Back to Journals » The Application of Clinical Genetics » Volume 14
GM1 Gangliosidosis: Mechanisms and Management
Authors Rha AK, Maguire AS, Martin DR
Received 11 November 2020
Accepted for publication 15 January 2021
Published 9 April 2021 Volume 2021:14 Pages 209—233
DOI https://doi.org/10.2147/TACG.S206076
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Allisandra K Rha,1 Anne S Maguire,1,2 Douglas R Martin1,2
1Scott-Ritchey Research Center, Auburn University, Auburn, AL, 36849, USA; 2Department of Anatomy, Physiology, and Pharmacology, Auburn University College of Veterinary Medicine, Auburn, AL, 36849, USA
Correspondence: Douglas R Martin
Scott-Ritchey Research Center, Auburn University, 1265 H.C. Morgan Drive, Auburn, AL, 36849, USA
Tel +1 334 844 5951
Email [email protected]
Abstract: The lysosomal storage disorder, GM1 gangliosidosis (GM1), is a neurodegenerative condition resulting from deficiency of the enzyme β-galactosidase (β-gal). Mutation of the GLB1 gene, which codes for β-gal, prevents cleavage of the terminal β-1,4-linked galactose residue from GM1 ganglioside. Subsequent accumulation of GM1 ganglioside and other substrates in the lysosome impairs cell physiology and precipitates dysfunction of the nervous system. Beyond palliative and supportive care, no FDA-approved treatments exist for GM1 patients. Researchers are critically evaluating the efficacy of substrate reduction therapy, pharmacological chaperones, enzyme replacement therapy, stem cell transplantation, and gene therapy for GM1. A Phase I/II clinical trial for GM1 children is ongoing to evaluate the safety and efficacy of adeno-associated virus-mediated GLB1 delivery by intravenous injection, providing patients and families with hope for the future.
Keywords: GM1 gangliosidosis, gene therapy, biomarkers, chaperone, GLB1, LSD
Introduction
GM1 gangliosidosis (GM1) is an inherited autosomal recessive lysosomal storage disorder (LSD) affecting one in every 100,000 to 200,000 live births within the general population.1–4 However, a notably higher incidence of GM1 exists in Malta (1 in 3700 live births),5 Roma with a ~1 in 50 carrier rate of the general Roma population and up to 1 in 10 of the Rudari sub-isolate,6 Japan,7,8 and Cyprus where 1 in 12 residents of the village of Pelendri are carriers.9 Progressive neurodegeneration results from mutation of the GLB1 gene, which codes for the lysosomal hydrolase, acid beta-galactosidase (β-gal, EC 3.2.1.23).1,2,4 Absence or attenuation of β-gal activity leads to the accumulation of the β-linked galactose-containing glycolipids GM1 ganglioside and GA1 ganglioside, and other glycoconjugates, including N-linked and O-linked glycans.1–4,10–12 Substrates accumulate to toxic levels in lysosomes, causing profound dysfunction of the nervous system.1–4,10,11 Three clinical subtypes of GM1 gangliosidosis are classified by age of symptom onset: infantile (Type 1), late infantile/juvenile (Type 2), and adult/chronic (Type 3).2–4,13,14 The infantile form is the most common and severe of the three subtypes, with symptom onset by six months and death by two to four years of age.2 Late infantile/juvenile GM1 presents as early as 7 months and as late as five years, with death occurring in mid-childhood to early adulthood.3 Adult GM1 represents the least severe form with slower disease progression, symptom onset from early childhood to the late teens, and in many cases affects individuals of Japanese descent.13,15,16 Variation in the time of symptom onset has been attributed, at least partially, to the degree of residual β-gal activity.1–3 Whether GM1 should be classified as four subtypes with late infantile and juvenile represented independently, or as a spectrum disorder based on level of β-gal activity, remains a topic for discussion.
Typical treatments for patients with GM1 are symptomatic and palliative in nature.17,18 Patients with GM1 often receive anti-convulsants to control seizures, gastrostomy tubes to ensure proper nutrition and hydration, and procedures to maintain an open airway. However, symptomatic treatment does not alter the clinical course substantially. Here we describe the clinical presentation of patients with GM1, animal models being used for the development of effective treatments, the pathophysiology of the disease, and current therapeutic approaches, including those in clinical trials.
Clinical Presentation
While the infantile form of GM1 has a relatively homogeneous clinical presentation, symptom variability increases with the age of disease onset.2–4 In other words, the less severe the disease, the more variable is the clinical phenotype and the more delayed is the diagnosis.18–20 Patients with one of the three subtypes of GM1 may present with any of the symptoms outlined below.
Type 1: Infantile patients first present with developmental (psychomotor) regression around three to four months of age, though symptoms may be apparent at birth. Parents may notice an exaggerated startle response and changes in appetite initially. About 50% of affected children have a cherry red spot on the macula of their retina, which is identified through ophthalmological examination and can lead to a faster diagnosis.2,21 As the child ages, other symptoms may become apparent, including coarse facial features, hepatosplenomegaly, skeletal abnormalities, profound intellectual disability, nystagmus, gingival hypertrophy, cardiomyopathy, joint stiffness, distended abdomen, muscular hypotonia, and deafness.2,14,22–27 Most patients with type 1 GM1 experience seizures, strabismus, corneal clouding, or vision loss. Infantile GM1 is terminal, resulting in death by two to four years of age.2
Type 2: Late infantile/Juvenile GM1 patients present with developmental regression as early as 7 months and as late as 5 years.3,22,27 Skills that may have been gained, such as crawling or walking, are eventually lost. Children have early locomotor problems, muscular hypotonia and atrophy, intellectual disability, seizures, difficulties with speech and swallowing, strabismus, and lethargy.3,4,28 Some patients may present with spasticity, scoliosis, hyperreflexia, or ataxia. Because of the wide range of symptom onset and reduced severity of disease progression, patients may live to late teens or early adulthood.14,22,23,28–31
Type 3: Adult/Chronic GM1 patients have a substantially slower disease progression compared with the other two subtypes and are often of Japanese descent.16,32 Most adult/chronic GM1 patients experience symptom onset as teens or in early adulthood. Clinically, patients present with mild vertebral abnormalities, muscular atrophy, hypotonia, corneal clouding, slurred speech, and short stature.16,32–34 Adult patients rarely have cherry red spots, a characteristic symptom of severe lipid storage disorders.15,16,30,33–35 The adult subtype is characterized clinically by dystonia, which rarely presents in the other GM1 subtypes. Adult/chronic GM1 patients may have all or few of the symptoms described in subtypes 1 and 2. For all three GM1 subtypes, bronchopneumonia from recurrent aspiration may be fatal.
Diagnosis
Diagnosis of all subtypes of GM1 gangliosidosis often occurs by assay of β-galactosidase enzyme activity.17,36 Genetic testing through whole-genome sequencing or sequencing of the GLB1 gene is also a route for diagnosis. Most patients are diagnosed by enzyme assay because of the cost and uncertainty of genetic testing.17 With more than 200 pathogenic GLB1 mutations, and more than 40,000 allele combinations, the data from genetic testing are not clear-cut.4,17,37 However, preimplantation genetic diagnosis of GM1 to detect known pathogenic alleles from parents is possible during in vitro fertilization procedures, maximizing positive pregnancy outcomes.38 Exome sequencing, which enables sequencing of all genes in a single test, was also used previously to diagnose GM1 in a teenage patient with an unknown neurodegenerative condition.19 Further, prenatal diagnosis is possible via amniocentesis for families with a history of GM1 or if genetic testing reveals both parents are carriers of a pathogenic allele.39–41 Neonatal congenital ascites identified by routine ultrasound can also indicate an LSD, though final diagnosis must be made by other means.26,39 Although few LSDs are included on newborn screening cards, dried blood spots can be used for diagnosis by enzyme assay for at least 13 months post birth and by detection of GM1 ganglioside content.17,42,43
Animal and Cell Culture Models
Naturally-occurring and laboratory-generated animal models of GM1 gangliosidosis are of pre-clinical significance and provide insight into target pathways for the development of therapeutics. GM1 gangliosidosis naturally occurs and has been clinically and pathologically characterized for feline, canine, bovine, and ovine models.4,44–49 However, only a few “large animal” models of GM1 are maintained in a research setting (feline, ovine), and no known small laboratory animals present with naturally-occurring GM1. GM1 gangliosidosis mouse models have been generated through knockout of the glb1 gene and complete loss of β-gal activity.50–53 Gross abnormalities are absent in mice until about 4–5 months of age, at which point, affected mice develop severe clinical signs including tremor, ataxia, and abnormal gait.50,52 In the recent literature, a CRISPR/Cas-generated 20 bp deletion in exon 8 of glb1 produces a GM1 mouse that reaches humane endpoint at 7–10 months with no detectable β-gal activity.54 Transgenic mouse models expressing relevant human mutations and residual β-gal activity may have mild clinical signs with a slightly shortened life span.55 However, they represent important pre-clinical models for the application of genome editing or enzyme enhancement therapy. For example, a mouse model with hemizygous expression of human β-gal having the mutation R201C,55 most often associated with juvenile GM1, may be used to demonstrate the clinical efficacy of genome editing complexes incorporating base editors. In addition to the lissencephalic mouse models, gyrencephalic animals have been integral to our understanding of disease pathogenesis and the translation of efficacious treatments to humans.46,47,56 Adeno-associated virus (AAV)-mediated GLB1 gene therapy experiments in a naturally-occurring feline model that closely mimics juvenile GM1 and in β-gal-/- mouse models were critical to the initiation of ongoing gene therapy clinical trials, in children with GM1, discussed in more detail below.57–63
Latour et al (2019) defined a novel three-dimensional cerebral organoid cell culture model for the study of GM164. CRISPR/Cas-mediated knockout of GLB1 in human induced pluripotent stem cells (iPSC) and subsequent generation of cerebral organoids provided a relevant pre-clinical model of human brain tissue to support the ongoing AAV gene therapy trial. GM1 cerebral organoids have progressive GM1 ganglioside accumulation and <5% β-gal activity.64
Molecular Mechanisms/Pathophysiology
β-Galactosidase and GM1 Ganglioside
Gangliosides are glycosphingolipids (GSL) that reside at the plasma membrane and are responsible for the modulation of signal transduction.44,65–72 There are more than 180 gangliosides identified in vertebrates with each classified based on number of sialic acid moieties.67,68,70–75 GM1 ganglioside has a single sialic acid (or n-acetylneuraminic acid) and is therefore classified as a monosialo (M) ganglioside.67,68,72,74,76 Gangliosides are abundant in the brain, accounting for up to 10% of the lipid content, but are also expressed in many peripheral tissues supporting their role in cell physiology.71,72,76 These GSLs are critical for neurodevelopment, neuritogenesis, and neuronal plasticity.44,65–68,71,72,76 GSL biosynthesis is a multi-compartment process that occurs within the endoplasmic reticulum (ER) and the Golgi apparatus and begins with the synthesis of ceramide at the ER membrane.67,68,70–73,76,77 Ceramide glucosyltransferase, which is a target of inhibition for substrate reduction strategies in GSL storage diseases, transfers a glucose residue to ceramide on the cytosolic side of the Golgi, forming glucosylceramide.70,73,76 Subsequent addition of galactose within the lumen of the Golgi by beta-1,4-galactosyltransferase 6 leads to the formation of lactosylceramide.69–71,76–78 Distinct transferases then convert lactosylceramide to several different precursor glycolipids, fated for specific terminal pathways. For ganglioside biosynthesis, ST3 beta-galactoside alpha-2,3-sialyltransferase 5 catalyzes the formation of GM3 ganglioside from lactosylceramide through transfer of a single sialic acid moiety to the galactose group of lactosylceramide within the Golgi lumen.69,70,73,76 GM2 ganglioside results from the addition of N-acetylgalactosamine (GalNAc) to the galactose moiety of GM3 by GalNAc transferase.65,70,71,73,76,77 The final step of GM1 ganglioside biosynthesis is the addition of galactose to GalNAc by beta-1,3-galactosyltransferase at the Golgi.65,70,71,73,76,77 Within the secretory pathway, GM1 ganglioside is trafficked to the plasma membrane via vesicular transport where it associates with lipid microdomains/rafts and contributes to several processes including membrane organization, protein anchoring, and signal transduction.44,65–68,70–72,76
Degradation of gangliosides occurs at the lysosome and requires a sophisticated cellular mechanism.70,72–74,77,79 Gangliosides are endocytosed and delivered to the lysosomal membrane where sphingolipid activator proteins deliver the highly hydrophobic glycolipids to the glycosidases of the lysosome for catabolism.70,73,74,77,79 In GM1, deficiency or absence of acid β-gal prevents the efficient degradation of GM1 ganglioside and recycling of its subunits to the salvage pathways.1,4,10,72–74,77,80 To correct the cellular pathogenesis, therapeutics seek to promote proper GM1 ganglioside catabolism or reduce substrate (GM1 ganglioside) accumulation. Targeted restoration of GM1 ganglioside catabolism occurs through gene therapy,57,61–63,81,82 stem cell transplantation,83–86 enzyme replacement therapy (ERT),56,87–90 and treatment with pharmacological chaperones,55,91–98 while substrate reduction is mediated through inhibition of ceramide glucosyltransferase,30,99–102 the enzyme that catalyzes the first committed step of GSL biosynthesis. For treatment of symptomatic individuals, substrate reduction therapy relies on residual β-gal activity to remove ganglioside that has already accumulated.30 Thus, it likely would not be therapeutic for symptomatic infantile GM1 patients, who have little or no residual enzyme activity. These strategies are discussed in more detail below.
GLB1 Mutations
More than 200 pathogenic or likely pathogenic mutations have been characterized for the GLB1 gene (HGMD professional database: https://portal.biobase-international.com/cgi-bin/portal/login.cgi; ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/), which maps to the short arm of chromosome 3 and codes for the lysosomal hydrolase β-galactosidase.1,4,103–105 Of those mutations, single nucleotide polymorphisms account for ~75%, with insertions and deletions representing ~25%.1,4,103 Combinations of different allelic mutations contribute to the clinical heterogeneity and the difficulty in diagnosis by GLB1 sequencing.1,4,14,103,104,106–110 Pathogenic mutations of GLB1 are dispersed across its 16 exons and at sites critical to splicing. However, the majority are found in exons 2, 6, and 154. The infrequent occurrence of GM1 coupled with the considerable number of allelic combinations makes the probability of having a nonconsanguineous homozygous combination unlikely, but not impossible. Some alleles have a higher frequency in specific populations.6,8,9,106,107,109 For example, 601C>T and 152T>C frequently present as juvenile and adult GM1 in Japanese patients, respectively.107,111 In Brazil, a study of 65 patients identified c.1622–1627insG as the most frequent allele.109
Assigning any one mutation to a specific GM1 subtype is also difficult because of the number of allelic combinations. Mutations occurring in cis contribute further to this challenge.1,112 Overall, the degree of β-gal dysfunction is directly related to the impact of the mutation on the enzyme structure.108 Mutations resulting in an early termination codon are processed by nonsense-mediated decay or lead to the production of a truncated enzyme, which is often degraded.37,108 However, aggregation of misfolded or truncated proteins can also lead to ER stress and the UPR.113,114 Affected individuals with alleles harboring an early termination codon usually have a complete loss of β-gal activity and are not candidates for certain types of therapy. Knowing a patient’s genotype assists in the identification of the most efficacious treatments.
GM1 Gangliosidosis Disease Mechanisms
UPR-Mediated Apoptosis
The unquestionable cause of GM1 gangliosidosis is attenuation or absence of β-gal activity. However, how accumulation of GM1 ganglioside and other GSLs leads to neurodegeneration is not fully understood. Tessitore et al (2004) defined one of the first disease mechanisms where GM1-ganglioside depletion of ER Ca2+ stores induced the unfolded protein response (UPR) and subsequent neuronal apoptosis in β-gal-/- mice.113 Significantly more TUNEL+ cells were identified throughout the spinal cord of β-gal-/- mice compared to wild-type, which aligned with the observed age-dependent activation of the UPR and neurological decline. ER-stress-mediated apoptosis was supported by the transcriptional upregulation of BiP and CHOP, and the reproducible induction of Jnk2 and caspase-12 in GM1-loaded neurospheres from WT mice. ER-stress specific cleavage of caspase-12 to the p42 and p20 forms associated with apoptosis further supported the defined mechanism. In GM1-loaded WT and β-gal-/- murine embryonic fibroblasts (MEF), a substantial pool of GM1 ganglioside co-localized with the ER-marker calnexin. A concurrent depletion of Ca2+ from the ER was confirmed following thapsigargin treatment and use of an ER-targeted calmodulin-based FRET construct, establishing GM1 ganglioside as a causative agent in the induction of apoptosis.113
A similar elevation of BiP was detected in the naturally-occurring feline model of GM1, where the p.R483P β-gal mutation (analogous to p.R482H/C of humans) was identified as causative. In GM1 cats, β-gal mRNA levels were normal throughout the cerebral cortex, but trafficking of mutant β-gal to the lysosome was impaired. Restriction to the ER, as determined by β-gal and protein disulfide isomerase (PDI) co-localization, suggested defective targeting to lysosomes. The accumulation of mutant β-gal within the ER represents a second possible trigger for the induction of the UPR in GM1114.
