Back to Journals » International Journal of Nanomedicine » Volume 13
Gold nanoparticle-based colorimetric method for the detection of prostate-specific antigen
Authors Xia N, Deng D, Wang Y, Fang C, Li S
Received 13 October 2017
Accepted for publication 24 January 2018
Published 24 April 2018 Volume 2018:13 Pages 2521—2530
DOI https://doi.org/10.2147/IJN.S154046
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Ning Xia,1 Dehua Deng,1,2 Yiru Wang,1 Chao Fang,3 Su-Juan Li1
1Henan Province of Key Laboratory of New Optoelectronic Functional Materials, College of Chemistry and Chemical Engineering, Anyang Normal University, Anyang, Henan, People’s Republic of China; 2College of Chemistry and Chemical Engineering, Henan University, Kaifeng, Henan, People’s Republic of China; 3School of Chemical Engineering, Xiamen University Malaysia, Sepang, Selangor, Malaysia
Background: Prostate-specific antigen (PSA), a serine protease, is a biomarker for preoperative diagnosis and screening of prostate cancer and monitoring of its posttreatment.
Methods: In this work, we reported a colorimetric method for clinical detection of PSA using gold nanoparticles (AuNPs) as the reporters. The method is based on ascorbic acid (AA)-induced in situ formation of AuNPs and Cu2+-catalyzed oxidation of AA. Specifically, HAuCl4 can be reduced into AuNPs by AA; Cu2+ ion can catalyze the oxidation of AA by O2 to inhibit the formation of AuNPs. In the presence of the PSA-specific peptide (DAHSSKLQLAPP)-modified gold-coated magnetic microbeads (MMBs; denoted as DAHSSKLQLAPP-MMBs), complexation of Cu2+ by the MMBs through the DAH–Cu2+ interaction depressed the catalyzed oxidation of AA and thus allowed for the formation of red AuNPs. However, once the peptide immobilized on the MMB surface was cleaved by PSA, the DAHSSKLQ segment would be released. The resultant LAPP fragment remaining on the MMB surface could not sequestrate Cu2+ to depress its catalytic activity toward AA oxidation. Consequently, no or less AuNPs were generated.
Results: The linear range for PSA detection was found to be 0~0.8 ng/mL with a detection limit of 0.02 ng/mL. Because of the separation of cleavage step and measurement step, the interference of matrix components in biological samples was avoided.
Conclusion: The high extinction coefficient of AuNPs facilitates the colorimetric analysis of PSA in serum samples. This work is helpful for designing of other protease biosensors by matching specific peptide substrates.
Keywords: colorimetric assay, gold nanoparticles, prostate-specific antigen, ascorbic acid, Cu2+ ion
Introduction
Prostate cancer is one of the most prevalent forms of silent killers among men worldwide. It accounts for up to 29% of all cancer cases and 13% of deaths.1 Prostate-specific antigen (PSA) is a 33-kDa androgen-regulated serine protease of the kallikrein family that is secreted by the epithelial cells of the prostate gland. Normally, a healthy prostate releases <4 ng/mL PSA into the circulatory system.1,2 However, the PSA concentrations are often elevated in the presence of prostate cancer or other prostate disorders. Thus, PSA has been believed to be the current gold standard biomarker for preoperative diagnosis and screening of prostate cancer and monitoring of its posttreatment.1 For example, in the USA, the US Food and Drug Administration has approved the PSA test for annual screening of prostate cancer in men aged ≥50 years. The patient needs to be informed of the risks and benefits of PSA testing prior to performing the test. However, the conventional methods for PSA detection are time-consuming, laborious, and expensive and require fluorescent- or enzyme-labeling and complicated instruments, including enzyme-linked immunosorbent assays, fluorescence immunoassays, electrochemical immunoassays, colorimetric immunoassays, and chemiluminescence immunoassays.1,3–6 Therefore, simple, rapid, cost-effective, and high-throughput methods for PSA detection are desired.
PSA is a serine protease to hydrolyze the peptide bond. Since the discovery of a PSA-specific peptide substrate with a core region of HSSKLQ,7 a few novel methods based on the proteolysis reaction have been developed for PSA detection, including fluorescence,8,9 electrochemistry,10–15 surface-enhanced Raman spectroscopy,16 colorimetry,17 and electrogenerated chemiluminescence.18,19 Among these techniques, gold nanoparticles (AuNPs)-based colorimetric assays are in particular prevalent in the past 20 years for biomarker detection because AuNPs show a high extinction coefficient, and the color change can be easily detected with naked eye or spectrometry for quantitative analysis.20–24 For protease detection, the peptide substrates are usually functionalized with one or more cysteine residues or positively charged amino acid residues including Lys and Arg.25–33 Cleavage of a single peptide into two shorter fragments by the specific protease prevents the peptide substrate-induced aggregation or facilitates the fragment-triggered aggregation of AuNPs based on the Au–S and/or electrostatic interactions. Nonetheless, to trigger the AuNP aggregation, the peptide substrate must be well designed. The thiol groups in substrate peptide may inhibit protease activity by breaking disulfide bonds in the enzyme molecule or be oxidized to form the disulfide bonds between the substrate.28 Moreover, some matrix components in biological fluids such as serums may affect the stability of unmodified AuNPs. For these reasons, the practical applications of AuNP-based colorimetric assays for the clinical detection of proteases including PSA are still limited.
