmol/L in infants, children, and adults and >100 μmol/L in newborns.25,45 Other authors have described the normal reference range as follows: up to 7 days: 94 μmol/L, 8–30 days: 80 μmol/L, 1 month–15 years: 48 μmol/L, and >15 years: 26 μmol/L.46 It should be highlighted that the values presented here should be considered as decision limits; the normal reference ranges of individual laboratories should be used for clinical interpretation. An alert system for nurses and clinicians should be active in the lab in case ammonia is above the reference range and this must be reported immediately to the health care professional in charge of the patient.
Statement #2: strong recommendation, low quality evidence (1C)
The normal reference ranges of individual laboratories should be used for clinical interpretation. In general, hyperammonemia requires further action, if plasma ammonia is >50 μmol/L in infants, children, and adults and >100 μmol/L in neonates. The given limits of plasma ammonia are decision limits (Table 1).
What precaution must be taken for testing of ammonia?
False-positive hyperammonemia is not uncommon; therefore, several precautions should be taken into consideration when collecting blood samples to measure ammonia:
- A free-flowing venous (or arterial) blood sample without tourniquet should be collected into a tube containing an anticoagulant (eg, lithium or heparin).
- The sample should be placed in ice water, transported to the laboratory, and analyzed immediately. Results should be available within 60 minutes of drawing the samples.
Statement #3: strong recommendation, moderate quality evidence (1B)
Ammonia should be measured in an emergency setting with results available in 60 minutes.
Statement #4: strong recommendation, low quality evidence (1C)
It is crucial to take blood for plasma amino acids and urine for organic acids and orotic acid before starting the treatment; however, urine sampling must not delay start of treatment.
What are the key elements of the initial management once hyperammonemia is identified?
Reversal of catabolism
- Stop all sources of protein temporarily (for a maximum of 24–48 h).
- Call the pharmacy to prepare the medications and glucose/lipid infusions (see dosages in Table 4).
- Check blood glucose regularly (GlucoCheck).
- Start high caloric intake in form of IV dextrose 10%, or higher if appropriate, at the rate of 1.5 times to double of maintenance requirements, with age-dependent glucose requirement (in mg/kg/min for 0–1 years: 8–10, 1–3 years: 7–8, 4–6 years: 6–7, 7–12 years: 5–6, adolescents: 4–5, adults: 3–4).47
- If fatty acid oxidation defects are excluded, intralipid infusions should be started at 2–3 g/kg/day to give additional calories.17,48
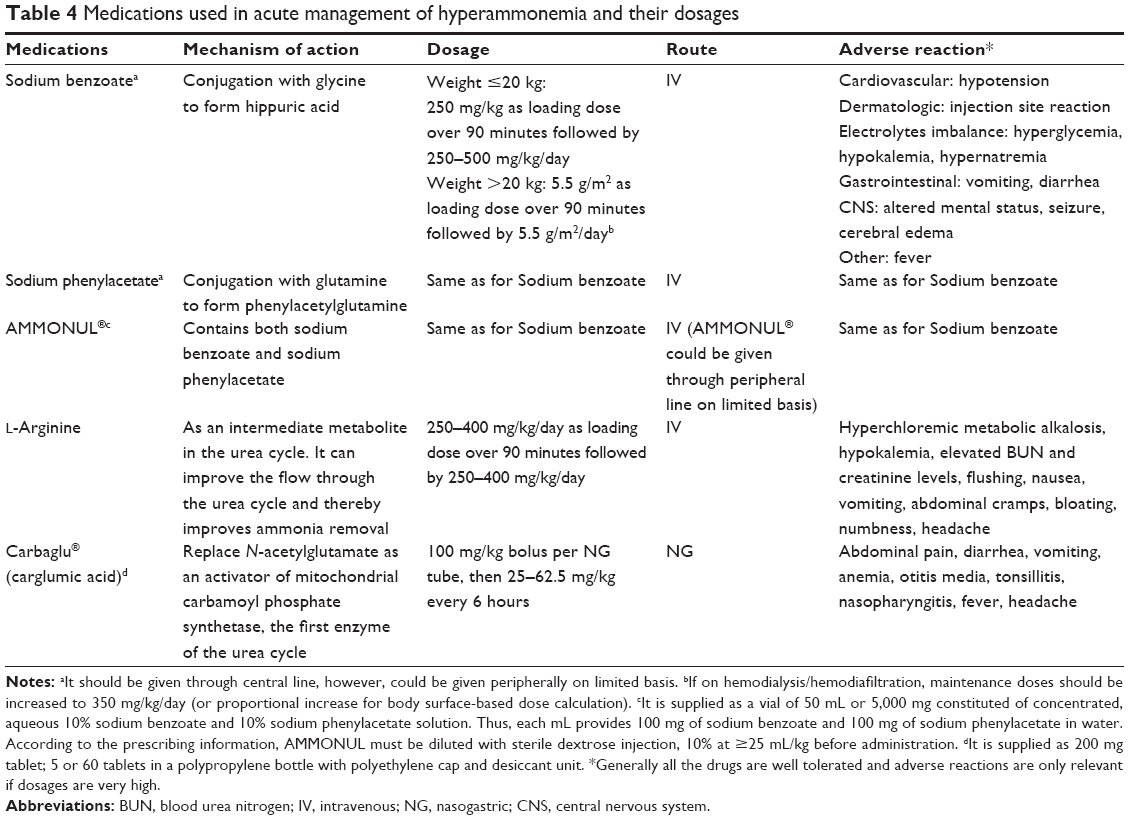 | Table 4 Medications used in acute management of hyperammonemia and their dosages |
What do we mean by high caloric intake?