Neurotransmission
GM1 ganglioside’s role in regulating calcium homeostasis may explain the global dysregulation of calcium levels observed in GM1.45,71,113,115–118 In synaptosomes isolated from GM1 cats, calcium efflux and influx was significantly reduced compared to that of normal controls. Koenig et al (1987) identified two distinct phases of calcium influx, fast and slow, and attributed the differences to unique calcium channels.115 For GM1 cats, the Ca2+ influx through the fast-phase channels was half that of normal cats. Calcium influx through slow-phase channels was also significantly reduced, with more than three times the calcium concentration required to reach half maximal influx. Similar defects were noted for the Na+-dependent efflux of Ca,2+ with significantly lower values recorded for GM1 cats compared to controls.115
Synaptosomes from GM1 cats further revealed deficits in neurotransmitter uptake and reduced membrane fluidity. The uptake of 3H-glutamate,3H-GABA, and3H-norepinephrine in GM1 synaptosomes was reduced by up to 75% of normal values, while the overall concentration of neurotransmitters and activity of synthetic enzymes were not significantly altered. Singer et al (1982) concluded that neuronal dysfunction resulted from disturbances to the synaptic glycolipid composition and the associated impairment of neurotransmitter uptake mechanisms.119 Indeed, abnormal synaptic membrane structure and function is a consequence of deficient GM1 ganglioside catabolism. Attenuated β-gal activity leads to increased GM1 ganglioside and cholesterol at the synaptosomal membrane and as a consequence, detergent-resistant lipid microdomains.120 Further, inhibitory postsynaptic potentials (IPSP) evoked in pyramidal neurons of GM1 cats, differed markedly in their duration and rise time. For GM1, the duration of IPSP was significantly reduced at 7–40ms compared to that of normal neurons (80–150ms). A reduction in rise time was also noted, with GM1 rise time approximately one-quarter of normal IPSP. These measurements were consistently recorded from ganglioside-laden pyramidal neurons regardless of meganeurite formation.121 However, the altered electrical properties may be a consequence of disruptions to neuronal circuitry caused by meganeurites and ectopic dendrites.44,122
Autophagy
In the GM1 mouse, enhanced autophagy and mitochondrial dysfunction aligned with disease progression.123 The autophagosomal marker LC3II was significantly increased at 10 months in the GM1 mouse compared to WT controls. A concurrent increase in beclin-1, a protein critical to autophagosome assembly, along with Akt, Erk, and mTOR phosphorylation was also noted for GM1 mice at 10 months. In GM1 mice, mitochondria were smaller, fragmented, or circular with decreased membrane potential and cytochrome c oxidase activity. In GM1 astrocytes, a significantly higher percentage of dead cells was detected following oxidative stress compared to WT. Toxicity from oxidative stress was suppressed with the addition of ATP, 3-MA (an inhibitor of autophagy) or z-VAD-fmk (a pan-caspase inhibitor), while rapamycin treatment had no impact on cell survival. Parallel analyses of WT astrocytes suggested autophagosome-lysosome fusion underlies the accumulation of fragmented mitochondria.123 The cellular events that lead to initial mitochondrial dysfunction have not yet been defined, but enhanced autophagy likely contributes to the induction of these phenomena.
Neurotrophic Factor Activity
GM1 ganglioside enhances the nerve growth factor (NGF)-induced autophosphorylation of the tropomyosin receptor kinase (Trk) and receptor dimerization.66 In GM1, increased associations between Trk and GM1 ganglioside lead to significant elevation of neurotrophic activity.96 Increases in phosphorylated Trk (pTrk) in β-gal-/- mice are in part attributed to the higher concentration of GM1 ganglioside at cell membrane lipid rafts. Ubiquitination of Trk, which mediates receptor protein levels, was significantly elevated in β-gal-/- mice. Internalized and ubiquitinated Trk co-localized with lysosomal associated membrane protein 2 (LAMP2) in the cerebral cortex, and cell fractionation defined the accumulation and redistribution of Trk to the endo-lysosomal network (ELN). GM1 ganglioside had a similar pattern, with impaired catabolism contributing to its storage within the ELN of β-gal-/- mice.96 In the p.R201C GM1 mouse, the pharmacological chaperone N-octyl-4-epi-β-valienamine (NOEV) reduced the accumulation of pTrk and GM1 ganglioside, highlighting the utility of chemical chaperones in correcting neuropathology.92,96
Neuroinflammation
Central nervous system (CNS) inflammation plays a prominent role in the pathogenesis of the gangliosidoses.124 In the symptomatic β-gal-/- mouse, MHC class II staining was more pronounced in the thalamus and brain stem than the cerebral cortex, which aligned with areas of significant neuropathology. Fas staining was detected at moderate levels and supported the defined apoptotic events underlying neurodegeneration.113,124 Further, analysis of nitrotyrosine positive cells throughout the β-gal-/- mouse brain supported complementary analyses of mitochondrial dysfunction.123,124 Other markers of neuroinflammation including expression of TNFα, TNFβ1, and IL1β, microgliosis and astrogliosis occurred concurrently with neurological decline. In the β-gal-/- mouse, blood-brain barrier integrity was also significantly altered, and permeability changes were attributed to CNS immune activation.124 Defects in phagocytosis and ganglioside pro-inflammatory properties may dictate CNS immune activation, with clearance of apoptotic neurons potentially initiating pathogenesis.
In a novel cell culture model based on induced pluripotent stem cell (iPSC) technology, neural progenitor cells from GM1 patient-derived iPSCs demonstrated GM1 ganglioside-associated activation of inflammasomes.125 Transcriptional upregulation of inflammasome components including inflammatory caspases and pro-inflammatory cytokines was partially suppressed upon treatment with a caspase-1 inhibitor or an IL1β antagonist. Both inhibitors reduced morphological abnormalities and promoted rounding and enlargement of GM1 neurospheres. These studies define a novel target pathway and highlight the evolving complexity of GM1 cellular pathogenesis.123,125
Histopathology
Accumulation of GM1 ganglioside in lysosomes leads to lysosomal swelling and cellular dysfunction. Universal histopathological examination of the brain and visceral organs of GM1 patients reveals enlarged neurons characterized by cytoplasmic inclusions, periodic acid Schiff (PAS) staining, and hepatocellular vacuolation.24,126–130 The most prominent cytoplasmic inclusions are swollen lysosomes or lysosome residual bodies termed membranous cytoplasmic bodies (MCBs) and/or granular storage, which displace normal organelles.46,131–134 PAS staining detects the oligosaccharide chain of gangliosides as well as other polysaccharides, glycoproteins, and glycolipids, and is used as a reproducible qualitative method for the detection of storage in the gangliosidoses.
Within neurons, Nissl substance is condensed in the perinuclear region and ultrastructural analysis reveals ER compression.113,127,134,135 Pyramidal and medium spiny neurons both exhibit meganeurites (enlargements of the axon hillock) while ectopic dendritogenesis is restricted to specific populations of excitatory neurons and neuroaxonal dystrophy predominantly affects GABAergic neurons.121,122,132,136,137 In most neurons, storage is confined to the perikarya, while extension of storage into meganeurites of cortical pyramidal neurons and dendrites of Purkinje cells also occurs and may be a reflection of the amount of storage in each cell type.127,132,135,137,138 Functional impairments in synaptic transmission, myelin deficits (discussed below), and neuron-specific pathology likely underlie neurodegeneration and the emergence of clinical neurological features.121,122,128,139,140
Consistent with neuroimaging (below), pathology is prominent in the basal ganglia. Atrophy of the caudate nucleus and putamen, with conspicuous swollen neurons by microscopic examination, was consistent with neuron loss and gliosis.16,27,32,127,141 Pathology in this region was most pronounced in the posterior portion of the caudate nuclei in three patients with adult/chronic GM1.127,142,143
Myelin Pathology
Recent literature has addressed the role of myelin pathology in GM1 and other neuropathic LSDs. Studies involving children,24,126,128 dogs,144 mice,145 and cats146 with GM1 have universally reported decreased myelin in the CNS, with a reduction in Luxol Fast Blue staining being the most commonly reported evidence of this pathology.24,126,146 Some studies have measured myelin using X-ray diffraction145 or noted ultrastructural abnormalities with transmission electron microscopy (TEM) such as fewer myelinated axons126 and unraveling of myelin sheaths (Figure 1A and B).146
 |
Figure 1 Transmission Electron Microscopy (TEM) images of feline CNS tissue. Normal axon in cross-section surrounded by thick myelin sheath (A). Multiple axons in cross-section of GM1 white matter, surrounded by disjointed and unravelling myelin layers (B). Normal oligodendrocyte with round nuclear envelope and prominent cytocavitary network (C). GM1 oligodendrocyte with multiple swollen mitochondria (black arrows) and irregularly-shaped nuclear envelope (white arrow) (D). Scale bars are 1μm. Transmission electron micrographs in panels A, B, and D, reproduced with permission Gray-Edwards HL, Maguire AS, Salibi N, et al. 7T MRI predictsamelioration of neurodegeneration in the brain after AAV genetherapy. Mol Ther - Methods Clin Dev. 2020;17:258–270.146 © 2019 Elsevier. |
The mechanism(s) behind myelin abnormalities and their effects on disease pathogenesis is an emerging discussion topic in the literature, with three main hypotheses considered: dysmyelinogenesis (failure to form myelin properly), primary demyelination (dysfunction or destruction of myelin after proper formation), or secondary demyelination (due to axonal degeneration). Since neuronal storage is an obvious and severe lesion, secondary demyelination resulting from neuroaxonal pathology was formerly the sole explanation.126 This is further supported by the near-universal agreement that oligodendrocytes do not store GM1 ganglioside24,126,146 and the resulting hypothesis that unaffected oligodendrocytes would produce healthy myelin that is destroyed by neuronal pathology. However, other signs of severe oligodendrocyte pathology have become evident with ultrastructural (Figure 1C and D) and histological evaluation, including decreased numbers of mature oligodendrocytes24,126 and increased cell death (TUNEL-positive).24 Most authors now hypothesize that dysmyelinogenesis plays a more prominent role in myelin pathology than previously thought, although its extent is still unknown. A confounding factor is that myelination in normal organisms occurs postnatally, during the same developmental stages that GM1 storage causes major deficits. Similar white matter pathology has been documented in closely related LSDs (Sandhoff disease,147 Niemann Pick A148, Niemann Pick C149, and Krabbe disease150), so detailed comparison among these diseases may provide valuable insight.
Biomarkers
Noninvasive or minimally invasive methods to track GM1 disease progression include blood-based biomarkers, cerebrospinal fluid biomarkers, magnetic resonance spectroscopy, and neuroimaging.3,22,25,59,60,146,151,152 For neuropathic diseases, analysis of biomarkers can provide a picture of disease progression and insight into the efficacy of therapeutics.
Serum and CSF Biomarkers
Feline and canine GM1 models have been used to identify biomarkers in the CSF and serum that align with disease progression.60,153 In Shiba dogs with GM1, aspartate aminotransferase (AST), lactate dehydrogenase (LDH), GM1 ganglioside, neuron-specific enolase (NSE), and myelin basic protein (MBP) were all significantly elevated in the CSF. Though some of the markers eventually reached a plateau, they increased concordant with disease progression, supporting their use as indicators of neurological decline.153 In cats with GM1, AAV-mediated GLB1 delivery restored elevated AST and LDH to normal values in the CSF of animals two to three years post-treatment. Similar elevation of AST was observed in the serum of both GM1 cats and late-infantile GM1 human patients. Other serum biomarkers identified included calcium, creatinine, and albumin, all of which were significantly decreased in cats with GM1. The levels of calcium, creatinine, and albumin were variable between GM1 subtypes of human patients. Hypocalcemia was present in infantile GM1 patients, while late-infantile and juvenile patients did not have a significant change in their calcium levels.60 A single adult GM1 patient also presented with reduced calcium levels;141 however, analysis of adult GM1 patient biomarkers in the literature is incomplete. Creatinine showed the inverse pattern, with significantly lower levels in the serum of late-infantile and juvenile patients relative to infantile GM1. Albumin levels were decreased to the greatest extent in infantile GM1 patients and appeared to correspond with disease severity. These analytes identified in CSF and serum represent valuable biomarkers that can be easily measured using clinically available tests, providing a clearer picture of disease progression (Table 1).60