Ascorbic acid (AA) is a commonly used reducing agent for the growth of metal (eg, Au and Ag) nanoparticles.34,35 Free Cu2+ ion can initiate the catalyzed oxidation of AA in the presence of O2.36 In this process, AA is oxidized into dehydroascorbate by free Cu2+ ion; then, the resultant Cu+ ion is subsequently oxidized into Cu2+ by O2. Thus, the oxidation of AA is promoted by the Cu2+-initiated redox cycling, which can be described as follows:
|
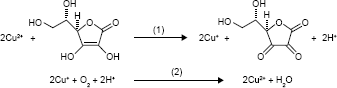
However, the complexation of Cu2+ ion by peptide may change its catalytic activity. For example, amino terminal copper and nickel (ATCUN)-binding peptide with a structural characterization comprising of a free NH2-terminus, a histidine (His) residue in the third position and two intervening peptide nitrogens (denoted as NH2–X–X–His sequence) exhibits high affinity to Cu2+ ion.37–40 The formation of ATCUN–Cu2+ complex depresses Cu2+-initiated redox cycling for AA oxidation.40 In this work, we demonstrated that the PSA substrate peptide (DAHSSKLQLAPP) that contains an ATCUN motif of DAH can sequestrate Cu2+ by the formation of ATCUN–Cu2+ complex (Figure 1A), thus depressing the Cu2+-catalyzed oxidation of AA. Based on this fact, we developed a colorimetric method for the detection of PSA in human serums by employing magnetic microbeads (MMBs, Figure 1B). Figure 1C shows the detection principle. HAuCl4 can be reduced into AuNPs by AA; Cu2+ ion promotes the oxidation of AA, thus preventing the production of AuNPs. The peptide immobilized on the MMB surface can sequestrate Cu2+ ion, thus inhibiting the Cu2+-catalytic oxidation of AA and facilitating the generation of AuNPs. However, once the peptide on the MMB surface is cleaved by PSA, the ATCUN-containing fragment of DAHSSKLQ would be released. The resultant MMBs cannot inhibit the Cu2+-catalytic oxidation of AA, thus disfavoring the formation of AuNPs. The high extinction coefficient of the generated AuNPs enables the quantitative and sensitive colorimetric analysis of PSA in serums.
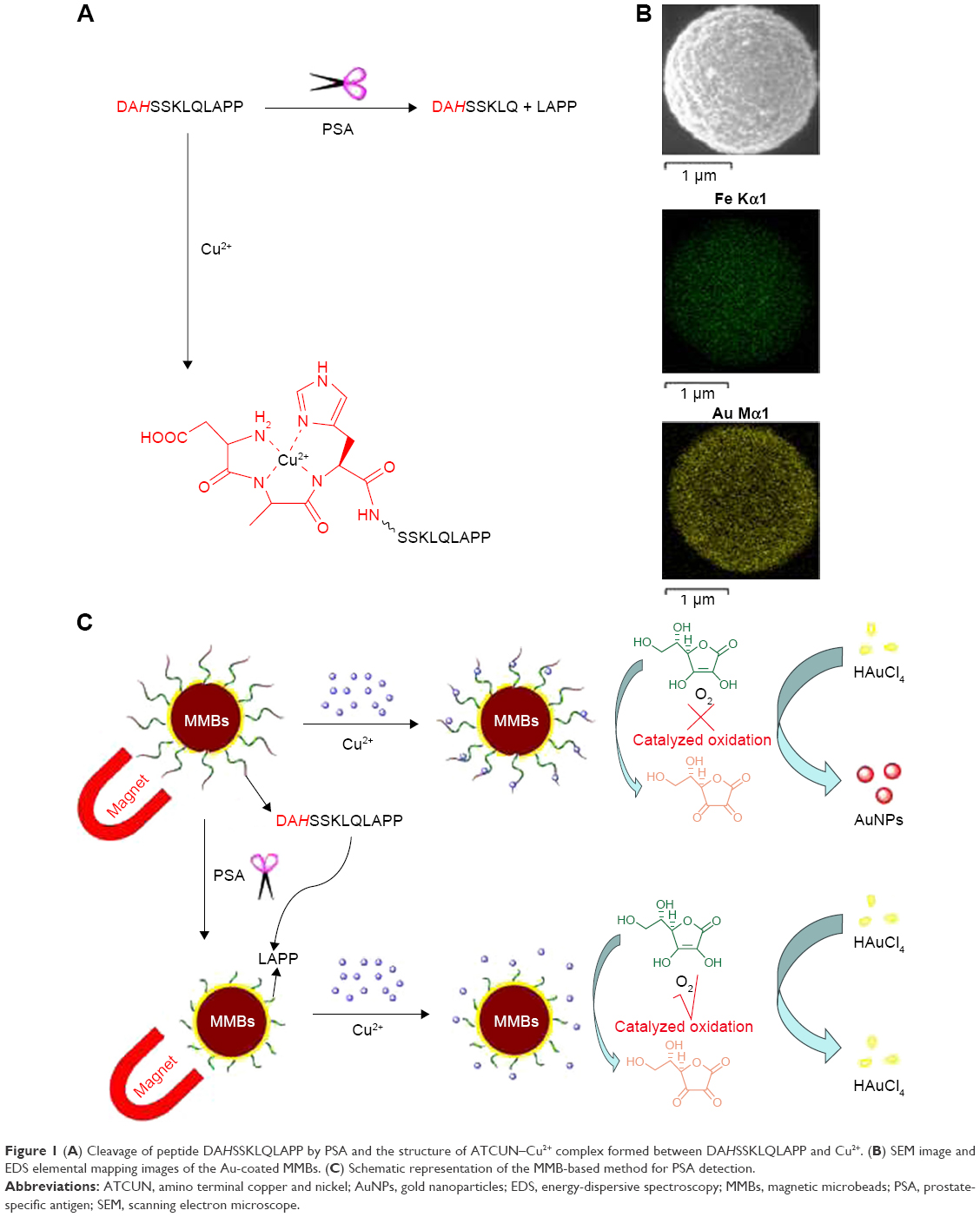 | Figure 1 (A) Cleavage of peptide DAHSSKLQLAPP by PSA and the structure of ATCUN–Cu2+ complex formed between DAHSSKLQLAPP and Cu2+. (B) SEM image and EDS elemental mapping images of the Au-coated MMBs. (C) Schematic representation of the MMB-based method for PSA detection. |
Materials and methods
Materials
Peptides were synthesized and purified by China Peptides Co. Ltd. (Shanghai, People’s Republic of China). Trypsin, hemoglobin, and AA were obtained from Sangon Biotech Co. Ltd. (Shanghai, People’s Republic of China). Bovine serum albumin, thrombin, K2HPO4, KH2PO4, and 6-mercapto-1-hexanol were purchased from Sigma-Aldrich Co. (Shanghai, People’s Republic of China). Hexadecyltrimethylammonium chloride (CTAC) and other reagents were of analytical grade and obtained from the Aladdin Reagent Company (Shanghai, People’s Republic of China). All solutions were prepared freshly with deionized water. The serum samples were supplied by the Anyang Tumor Hospital, People’s Republic of China.