It means covering at least 110% of the recommended daily allowance in order to shut down endogenous protein breakage (recommended daily allowance: for newborn and infant: 110–120 kcal/kg/day, for 1–3 years: 100 kcal/kg/day, for 4–6 years: 90 kcal/kg/day, for 7–10 years: 70 kcal/kg/day, for 11–14 years: 50–55 kcal/kg/day).47
Statement #5: strong recommendation, low quality evidence (1C)
High index and early clinical suspicion as well as prompt diagnosis of hyperammonemia are vital to have a good outcome. The treatment of hyperammonemia should be started without delay unless a decision for withdrawal of treatment and for palliative care is made.
What drugs are used to treat hyperammonemia?
Sodium benzoate and sodium phenylacetate are ammonia scavengers that bypass the urea cycle by conjugation of benzoate with glycine to generate hippurate, and of phenylacetate with glutamine to generate phenylacetylglutamine, which are then excreted in urine. L-Arginine is a metabolite within the urea cycle and can help the urea cycle to run again if it is interrupted due to arginine deficiency resulting from a block within the urea cycle. Carbaglu® (N-carbamyl-L-glutamate) can replace N-acetylglutamate as an activator of mitochondrial carbamoyl phosphate synthetase 1, the first enzyme of the urea cycle.17,49,50 The dosages of medications, mechanism of action, route, and side effects are summarized in Table 4.
Dialysis
What is the indication of dialysis?
Neonates and children
Continuous venovenous hemodiafiltration should be started in neonates and children who have ammonia levels of >500 μmol/L or even at lower levels if there has been an inadequate response to medical management after 4 hours, which is the estimated time for preparing dialysis, including vascular access.51
Statement #6: strong recommendation, low quality evidence (1C)
Dialysis is indicated in neonates and children with ammonia levels 300–500 μmol/L. Dialysis should be strongly considered in neonates and children with ammonia >500 μmol/L or when there is no response to the treatment. Consult intensive care unit and nephrology team, if you anticipate starting dialysis in the next few hours.
Adults
In adults, hemodialysis (HD) or continuous renal replacement therapy (CRRT) in the form of continuous venovenous hemofiltration is the first-line therapy in case of acute hyperammonemia. Dialysis should be started as soon as possible when ammonia exceeds 200 μmol/L.17
Statement #7: strong recommendation, low quality evidence (1C)
Dialysis is the first-line treatment in acute hyperammonemia decompensations in adults.
What is the method of choice for dialysis?
Dialysis choices usually include HD and CRRT. Alternatively, but only in centers that lack the ability or expertise to perform extracorporeal therapy, peritoneal dialysis (PD) can be utilized. However, PD clears ammonia at a lower rate than hemodialysis modalities. HD is intermittent and gives highest ammonia clearance.18 However, in neonates, HD is difficult to perform due to technical challenges and a high risk of complications, for example, concerning the maintenance of an adequate blood pressure. Therefore, CRRT including continuous venovenous hemofiltration or continuous venovenous hemodiafiltration is the preferred method.17,18,52 It is very important to emphasize that at this stage, fast action is required to prevent brain injury; therefore, the line for dialysis should be inserted immediately. Every place should develop its own protocol for who should insert the dialysis line depending on the local resources. The catheter of dialysis can be removed after the plasma ammonia level has been stable in the normal range for at least a day. However, keeping the catheter in place should be balanced with the risk of maintaining the patient in an anticoagulated state.