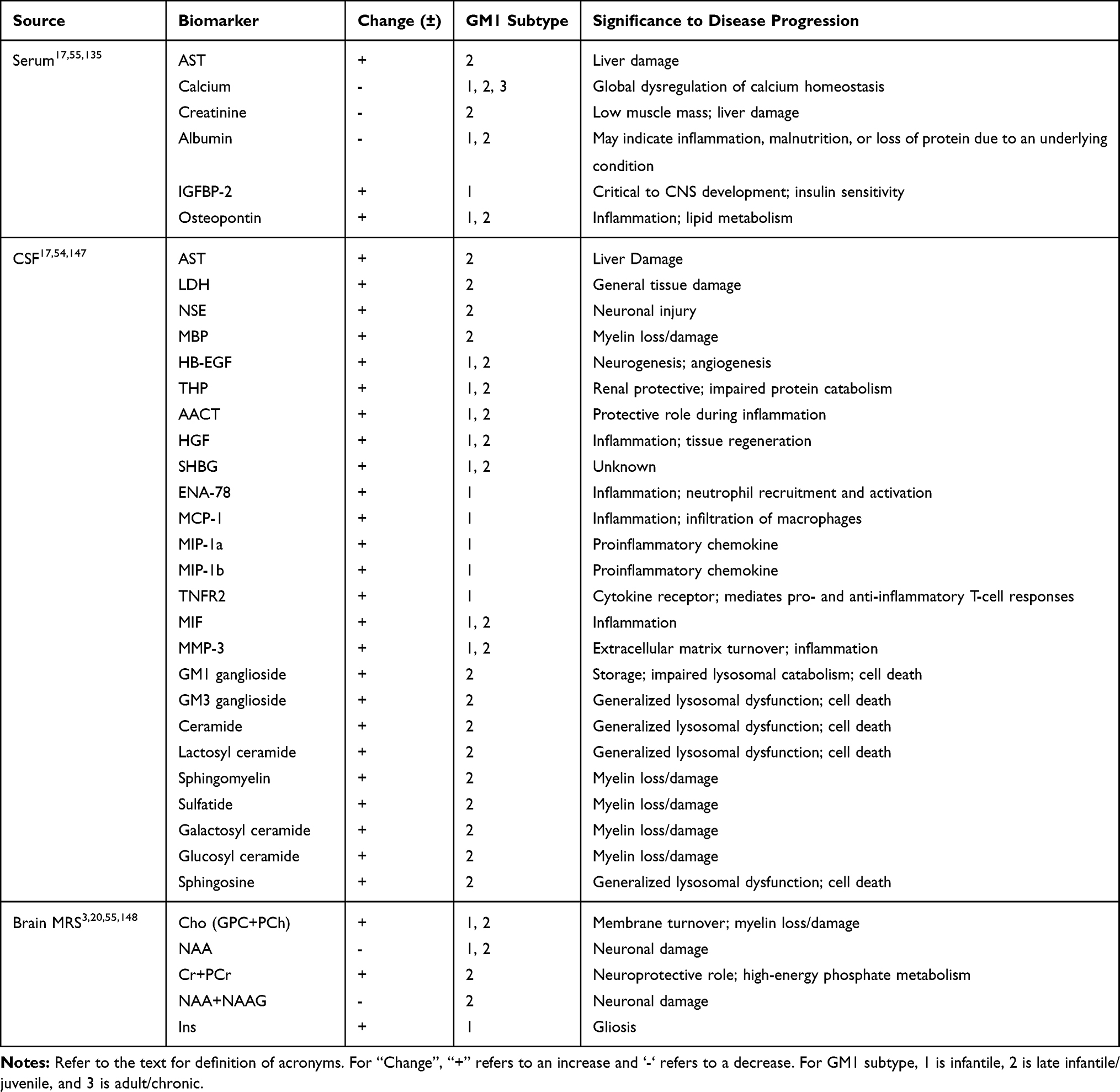 |
Table 1 Summary of GM1 Biomarkers Identified in the Serum, CSF, and by Brain MRS of Animal Models and Human Patients |
Other biomarkers identified through complex lipidomics are not easily quantified by routine laboratory assays. However, their inclusion contributes to our understanding of disease progression and the efficacy of therapeutic regimens. Gray-Edwards et al (2017) examined 36 sphingolipids and their derivatives in the CSF of GM1 cats at humane endpoint, in AAV-treated GM1 cats, and in normal age-matched controls.59 Of the lipids examined, 16 had significant alterations. Elevated lipids included GM1 ganglioside, GM3 ganglioside, ceramide, lactosyl ceramide, sphingomyelin, sulfatide, galactosyl ceramide, glucosyl ceramide, and sphingosine (Table 1). For many metabolites, AAV treatment restored values to within the normal range, highlighting the efficacy of gene therapy in the treatment of neuropathic lysosomal storage diseases and the utility of lipidomics in assessing pathophysiology.59
Utz et al (2015) quantified 72 analytes from CSF and serum of human patients with infantile, late-infantile, and juvenile GM1 gangliosidosis.22 From this analysis, twelve CSF and two serum candidate biomarkers were identified. CSF from infantile GM1 patients had elevated heparin-binding EGF-like growth factor (HB-EGF), Tamm-Horsfall urinary glycoprotein (THP), alpha-1-antichymotrypsin (AACT), hepatocyte growth factor (HGF), sex hormone-binding globulin (SHBG), epithelial-derived neutrophil-activating protein 78 (ENA-78), monocyte chemotactic protein 1 (MCP-1), macrophage inflammatory protein-1 alpha (MIP-1α), macrophage inflammatory protein-1 beta (MIP-1β), tumor necrosis factor receptor 2 (TNFR2), macrophage migration inhibitory factor (MIF), and matrix metalloproteinase-3 (MMP-3), while a single late-infantile GM1 patient showed elevation only in HB-EGF, THP, AACT, HGF, SHBG, MIF, and MMP-3. In serum samples, infantile GM1 patients had elevated insulin-like growth factor-binding protein 2 (IGFBP-2) and osteopontin, while the single late-infantile patient had a significant increase only in osteopontin. These CSF and serum biomarkers are summarized in Table 1. Elevation of inflammatory markers in GM1 reflects disease severity and highlights the role of inflammation in CNS pathology.22
Brain MRS
Magnetic resonance spectroscopy (MRS) has emerged as a minimally invasive analytical tool for the assessment of disease progression in the brains of living patients (Figure 2). With this spectroscopic method, which is coupled to magnetic resonance imaging (MRI), suppression of brain water and lipid signals allows for the quantification of metabolites in specific brain regions, localizing dysfunctional processes biochemically and providing a picture of disease severity (Table 1).3,25,60,152,154
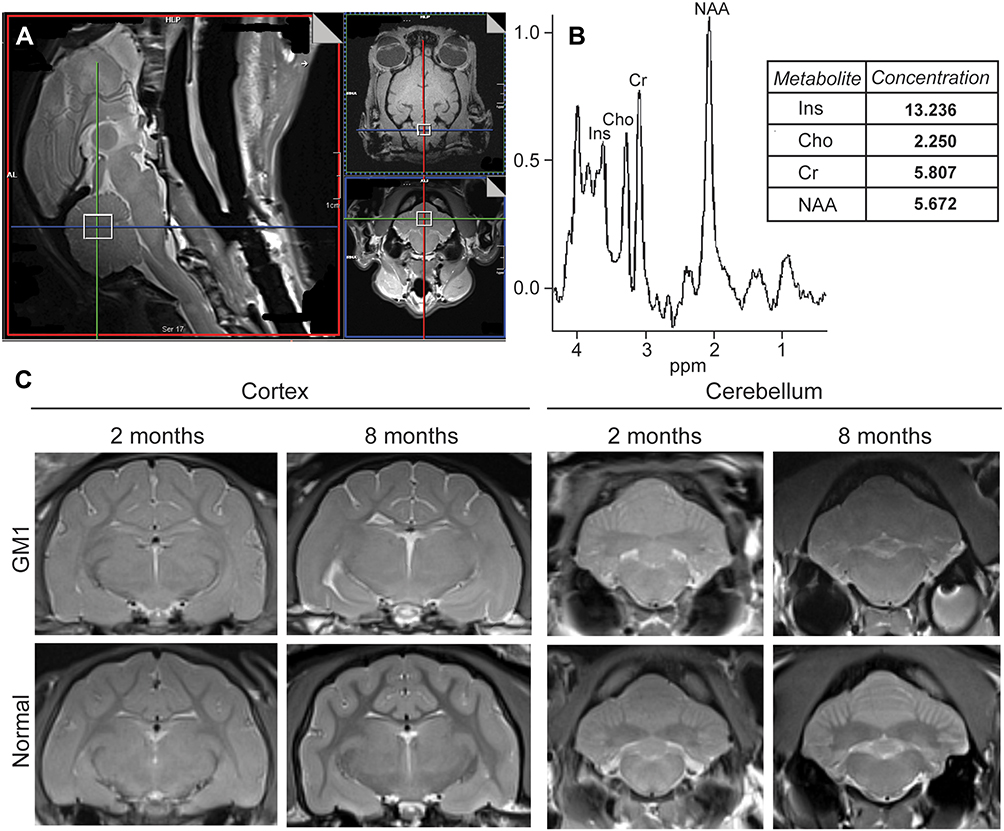 |
Figure 2 An example of data collected from Magnetic Resonance Spectroscopy (MRS) and MRI of normal and GM1 cats. Cerebellar voxel defined in the cat for measurement of 1H MRS (open white square) (A). Example 1H spectrum with metabolites labeled and data output for the respective metabolites (B). Transverse T2w images of the cerebral cortex and cerebellum in normal and GM1 cats at 2 and 8 months of age. The isointensity of gray and white matter is prominent at 8 months for both the cerebral cortex and cerebellum of GM1 cats compared to normal controls. In GM1 cats, atrophy is mild and unilateral ventriculomegaly is apparent in the provided example (also occurs in normal controls) (C). |
Erol et al (2006) first described the use of MRS in an infantile GM1 patient noting an increase in choline (Cho), which includes glycerophosphocholine (GPC) and phosphocholine (PCh), and a decrease in N-acetylaspartate (NAA) when normalized to creatine (Cr) in the thalamus relative to normal controls.25 (When normalizing NAA to Cr, it is important to verify that Cr itself does not to change with disease progression, as in the feline GM1 model discussed in the following paragraph.) A study of a different infantile GM1 patient similarly identified a decrease in NAA, while also noting an increase in myo-inositol (Ins) in the basal ganglia.154 Regier et al (2016) performed a more comprehensive MRS analysis of fifteen patients having late infantile/juvenile GM1 by quantifying metabolites at four brain regions: superior cerebellar vermis, left thalamus, left centrum semiovale, and midline parietal gray matter. In all four regions, a significant decrease in NAA was measured, which continued to decrease over time in concordance with disease progression.3 The consistent decrease in NAA measured in infantile and late infantile/juvenile GM1 subtypes is suggestive of axonal damage or a decrease in neuroaxonal integrity.3,25 Other metabolic alterations, including elevation of Ins, are indicative of gliosis, which is supported by histopathological findings.154 Excess membrane turnover, or the inability to assemble choline-containing molecules in myelin, may explain the elevation in brain Cho and the overall isointensity of white and gray matter in advanced GM1.25 One challenge noted in the acquisition and comparison of data from affected individuals is the absence of values from age-matched controls. Although MRS values for several metabolites from pediatric patients have been published, the brain region-specificity is noticeably absent.152 Regier et al (2016) attributed the absence of suitable reference samples to the difficulty in justifying “the risk of sedation for the purpose of studying normal children”.3
In the feline model of late-infantile/juvenile GM1, Gray-Edwards et al (2017) examined six brain regions and found metabolite levels consistent with previous human studies.60 In all regions, Cho was significantly elevated for GM1 cats at 8 months, which is at the time of humane endpoint. A significant decrease in NAA was noted in four out of the six regions, including the thalamus, temporal cortex, occipital cortex, and cerebellum. Overall, the regions with the most significant metabolic abnormalities were the thalamus and the cerebellum. In both structures, Ins and Cho were significantly elevated, while NAA and NAA+NAAG (N-acetylaspartylglutamate) were significantly decreased. The cerebellum also had some unique metabolic changes, with a significant increase in Cr+PCr (phosphocreatine).60
Analyses of brain metabolites in affected children using MRS represent a promising minimally invasive tool. However, prolonged sedation of any child for the purpose of collecting MRS data warrants ethical consideration. If the value of MRS data is rooted in the assessment of therapeutic efficacy, other biomarkers identified in the blood and CSF, neuroimaging strategies outlined below, and neurological assessments, represent alternative modalities, posing fewer risks to the patient. Nevertheless, MRS provides a minimally invasive, quantitative method of evaluating disease status directly in the brain itself.
Neuroimaging
Atrophy
While brain atrophy has been a common neuroimaging finding in case studies of all subtypes of GM1, two recent multi-patient studies have applied modern techniques of volumetric MRI analysis to quantify and localize it. Nestrasil et al (2018) found significantly decreased brain volumes across multiple regions in a longitudinal study of 4 infantile, 1 late infantile, and 2 juvenile patients.27 Regier et al (2016) also quantified MRI volume and found more substantial changes in late infantile than juvenile patients. Progressive atrophy of the cerebrum, cerebellum, and hippocampus was common in late infantile patients but much less frequent in juvenile patients.3 A detailed, qualitative MRI study of an adult patient with GM1 localized cerebral atrophy to the frontal and temporal cortices. Non-quantified generalized atrophy was also noted on CT and MRI in 3 other case studies of varying subtypes.13,16,31 Similar atrophic findings are present in a feline model of juvenile GM1 but are minimized by intracranial gene therapy treatment.60
Hypomyelination
A clear distinction regarding myelin loss exists between GM1 subtypes, in which studies of infantile and late infantile patients almost universally report hypomyelination, while studies of juvenile and adult/chronic patients either omit a discussion of white matter entirely or state that it is normal. Hypomyelinated white matter (defined for this review as simply a reduction in myelin content compared to normal controls) appears diffusely hyperintense on T2-weighted MRI, hypointense on T1-weighted MRI, or hypo-attenuated on CT. Most studies reported these changes in cerebral cortical white matter,3,25,128,154–161 though some also specified lesions in the cerebellar cortex154,158 or deep white matter.3,159 Similar alterations are found in canine and feline models of GM1,60,162 with normalization of white matter/gray matter MRI intensities after intracranial gene therapy in GM1 cats. Van der Voorn et al (2005) reported radial stripes of hypointensity within hyperintense cerebral white matter (antemortem T2-weighted and postmortem FLAIR MRI) in a 7-month-old infantile patient that correlated to areas of weak Luxol Fast Blue staining on histopathological evaluation.128 This pattern was also noted by Brunetti-Pierri et al (2008) in a 10-month-old infantile patient, though concurrent CNS histopathology was not discussed.154 While several authors proposed causes for hypomyelination such as demyelination,156 delayed myelination,157 or leukomalacia, current neuroimaging technology is unlikely to single-handedly elucidate this phenomenon, and a multi-modal approach is needed.
Thalamus
Similar to white matter lesions, abnormal findings in the thalamus have only been reported in patients with the infantile or late infantile subtypes of GM1 gangliosidosis. The thalamus appears hypointense on T2-weighted MRI,3,25,156,157,159–161 hyperintense on T1-weighted MRI,3,25,154,157,160,161 or hyper-attenuated on CT.25,157,160 Proposed causes of these lesions include calcium deposition,25,156,157 an excessively hydrophobic environment,25,156,157,161 or abnormal myelination.161 While the thalamic imaging abnormalities are opposite those in white matter (ie, hypointense on T2-weighted MRI rather than hyperintense), the strict occurrence in only early-onset patients as well as the thalamic function of integrating gray matter nuclei and white matter tracts could indicate myelin-related pathological processes.
Basal Ganglia
While neuroimaging abnormalities within the basal ganglia are common across all subtypes of GM1,3,13,16,29,31–33,154,156,157,159,160,163–166 a hallmark lesion is posterior putaminal hyperintensity on T2-weighted MRI scans.3,13,16,29,31,33,164–166 Proposed causes of this change include neuronal loss with secondary gliosis,164 iron deposition,165 and ganglioside storage,16 with Uyama et al (1992) postulating that this lesion could be the cause of severe dystonia in adult/chronic patients.16 Lesions of the individual basal ganglia appear to be independent from each other, since the caudate follows a similar intensity pattern to the putamen (hyperintense on T2-weighted and proton density images), but the globus pallidus and substantia nigra display the opposite pattern (hypointense on T2,29,31,32,159 susceptibility,29 and diffusion-weighted31 scans, hyperintense on T1-weighted scans31). An explanation for these differences has yet to be offered, which could be partially due to the tendency of some studies to generalize observed intensity changes as affecting “basal ganglia”, rather than specifying individual structures. Because this group of nuclei is functionally diverse and GM1 gangliosidosis causes different neuroimaging changes across patients, future studies should distinguish these structures from each other when reporting lesions.
Management
There are no current effective FDA-approved treatments for GM1, though advances in gene therapy are rapidly gaining traction with human clinical trials underway. Targeted research approaches for the treatment of GM1 typically align with one of the following areas: substrate reduction therapy (SRT),30,99–102,167,168 enzyme enhancement therapy (EET),55,91,93,94,169–171 stem cell transplantation,83–86 enzyme replacement therapy (ERT),56,87,89,90 or gene therapy48,57,60–64 (Figure 3). These therapeutic approaches aim to slow clinical progression, increase quality of life, and extend life expectancy through reduction of GM1 ganglioside content, enhancement of residual β-gal activity, or introduction of an exogenous active β-gal cDNA or enzyme. Developing therapeutics for CNS diseases is challenging because of the semi-permeable blood-brain barrier (BBB), which must be penetrated for any potential treatment to be effective.