Kinetic measurements
The kinetic measurements for probing of AA oxidation were conducted on a Cary 50 ultraviolet–visible (UV–Vis) spectrophotometer with a fixed wavelength of 265 nm. To make sure that the amount of free Cu2+ remaining in solution is negligible, the peptide/Cu2+ concentration ratio was kept at 1.5:1. The stock AA concentration was determined with an extinction coefficient (ε) of 1.5×104 M−1cm−1 at 265 nm.41
AA-induced growth of AuNPs
To demonstrate the formation of AuNPs with AA as the reducing reagent, 250 μL of different concentrations of AA in a phosphate buffer (10 mM, pH =7.0) was first mixed with 200 μL of 500 μM CTAC solution. This was followed by the addition of 50 μL of 2 mM HAuCl4 in batches (5 μL per drop). After incubation for 2 minutes, the photographic images were taken by a mobile phone, and the absorption spectra were recorded with the UV–Vis spectrophotometer. To demonstrate the effect of Cu2+ species on the AA-induced growth of AuNPs, the AA stock solution was preincubated with Cu2+ in the absence or presence of DAHSSKLQLAPP-functionalized MMBs at ambient temperature for 30 minutes.
Preparation of peptide-functionalized MMBs
The Au-coated MMBs were prepared with the procedure in the previous report,42 which were characterized by scanning electron microscope (JSM-7800F; JEOL, Tokyo, Japan) and energy-dispersive spectroscopy (NS7; Thermo Fisher Scientific, Waltham, MA, USA; Figure 1A). Immobilization of the thiolated peptide (DAHSSKLQLAPPC) was performed by mixing 50 mg Au-coated MMBs with 0.5 mL of 1 mM peptide in phosphate buffer for 3 hours. The peptide was attached onto the Au-coated MMB surface through the Au–S interaction. After magnetic separation, the content of free peptide in the residue liquid was determined by LCT Premier XE mass spectrometry (Waters, Milford, MA, USA). From the standard curve method, the absorption capability of Au-coated MMBs to peptide was found to be 7.8 nmol/mg. To block the unreacted gold surface, the peptide-covered Au-coated MMBs were incubated with 1 mM 6-mercapto-1-hexanol for 15 minutes, followed by magnetic separation and washing with 50% ethanol.
PSA detection
For PSA detection, 4 mg of peptide-functionalized MMBs was incubated with 200 μL of PSA solution or serum sample in the phosphate buffer for 40 minutes at 30°C. After magnetic separation and washing with acetic acid (pH=4.0) and water, the resultant MMBs were dispersed in 150 μL of 3 μM Cu2+ in the phosphate buffer. This was followed by the addition of 150 μL of 400 μM AA and incubation for 30 minutes. Then, 250 μL of the suspension was taken out and added to 200 μL of 500 μM CTAC solution, which was followed by the addition of 2 mM HAuCl4 in batches. The change of solution color was observed with naked eyes, and the absorption spectra were recorded by using the UV–Vis spectrophotometer. The photographic images were taken by using the camera in a mobile phone. The absorption spectra were collected on the UV–Vis spectrophotometer.
Results and discussion
Cu2+-catalyzed oxidation of AA
To demonstrate the catalyzed activity of Cu2+ toward AA oxidation in the absence and presence of different peptides, the kinetics of AA consumption by O2 was examined by monitoring the change of AA absorbance at 265 nm. As shown in Figure 2A, the absorbance intensity of AA itself decreased slightly, indicating that the autoxidation of AA is slow. However, in the presence of Cu2+, AA became rapidly consumed, demonstrating that the oxidation of AA was promoted by Cu2+. Interestingly, in the presence of ATCUN peptide (DAHSSKLQLAPP), no apparent decrease in the absorbance intensity was observed. This result indicated that the catalytic activity of Cu2+ toward AA oxidation was depressed by DAHSSKLQLAPP. The result is acceptable since the ATCUN–Cu2+ complex can depress the Cu2+-initiated redox cycling for AA oxidation.40 However, the presence of LAPP (one of the cleavage products) did not inhibit the Cu2+-catalytic oxidation of AA. In addition, we found that the absorption intensity of AA almost decreased to the background level after incubation with the mixture of Cu2+ and LAPP-functionalized MMBs for 30 minutes (Figure 2B). Nevertheless, no significant decrease in the absorption intensity was observed when AA was incubated with the mixture of Cu2+ and DAHSSKLQLAPP-functionalized MMBs. These results demonstrated that the proposed strategy can be used to develop PSA biosensor based on the ATCUN–Cu2+ interaction and the catalytic oxidation of AA.