Exchange transfusions should be avoided.17 Ammonia scavenger therapy needs to be continued during dialysis.
Statement #8: strong recommendation, moderate quality evidence (1B)
The method of choice for dialysis is CRRT, preferably hemodiafiltration. Peritoneal dialysis is a far less effective method. Exchange transfusions should be avoided.
Statement #9: strong recommendation, low quality evidence (1C)
The line for dialysis should be inserted immediately by the most experienced physician (depending on the local resources).
Statement #10: weak recommendation, low quality evidence (2C)
Ammonia scavenger therapy needs to be continued during dialysis.
Nutrition
The metabolic dietitian should be involved immediately after diagnosis of a hyperammonemic patient. Initially, all protein intakes must be stopped and high calories (IV) must be provided as soon as possible. Additional calories may be given by nasogastric (NG) tube using glucose polymers, or protein-free formulas (eg, Polycose, Maxijul, or Pro-phree formulas) if not contraindicated.
Protein should be reintroduced as soon as ammonia level is 80–100 μmol/L to maximize caloric intake and this should not be delayed more than 24–48 hours. If there is hyperglycemia, start insulin drip and do not decrease the rate of dextrose infusion.
Statement #11: strong recommendation, low quality evidence (1C)
In management of acute hyperammonemia, it is essential to achieve high caloric intake by giving IV dextrose 10% or higher if appropriate with age-dependent glucose requirement and IV lipid emulsion (if a fatty acid oxidation disorder has been excluded).
Protein should be reintroduced when ammonia returns to 80–100 μmol/L and this should not be delayed more than 24–48 hours. Protein should be reintroduced after 48 hours even if ammonia is still >100 μmol/L.
Practical issues that may arise during management of acute hyperammonemia
Statement #12:
- Ondansetron (0.15 mg/kg) could be given to avoid vomiting when boluses of the ammonia scavengers are given,53 strong recommendation, low quality evidence (1C).
- Laxative: Treat constipation aggressively since ammonia is also produced from urea breakdown by intestinal bacteria,54,55 strong recommendation, low quality evidence (1C).
- If patient not diagnosed, consider additional administration of carnitine 100 mg/kg IV, hydroxycobalamin 1 mg intramuscular/intravenous, and biotin 10 mg IV/peroral,17 strong recommendation, moderate quality evidence (1B).
- Antibiotics: Preferred to continue or initiate them as prophylaxis, strong recommendation, low quality evidence (1C).
- Hyperventilation: When patient is ventilated, it is recommended to perform moderate hyperventilation to counteract cerebral edema,43 strong recommendation, low quality evidence (1C).
- Mannitol: It has not been demonstrated to be effective in managing cerebral edema caused by hyperammonemia and therefore, it should not be given,43 strong recommendation, low quality evidence (1C).
- Steroids should be avoided as they increase the amount of protein turnover and hence increase the nitrogen load,43 strong recommendation, low quality evidence (1C).
- Glucose and insulin can serve as suppressors of catabolism, but their use require care. Consider insulin if hyperglycemia develops (blood glucose level >10 mmol/L) at dose of 0.05–0.1 unit/kg/h and titrate up until blood glucose controlled. The presence of glycosuria is an indication for continued administration of IV regular insulin at a rate that keeps glucose levels between 6.5 and 10 mmol/L, weak recommendation, low quality evidence (2C).
- Valproic acid should be avoided in any patient who has known risk for hyperammonemia. It is known to decrease urea cycle function by inhibition of N-acetylglutamate synthase,56 strong recommendation, low quality evidence (1C).
- NaHCO3: Not recommended except if there is refractory acidosis, weak recommendation, low quality evidence (2C).
- Citrulline: A clear diagnosis should be made before supplementing the patient with citrulline. Patients with argininosuccinate synthetase and argininosuccinate lyase deficiency have elevated citrulline,43 strong recommendation, moderate quality evidence (1B).