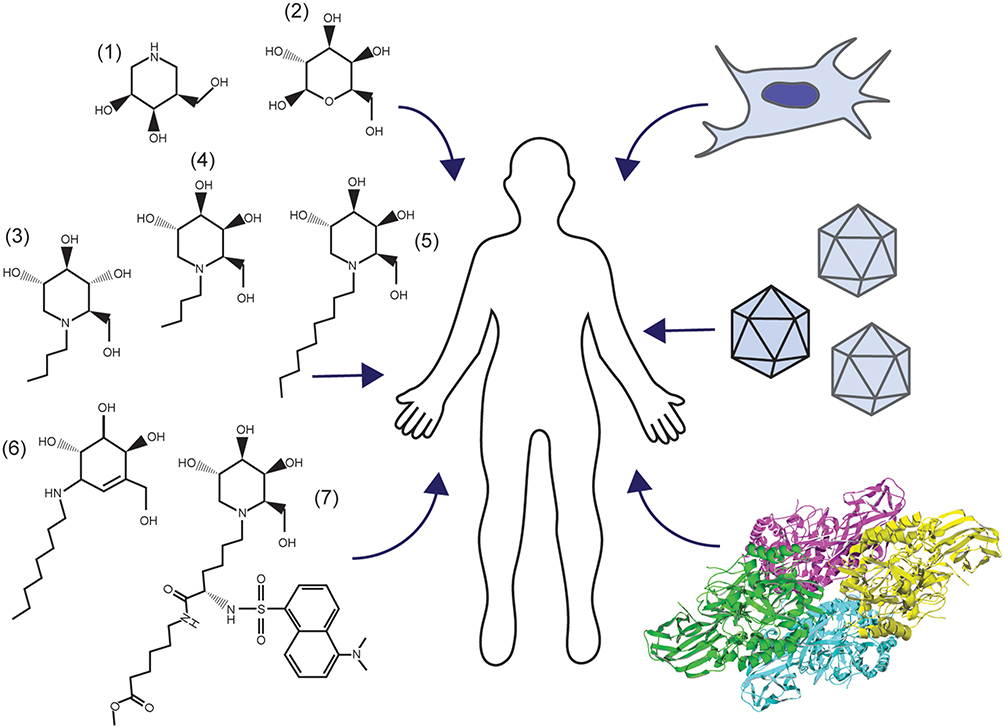 |
Figure 3 Management of GM1 Gangliosidosis. Clockwise, beginning on the left: Pharmacological chaperones for enzyme enhancement and small molecules for substrate reduction therapy (1–7), stem cell transplantation, gene therapy, and enzyme replacement (RCSB PDB ID: 3THC).12 |
Substrate Reduction Therapy
In GM1, SRT relies on potent inhibitors of GSL biosynthesis.99–102 However, inhibiting ganglioside synthesis without a means of degrading previously accumulated substrate may minimize the impact of SRT on clinical disease. Thus, some degree of residual β-gal activity is thought to be essential, limiting the utility of SRT to non-infantile patients.30 The N-alkylated imino-sugar derivatives N-butyl-deoxynojirimycin (NB-DNJ), which is FDA approved for the treatment of Gaucher disease type,100,172,173 and N-butyl-deoxygalactonojirimycin (NB-DGJ)102 are promising candidate inhibitors of glucosylceramide synthase, which catalyzes the first committed step of GSL biosynthesis.100,173
Miglustat, N-Butyl-Deoxynojirimycin (NB-DNJ)
NB-DNJ is known commercially as Miglustat and marketed under the trade name Zavesca.100 Identified as an inhibitor of the N-linked oligosaccharide processing enzymes α-glucosidase I and II within the ER, NB-DNJ is also a potent inhibitor of HIV replication and syncytium formation.174 As an inhibitor of glucosylceramide synthase, NB-DNJ effectively blocks the synthesis of glucosylceramide, a target substrate of glucocerebrosidase.99,100,173 In patients with Gaucher disease Type 1, deficiency of glucocerebrosidase leads to lysosomal storage of glucosylceramide in peripheral tissues.99,100 Interestingly, NB-DNJ effectively crosses the BBB, making it a candidate for the treatment of other LSDs with GSL and CNS involvement, including GM1.100,101,168,175–177 β-gal-/- mice treated with NB-DNJ showed no significant increase in survival compared with untreated mice due to exacerbation of intrinsic gastrointestinal tract dysfunction.101 However, there was significant functional improvement and a decrease in neuroinflammation.101,124 In three Italian patients with juvenile or adult forms of GM1 gangliosidosis, NB-DNJ produced clear improvements in clinical presentation suggesting that the reversal of disease progression is possible in certain populations.30
Despite these clear positive outcomes, the exact therapeutic mechanism remains under investigation. Whether NB-DNJ solely acts to inhibit GSL biosynthesis or has a secondary supportive role as a pharmacological chaperone for defective β-gal is unknown.172 Treatment of individuals, animals, or cell lines of varying genotype with Miglustat has not been explored comprehensively. Therefore, whether Miglustat is effective for all patients with attenuated forms of GM1 remains an area of active investigation. Nevertheless, as foreshadowed by the mouse studies, gastrointestinal side effects are common in patients, causing many to discontinue use of Miglustat even if mild efficacy is achieved.
N-Butyl-Deoxygalactonojirimycin (NB-DGJ)
NB-DGJ, a galactose derivative, similarly inhibits glucosylceramide synthase. β-gal‐/‐ mice treated with NB-DGJ had a dose-dependent depletion of GSLs in visceral organs following treatment for 10 days. Additionally, β-gal‐/‐ mice had a 50% increase in life span compared with untreated controls and a significant decrease in neuroinflammation.101 Neonatal mice treated with NB-DGJ had a significant reduction in brain GM1 ganglioside.102,168 These results highlight the potential of SRT as an effective early intervention for GM1 gangliosidosis.
Pharmacological Chaperones
Pharmacological chaperones were originally designed as competitive inhibitors of β-gal, promoting proper folding and stabilizing the enzyme in its active state for safe transport to the lysosome.91,93,178–180 Unstable mutant enzymes in the absence of PCs are rapidly processed by ER-associated protein degradation. In some cases, misfolding and aggregation of mutant β-gal leads to ER stress, the UPR, and apoptosis.113 However, certain mutations to the GLB1 gene result in early termination and nonsense-mediated decay, or protein structures incapable of stabilization.1,4 Also, chaperone effects are often mutation-specific, so evaluation of each chaperone in the context of each genotype is critical (Table 2).55,93–95,97,98,178,181
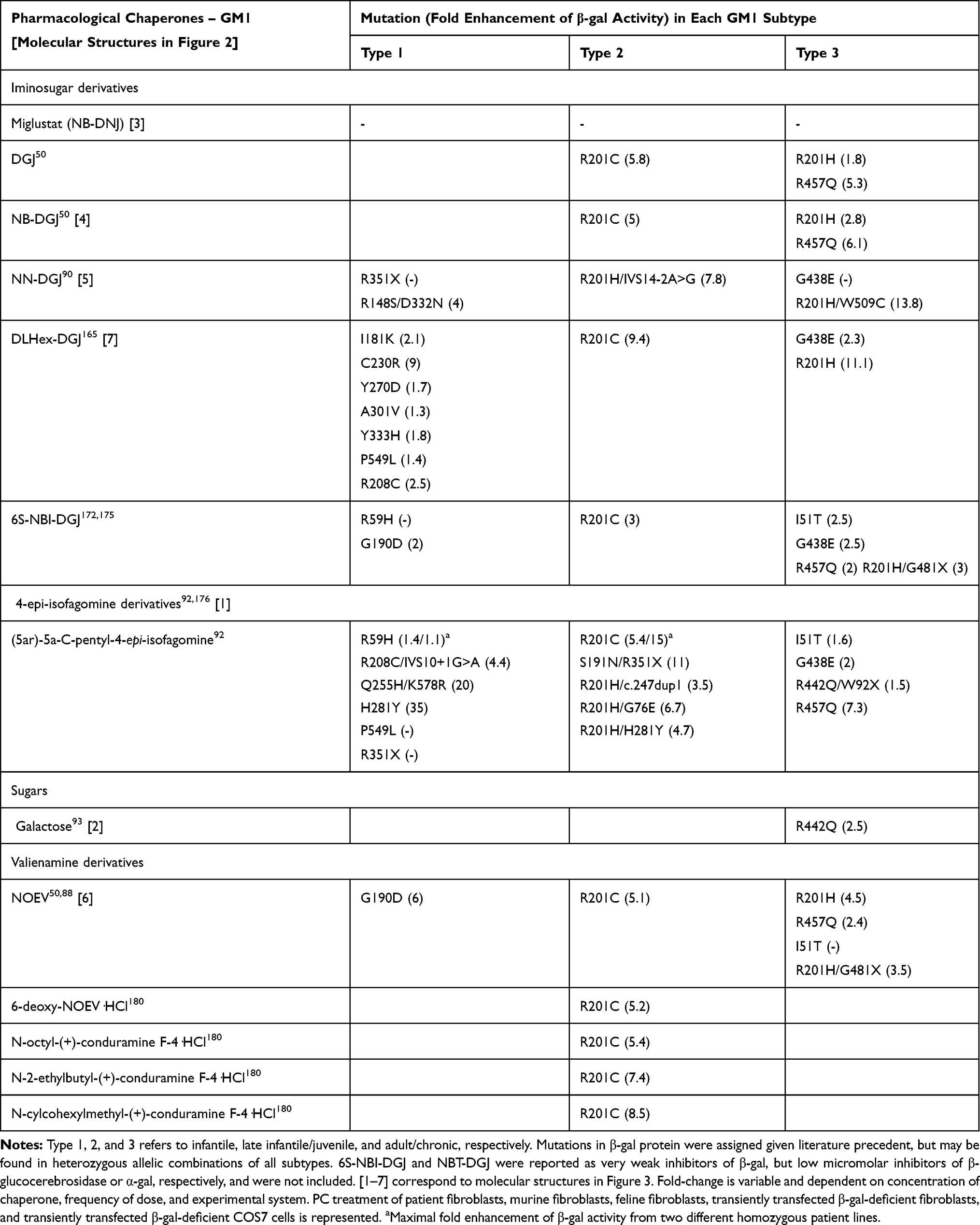 |
Table 2 Pharmacological Chaperone Influence on Mutant β-Gal Activity |
Most chaperones are imino-sugar, 4-epi-isofagomine, or valienamine derivatives.91,92,97,101,182 The most heavily studied small molecules, NB-DNJ and NB-DGJ, are imino-sugar derivatives of glucose and galactose, respectively.30,183 Although NB-DNJ and NB-DGJ have been marketed for SRT through inhibition of glucosylceramide synthase, β-gal enhancement may more clearly define the decrease in ganglioside accumulation.30,101,102,172,183 Indeed, NB-DNJ treatment of COS7 cells transiently transfected with different Gaucher mutations revealed significant β-glucosidase enhancement.172 In a study examining the chaperone capabilities of NB-DNJ stereoisomers, D-NB-DNJ (Miglustat) was highlighted as a weak inhibitor of β-gal.184 NB-DGJ treatment of mouse fibroblasts expressing human β-galactosidase mutations associated with attenuated forms of GM1 resulted in significant increases in β-gal activity and affirmed its utility in both SRT and EET.55 A thorough analysis of the inhibitory and enzyme enhancement potential of these imino-sugar derivatives in the context of mutant lysosomal hydrolases is warranted given the structural similarities to known competitive inhibitors of β-gal including the N-alkylated imino-sugar derivative N-nonyl-deoxygalactonojirimycin (NN-DGJ).95
Galactose
Treatment of p.T329A/p.R442Q GM1 patient fibroblasts with galactose, which is bound to the active site of β-gal during GM1 ganglioside cleavage, led to a 2.5-fold increase in β-gal activity. Transient transfection of β-gal-deficient COS7 cells with GLB1 having either mutation revealed that p.R442Q was the target of enzyme enhancement. Interestingly, normal fibroblasts treated with galactose had a significant reduction in β-gal activity to 7.8% of normal values.98 Though the effect of galactose in GM1 patients is unknown, it provided substantial improvement in cardiac function in a patient with Fabry disease (α-galactosidase A deficiency). Cardiac pathology was so severe in this patient that he was awaiting a heart transplant. Intravenous infusion of galactose increased enzyme activity 2.8-fold over baseline, decreased cardiac mass by 20%, and improved left ventricular ejection fraction from 32% to 55%. The patient was able to return to full-time work as a bus driver due to galactose infusion therapy.185
Sugar Mimetics as Chaperones
N-Nonyl-Deoxygalactonojirimycin (NN-DGJ)
The N-alkylated imino-sugar derivative NN-DGJ was identified as a potent inhibitor of β-gal in vitro with an IC50 value of 0.12μM, nearly 30 times lower than NB-DGJ and more than 150 times lower than deoxygalactonojirimycin (DGJ).95 Though therapeutic efficacy was achieved at concentrations ~15 fold more than the IC50, a foundational hypothesis of enzyme enhancement therapy is that, once the chaperone:enzyme complex reaches the lysosome, supranormal concentrations of the native substrate will displace the chaperone.
In GM1 fibroblasts from cats expressing p.R483P mutant β-gal, analogous to p.R482H in humans, a 6-fold increase in β-gal activity and effective lysosomal targeting of feline β-gal was detected after NN-DGJ treatment. The enhancement of other lysosomal enzymes was not detected, suggesting NN-DGJ acted as a β-gal-specific chaperone. These results described the first large animal model with a responsive genotype to EET and clinical phenotype consistent with GM1 disease progression, supporting in vivo studies for EET validation.95
Methyl 6-{[N2-(Dansyl)-N6-(1,5-Diyl)-L-Lysyl]amino} Hexanoate 1-Deoxygalactonojirimycin (DLHex-DGJ)
DLHex-DGJ, an imino-sugar derivative, was synthesized and analyzed comprehensively for PC activity of mutant β-gal in GM1 fibroblasts. Of the 13 fibroblast lines examined, four had a significant increase in β-gal catalytic activity, normalization of β-gal transport to the lysosome, and efficient lysosomal processing of β-gal precursors.171
5N,6S-(N’-Butyliminomethylidene)-6-Thio-1-Deoxygalactonojirimycin (6S-NBI-DGJ)
The sp2-iminosugar inhibitor 6S-NBI-DGJ, which has a rigid bicyclic core and a hydrophobic aglycone substituent, binds specifically to the active site of β-gal with an IC50 of 32μM181. Fibroblasts from human GM1 patients expressing p.R201C or p.R458Q mutant β-gal had significantly increased enzymatic activity after treatment. In contrast to NOEV (discussed below), 6S-NBI-DGJ significantly enhanced β-gal activity in fibroblasts also expressing p.I51T or p.G438E mutations.181 Of the 88 mutations examined by transient transfection of β-gal-deficient COS7 cells, 24 responded positively to EET with 6S-NBI-DGJ. Further, studies in mice expressing human p.R201C mutant β-gal showed that 6S-NBI-DGJ is capable of crossing the BBB, enhancing β-gal activity throughout the CNS, and reducing GM1 ganglioside content.178,181
Valienamine Derivatives as Chaperones
N-Octyl-4-Epi-β -Valienamine (NOEV)92
Modification of a glucocerebrosidase inhibitor through removal of its ceramide moiety and substitution with aliphatic chains led to the identification of N-octyl-4-epi-β-valienamine (NOEV) as a potent competitive inhibitor of β-gal and a candidate for EET. In human and mouse GM1 fibroblasts having p.R201H or p.R201C mutations, respectively, NOEV administration led to a marked increase in β-gal activity and subsequent reduction in GM1 ganglioside content.55 β-gal activity was normalized to a lesser extent in p.R457Q, p.W273L, and p.Y38H GM1 patient cell lines. Perhaps most remarkably, NOEV achieved similar restorative effects at 2,500-fold lower concentrations than DGJ and NB-DGJ.55 NOEV treatment of β-gal-deficient fibroblasts transiently transfected with each of 94 different human GLB1 mutations, identified 22 missense mutations that responded therapeutically.93,178 Oral administration of NOEV for one week to a mouse model of late infantile/juvenile GM1 gangliosidosis (p.R201C human β-gal mutation) revealed efficient distribution throughout the CNS and peripheral tissues with a congruent increase in β-gal activity.55 Long-term oral administration of NOEV to p.R201C mice increased β-gal activity throughout the brain and viscera, reduced GM1 ganglioside content, and prevented neurological deficits. No adverse effects were identified in long-term studies, supporting NOEV as a candidate for EET in humans.91,94,96
N-Substituted (+)-Conduramine F-4 Derivatives
N-substituted (+)-conduramine F-4 derivatives were synthesized from NOEV and provide moderate inhibition of β-gal. Treatment of mouse fibroblasts expressing human p.R201C mutant β-gal had variable levels of enzyme enhancement. Several of these candidate chaperones are included in Table 2.186
4-Epi-Isofagomine Derivatives as Chaperones
Derivatives of 4-epi-isofagomine exhibit inhibitory activity for multiple human glycosidases.97,182 (5aR)-5a-C-pentyl-4-epi-isofagomine, an exceptional inhibitor of lysosomal β-galactosidase, was found to be highly selective for β-gal, with only weak inhibition of β-glucosidase.97 Other 5a-C-alkyl derivatives of 4-epi-isofagomine had similar inhibitory profiles. 5a-C-2-phenylethyl-4-epi-isofagomine and 5a-C-nonyl-4-epi-isofagomine were identified as strong inhibitors of lysosomal β-gal, with distinct inhibitory potential recorded for β-glucosidase.182 The nonyl analog was a more potent inhibitor of β-gal but was also a strong inhibitor of β-glucosidase. In contrast, 5a-C-methyl-4-epi-isofagomine only moderately inhibited β-gal with an IC50 of 250nM. Further, 5a-C-pentyl-4-epi-isofagomine significantly enhanced mutant β-gal activity in 11 out of 21 human fibroblast lines from GM1 gangliosidosis patients.97 For some β-gal mutants, treatment with 5a-C-pentyl-4-epi-isofagomine led to a greater than 20-fold increase in activity. Studies assessing the ability of 5a-C-pentyl-4-epi-isofagomine to traverse the BBB will be critical to its therapeutic efficacy in human GM1 patients.
Despite efficacious results in vitro, EET remains a controversial therapeutic strategy. Treatment of small and large animal models that recapitulate aspects of GM1 disease progression clinically, and possess mutant β-gal amenable to EET, will be critical for translation of PCs to human application.