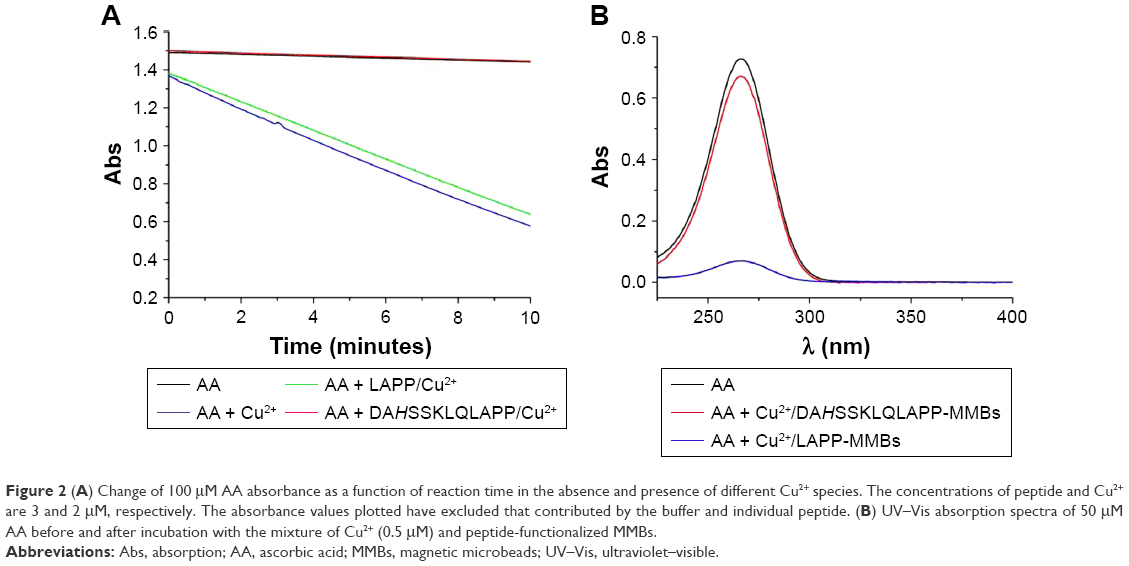 | Figure 2 (A) Change of 100 μM AA absorbance as a function of reaction time in the absence and presence of different Cu2+ species. The concentrations of peptide and Cu2+ are 3 and 2 μM, respectively. The absorbance values plotted have excluded that contributed by the buffer and individual peptide. (B) UV–Vis absorption spectra of 50 μM AA before and after incubation with the mixture of Cu2+ (0.5 μM) and peptide-functionalized MMBs. |
Formation of AuNPs
With CTAC as the stabilizer, HAuCl4 can be reduced into AuNPs with neurotransmitters as active reducing agents.43 The formation of AuNPs does not need any nanoparticle seeds. Based on the optical property of the generated AuNPs, the quantitative analysis of the neurotransmitters has been performed. Herein, we found that HAuCl4 can also be reduced into AuNPs by AA with CTAC as the stabilizer. As shown in Figure 3A, the mixed solution of AA and CTAC became red after the addition of HAuCl4 (tube a) and exhibited an absorption peak at ~530 nm (curve a). The absorption peak is ascribed to the surface plasmon resonance of the generated AuNPs. The transmission electron microscope image indicated that the AuNPs are nearly monodispersed, spherical nanoparticles within very similar particle dimensions (Figure 3B). The size of AuNPs is in agreement with that obtained by using dopamine as the reducing reagent.43 We also found that the addition of HAuCl4 in batches into different concentrations of AA solution did not cause significant difference in the size of AuNPs. When AA was preincubated with the mixture of Cu2+ and DAHSSKLQLAPP-functionalized MMBs for 30 minutes, a strong absorption peak and a red color were still observed after adding HAuCl4 into the above mixed solution (curve/tube b). No difference in the size of the generated AuNPs was observed when AA was incubated with and without DAHSSKLQLAPP-functionalized MMBs (Figure 3C). The result indicated that DAHSSKLQLAPP-functionalized MMBs/Cu2+ did not depress the generation of AuNPs. However, when the HAuCl4 solution was added into the mixed solution of AA and LAPP-functionalized MMBs/Cu2+ under the same condition, a colorless solution was obtained, and a lower absorption peak was observed. This implied that no or less AuNPs were generated. This is understandable since the oxidized AA (dehydroascorbate) cannot reduce HAuCl4 into AuNPs. The asymmetric absorbance in this case is attributed to the absorbance of yellowish HAuCl4 and CTAC. Thus, the formation of AuNPs is dependent upon the interaction of Cu2+ and peptide-functionalized MMBs and the Cu2+-catalyzed oxidation of AA.
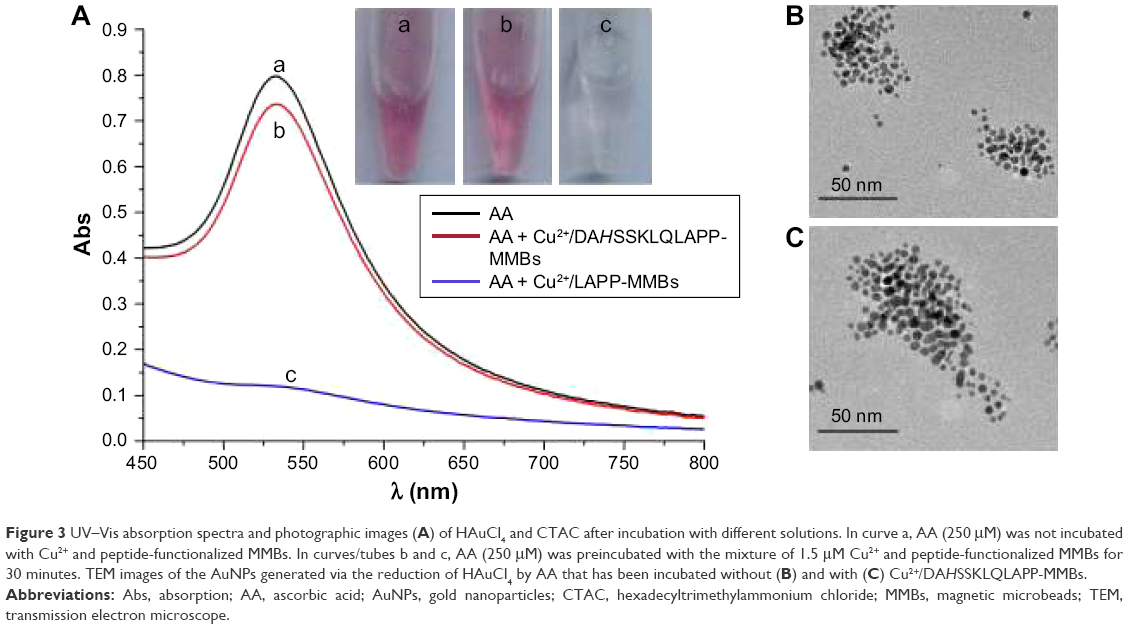 | Figure 3 UV–Vis absorption spectra and photographic images (A) of HAuCl4 and CTAC after incubation with different solutions. In curve a, AA (250 μM) was not incubated with Cu2+ and peptide-functionalized MMBs. In curves/tubes b and c, AA (250 μM) was preincubated with the mixture of 1.5 μM Cu2+ and peptide-functionalized MMBs for 30 minutes. TEM images of the AuNPs generated via the reduction of HAuCl4 by AA that has been incubated without (B) and with (C) Cu2+/DAHSSKLQLAPP-MMBs. |
Optimization of experimental conditions
In this method, AA was used as the reducing reagent for the formation of AuNPs. Thus, its concentration plays a decisive role in the formation of AuNPs. As shown in Figure 4A, the absorption intensity of the generated AuNPs at 530 nm increased with the increase of AA concentration in the range of 10–250 μM and began to level off beyond 200 μM. AA at the used concentration of 10 μM can be readily determined. In addition, Cu2+ concentration has a profound influence on the catalyzed oxidation of AA. Thus, we also investigated the effect of Cu2+ concentration on the absorption intensity. When AA was preincubated with different concentrations of Cu2+ for 30 minutes, the absorbance value decreased with increasing concentration of Cu2+ (Figure 4B). This demonstrated that high concentration of Cu2+ facilitated the oxidation of AA and thus disfavored the generation of AuNPs. As depicted in Figure 3, DAHSSKLQLAPP-functionalized MMBs can sequestrate Cu2+ and depress its catalytic capability of oxidizing AA, thus favoring the formation of AuNPs. We also found that the absorbance value increased with increasing concentration of DAHSSKLQLAPP-functionalized MMBs and reached to the maximum at 4 mg (Figure 4C). The good dependence of the absorption intensity upon the MMB concentration demonstrated that it is possible to determine PSA by removing the ATCUN-containing peptide fragment (DAHSSKLQ) from the MMB surface.