- If the patient is on combined sodium benzoate and phenylacetate (AMMONUL®) or arginine give KCl 40 meq/L because they cause hyperchloremic hypokalemic metabolic acidosis. KCl can be given through peripheral line up to 60 meq/L, rate must not exceed 0.125 meq/kg/h, weak recommendation, low quality evidence (2C).
- KCl can be given in the same line with lipid and ammonia scavengers, weak recommendation, low quality evidence (2C).
- Combined sodium benzoate and sodium phenylacetate (AMMONUL®) can be given through peripheral lines, if no central line can be established, strong recommendation, low quality evidence (1C).
- Reloading of ammonia scavengers should only be considered in neonates with severe hyperammonemic decompensation or those who are undergoing dialysis, and should be given with great caution to avoid adverse effects due to increased serum drug concentrations,43 strong recommendation, low quality evidence (1C).
Prognosis
Poor prognostic factors in such cases include the following:
- Hyperammonemic coma has lasted more than 3 days.17,57
- Intracranial pressure is clearly increased.17
- High level and long duration of hyperammonemia.17
Statement #13: strong recommendation, low quality evidence (1C)
Neurodevelopmental prognosis is mainly dependent on total duration of coma, peak ammonia levels, and number of episodes of hyperammonemia. Additional studies are needed to identify other potential contributing factors.
Conclusion
In summary, there are several challenges and obstacles that clinicians face as they try to select the appropriate management protocol for acute hyperammonemia. These suggested guidelines aim to ease these challenges. However, the rarity of these diseases has resulted in mostly low evidence level for the statements made here, which corresponds to inferences derived from none analytical studies, such as case reports or case series or from expert opinion. Therefore, the recommendations contained herein should not be considered infallible or absolute.
Acknowledgments
The authors would like to thank Erik Brouwer from SOBI, Swedish Orphan Biovitrum for his valuable contribution. The Middle East Hyperammonemia and Urea Cycle Disorders Scientific Group meetings were organized with the help and financial support of SOBI, Swedish Orphan Biovitrum, but the company has no influence on the scientific contents of these guidelines.
Disclosure
The authors report no conflicts of interest in this work.
References
Auron A, Brophy PD. Hyperammonemia in review: pathophysiology, diagnosis, and treatment. Pediatr Nephrol. 2012;27(2):207–222. | ||
Haberle J. Clinical practice: the management of hyperammonemia. Eur J Pediatr. 2011;170(1):21–34. | ||
Braissant O. Current concepts in the pathogenesis of urea cycle disorders. Mol Genet Metab. 2010;100(Suppl 1):S3–S12. | ||
Braissant O, McLin VA, Cudalbu C. Ammonia toxicity to the brain. J Inherit Metab Dis. 2013;36(4):595–612. | ||
Gropman AL, Summar M, Leonard JV. Neurological implications of urea cycle disorders. J Inherit Metab Dis. 2007;30(6):865–879. | ||
Kido J, Nakamura K, Mitsubuchi H, et al. Long-term outcome and intervention of urea cycle disorders in Japan. J Inherit Metab Dis. 2012;35(5):777–785. | ||
Msall M, Batshaw ML, Suss R, Brusilow SW, Mellits ED. Neurologic outcome in children with inborn errors of urea synthesis. Outcome of urea-cycle enzymopathies. N Engl J Med. 1984;310(23):1500–1505. | ||
Picca S, Dionisi-Vici C, Abeni D, et al. Extracorporeal dialysis in neonatal hyperammonemia: modalities and prognostic indicators. Pediatr Nephrol. 2001;16(11):862–867. | ||
Al-Shamsi A, Hertecant JL, Al-Hamad S, Souid AK, Al-Jasmi F. Mutation spectrum and birth prevalence of inborn errors of metabolism among Emiratis: a study from Tawam Hospital Metabolic Center, United Arab Emirates. Sultan Qaboos Univ Med J. 2014;14(1):e42–e49. | ||