Stem Cell Transplantation
Bone marrow transplantation (BMT) is an efficacious procedure for several LSDs with primary involvement of peripheral organs, including Gaucher disease, mucopolysaccharidosis I (MPS-I), and MPS-II.85 BMT in patients with neurological symptoms did not slow or reverse disease progression. However, visceral features, including hepatosplenomegaly and cardiac hypertrophy, were markedly improved following BMT.85 These results suggest treatment of GM1 gangliosidosis by unmodified BMT may reduce visceral features, but the correction of neurological symptoms would require an alternative approach. The risk of mortality following BMT should be considered in the context of each patient case.
BMT from a fully HLA-matched (older sister) donor to an asymptomatic 7 month old with juvenile GM1 had no effect on neurological disease progression, in agreement with earlier studies examining other neuropathic LSDs.83,85 The child developed normally until 20–25 months, at which point regression was noted.83 Unlike the outcome for the human patient, β-gal‐/‐ mice treated with bone marrow derived cells transfected with a therapeutic retroviral vector developed β-gal activity in several brain regions, including the cerebellum, brain stem, hippocampus, thalamus, and cortex.86 These regions had a decrease in inflammation and UPR-induced apoptosis, and treated mice had improved function in rotarod and open field testing. The authors concluded that BMCs travelled to the CNS on a chemokine gradient initiated by neuroinflammation.86 Based on these results, BMT may be more effective in symptomatic GM1 patients. However, it is also important to consider that, in contrast to patients, GM1 mice were treated with bone marrow transduced with a retroviral vector that over-expressed β-gal, increasing the amount of secreted enzyme available for cross-correction. BMT in GM1 patients may be effective if similar strategies of over-expressing β-gal are employed. Pre-clinical and clinical studies in metachromatic leukodystrophy, a related LSD, have demonstrated the importance of enzyme over-expression for treatment of neurological disease features.187–189
Transplantation of fetal brain cells (FBC) or bone-marrow-derived mesenchymal stem cells (MSC) from mice over-expressing β-gal, was also able to effectively treat neurological disease short term for β-gal‐/‐ mice.84 However, short-term efficacy did not result in long-term benefit. Grafted cells were not detected at six months post treatment for mice administered MSCs, or a mixture of MSCs and FBCs, and FBC number significantly decreased over time.84
Enzyme Replacement Therapy
Early enzyme replacement therapy (ERT) strategies for GM1 involved the use of liposomes as vehicles for β-gal delivery to affected feline fibroblasts in vitro. These studies highlighted the necessity of carriers for enzyme delivery and the value of liposomes in the treatment of LSDs.56 In recent years, strategies for enzyme delivery have evolved to address the cell-specific challenges of cellular uptake mechanisms. Through an attempt to identify new delivery strategies for ERT in patients with GM1, an alternative cellular-uptake method was proposed: adsorptive-mediated endocytosis. Fusions of a plant galactose/galactosamine-binding lectin (RTB) and human β-gal were effectively endocytosed by GM1 patient fibroblasts with activities matching normal mammalian cell-derived β-gal. These RTB-β-gal fusions were processed in the cell, which rapidly cleaved and degraded the RTB lectin. The detection of active β-gal 48 hours post-treatment suggested protective cellular interactions with the enzyme.87 Use of this system in GM1 represents an alternative uptake mechanism that, though yet unproven in vivo, may be effective in the delivery of active enzyme to the CNS.
Other delivery methods for ERT have focused on the use of co-polymeric nanoparticles. In one study, Gupta et al (2017) delivered β-gal to affected mouse fibroblasts using arginase-responsive dextran sulfate/poly-L-arginine polymer capsules.88 To load active β-gal into the capsules, a step-wise process and hierarchical assembly was required. β-gal first bound calcium carbonate templates, which was followed by alternating the assembly of dextran sulfate and poly-L-arginine layers. The resulting multi-lamellar capsule relies heavily on electrostatic interactions for structural integrity, and its mechanism of delivery remains unclear.88
Poly(ethylene glycol)-b-poly(lactic acid) (PEGPLA) polymersomes have also been investigated as effective carriers of β-gal. Kelly et al (2017) described efficient loading and release of β-gal from PEGPLA nanoparticles.89 Though the mechanism of cellular uptake remains uncharacterized, β-gal was internalized and transported to the lysosome of fibroblasts from GM1 cats. Treated fibroblasts from GM1 cats had a significant increase in β-gal activity relative to untreated controls, supporting the use of polymersomes in ERT of LSDs. To target the CNS, the PEG moiety was modified with ApoE, a candidate for transcytosis across the BBB. ApoE-modified polymersomes efficiently delivered 22-fold more β-gal to fibroblasts than the same dose of unencapsulated β-gal.89 These results served as a proof-of-concept approach to CNS delivery, but in vivo delivery of β-gal polymersomes to GM1 animal models will be crucial to determining the potential efficacy of this ERT in patients.
Recent work by Chen et al (2019) showcased the cation-independent mannose-6-phosphate receptor-mediated endocytosis of recombinant human beta-gal (rhBeta-Gal) in a vehicle-free treatment of both human fibroblasts and β-gal‐/‐ mice.90 In fibroblasts, a single 3nM dose of rhBeta-Gal was sufficient to normalize enzyme activity for up to six weeks. Intracerebroventricular (ICV) injection of rhBeta-Gal to β-gal‐/‐ mice over the course of eight weeks revealed normalization of secondary neuropathology including LAMP2 expression, astrogliosis, and microgliosis. Broad bilateral distribution of rhBeta-Gal to critical regions of GM1 pathology was detected in β-gal‐/‐ mouse cohorts administered a single dose, doses twice weekly for two weeks, and a single dose weekly for eight weeks. RhBeta-Gal was detected in the vasculature of all cohorts and in the liver and bone marrow of mice treated for eight weeks, further supporting ICV administration of rhBeta-Gal as an ERT strategy for GM1 CNS and peripheral pathology.90
Gene Therapy
Gene therapy represents a promising therapeutic approach for monogenic diseases including GM1. However, in genetic diseases with neuropathology, achieving global distribution of viral genomes or diffusion of active enzyme throughout the CNS is challenging. Not only does the BBB impede intravenous delivery of therapeutics to the brain, but methods of bypassing the BBB by intrathecal or intracranial injection are invasive.57,61–63 Despite these challenges, gene therapy has the potential to alleviate symptoms and halt disease progression with a single treatment. For GM1, studies in mice and cats have led to the initiation of an ongoing phase I/II clinical trial for children with GM157,61–63,190 (ClinicalTrials.gov Identifier NCT03952637) and hope for a future.
In early gene therapy studies, retroviral delivery of GLB1 to human infantile GM1 patient fibroblasts resulted in complete correction of β-gal deficiency. However, the newly synthesized human β-gal was transferred to untreated cells at low efficiency during cross-correction.81 An in vivo study of adenovirus-mediated intravenous delivery of mouse β-gal to β-gal‐/‐ mice resulted in a significant increase in β-gal activity to above normal values in peripheral tissues and to 10–20% throughout the brain.82 Mice were injected via the superficial temporal vein 24 to 48 hours after birth, at which point the BBB is incompletely formed. This experimental design promoted distribution of vector throughout the brain and highlighted the potential for intravenous gene therapy as an early therapeutic strategy.82 A significant attenuation of GM1 accumulation in the cerebrum at 30 days post-treatment was also identified. However, by 60 days post-treatment, GM1 ganglioside content in all treated mice was above normal values.82 Most individuals with GM1 are diagnosed after symptom onset when the BBB may be fully intact,124 presenting a challenge for adenovirus-based therapies administered intravenously. Additional challenges arise from immune responses to adenovirus vectors, which are derived from the virus for the common cold to which many have pre-existing immunity.
To target GM1 neuropathology directly, Broekman et al (2007) treated neonatal β-gal‐/‐ mice using bilateral ICV delivery of an adeno-associated viral serotype 1 (AAV1) vector encoding mouse β-gal.61 High levels of β-gal (7 to 65 times normal) were detected throughout the brain of treated mice at three months of age, and brain GSL levels were normalized. However, β-gal activity in the liver was not significantly increased relative to untreated β-gal‐/‐ mice.61
In a follow-up study, Broekman et al (2009) performed intraparenchymal injection of the hippocampus to identify routes of brain and systemic distribution of AAV vector genomes.62 Mouse β-gal was deteted in both physically and synaptically connected areas of the brain, supporting three methods of distribution: 1) diffusion, 2) axonal transport from neurons at the site of injection, and 3) CSF flow in the Virchow-Robin perivascular space. Further, evidence of axonal transport of vector encoded mRNA suggested interconnected brain regions were treated the most effectively.62 In the translation of these findings to future studies and clinical trials, the authors urged careful consideration of the routes of administration and target brain regions.
To identify the optimal intracranial injection sites for complete CNS distribution, Baek et al (2010) chose sites of high connectivity within the cerebrum and cerebellum.63 Two cohorts of six to eight-week-old mice were injected bilaterally with AAV1 encoding mouse β-gal to the thalamus alone or the thalamus and deep cerebellar nuclei (DCN). Both cohorts had a significant increase in survival compared to the 38 week lifespan of untreated mice. Mice that received only thalamic injections had a median lifespan of 45 weeks, while almost all mice treated in the thalamus and DCN survived to the 52 week experimental endpoint. Though the lifespan of GM1 mice clearly was extended by treatment, little functional improvement was observed compared to untreated mice.63
AAV-mediated GLB1 delivery in a feline model of GM1 gangliosidosis provided important support of potential therapeutic efficacy in humans. Not only are cats gyrencephalic, but the cat brain is only ~20 times smaller than the human infant brain while the mouse brain is ~1000 times smaller. AAV1 or AAVrh8 vectors expressing a feline β-gal cDNA were injected bilaterally to the thalamus and DCN of GM1 cats. β-gal activity was restored to ~1-5 fold normal throughout the brain and spinal cord, and neurological function/quality of life improved dramatically. As of the current publication, mean survival of AAV-treated GM1 cats is 5.4 years compared to an untreated lifespan of 8.0±0.6 months, and two animals remain alive and in good health at 9.9 and 10.8 years of age57,60 (and unpublished data). Peripheral disease was not detected and may be due to the leakage of AAV into the blood during the initial injection or following CSF reabsorption into the circulatory system. Seizures were present in one-half of the treated cats and attributed to the suboptimal restoration of β-gal activity throughout the temporal lobe. Both serotypes of AAV had similar clinical outcomes.57 Despite the invasiveness of intracranial injections, these findings support human clinical application of intracranial gene therapy for this fatal neurologic condition.
With the report in 2009 that AAV serotype 9 efficiently crosses the BBB following IV injection, a new era of CNS gene therapy began.191 Additionally, an earlier study concluded that AAV9 vector genomes undergo axonal transport to distant neuron cell bodies.192 To examine the efficacy of intravenous gene therapy for GM1, an AAV9 vector expressing mouse βgal was injected via the tail vein of six-week-old β-gal‐/‐ mice at a dose of 1.5×1013 vg/kg body weight.190 Treated mice survived 90% longer than untreated control mice. A gender effect was observed, with treated males surviving 59% longer and treated females surviving 118% longer than controls (p<0.0003 for both cohorts). Survival increases were accompanied by improved neuromuscular function and cognitive behavior, as measured by rotarod, inverted screen, and home cage testing. β-gal activity in the cerebrum and cerebellum was restored to ~16-24% of normal after treatment. Also, β-gal activity ranged from 22.7% (males) to 51.7% (females) of normal in the brainstem and from 79.1% (males) to 242.3% (females) of normal in the spinal cord. It is thought that lower liver transduction in GM1 females led to higher transduction of the brain and thus increased longevity. Restoration of β-gal activity in the CNS led to corresponding decreases in GM1 ganglioside storage in the cerebrum (47–80%), cerebellum (38–52%), brainstem (69–72%), and spinal cord (88–100%). Astrogliosis, a prominent feature of untreated GM1 mice, was substantially reduced, providing further evidence of therapeutic benefit.190 Intravenous AAV9-mediated GLB1 delivery to GM1 cats produced results similar to those of mice. Normalization of GM1 ganglioside content in most brain regions was notable in treated cats at >40 months of age, and β-gal activity was significantly increased throughout the brain, reaching normal levels in several regions (unpublished data). Strong and consistently positive results from pre-clinical gene therapy studies have led to clinical trials currently underway or nearing initiation.
Clinical Trials
Initial studies to treat GM1 gangliosidosis by gene therapy focus on AAV9 for intravenous delivery (ClinicalTrials.gov Identifier NCT03952637) or AAVrh10 or AAVhu68 for CSF-mediated delivery via the cisterna magna (NCT04273269 or NCT04713475, respectively). Vectors include a human GLB1 cDNA, and doses based on body weight range from 1.5-4.5×1013 vg/kg (IV) or 8.0×1012 vg/kg or greater (CSF). The trials will enroll infantile and late infantile/juvenile GM1 patients. At the time of publication, the clinical trial of CSF-administered AAVrh10 or AAVhu68 were preparing to begin recruiting patients while initial results of IV administration in a GM1 patient had been published in the form of a press release (Axovant, http://investors.axovant.com/news-releases/news-release-details/axovant-provides-clinical-program-update-axo-aav-gm1-novel). AAV9 treatment was generally well tolerated and, through six months post-treatment, the patient was reported to have had clinically significant improvements based on neurological exam, the Vineland-3 scale, Clinical Global Impression (CGI) assessments, and nutritional status. The patient reportedly regained her ability to swallow more effectively and had meaningful weight gain. While these clinical trials continue to follow treated patients for evidence of safety and efficacy, other gene therapy trials are in the planning stage. A separate clinical trial of Miglustat combined with a ketogenic diet is also underway (ClinicalTrials.gov Identifier NCT02030015). It is hoped and anticipated that these clinical trials will pave the way for effective treatment of GM1 gangliosidosis and other monogenic neuropathic diseases.
Conclusion
GM1 gangliosidosis incidence, etiology, and clinical features limit the resources available for the definition of relevant disease mechanisms and the development of efficacious therapeutic strategies. From the molecular examination of patients with GM1, we know that mutation of lysosomal acid β-galactosidase leads to attenuation or loss of enzyme activity and accumulation of its substrates in both the CNS and visceral organs. The defining neuropathic features of GM1 reflect a neurodegenerative disease process and are the focus of therapeutic design and development. Although β-gal deficiency and accumulation of undigested substrate is central to GM1 etiology, the impact of lysosomal storage on normal cell physiology is still unclear. However, it is clear that feedback inhibition does not prevent the biosynthesis of storage material, and no enzymatic redundancies exist for the catabolism of GM1 ganglioside or other β-linked galactose-containing glycoconjugates. Future studies examining how storage impacts lysosomal integrity, the endo-lysosomal network, and global catabolism of cellular substrates will be critical to identifying the most efficacious therapeutic strategies. Lysosomal or general cellular dysfunction may limit the efficacy of strategies seeking to restore β-gal activity in advanced disease states.
Advancements in the design and development of pharmacological chaperones will be best supported by pre-clinical models that recapitulate human disease progression. High-throughput or near-high-throughput approaches that examine all known genotypes in the context of distinct PCs will best identify those that respond efficaciously and propel translation to appropriate GM1 patients. A promising three-dimensional cell culture model that may serve as a pre-clinical substitute for animals in the future is the cerebral organoid. These iPSC-derived miniature organs (brains) in a dish have been used to recapitulate neurodevelopment and neuropathology for several diseases including GM164. CRISPR/Cas-editing of GLB1 in iPSCs to generate isogenic lines possessing distinct GM1 alleles for culture of cerebral organoids would support the pre-clinical assessment of PCs with high potential, including NOEV and several 4-epi-isofagomine derivatives.
To circumvent the accumulation or aggregation of residual mutant β-gal at the ER and its subsequent downstream impact on neuropathology, EET may be combined with other therapeutic strategies including SRT, BMT, ERT, or gene therapy. However, PC selection will depend on GM1 patient genotype. As a novel approach to GM1 treatment, genome editing targets the endogenous GLB1, possibly eliminating the need for the introduction or overexpression of exogenous active β-gal. Given the broad distribution of mutations across GLB1 and the various different types of mutations, approaches based on genome editing may be accessible only for certain patients in the near future. Nonetheless, optimized genome editing strategies that achieve broad distribution throughout the CNS and efficient mutation targeting, have the potential to be efficacious for monogenic diseases including GM1.