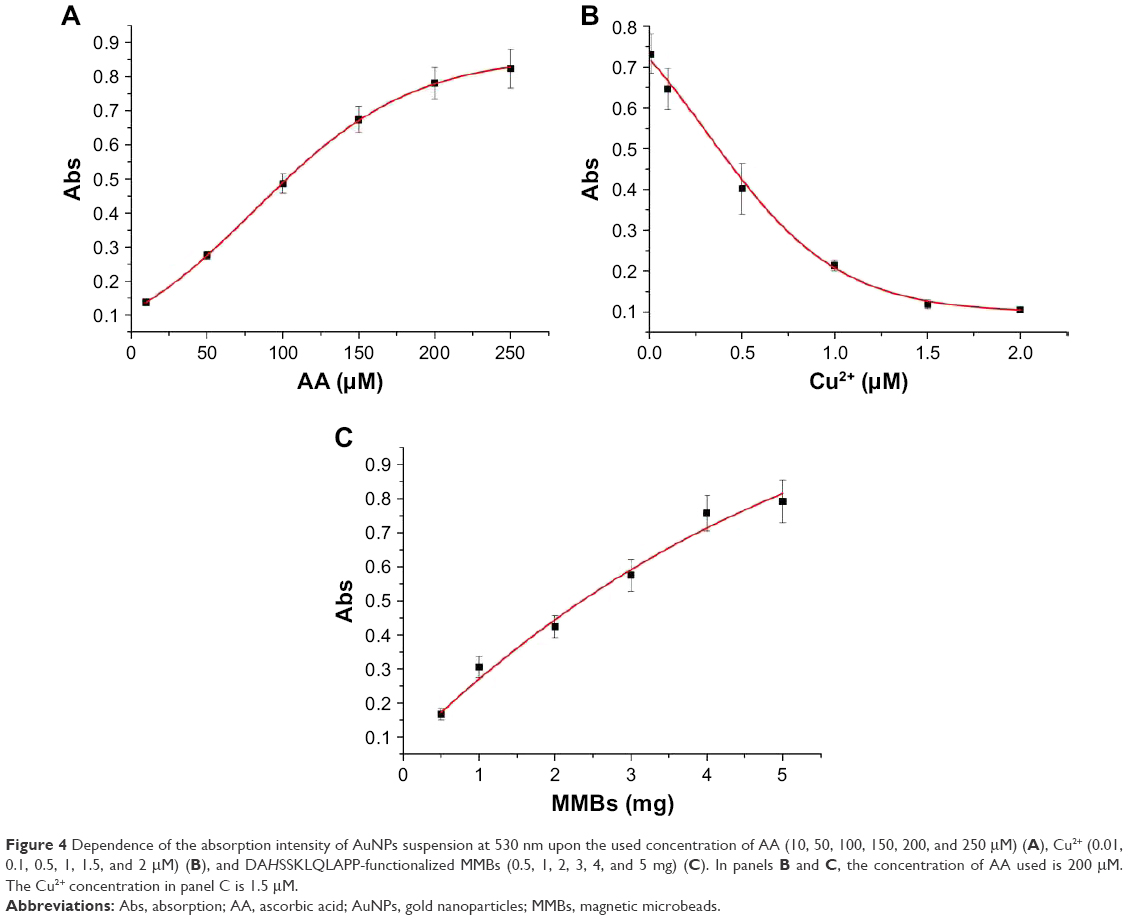 | Figure 4 Dependence of the absorption intensity of AuNPs suspension at 530 nm upon the used concentration of AA (10, 50, 100, 150, 200, and 250 μM) (A), Cu2+ (0.01, 0.1, 0.5, 1, 1.5, and 2 μM) (B), and DAHSSKLQLAPP-functionalized MMBs (0.5, 1, 2, 3, 4, and 5 mg) (C). In panels B and C, the concentration of AA used is 200 μM. The Cu2+ concentration in panel C is 1.5 μM. |
PSA detection
Under the optimal conditionals, the feasibility of the proposed method for PSA detection was demonstrated. Figure 5A represents the photographic images, and the UV–Vis absorption spectra when DAHSSKLQLAPP-functionalized MMBs were preincubated with different concentrations of PSA, followed by incubating the resultant MMBs with Cu2+ and AA before the addition of HAuCl4. With the increase in PSA concentration, the red color solution became colorless gradually. The result was also confirmed by the UV–Vis spectroscopy: the more higher the PSA concentration, the more lower the plasmon absorbance peak of AuNPs. The value decreased linearly with an increase in PSA concentration in the range of 0–0.8 ng/mL with an equation of Abs=0.697-0.673 (PSA) ng/mL (Figure 5B). The detection limit was estimated to be 0.02 ng/mL by detecting the smallest PSA concentration at which the response is clearly distinguishable from the background. This value is comparable to those achieved by other methods based on the PSA-induced cleavage of peptide (Table 1). The value is lower than that (4 ng/mL) of PSA released by a healthy prostate; thus, the colorimetric method is promising to determine PSA in a biological sample for preoperative diagnosis and screening of prostate cancer.