Moammar H, Cheriyan G, Mathew R, Al-Sannaa N. Incidence and patterns of inborn errors of metabolism in the Eastern Province of Saudi Arabia, 1983–2008. Ann Saudi Med. 2010;30(4):271–277. | ||
Saadallah AA, Rashed MS. Newborn screening: experiences in the Middle East and North Africa. J Inherit Metab Dis. 2007;30(4):482–489. | ||
Joshi SN, Hashim J, Venugopalan P. Pattern of inborn errors of metabolism in an Omani population of the Arabian Peninsula. Ann Trop Paediatr. 2002;22(1):93–96. | ||
El-Mouzan MI, Al-Salloum AA, Al-Herbish AS, Qurachi MM, Al-Omar AA. Regional variations in the prevalence of consanguinity in Saudi Arabia. Saudi Med J. 2007;28(12):1881–1884. | ||
Tadmouri GO, Al Ali MT, Al-Haj Ali S, Al Khaja N. CTGA: the database for genetic disorders in Arab populations. Nucleic Acids Res. 2006;34(Database issue):D602–D606. | ||
Tadmouri GO, Nair P, Obeid T, Al Ali MT, Al Khaja N, Hamamy HA. Consanguinity and reproductive health among Arabs. Reprod Health. 2009;6:17. | ||
Al-Owain M, Al-Zaidan H, Al-Hassnan Z. Map of autosomal recessive genetic disorders in Saudi Arabia: concepts and future directions. Am J Med Genet A. 2012;158A(10):2629–2640. | ||
Haberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. | ||
Baumgartner MR, Horster F, Dionisi-Vici C, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis. 2014;9:130. | ||
Rafique M. Emerging trends in management of propionic acidemia. Arq Bras Endocrinol Metabol. 2014;58(3):237–242. | ||
Lee HH, Poon KH, Lai CK, et al. Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome: a treatable genetic liver disease warranting urgent diagnosis. Hong Kong Med J. 2014;20(1):63–66. | ||
Van Leynseele A, Jansen A, Goyens P, et al. Early treatment of a child with NAGS deficiency using N-carbamyl glutamate results in a normal neurological outcome. Eur J Pediatr. 2014;173(12):1635–1638. | ||
Nakamura K, Yamane K, Shinohara K, et al. Hyperammonemia in idiopathic epileptic seizure. Am J Emerg Med. 2013;31(10):1486–1489. | ||
Vergano SA, Crossette JM, Cusick FC, Desai BR, Deardorff MA, Sondheimer N. Improving surveillance for hyperammonemia in the newborn. Mol Genet Metab. 2013;110(1–2):102–105. | ||
Tummolo A, Favia V, Bellantuono R, et al. Successful early management of a female patient with a metabolic stroke due to ornithine transcarbamylase deficiency. Pediatr Emerg Care. 2013;29(5):656–658. | ||
Haberle J. Clinical and biochemical aspects of primary and secondary hyperammonemic disorders. Arch Biochem Biophys. 2013;536(2):101–108. | ||
Lamb S, Aye CY, Murphy E, Mackillop L. Multidisciplinary management of ornithine transcarbamylase (OTC) deficiency in pregnancy: essential to prevent hyperammonemic complications. BMJ Case Rep. 2013;2013:bcr2012007416. | ||
Chan E, McQueen F. Valproate-induced hyperammonaemia superimposed upon severe neuropsychiatric lupus: a case report and review of the literature. Clin Rheumatol. 2013;32(3):403–407. | ||
Tranah TH, Vijay GK, Ryan JM, Shawcross DL. Systemic inflammation and ammonia in hepatic encephalopathy. Metab Brain Dis. 2013;28(1):1–5. | ||
Zhang Y, Landau YE, Miller DT, Marsden D, Berry GT, Kellogg MD. Recurrent unexplained hyperammonemia in an adolescent with arginase deficiency. Clin Biochem. 2012;45(18):1583–1586. | ||
Mehta N, Kirk PC, Holder R, Precheur HV. Urea cycle disorder – argininosuccinic lyase deficiency. Spec Care Dentist. 2012;32(4):155–159. | ||
Oliva Rodriguez R, Suleiman Martos Y, Pereira Cunnil JL, Serrano Aguayo P, Garcia Alfaro C, Garcia Luna PP. Hyperammonemic encephalopathy: nutritional assessment and management of a case. Nutr Hosp. 2012;27(1):303–305. | ||
Nagamani SC, Erez A, Lee B. Argininosuccinate lyase deficiency. Genet Med. 2012;14(5):501–507. | ||
Iyer H, Sen M, Prasad C, Rupar CA, Lindsay RM. Coma, hyperammonemia, metabolic acidosis, and mutation: lessons learned in the acute management of late onset urea cycle disorders. Hemodial Int. 2012;16(1):95–100. | ||