Research assessing the management of GM1 by SRT, ERT, and BMT is limited. However, each of these strategies has demonstrated success in the treatment of other LSDs. Gene therapy appears to be the leading strategy for GM1 treatment, which may come as no surprise given that the causative mutations are localized to a single gene, GLB1. Ongoing clinical trials enrolling infantile and late infantile/juvenile GM1 patients will provide insight into the safety and efficacy of AAV-mediated gene delivery in humans. Pre-clinical studies were limited to the treatment of mice and cats prior to the onset of clinical symptoms. Therefore, whether gene therapy will simply halt disease progression or ameliorate symptoms will be of great interest. For any therapeutic strategy, determining optimal windows of efficacy is of utmost importance for patient quality of life. For GM1, prevention of irreversible neurological deficits is crucial and will require expeditious diagnosis and early disease management.
Disclosure
Dr Douglas R Martin reports grants from National Institutes of Health, during the conduct of the study; stock options from Lysogene, and personal fees from Axovant, outside the submitted work. The authors report no other potential conflicts of interest for this work.
References
1. Caciotti A, Garman SC, Rivera-Colón Y, et al. GM1 gangliosidosis and Morquio B disease: an update on genetic alterations and clinical findings. Biochim Biophys Acta - Mol Basis Dis. 2011;1812(7):782–790. doi:10.1016/j.bbadis.2011.03.018
2. Jarnes Utz JR, Kim S, King K, et al. Infantile gangliosidoses: mapping a timeline of clinical changes. Mol Genet Metab. 2017;121(2):170–179. doi:10.1016/j.ymgme.2017.04.011
3. Regier DS, Kwon HJ, Johnston J, et al. MRI/MRS as a surrogate marker for clinical progression in GM1 gangliosidosis. Am J Med Genet Part A. 2016;170(3):634–644. doi:10.1002/ajmg.a.37468
4. Brunetti-Pierri N, Scaglia F. GM1 gangliosidosis: review of clinical, molecular, and therapeutic aspects. Mol Genet Metab. 2008;391–396. doi:10.1016/j.ymgme.2008.04.012
5. Lenicker HM, Agius PV, Young EP, Montalto SPA. Infantile generalized GM1 gangliosidosis: high incidence in the Maltese Islands. J Inherit Metab Dis. 1997;20(5):723–724. doi:10.1023/A:1005303332529
6. Sinigerska I, Chandler D, Vaghjiani V, et al. Founder mutation causing infantile GM1-gangliosidosis in the Gypsy population. Mol Genet Metab. 2006;88(1):93–95. doi:10.1016/j.ymgme.2005.12.009
7. Nishimoto J, Nanba E, Inui K, Okada S, Suzuki K. GM1-gangliosidosis (genetic β-galactosidase deficiency): identification of four mutations in different clinical phenotypes among Japanese patients. Am J Hum Genet. 1991;49(3):566–574.
8. Yoshida K, Oshima A, Sakuraba H, et al. GM1 gangliosidosis in adults: clinical and molecular analysis of 16 Japanese patients. Ann Neurol. 1992;31(3):328–332. doi:10.1002/ana.410310316
9. Georgiou T, Stylianidou G, Anastasiadou V, et al. The Arg482His mutation in the β-galactosidase gene is responsible for a high frequency of GM1 gangliosidosis carriers in a Cypriot village. Genet Test. 2005;9(2):126–132. doi:10.1089/gte.2005.9.126
10. Callahan JW. Molecular basis of GM1 gangliosidosis and Morquio disease, type B. Structure-function studies of lysosomal β-galactosidase and the non-lysosomal β-galactosidase-like protein. Biochim Biophys Acta - Mol Basis Dis. 1999;1455(2–3):85–103. doi:10.1016/S0925-4439(99)00075-7
11. Lawrence R, Van Vleet JL, Mangini L, et al. Characterization of glycan substrates accumulating in GM1 Gangliosidosis. Mol Genet Metab Reports. 2019;21(November):100524. doi:10.1016/j.ymgmr.2019.100524
12. Ohto U, Usui K, Ochi T, Yuki K, Satow Y, Shimizu T. Crystal structure of human β-galactosidase: structural basis of G M1 gangliosidosis and morquio B diseases. J Biol Chem. 2012;287(3):1801–1812. doi:10.1074/jbc.M111.293795
13. Inui K, Namba R, Ihara Y, et al. A case of chronic GM1 gangliosidosis presenting as dystonia: clinical and biochemical studies. J Neurol. 1990;237(8):491–493. doi:10.1007/bf00314770
14. Farrell DF, Ochs U. GM1 Gangliosidosis: phenotypic variation in a single family. Ann Neurol. 1981;9(3):225–231. doi:10.1002/ana.410090304
15. Yamamoto A. Localized ß-Galactosidase Deficiency. Arch Intern Med. 1974;134(4):627. doi:10.1001/archinte.1974.00320220029002
16. Uyama E, Terasaki T, Watanabe S, et al. Type 3 GM1 gangliosidosis: characteristic MRI findings correlated with dystonia. Acta Neurol Scand. 1992;86(6):609–615. doi:10.1111/j.1600-0404.1992.tb05497.x
17. Ferreira CR, Gahl WA. Lysosomal storage diseases. Transl Sci Rare Dis. 2017. doi:10.3233/TRD-160005
18. Tonin R, Caciotti A, Procopio E, et al. Pre-diagnosing and managing patients with GM1 gangliosidosis and related disorders by the evaluation of GM1 ganglioside content. Sci Rep. 2019;9:17684. doi:10.1038/s41598-019-53995-5
19. Pierson TM, Adams DA, Markello T, et al. Exome sequencing as a diagnostic tool in a case of undiagnosed juvenile-onset GM 1 -gangliosidosis. Neurology. 2012;79(2):123–126. doi:10.1212/WNL.0b013e31825f047a
20. Reuser AJ, Verheijen FW, Bali D, et al. The use of dried blood spot samples in the diagnosis of lysosomal storage disorders - Current status and perspectives. Mol Genet Metab. 2011;104:144–148. doi:10.1016/j.ymgme.2011.07.014
21. Emery JM, Green WR, Wyllie RG, Howell RR. GM1-gangliosidosis: ocular and pathological manifestations. Arch Ophthalmol. 1971;85(2):177–187. doi:10.1001/archopht.1971.00990050179011
22. Utz JRJ, Crutcher T, Schneider J, Sorgen P, Whitley CB. Biomarkers of central nervous system inflammation in infantile and juvenile gangliosidoses. Mol Genet Metab. 2015;114(2):274–280. doi:10.1016/j.ymgme.2014.11.015
23. Pinsky L, Powell E, Callahan J. GM1-gangliosidosis types 1 and 2: enzymatic differences in cultured fibroblasts. Nature. 1970;228:1093–1095. doi:10.1038/2281093b0
24. Van der Voorn JP, Kamphorst W, van der Knaap MS, Powers JM. The leukoencephalopathy of infantile GM1 gangliosidosis: oligodendrocytic loss and axonal dysfunction. Acta Neuropathol. 2004;107(6):539–545. doi:10.1007/s00401-004-0848-9
25. Erol I, Alehan F, Ali Pourbagher M, Canan O, Vefa Yildirim S. Neuroimaging findings in infantile GM1 gangliosidosis. Eur J Paediatr Neurol. 2006;10(5–6):245–248. doi:10.1016/j.ejpn.2006.08.005
26. Kaback MM, Sloan HR, Sonneborn M, Herndon RM, Percy AK. Gm1-gangliosidosis type I: in utero detection and fetal manifestations. J Pediatr. 1973;82(6):1037–1041. doi:10.1016/S0022-3476(73)80438-X
27. Nestrasil I, Ahmed A, Utz JM, Rudser K, Whitley CB, Jarnes-Utz JR. Distinct progression patterns of brain disease in infantile and juvenile gangliosidoses: volumetric quantitative MRI study. Mol Genet Metab. 2018;123(2):97–104. doi:10.1016/j.ymgme.2017.12.432
28. Goebel HH, Fix JD, Zeman W. Retinal pathology in GM1 ganglios1dosis, type II. Am J Ophthalmol. 1973;75(3):434–441. doi:10.1016/0002-9394(73)91152-5
29. Vieira JP, Conceição C, Scortenschi E. GM1 gangliosidosis, late infantile onset dystonia, and T2 hypointensity in the globus pallidus and substantia nigra. Pediatr Neurol. 2013;49(3):195–197. doi:10.1016/j.pediatrneurol.2013.02.003
30. Deodato F, Procopio E, Rampazzo A, et al. The treatment of juvenile/adult GM1-gangliosidosis with Miglustat may reverse disease progression. Metab Brain Dis. 2017;32(5):1529–1536. doi:10.1007/s11011-017-0044-y
31. De Grandis E, Di M, Pessagno RA, Veneselli E, Rossi A. MR Imaging Findings in 2 Cases of Late Infantile GM1 Gangliosidosis. AJNR Am J Neuroradiol. 2009;30:1325–1327. doi:10.3174/ajnr.A1508
32. Tanaka R, Momoi T, Yoshida A, et al. Type 3 GM I gangliosidosis: clinical and neuroradiological findings in an 11-year-old girl. J Neurol. 1995;242(5):299–303. doi:10.1007/BF00878872
33. Roze E, Paschke E, Lopez N, et al. Dystonia and Parkinsonism in GM1 type 3 gangliosidosis. Mov Disord. 2005;20(10):1366–1369. doi:10.1002/mds.20593
34. Roze E, Navarro S, Cornu P, Welter ML, Vidailhet M. Deep brain stimulation of the globus pallidus for generalized dystonia in GM1 type 3 gangliosidosis: technical case report. Neurosurgery. 2006;59(6):E1340. doi:10.1227/01.NEU.0000245620.24603.1B
35. Kobayashi T, Chronic SK. GM1 gangliosidosis presenting as Dystonia: II. Biochemical studies. Ann Neurol. 1980;9(5):476–483. doi:10.1002/ana.410090510
36. Patterson MC. Pediatric Neurology Part III - Gangliosidoses. In: Handbook of Clinical Neurology. Vol. 113. Elsevier;2013:1707–1708. doi:10.1016/B978-0-444-59565-2.00039-3
37. Hofer D, Paul K, Fantur K, et al. Phenotype determining alleles in GM1 gangliosidosis patients bearing novel GLB1 mutations. Clin Genet. 2010;78(3):236–246. doi:10.1111/j.1399-0004.2010.01379.x
38. Brezina PR, Benner A, Rechitsky S, et al. Single-gene testing combined with single nucleotide polymorphism microarray preimplantation genetic diagnosis for aneuploidy: a novel approach in optimizing pregnancy outcome. Fertil Steril. 2011;95:
39. Gillan JE, Lowden JA, Gaskin K, Cutz E. Congenital ascites as a presenting sign of lysosomal storage disease. J Pediatr. 1984;104(2):225–231. doi:10.1016/S0022-3476(84)80997-X
40. Lowden JA, Cutz E, Conen PE, Rudd N, Doran TA. Prenatal Diagnosis of GM1-Gangliosidosis. N Engl J Med. 1973;288(5):225–228. doi:10.1056/NEJM197302012880502
41. Warner TG, Robertson AD, Mock AK, Johnson WG, O’Brien JS. Prenatal diagnosis of GM1 gangliosidosis by detection of galactosyl-oligosaccharides in amniotic fluid with high-performance liquid chromatography. Am J Hum Genet. 1983;35:1034–1041.
42. Chamoles NA, Blanco MB, Iorcansky S, Gaggioli D, Spècola N, Casentini C. Retrospective diagnosis of GM1 gangliosidosis by use of a newborn-screening card. Clin Chem. 2001;47(11):2068. doi:10.1093/clinchem/47.11.2068
43. Polo G, Burlina AP, Ranieri E, et al. Plasma and dried blood spot lysosphingolipids for the diagnosis of different sphingolipidoses: A comparative study. Clin Chem Lab Med. 2019;57(12):1863–1874. doi:10.1515/cclm-2018-1301
44. Walkley SU, Zervas M, Wiseman S. Gangliosides as modulators of dendritogenesis in normal and storage disease-affected pyramidal neurons. Cereb Cortex. 2000;10(10):1028–1037. doi:10.1093/cercor/10.10.1028
45. Walkley SU. Neurobiology and cellular pathogenesis of glycolipid storage diseases. Philos Trans R Soc. 2003;358:893–904. doi:10.1098/rstb.2003.1276
46. Baker HJ, Lindsey JR. Animal model: feline GM1 gangliosidosis. Am J Pathol. 1974;74(3):649–652.
47. Baker HJ, Reynolds GD, Walkley SU, Cox NR, Baker GH. The gangliosidoses: comparative features and research applications. Vet Pathol. 1979;16(6):635–649. doi:10.1177/030098587901600602
48. Gurda BL, Vite CH. Large animal models contribute to the development of therapies for central and peripheral nervous system dysfunction in patients with lysosomal storage diseases. Hum Mol Genet. 2019;28(R1):R119–R131. doi:10.1093/hmg/ddz127
49. Murnane RD, Prieur DJ, Ahern‐Rindell AJ, Holler LD, Parish SM. Clinical and clinicopathologic characteristics of ovine GM‐1 gangliosidosis. J Vet Intern Med. 1994;8(3):221–223. doi:10.1111/j.1939-1676.1994.tb03220.x
50. Matsuda J, Suzuki O, Oshima A, et al. Beta-Galactosidase-Deficient Mouse as an Animal Model for GM1-Gangliosidosis. Glycoconj J. 1997;14(6):729–736.
51. Itoh M, Matsuda J, Suzuki O, et al. Development of lysosomal storage in mice with targeted disruption of the β-galactosidase gene: a model of human GM1-gangliosidosis. Brain Dev. 2001;23(6):379–384. doi:10.1016/S0387-7604(01)00244-3
52. Hahn CN, Del Pilar Martin M, Schröder M, et al. Generalized CNS disease and massive G(M1)-ganglioside accumulation in mice defective in lysosomal acid β-galactosidase. Hum Mol Genet. 1997;6(2):205–211. doi:10.1093/hmg/6.2.205
53. Eikelberg D, Lehmbecker A, Brogden G, et al. Axonopathy and reduction of membrane resistance: key features in a new murine model of human GM1-gangliosidosis. J Clin Med. 2020;9(4):1004. doi:10.3390/jcm9041004
54. Przybilla MJ, Ou L, Tăbăran AF, et al. Comprehensive behavioral and biochemical outcomes of novel murine models of GM1-gangliosidosis and Morquio syndrome type B. Mol Genet Metab. 2019;126(2):139–150. doi:10.1016/j.ymgme.2018.11.002
55. Matsuda J, Suzuki O, Oshima A, et al. Chemical chaperone therapy for brain pathology in GM1-Gangliosidosis.Proc Natl Acad Sci U S A. 2003;100(26):15912–7. doi:10.1073/pnas.2536657100.