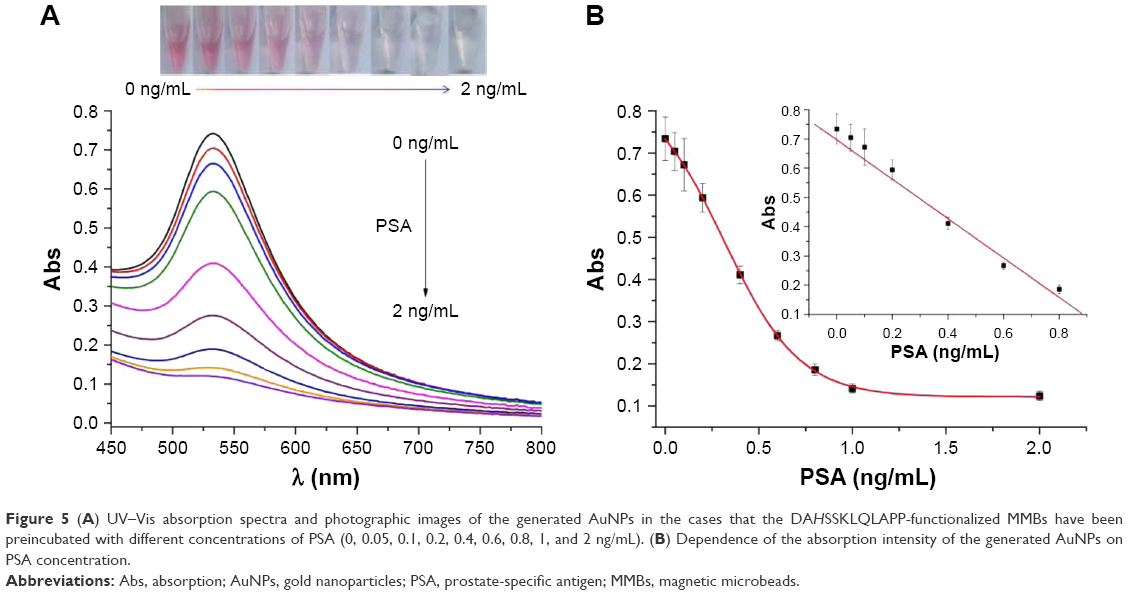 | Figure 5 (A) UV–Vis absorption spectra and photographic images of the generated AuNPs in the cases that the DAHSSKLQLAPP-functionalized MMBs have been preincubated with different concentrations of PSA (0, 0.05, 0.1, 0.2, 0.4, 0.6, 0.8, 1, and 2 ng/mL). (B) Dependence of the absorption intensity of the generated AuNPs on PSA concentration. |
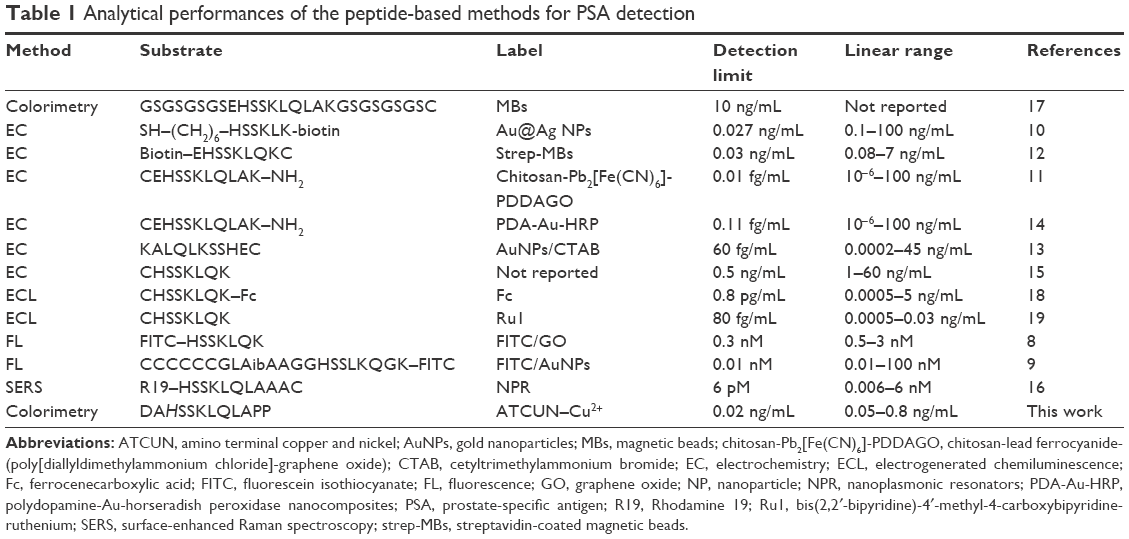 | Table 1 Analytical performances of the peptide-based methods for PSA detection |
Selectivity and real sample assays
To demonstrate the applicability of the sensor, we first examined its selectivity toward PSA by testing the influence of proteins (bovine serum albumin and hemoglobin), AA, and other proteases (thrombin and trypsin). As shown in Figure 6A, a lower absorption value was obtained for 0.8 ng/mL PSA, while negligible changes in the absorption intensity were observed in the presence of at least 10-fold higher concentrations of the tested proteins. The result is indicative of high selectivity of the method, which can be attributed to the high specificity of the peptide substrate (HSSKLQ) toward PSA.7,18,19 In addition, AA itself also did not induce any significant change. The result is understandable since the analytical detection was carried out by the magnetic separation of cleavage step and measurement step, thus reducing the interference of matrix components in biological samples.