Bergmann KR, McCabe J, Smith TR, Guillaume DJ, Sarafoglou K, Gupta S. Late-onset ornithine transcarbamylase deficiency: treatment and outcome of hyperammonemic crisis. Pediatrics. 2014;133(4):e1072–e1076. | ||
Cartagena A, Prasad AN, Rupar CA, et al. Recurrent encephalopathy: NAGS (N-acetylglutamate synthase) deficiency in adults. Can J Neurol Sci. 2013;40(1):3–9. | ||
Labarthe F, Tardieu M, de Parscau L, Lamireau D. Clinical presentation of inborn metabolic diseases in the neonatal period. Arch Pediatr. 2012;19(9):953–958. | ||
Gupta N, Kabra M. Acute management of sick infants with suspected inborn errors of metabolism. Indian J Pediatr. 2011;78(7):854–859. | ||
Picca S, Dionisi-Vici C, Bartuli A, et al. Short-term survival of hyperammonemic neonates treated with dialysis. Pediatr Nephrol. 2015;30(5):839–847. | ||
Andrews J, Guyatt G, Oxman AD, et al. GRADE guidelines: 14. Going from evidence to recommendations: the significance and presentation of recommendations. J Clin Epidemiol. 2013;66(7):719–725. | ||
Balshem H, Helfand M, Schunemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401–406. | ||
Guyatt GH, Norris SL, Schulman S, et al. Methodology for the development of antithrombotic therapy and prevention of thrombosis guidelines. Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):53S–70S. | ||
Terracciano L, Brozek J, Compalati E, Schunemann H. GRADE system: new paradigm. Curr Opin Allergy Clin Immunol. 2010;10(4):377–383. | ||
Summar M. Current strategies for the management of neonatal urea cycle disorders. J Pediatr. 2001;138(1 Suppl):S30–S39. | ||
Hazinski MF, Nolan JP, Billi JE, et al. Part 1: Executive summary: 2010 International Consensus on Cardiopulmonary Resuscitation and Emergency Cardiovascular Care Science with Treatment Recommendations. Circulation. 2010;122(16 Suppl 2):S250–S275. | ||
Colombo JP, Peheim E, Kretschmer R, Dauwalder H, Sidiropoulos D. Plasma ammonia concentrations in newborns and children. Clin Chim Acta. 1984;138(3):283–291. | ||
Nagamani SCS, Erez A, Lee B. Argininosuccinate lyase deficiency. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. SourceGeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2016. February 3, 2011. [updated February 2, 2012]. | ||
Association CD. Manual of Clinical Dietetics. 5 edition. Chicago, IL: American Dietetic Association; 1996. | ||
Chapman KA, Gropman A, MacLeod E, et al. Acute management of propionic acidemia. Mol Genet Metab. 2012;105(1):16–25. | ||
Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr. 1996;43:127–170. | ||
Leonard JV, Morris AA. Urea cycle disorders. Semin Neonatol. 2002;7(1):27–35. | ||
Picca S, Bartuli A, Dionisi-Vici C. Medical management and dialysis therapy for the infant with an inborn error of metabolism. Semin Nephrol. 2008;28(5):477–480. | ||
Hanudel M, Avasare S, Tsai E, Yadin O, Zaritsky J. A biphasic dialytic strategy for the treatment of neonatal hyperammonemia. Pediatr Nephrol. 2014;29(2):315–320. | ||
Batshaw ML, MacArthur RB, Tuchman M. Alternative pathway therapy for urea cycle disorders: twenty years later. J Pediatr. 2001;138(1 Suppl):S46–S54; discussion S54–S55. | ||
Romero-Gomez M, Jover M, Galan JJ, Ruiz A. Gut ammonia production and its modulation. Metab Brain Dis. 2009;24(1):147–157. | ||
Vince AJ, Burridge SM. Ammonia production by intestinal bacteria: the effects of lactose, lactulose and glucose. J Med Microbiol. 1980;13(2):177–191. | ||
Aires CC, van Cruchten A, Ijlst L, et al. New insights on the mechanisms of valproate-induced hyperammonemia: inhibition of hepatic N-acetylglutamate synthase activity by valproyl-CoA. J Hepatol. 2011;55(2):426–434. | ||
Krivitzky L, Babikian T, Lee HS, Thomas NH, Burk-Paull KL, Batshaw ML. Intellectual, adaptive, and behavioral functioning in children with urea cycle disorders. Pediatr Res. 2009;66(1):96–101. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.