56. Reynolds GD, Baker HJ, Reynolds RH. Enzyme replacement using liposome carriers in feline GM1 gangliosidosis fibroblasts. Nature. 1978;275(5682):754–755. doi:10.1038/275754a0
57. McCurdy VJ, Johnson AK, Gray-Edwards HL, et al. Sustained normalization of neurological disease after intracranial gene therapy in a feline model. Sci Transl Med. 2014;6(231):231ra48. doi:10.1126/scitranslmed.3007733
58. Gray-Edwards HL, Salibi N, Josephson EM, et al. High resolution MRI anatomy of the cat brain at 3 Tesla. J Neurosci Methods. 2014;227:10–17. doi:10.1016/j.jneumeth.2014.01.035
59. Gray-Edwards HL, Jiang X, Randle AN, et al. Lipidomic evaluation of feline neurologic disease after AAV gene therapy. Mol Ther - Methods Clin Dev. 2017;6:135–142. doi:10.1016/j.omtm.2017.07.005
60. Gray-Edwards HL, Regier DS, Shirley JL, et al. Novel biomarkers of human GM1 gangliosidosis reflect the clinical efficacy of gene therapy in a feline model. Mol Ther. 2017;25(4):892–903. doi:10.1016/j.ymthe.2017.01.009
61. Broekman MLD, Baek RC, Comer LA, Fernandez JL, Seyfried TN, Sena-Esteves M. Complete correction of enzymatic deficiency and neurochemistry in the GM1-gangliosidosis mouse brain by neonatal adeno-associated virus–mediated gene delivery. Mol Ther. 2007;15(1):30–37. doi:10.1038/sj.mt.6300004
62. Broekman MLD, Tierney LA, Benn C, Chawla P, Cha JH, Sena-Esteves M. Mechanisms of distribution of mouse β-galactosidase in the adult GM1-gangliosidosis brain. Gene Ther. 2009;16(2):303–308. doi:10.1038/gt.2008.149
63. Baek RC, Boekman MLD, Leroy SG, et al. AAV-mediated gene delivery in adult GM1-gangliosidosis mice corrects lysosomal storage in CNS and improves survival. PLoS One. 2010;5(10):e13468. doi:10.1371/journal.pone.0013468
64. Latour YL, Yoon R, Thomas SE, et al. Human GLB1 knockout cerebral organoids: a model system for testing AAV9-mediated GLB1 gene therapy for reducing GM1 ganglioside storage in GM1 gangliosidosis. Mol Genet Metab Reports. 2019;21(May):100513. doi:10.1016/j.ymgmr.2019.100513
65. Chiricozzi E, Lunghi G, Di Biase E, Fazzari M, Sonnino S, Mauri L. GM1 ganglioside is a key factor in maintaining the mammalian neuronal functions avoiding neurodegeneration. Int J Mol Sci. 2020;21(3):868. doi:10.3390/ijms21030868
66. Chiricozzi E, Pomè DY, Maggioni M, et al. Role of the GM1 ganglioside oligosaccharide portion in the TrkA-dependent neurite sprouting in neuroblastoma cells. J Neurochem. 2017;143(6):645–659. doi:10.1111/jnc.14146
67. Mocchetti I. Exogenous gangliosides, neuronal plasticity and repair, and the neurotrophins. Cell Mol Life Sci. 2005;62:2283–2294. doi:10.1007/s00018-005-5188-y
68. Head BP, Patel HH, Insel PA. Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function: membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim Biophys Acta - Biomembr. 2014;1838(2):532–545. doi:10.1016/J.BBAMEM.2013.07.018
69. Jung J-U, Ko K, Lee D-H, Ko K, Chang K-T, Choo Y-K. The roles of glycosphingolipids in the proliferation and neural differentiation of mouse embryonic stem cells. Exp Mol Med. 2009;41(12):935–945. doi:10.3858/emm.2009.41.12.099
70. Kolter T, Proia RL, Sandhoff K. Combinatorial ganglioside biosynthesis. J Biol Chem. 2002;277(29):25859–25862. doi:10.1074/jbc.R200001200
71. Yu RK, Tsai Y-T, Ariga T, Yanagisawa M. Structures, biosynthesis, and functions of gangliosides—An overview. J Oleo Sci. 2011;60(10):537–544. 10.5650/jos.60.537.
72. Posse de Chaves E, Sipione S. Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett. 2010;584(9):1748–1759. doi:10.1016/J.FEBSLET.2009.12.010
73. Kolter T. Ganglioside Biochemistry. Int Sch Res Netw Biochem. 2012;2012:1–36. doi:10.5402/2012/506160
74. Sano R, Trindade VMT, Tessitore A, et al. GM1-ganglioside degradation and biosynthesis in human and murine GM1-gangliosidosis. Clin Chim Acta. 2005;354(1–2):131–139. doi:10.1016/j.cccn.2004.11.035
75. Yu RK, Yanagisawa M, Ariga T. Glycosphingolipid Structures. Compr Glycosci from Chem to Syst Biol. 2007;73–122. doi:10.1016/B978-044451967-2/00003-9
76. Breiden B, Sandhoff K. Ganglioside Metabolism and Its Inherited Diseases. Methods Mol Biol.2018;1804:97–141 doi:10.1007/978-1-4939-8552-4_5
77. Sandhoff K, Harzer K. Gangliosides and gangliosidoses: principles of molecular and metabolic pathogenesis. J Neurosci. 2013;33(25):10195–10208. doi:10.1523/JNEUROSCI.0822-13.2013
78. Yoshihara T, Satake H, Nishie T, et al. Lactosylceramide synthases encoded by B4galt5 and 6 genes are pivotal for neuronal generation and myelin formation in mice. PLoS Genet. 2018;14(8):e1007545. doi:10.1371/journal.pgen.1007545
79. Yu RK, Ariga T, Yanagisawa M, Zeng G. Gangliosides in the nervous system: biosynthesis and degradation. Glycoscience. 2008;1671–1695. doi:10.1007/978-3-540-30429-6_41
80. Tettamanti G, Bassi R, Viani P, Riboni L. Salvage pathways in glycosphingolipid metabolism. Biochimie. 2003;85:423–437. doi:10.1016/S0300-9084(03)00047-6
81. Sena-Esteves M, Camp SM, Alroy J, Breakefield XO, Kaye EM. Correction of acid β-galactosidase deficiency in GM1 gangliosidosis human fibroblasts by retrovirus vector-mediated gene transfer: higher efficiency of release and cross-correction by the murine enzyme. Hum Gene Ther. 2000;11(5):715–727. doi:10.1089/10430340050015617
82. Takaura N, Yagi T, Maeda M, et al. Attenuation of ganglioside GM1 accumulation in the brain of GM1 gangliosidosis mice by neonatal intravenous gene transfer. Gene Ther. 2003;10:1487–1493. doi:10.1038/sj.gt.3302033
83. Shield JPH, Stone J, Steward CG. Bone marrow transplantation correcting β-galactosidase activity does not influence neurological outcome in juvenile GM1-gangliosidosis. J Inherit Metab Dis. 2005;28(5):797–798. doi:10.1007/s10545-005-0089-7
84. Sawada T, Tanaka A, Higaki K, et al. Intracerebral cell transplantation therapy for murine GM1 gangliosidosis. Brain Dev. 2009;31(10):717–724. doi:10.1016/j.braindev.2008.11.004
85. Hoogerbrugge PM, Brouwer OF, Bordigoni P, et al. Allogeneic bone marrow transplantation for lysosomal storage diseases. Lancet. 1995;345(8962):1398–1402. doi:10.1016/S0140-6736(95)92597-X
86. Sano R, Tessitore A, Ingrassia A, D’Azzo A. Chemokine-induced recruitment of genetically modified bone marrow cells into the CNS of GM1-gangliosidosis mice corrects neuronal pathology. Blood. 2005;106(7):2259–2268. doi:10.1182/blood-2005-03-1189
87. Condori J, Acosta W, Ayala J, et al. Enzyme replacement for GM1-gangliosidosis: uptake, lysosomal activation, and cellular disease correction using a novel β-galactosidase: RTB lectin fusion. Mol Genet Metab. 2016;117(2):199–209. doi:10.1016/j.ymgme.2015.12.002
88. Gupta M, Pandey H, Sivakumar S. Intracellular Delivery of β-galactosidase enzyme using arginase-responsive dextran sulfate/Poly- l -arginine capsule for lysosomal storage disorder. ACS Omega. 2017;2:9002–9012. doi:10.1021/acsomega.7b01230
89. Kelly JM, Gross AL, Martin DR, Byrne ME. Polyethylene glycol-b-poly(lactic acid) polymersomes as vehicles for enzyme replacement therapy. Nanomedicine. 2017;12(23):2591–2606. doi:10.2217/nnm-2017-0221
90. Chen JC, Luu AR, Wise N, et al. Intracerebroventricular enzyme replacement therapy with Beta-Galactosidase reverses brain pathologies due to GM1 gangliosidosis in mice. J Biol Chem. 2019;295(39):13532–13555. doi:10.1074/jbc.RA119.009811
91. Suzuki Y, Ichinomiya S, Kurosawa M, et al. Chemical chaperone therapy: clinical effect in murine G M1 -gangliosidosis. Ann Neurol. 2007;62(6):671–675. doi:10.1002/ana.21284
92. Suzuki Y. Chemical chaperone therapy for GM1-gangliosidosis. Cell Mol Life Sci. 2008;65:351–353. doi:10.1007/s00018-008-7470-2
93. Higaki K, Li L, Bahrudin U, et al. Chemical chaperone therapy: chaperone effect on mutant enzyme and cellular pathophysiology in β-galactosidase deficiency. Hum Mutat. 2011;32(7):843–852. doi:10.1002/humu.21516
94. Suzuki Y, Ichinomiya S, Kurosawa M, et al. Therapeutic chaperone effect of N-Octyl 4-Epi-β-valienamine on murine GM1-gangliosidosis. Mol Genet Metab. 2012;106:92–98. doi:10.1016/j.ymgme.2012.02.012
95. Rigat BA, Tropak MB, Buttner J, et al. Evaluation of N-nonyl-deoxygalactonojirimycin as a pharmacological chaperone for human GM1 gangliosidosis leads to identification of a feline model suitable for testing enzyme enhancement therapy. Mol Genet Metab. 2012;107(1–2):203–212. doi:10.1016/j.ymgme.2012.06.007
96. Takamura A, Higaki K, Ninomiya H, et al. Lysosomal accumulation of Trk protein in brain of GM1- gangliosidosis mouse and its restoration by chemical chaperone. J Neurochem. 2011;118:399–406. doi:10.1111/j.1471-4159.2011.07310.x
97. Front S, Biela-Banaś A, Burda P, et al. (5aR)-5a-C-Pentyl-4-epi-isofagomine: a powerful inhibitor of lysosomal β-galactosidase and a remarkable chaperone for mutations associated with GM1-gangliosidosis and Morquio disease type B. Eur J Med Chem. 2017;126:160–170. doi:10.1016/j.ejmech.2016.09.095
98. Caciotti A, Donati MA, D’Azzo A, et al. The potential action of galactose as a “chemical chaperone”: increase of beta galactosidase activity in fibroblasts from an adult GM1-gangliosidosis patient. Eur J Paediatr Neurol. 2009;13:160–164. doi:10.1016/j.ejpn.2008.03.004
99. Coutinho MF, Santos JI, Alves S. Less is more: substrate reduction therapy for lysosomal storage disorders. Int J Mol Sci. 2016;17:7. doi:10.3390/ijms17071065
100. Aerts JMFG, Hollak CEM, Boot RG, Groener JEM, Maas M. Substrate reduction therapy of glycosphingolipid storage disorders. J Inherit Metab Dis. 2006;29:449–456. doi:10.1007/s10545-006-0272-5
101. Elliot-Smith E, Speak AO, Lloyd-Evans E, et al. Beneficial effects of substrate reduction therapy in a mouse model of GM1 gangliosidosis. Mol Genet Metab. 2008;94:204–211. doi:10.1016/j.ymgme.2008.02.005
102. Kasperzyk JL, El-Abbadi MM, Hauser EC, D’Azzo A, Platt FM, Seyfried TN. N-butyldeoxygalactonojirimycin reduces neonatal brain ganglioside content in a mouse model of GM1 gangliosidosis. J Neurochem. 2004;89:645–653. doi:10.1046/j.1471-4159.2004.02381.x
103. Kwak JE, Son M-Y, Son YS, Son MJ, Cho YS. Biochemical and molecular characterization of novel mutations in GLB1 and NEU1 in patient cells with lysosomal storage disorders. Biochem Biophys Res Commun. 2015;457:554–560. doi:10.1016/j.bbrc.2015.01.023
104. Galjaard H, Hoogeveen A, Keijzer W, et al. Genetic heterogeneity in GM1-gangliosidosis. Nature. 1975;257:60–62. doi:10.1038/257060a0
105. Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–D1067. doi:10.1093/nar/gkx1153
106. Santamaria R, Chabás A, Coll MJ, Miranda CS, Vilageliu L, Grinberg D. Twenty-one novel mutations in the GLB1 gene identified in a large group of GM1-gangliosidosis and Morquio B patients: possible common origin for the prevalent p.R59H mutation among gypsies. Hum Mutat. 2006;27(10):1060. doi:10.1002/humu.9451
107. Yoshida K, Oshima A, Shimmoto M, et al. Human beta-galactosidase gene mutations in GM1-gangliosidosis: a common mutation among Japanese adult/chronic cases. Am J Hum Genet. 1991;49(2):435–442.
108. Hofer D, Paul K, Fantur K, et al. GM1 gangliosidosis and Morquio B disease: expression analysis of missense mutations affecting the catalytic site of acid β-galactosidase. Hum Mutat. 2009;30(8):1214–1221. doi:10.1002/humu.21031
109. Sperb F, Vairo F, Burin M, Mayer FQ, Matte U, Giugliani R. Genotypic and phenotypic characterization of Brazilian patients with GM1 gangliosidosis. Gene. 2013;512:113–116. doi:10.1016/j.gene.2012.09.106
110. Caciotti A, Donati MA, Procopio E, et al. GM1 gangliosidosis: molecular analysis of nine patients and development of an RT-PCR assay for GLB1 gene expression profiling. Hum Mutat. 2007;28(2):204. doi:10.1002/humu.9475
111. Morita M, Saito S, Ikeda K, et al. Structural bases of GM1 gangliosidosis and Morquio B disease. J Hum Genet. 2009;54(9):510–515. doi:10.1038/jhg.2009.70
112. Caciotti A, Bardelli T, Cunningham J, D’Azzo A, Zammarchi E, Morrone A. Modulating action of the new polymorphism L436F detected in the GLB1 gene of a type-II GM1 gangliosidosis patient. Hum Genet. 2003;113:44–50. doi:10.1007/s00439-003-0930-8
113. Tessitore A, Martin MDP, Sano R, et al. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol Cell. 2004;15(5):753–766. doi:10.1016/j.molcel.2004.08.029
114. Martin DR, Rigat BA, Foureman P, et al. Molecular consequences of the pathogenic mutation in feline G M1 gangliosidosis. Mol Genet Metab. 2008;94:212–221. doi:10.1016/j.ymgme.2008.02.004
115. Koenig ML, Jope RS, Baker HJ, Lally KM. Reduced Ca2+ flux in synaptosomes from cats with GM1 gangliosidosis. Brain Res. 1987;424(1):169–176. doi:10.1016/0006-8993(87)91207-8
116. Spoerri PE, Dozier AK, Roisen FJ. Calcium regulation of neuronal differentiation: the role of calcium in GM1-mediated neuritogenesis. Dev Brain Res. 1990;56:177–188. doi:10.1016/0165-3806(90)90080-I
117. Wu G, Lu Z‐H, Ledeen RW. GM1 ganglioside in the nuclear membrane modulates nuclear calcium homeostasis during neurite outgrowth. J Neurochem. 1995;65(3):1419–1422. doi:10.1046/j.1471-4159.1995.65031419.x
118. Ledeen RW, Wu G. Ganglioside function in calcium homeostasis and signaling. Neurochem Res. 2002;27(7–8):637–647. doi:10.1023/A:1020224016830
119. Singer HS, Coyle JT, Weaver DL, Kawamura N, Baker HJ. Neurotransmitter chemistry in feline GM1 gangliosidosis: A model for human ganglioside storage disease. Ann Neurol. 1982;12(1):37–41. doi:10.1002/ana.410120107
120. Wood PA, McBride MR, Baker HJ, Christian ST. Fluorescence Polarization Analysis, Lipid Composition, and Na+, K+‐ATPase Kinetics of Synaptosomal Membranes in Feline GM1 and GM2 Gangliosidosis. J Neurochem. 1985;44:947–956. doi:10.1111/j.1471-4159.1985.tb12909.x
121. Karabelas AB, Walkley SU. Altered patterns of evoked synaptic activity in cortical pyramidal neurons in feline ganglioside storage disease. Brain Res. 1985;339:329–336. doi:10.1016/0006-8993(85)90099-X
122. Walkley SU, Wurzelmann S. Alterations in synaptic connectivity in cerebral cortex in neuronal storage disorders. Ment Retard Dev Disabil Res Rev. 1995;1:183–192. doi:10.1002/mrdd.1410010306
123. Takamura A, Higaki K, Kajimaki K, et al. Enhanced autophagy and mitochondrial aberrations in murine GM1-gangliosidosis. Biochem Biophys Res Commun. 2008;367:616–622. doi:10.1016/j.bbrc.2007.12.187
124. Jeyakumar M, Thomas R, Elliot-Smith E, et al. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain. 2003;126:974–987. doi:10.1093/brain/awg089
125. Son MY, Kwak JE, Seol B, Lee DY, Jeon H, Cho YS. A novel human model of the neurodegenerative disease GM1 gangliosidosis using induced pluripotent stem cells demonstrates inflammasome activation. J Pathol. 2015;237(1):98–110. doi:10.1002/path.4551
126. Folkerth RD, Alroy J, Bhan I, Kaye EM. Infantile G(M1) gangliosidosis: complete morphology and histochemistry of two autopsy cases, with particular reference to delayed central nervous system myelination. Pediatr Dev Pathol. 2000;3:73–86. doi:10.1007/s100240050010
127. Suzuki K. Neuropathology of late onset gangliosidoses. Dev Neurosci. 1991;13:205–210. doi:10.1159/000112161
128. Van der Voorn JP, Pouwels PJW, Kamphorst W, et al., Histopathologic correlates of radial stripes on MR images in lysosomal storage disorders. Am J Neuroradiol. 2005;26:442–446.