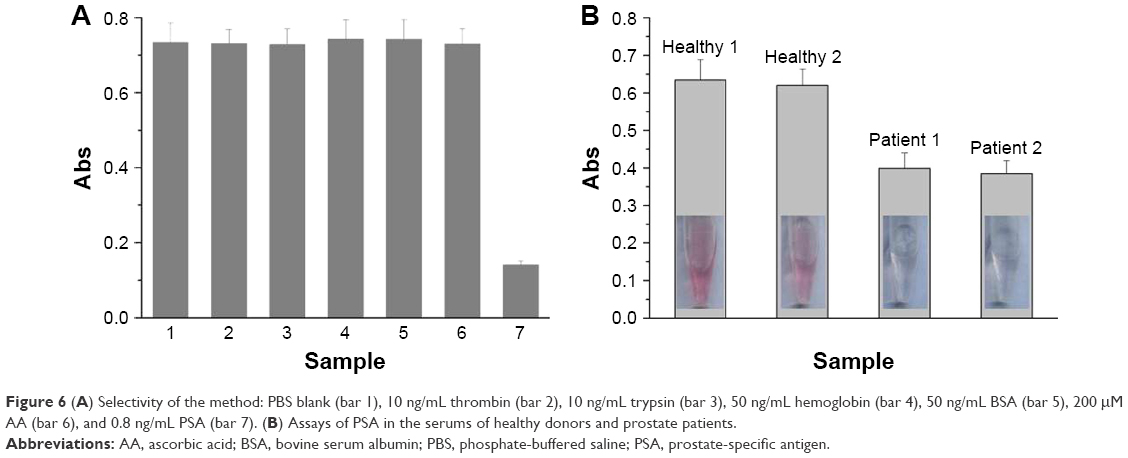 | Figure 6 (A) Selectivity of the method: PBS blank (bar 1), 10 ng/mL thrombin (bar 2), 10 ng/mL trypsin (bar 3), 50 ng/mL hemoglobin (bar 4), 50 ng/mL BSA (bar 5), 200 μM AA (bar 6), and 0.8 ng/mL PSA (bar 7). (B) Assays of PSA in the serums of healthy donors and prostate patients. |
To demonstrate the viability of the method for clinical assays, quantifications of PSA in serum samples from two healthy donors and two prostate patients were carried out. It can be clearly observed that the solution color is red for healthy controls, but it is colorless for patients (Figure 6B). By diluting the two healthy sample solutions for 10 times, the PSA concentrations were found to be 0.94 ng/mL and 1.14 ng/mL (n=3) according to the above established standard curve. However, for 10 times dilution, we found that the levels of PSA in the patient serums were beyond the linear range. Thus, the quantification of PSA for patients was performed by further diluting the samples with 20-fold. It was found that PSA concentrations in the two patients were 8.86 ng/mL and 9.30 ng/mL (n=3), which were much higher than those in the healthy controls. The result indicated that the colorimetric method is feasible for distinguishing prostate patients from healthy controls.
Conclusion
In this work, we developed a colorimetric method for PSA detection by inhibition of the formation of AuNPs. The separation of cleavage step and measurement step by peptide-functionalized MMBs facilitates the selective detection of PSA in serum samples. The method is fundamentally distinct from the existing peptide-based sensing strategies for PSA detection. It is very promising to be used in laboratory investigation and clinical diagnosis because it requires a simple sample handling procedure and a minimum instrumental investment. Moreover, this work is valuable for designing of high-throughput protease biosensors and likely finds many applications in other fields, including screening of protease inhibitor and probing of disease progression.
Acknowledgments
We acknowledge the financial support of the National Natural Science Foundation of China (21305004), the Program for Science and Technology Innovation Talents at the University of Henan Province (18HASTIT005), the Joint Fund for Fostering Talents of National Natural Science Foundation of China and Henan Province (U1304205), the Science & Technology Foundation of Henan Province (17A150001), and the Program for Innovative Research Team of Science and Technology in the University of Henan Province (16IRT-STHN003).
Disclosure
The authors report no conflicts of interest in this work.
References
Ibau C, Md Arshad MK, Gopinath SCB. Current advances and future visions on bioelectronic immunosensing for prostate-specific antigen. Biosens Bioelectron. 2017;98:267–284. | ||
Chen N, Rong M, Shao X, et al. Surface-enhanced Raman spectroscopy of serum accurately detects prostate cancer in patients with prostate-specific antigen levels of 4–10 ng/mL. Int J Nanomedicine. 2017;12:5399–5407. | ||
Liu D, Yang J, Wang HF, et al. Glucose oxidase-catalyzed growth of gold nanoparticles enables quantitative detection of attomolar cancer biomarkers. Anal Chem. 2014;86:5800–5806. | ||
Gao Z, Xu M, Hou L, Chen G, Tang D. Magnetic bead-based reverse colorimetric immunoassay strategy for sensing biomolecules. Anal Chem. 2013;85:6945–6952. | ||
Suaifan GA, Shehadeh M, Al-Ijel H, Ng A, Zourob M. Recent progress in prostate-specific antigen and HIV proteases detection. Expert Rev Mol Diagn. 2013;13:707–718. | ||
Zhang Q, Wu L, Wong TI, et al. Surface plasmon-enhanced fluorescence on Au nanohole array for prostate-specific antigen detection. Int J Nanomedicine. 2017;12:2307–2314. | ||
Denmeade SR, Lou W, Lövgren J, Malm J, Lilja H, Isaacs JT. Specific and efficient peptide substrates for assaying the proteolytic activity of prostate-specific antigen. Cancer Res. 1997;57:4924–4930. | ||
Feng T, Feng D, Shi W, Li X, Ma H. A graphene oxide-peptide fluorescence sensor for proteolytically active prostate-specific antigen. Mol Biosyst. 2012;8:1441–1445. | ||
Choi JH, Kim HS, Choi JW, Hong JW, Kim YK, Oh BK. A novel Au-nanoparticle biosensor for the rapid and simple detection of PSA using a sequence-specific peptide cleavage reaction. Biosens Bioelectron. 2013;49:415–419. | ||
Parnsubsakul A, Safitri RE, Rijiravanich P, Surareungchai W. Electrochemical assay of proteolytically active prostate specific antigen based on anodic stripping voltammetry of silver enhanced gold nanoparticle labels. J Electroanal Chem. 2017;785:125–130. | ||
Tang Z, Fu Y, Ma Z. Bovine serum albumin as an effective sensitivity enhancer for peptide-based amperometric biosensor for ultrasensitive detection of prostate specific antigen. Biosens Bioelectron. 2017;94:394–399. | ||
Hun X, Xu Y, Luo X. Peptide-based biosensor for the prostate-specific antigen using magnetic particle-bound invertase and a personal glucose meter for readout. Microchim Acta. 2015;182:1669–1675. | ||
Wang D, Zheng Y, Chai Y, Yuan Y, Yuan R. Target protein induced cleavage of a specific peptide for prostate-specific antigen detection with positively charged gold nanoparticles as signal enhancer. Chem Commun (Camb). 2015;51:10521–10523. | ||