129. Müller G, Alldinger S, Moritz A, et al. GM1-gangliosidosis in Alaskan huskies: clinical and pathologic findings. Vet Pathol. 2001;38:281–290. doi:10.1354/vp.38-3-281
130. Read DH, Harrington DD, Keenan TW, Hinsman EJ. Neuronal-Visceral GM1 Gangliosidosis in a dog with beta-galactosidase deficiency. Science. 1976;194:442–445.
131. Hanna RB, Pappas GD, Purpura DP. Freeze-fracture study of membranous cytoplasmic bodies of cortical neurons in feline GM1-ganglioside storage disease. Brain Res. 1982;252:172–176. doi:10.1016/0006-8993(82)90993-3
132. Walkley SU, Baker HJ, Rattazzi MC, Haskins ME, Wu JY. Neuroaxonal dystrophy in neuronal storage disorders: evidence for major GABAergic neuron involvement. J Neurol Sci. 1991;104:1–8. doi:10.1016/0022-510X(91)90208-O
133. Novikoff AB. Lysosomes: A personal Account. In: Lysosomes and Storage Diseases. 1973:1–41.
134. Read DH, Harrington DD, Keenan TW, Hinsman EJ. Neuronal-Visceral GM1 gangliosidosis in a dog with beta-galactosidase deficiency. Science. 1976;194(4263):442–445.
135. Müller G, Alldinger S, Moritz A, et al. GM1-gangliosidosis in Alaskan huskies: clinical and pathologic findings. Vet Pathol. 2001;38(3):281–290. doi:10.1354/vp.38-3-281
136. Purpura DP, Pappas GD, Baker HJ. Fine structure of meganeurites and secondary growth processes in feline GM1-gangliosidosis. Brain Res. 1978;143:1–12. doi:10.1016/0006-8993(78)90748-5
137. Walkley SU. Cellular pathology of lysosomal storage disorders. Brain Pathol. 1998;8:175–193. doi:10.1111/j.1750-3639.1998.tb00144.x
138. Walkley SU, Wurzelmann S, Purpura DP. Ultrastructure of neurites and meganeurites of cortical pyramidal neurons in feline gangliosidosis as revealed by the combined Golgi-EM technique. Brain Res. 1981;211:393–398. doi:10.1016/0006-8993(81)90711-3
139. Bahr BA, Bendiske J. The neuropathogenic contributions of lysosomal dysfunction. J Neurochem. 2002;83:481–489. doi:10.1046/j.1471-4159.2002.01192.x
140. Pará C, Bose P, Pshezhetsky AV. Neuropathophysiology of lysosomal storage diseases: synaptic dysfunction as a starting point for disease progression. J Clin Med. 2020;9:616. doi:10.3390/jcm9030616
141. Muthane U, Chickabasaviah Y, Kaneski C, et al. Clinical features of adult G M1 gangliosidosis: report of three Indian patients and review of 40 cases. Mov Disord. 2004;19(11):1334–1341. doi:10.1002/mds.20193
142. Goldman JE, Katz D, Rapin I, Purpura DP, Suzuki K. Chronic GM1 gangliosidosis presenting as dystonia: I. Clinical and pathological features. Ann Neurol. 1981;9(5):465–475. doi:10.1002/ana.410090509
143. Wenger DA, Sattler M, Mueller OT, Myers GG, Schneiman RS, Nixon GW. Adult GM1 gangliosidosis: clinical and biochemical studies on two patients and comparison to other patients called variant or adult Gm1 gangliosidosis. Clin Genet. 1980;17:323–334. doi:10.1111/j.1399-0004.1980.tb00158.x
144. Kaye EM, Alroy J, Raghavan SS, et al. Dysmyelinogenesis in animal model of GM1 gangliosidosis. Pediatr Neurol. 1992;8(4):255–261. doi:10.1016/0887-8994(92)90361-2
145. Heinecke KA, Luoma A, D’Azzo A, Kirschner DA, Seyfried TN. Myelin abnormalities in the optic and sciatic nerves in mice with GM1-gangliosidosis. ASN Neuro. 2015;7:1–15. doi:10.1177/1759091415568913
146. Gray-Edwards HL, Maguire AS, Salibi N, et al. 7T MRI predicts amelioration of neurodegeneration in the brain after AAV gene therapy. Mol Ther - Methods Clin Dev. 2020;17:258–270. doi:10.1016/j.omtm.2019.11.023
147. M-B C-G, Wang SZ, Ziegler R, Cheng SH, Cox TM. Reversibility of neuropathology in Tay-Sachs-related diseases. Hum Mol Genet. 2014;23:730–748. doi:10.1093/hmg/ddt459
148. Buccinnà B, Piccinini M, Prinetti A, et al. Alterations of myelin-specific proteins and sphingolipids characterize the brains of acid sphingomyelinase-deficient mice, an animal model of Niemann-Pick disease type A. J Neurochem. 2009;109:105–115. doi:10.1111/j.1471-4159.2009.05947.x
149. Yan X, Lukas J, Witt M, et al. Decreased expression of myelin gene regulatory factor in Niemann-Pick type C 1 mouse. Metab Brain Dis. 2011;26(4):299–306. doi:10.1007/s11011-011-9263-9
150. Svennerholm L, Vanier MT, Mansson JE. Krabbe disease: a galactosylsphingosine (psychosine) lipidosis. J Lipid Res. 1980;21(1):53–64. doi:10.1016/S0022-2275(20)39839-4
151. Yamato O, Masuoka Y, Yonemura M, et al. Clinical and clinico-pathologic characteristics of Shiba dogs with a deficiency of lysosomal acid β-galactosidase: a canine model of human GM1 gangliosidosis. J Vet Med Sci. 2003;65(2):213–217. doi:10.1292/jvms.65.213
152. Moore GJ. Proton magnetic resonance spectroscopy in pediatric neuroradiology. Pediatr Radiol. 1998;28(11):805–814. doi:10.1007/s002470050470
153. Satoh H, Yamato O, Asano T, et al. Cerebrospinal fluid biomarkers showing neurodegeneration in dogs with GM1 gangliosidosis: possible use for assessment of a therapeutic regimen. Brain Res. 2007;1133:200–208. doi:10.1016/j.brainres.2006.11.039
154. Brunetti-Pierri N, Bhattacharjee MB, Wang ZJ, et al. Brain proton magnetic resonance spectroscopy and neuromuscular pathology in a patient with GM1 gangliosidosis. J Child Neurol. 2008;23(1):73–78. doi:10.1177/0883073807307088
155. Ong MT, Chow GCS, Morton RE. Dual diagnosis of dihydropyrimidine dehydrogenase deficiency and GM 1 gangliosidosis. Pediatr Neurol. 2012;46:178–181. doi:10.1016/j.pediatrneurol.2011.12.005
156. Al-Essa MA, Bakheet SM, Patay ZJ, Nounou RM, Ozand PT. Cerebral fluorine-18 labeled 2-fluoro-2-deoxyglucose positron emission tomography (FDG PET), MRI, and clinical observations in a patient with infantile G(M1) gangliosidosis. Brain Dev. 1999;21(8):559–562. doi:10.1016/s0387-7604(99)00073-x
157. Chen CY, Zimmerman RA, Lee CC, Chen FH, Yuh YS, Hsiao HS. Neuroimaging findings in late infantile GM1 gangliosidosis. Am J Neuroradiol. 1998;19(9):1628–1630. doi:10.3174/ajnr.a1508
158. Di Rocco M, Rossi A, Parenti G, et al. Different molecular mechanisms leading to white matter hypomyelination in infantile onset lysosomal disorders. Neuropediatrics. 2005;36(4):265–269. doi:10.1055/s-2005-865863
159. Ji H, Li D, Wu Y, et al. Hypomyelinating disorders in China: the clinical and genetic heterogeneity in 119 patients. PLoS One. 2018;13(2):e0188869. doi:10.1371/journal.pone.0188869
160. Kobayashi O, Takashima S. Thalamic hyperdensity on CT in infantile GM1-gangliosidosis. Brain Dev. 1994;16(6):472–474. doi:10.1016/0387-7604(94)90011-6
161. Lin HC, Tsai FJ, Shen WC, Tsai CH, Peng CT. Infantile form GM1 gangliosidosis with dilated cardiomyopathy: a case report. Acta Paediatr Int J Paediatr. 2000;89(7):880–883. doi:10.1111/j.1651-2227.2000.tb00398.x
162. Hasegawa D, Tamura S, Nakamoto Y, et al. Magnetic resonance findings of the corpus callosum in canine and feline lysosomal storage diseases. PLoS One. 2013;8(12):e83455. doi:10.1371/journal.pone.0083455
163. Steenweg ME, Vanderver A, Blaser S, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain. 2010;133:2971–2982. doi:10.1093/brain/awq257
164. Campdelacreu J, Muñoz E, Gómez B, Pujol T, Chabás A, Tolosa E. Generalised dystonia with an abnormal magnetic resonance imaging signal in the basal ganglia: a case of adult-onset GM1 gangliosidosis. Mov Disord. 2002;17(5):1095–1097. doi:10.1002/mds.10247
165. Malik P, Muthusamy K, C M D, Sudhakar SV. Teaching NeuroImages: wishbone pattern of iron accumulation: a characteristic imaging sign in GM1 gangliosidosis. Neurology. 2019;92(18):e2176–e2177. doi:10.1212/WNL.0000000000007406
166. Marangi A, Tagliapietra M, Vicenzi V, Pasquin I, Salviati A. Teaching NeuroImages: brain MRI and DaT-SPECT imaging in adult GM1 gangliosidosis. Neurology. 2018;91(2):e187–e188. doi:10.1212/WNL.0000000000005775
167. Platt FM, Butters TD. Substrate Reduction Therapy. Editors, John A. Barranger and Mario A. Cabrera-Salazar. In: Lysosomal Storage Disorders. Boston, MA: Springer US;2007:153–168. doi:10.1007/978-0-387-70909-3_11
168. Kasperzyk JL, D’Azzo A, Platt FM, Alroy J, Seyfried TN. Substrate reduction reduces gangliosides in postnatal cerebrum-brainstem and cerebellum in GM1 gangliosidosis mice. J Lipid Res. 2005;46:744–751. doi:10.1194/jlr.M400411-JLR200
169. RFG F, Furneaux RH, Mahuran DJ, et al. 1-Deoxy-d-galactonojirimycins with dansyl capped N-substituents as β-galactosidase inhibitors and potential probes for GM1 gangliosidosis affected cell lines. Carbohydr Res. 2011;346(12):1592–1598. doi:10.1016/j.carres.2011.05.010
170. Thonhofer M, Gonzalez Santana A, Fischer R, et al. 5-Fluoro derivatives of 4-epi-isofagomine as d-galactosidase inhibitors and potential pharmacological chaperones for GM1-gangliosidosis as well as Fabry’s disease. Carbohydr Res. 2015;420:6–12. doi:10.1016/j.carres.2015.10.009
171. Fantur K, Hofer D, Schitter G, et al. DLHex-DGJ, a novel derivative of 1-deoxygalactonojirimycin with pharmacological chaperone activity in human GM1-gangliosidosis fibroblasts. Mol Genet Metab. 2010;100(3):262–268. doi:10.1016/j.ymgme.2010.03.019
172. Alfonso P, Pampín S, Estrada J, et al. Miglustat (NB-DNJ) works as a chaperone for mutated acid β-glucosidase in cells transfected with several Gaucher disease mutations. Blood Cells, Mol Dis. 2005;35(2):268–276. doi:10.1016/j.bcmd.2005.05.007
173. Platt FM, Neises GR, Dwek RA, Butters TD. N-butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. J Biol Chem. 1994;269(11):8362–8365. doi:10.1016/S0021-9258(17)37202-2
174. Fischer PB, Collin M, Karlsson GB, et al. The alpha-glucosidase inhibitor N-butyldeoxynojirimycin inhibits human immunodeficiency virus entry at the level of post-CD4 binding. J Virol. 1995;69(9):5791–5797. doi:10.1128/jvi.69.9.5791-5797.1995
175. Jeyakumar M, Butters TD, Cortina-Borja M, et al. Delayed symptom onset and increased life expectancy in Sandhoff disease mice treated with N-butyldeoxynojirimycin. Proc Natl Acad Sci U S A. 1999;96(11):6388–6393. doi:10.1073/pnas.96.11.6388
176. Jeyakumar M, Dwek RA, Butters TD, Platt FM. Storage solutions: treating lysosomal disorders of the brain. Nat Rev Neurosci. 2005;6(9):713–725. doi:10.1038/nrn1725
177. Platt FM, Neises GR, Reinkensmeier G, et al. Prevention of lysosomal storage in Tay-Sachs mice treated with N- butyldeoxynojirimycin. Science. 1997;276(5311):428–431. doi:10.1126/science.276.5311.428
178. Higaki K, Ninomiya H, Suzuki Y, Nanba E. Candidate molecules for chemical chaperone therapy of GM1- gangliosidosis. Future Med Chem. 2013;5(13):1551–1558. doi:10.4155/fmc.13.123
179. Parenti G. Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics. EMBO Mol Med. 2009;1(5):268–279. doi:10.1002/emmm.200900036
180. Suzuki Y, Ogawa S, Sakakibara Y. Chaperone therapy for neuronopathic lysosomal diseases: competitive inhibitors as chemical chaperones for enhancement of mutant enzyme activities. Perspect Medicin Chem. 2009;3:7–19. doi:10.4137/pmc.s2332
181. Takai T, Higaki K, Aguilar-Moncayo M, et al. A bicyclic 1-deoxygalactonojirimycin derivative as a novel pharmacological chaperone for GM1 gangliosidosis. Mol Ther. 2013;21(3):526–532. doi:10.1038/mt.2012.263
182. Front S, Almeida S, Zoete V, et al. 4-epi-Isofagomine derivatives as pharmacological chaperones for the treatment of lysosomal diseases linked to β-galactosidase mutations: improved synthesis and biological investigations. Bioorganic Med Chem. 2018;26(20):5462–5469. doi:10.1016/j.bmc.2018.09.023
183. Andersson U, Butters TD, Dwek RA, Platt FM. N-butyldeoxygalactonojirimycin: A more selective inhibitor of glycosphingolipid biosynthesis than N-butyldeoxynojirimycin, in vitro and in vivo. Biochem Pharmacol. 2000;59(7):821–829. doi:10.1016/S0006-2952(99)00384-6
184. D’Alonzo D, De Fenza M, Porto C, et al. N-Butyl-l-deoxynojirimycin (l-NBDNJ): synthesis of an allosteric enhancer of α-glucosidase activity for the treatment of pompe disease. J Med Chem. 2017;60(23):9462–9469. doi:10.1021/acs.jmedchem.7b00646
185. Frustaci A, Chimenti C, Ricci R, et al. Improvement in cardiac function in the cardiac variant of fabry’s disease with galactose-infusion therapy. N Engl J Med. 2001;345:25–32. doi:10.1056/NEJM200107053450104
186. Kuno S, Higaki K, Takahashi A, Nanba E, Ogawa S. Potent chemical chaperone compounds for G M1-gangliosidosis: N-substituted (+)-conduramine F-4 derivatives. Med Chem Commun. 2015;6:306–310. doi:10.1039/c4md00270a
187. Biffi A, De Palma M, Quattrini A, et al. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J Clin Invest. 2004;113(8):1118–1129. doi:10.1172/jci19205
188. Biffi A, Montini E, Lorioli L, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341(6148):1233158. doi:10.1126/science.1233158
189. Sessa M, Lorioli L, Fumagalli F, et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, Phase 1/2 trial. Lancet. 2016;388(10043):476–487. doi:10.1016/S0140-6736(16)30374-9
190. Weismann CM, Ferreira J, Keeler AM, et al. Systemic AAV9 gene transfer in adult GM1 gangliosidosis mice reduces lysosomal storage in CNS and extends lifespan. Hum Mol Genet. 2015;24(15):4353–4364. doi:10.1093/hmg/ddv168
191. Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi:10.1038/nbt.1515
192. Cearley CN, Wolfe JH. Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and Rh10 in the mouse brain. Mol Ther. 2006;13(3):528–537. doi:10.1016/j.ymthe.2005.11.015
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.