Tang Z, Wang L, Ma Z. Triple sensitivity amplification for ultrasensitive electrochemical detection of prostate specific antigen. Biosens Bioelectron. 2017;92:577–582. | ||
Deng D, Shi Y, Feng H, Chen Q, Li D, Liu L. Label-free electrochemical sensing platform for the detection of protease. Int J Electrochem Sci. 2013;8:6933–6940. | ||
Sun C, Su KH, Valentine J, et al. Time-resolved single-step protease activity quantification using nanoplasmonic resonator sensors. ACS Nano. 2010;4:978–984. | ||
Suaifan GA, Esseghaier C, Ng A, Zourob M. Ultra-rapid colorimetric assay for protease detection using magnetic nanoparticle-based biosensors. Analyst. 2013;138:3735–3739. | ||
Qi H, Li M, Dong M, Ruan S, Gao Q, Zhang C. Electrogenerated chemiluminescence peptide-based biosensor for the determination of prostate-specific antigen based on target-induced cleavage of peptide. Anal Chem. 2014;86:1372–1379. | ||
Zhang J, Qi H, Li Z, Zhang N, Gao Q, Zhang C. Electrogenerated chemiluminescence bioanalytic system based on biocleavage of probes and homogeneous detection. Anal Chem. 2015;87:6510–6515. | ||
Deng HH, Li GW, Liu AL, Chen W, Lin XH, Xia XH. Thermally treated bare gold nanoparticles for colorimetric sensing of copper ions. Microchim Acta. 2014;181:911–916. | ||
Guo JF, Rahme K, He Y, Li LL, Holmes JD, O’Driscoll CM. Gold nanoparticles enlighten the future of cancer theranostics. Int J Nanomedicine. 2017;12:6131–6152. | ||
Thavanathan J, Huang NM, Thong KL. Colorimetric biosensing of targeted gene sequence using dual nanoparticle platforms. Int J Nanomedicine. 2015;10:2711–2722. | ||
Saha K, Agasti SS, Kim C, Li X, Rotello VM. Gold nanoparticles in chemical and biological sensing. Chem Rev. 2012;112:2739–2779. | ||
Giljohann DA, Mirkin CA. Drivers of biodiagnostic development. Nature. 2009;462:461–464. | ||
Li Y, Li W, He KY, et al. A biomimetic colorimetric logic gate system based on multi-functional peptide-mediated gold nanoparticle assembly. Nanoscale. 2016;8:8591–8599. | ||
Chen G, Xie Y, Zhang H, et al. A general colorimetric method for detecting protease activity based on peptide-induced gold nanoparticle aggregation. RSC Adv. 2014;4:6560–6563. | ||
Xue W, Zhang G, Zhang D. A sensitive colorimetric label-free assay for trypsin and inhibitor screening with gold nanoparticles. Analyst. 2011;136:3136–3141. | ||
Kim CJ, Lee DI, Kim C, Lee K, Lee CH, Ahn IS. Gold nanoparticles-based colorimetric assay for Cathepsin B activity and the efficiency of its inhibitors. Anal Chem. 2014;86:3825–3833. | ||
Kim GB, Kim KH, Park YH, Ko S, Kim YP. Colorimetric assay of matrix metalloproteinase activity based on metal-induced self-assembly of carboxy gold nanoparticles. Biosens Bioelectron. 2013;41:833–839. | ||
Ding X, Ge D, Yang KL. Colorimetric protease assay by using gold nanoparticles andoligopeptides. Sensor Actuat B Chem. 2014;201:234–239. | ||
Kim GB, Lee JO, Kim YP. Graying the self-assembly of gold nanoparticles for improved enzyme activity assays. Sensor Actuat B Chem. 2017;246:271–277. | ||
Chen CK, Huang CC, Chang HT. Label-free colorimetric detection of picomolar thrombin in blood plasma using a gold nanoparticle-based assay. Biosens Bioelectron. 2010;25:1922–1927. | ||
Guarise C, Pasquato L, De Filippis V, Scrimin P. Gold nanoparticles-based protease assay. Proc Natl Acad Sci U S A. 2006;103:3978–3982. | ||
Ding X, Yang KL. Enzymatic deposition of silver particles for detecting protease activity. Part Part Syst Charact. 2014;31:1300–1306. | ||
Rastogi L, Dash K, Ballal A. Selective colorimetric/visual detection of Al3+ in ground water using ascorbic acid capped gold nanoparticles. Sens Actuat B Chem. 2017;248:124–132. | ||
Liu L, Jiang D, McDonald A, Hao Y, Millhauser GL, Zhou F. Copper redox cycling in the prion protein depends critically on binding mode. J Am Chem Soc. 2011;133:12229–12237. | ||
Harford C, Sarkar B. Amino terminal Cu(II)- and Ni(II)-binding (ATCUN) motif of proteins and peptides: metal binding, DNA cleavage, and other properties. Acc Chem Res. 1997;30:123–130. | ||
Jin Y, Lewis MA, Gokhale NH, Long EC, Cowan JA. Influence of stereochemistry and redox potentials on the single- and double-strand DNA cleavage efficiency of Cu(II) and Ni(II) Lys-Gly-His-derived ATCUN metallopeptides. J Am Chem Soc. 2007;129:8353–8361. | ||
Ding X, Yang KL. Quantitative serine protease assays based on formation of copper(II)–oligopeptide complexes. Analyst. 2015;140:340–345. | ||
Folk DS, Franz KJ. A prochelator activated by β-secretase inhibits Aβ aggregation and suppresses copper-induced reactive oxygen species formation. J Am Chem Soc. 2010;132:4994–4995. | ||
Scarpa M, Vianello F, Signor L, Zennaro L, Rigo A. Ascorbate oxidation catalyzed by bis(histidine)copper(II). Inorg Chem. 1996;35:5201–5206. | ||
Shiigi H, Kinoshita T, Shibutani N, Nishino T, Nagaoka T. Efficient collection and sensitive detection using conducting magnetic microbeads. Anal Chem. 2014;86:4977–4981. | ||
Baron R, Zayats M, Willner I. Dopamine-, L-DOPA-, adrenaline-, and noradrenaline-induced growth of Au nanoparticles: assays for the detection of neurotransmitters and of tyrosinase activity. Anal Chem. 2005;77:1566–1571. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.