Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Glucocorticoid-Induced Fatty Liver Disease
Authors Rahimi L ![]() , Rajpal A
, Rajpal A ![]() , Ismail-Beigi F
, Ismail-Beigi F
Received 27 January 2020
Accepted for publication 27 March 2020
Published 16 April 2020 Volume 2020:13 Pages 1133—1145
DOI https://doi.org/10.2147/DMSO.S247379
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Antonio Brunetti
Leili Rahimi,1 Aman Rajpal,1,2 Faramarz Ismail-Beigi1,2
1Department of Medicine, Case Western Reserve University, University Hospitals Cleveland Medical Center, Cleveland, OH, USA; 2Cleveland VA Medical Center, Cleveland, OH, USA
Correspondence: Faramarz Ismail-Beigi
Department of Medicine, Case Western Reserve University, 10900 Euclid Ave, Cleveland, OH 44106, USA
Tel + 12163686122
Fax +12163685824
Email [email protected]
Abstract: Glucocorticoids (GCs) are commonly used at high doses and for prolonged periods (weeks to months) in the treatment of a variety of diseases. Among the many side effects are increased insulin resistance with disturbances in glucose/insulin homeostasis and increased deposition of lipids (mostly triglycerides) in the liver. Here, we review the metabolic pathways of lipid deposition and removal from the liver that become altered by excess glucocorticoids. Pathways of lipid deposition stimulated by excess glucocorticoids include 1) increase in appetite and high caloric intake; 2) increased blood glucose levels due to GC-induced stimulation of gluconeogenesis; 3) stimulation of de novo lipogenesis that is augmented by the high glucose and insulin levels and by GC itself; and 4) increased release of free fatty acids from adipose stores and stimulation of their uptake by the liver. Pathways that decrease hepatic lipids affected by glucocorticoids include a modest stimulation of very-low-density lipoprotein synthesis and secretion into the circulation and inhibition of β-oxidation of fatty acids. Role of 11β-hydroxysteroid dehydrogenases-1 and -2 and the reversible conversion of cortisol to cortisone on intracellular levels of cortisol is examined. In addition, GC control of osteocalcin expression and the effect of this bone-derived hormone in increasing insulin sensitivity are discussed. Finally, research focused on gaining a better understanding of the dose and duration of treatment with glucocorticoids, which leads to increased triglyceride deposition in the liver, and the reversibility of the condition is highlighted.
Keywords: obesity, Cushing’s disease, fatty liver, metabolic syndrome, hyperphagia, LPL, hormone-sensitive lipase
Introduction
Adrenocorticotropic hormone (ACTH) secreted by the pituitary gland stimulates the synthesis and secretion of cortisol, the important glucocorticoid hormone in humans. Cortisol is a major stress hormone and is essential for survival in humans. Many physiological actions of cortisol that are discussed below are mediated by glucocorticoid receptors (GR) that are expressed in virtually all cells. Multiple isoforms of the receptor exist due to inclusion or exclusion of different exons.1 GRα mediates most actions of the hormone while GRβ is devoid of a ligand-binding domain and functions as a natural inhibitor.1 Upon binding of the hormone, the receptor-hormone complex is translocated to the nucleus where it participates in the transcriptional regulation of various genes. In addition, some actions of glucocorticoids are non-receptor mediated.2
At physiological concentrations, cortisol has positive effects on metabolism, control of inflammation, and mediation of immune response. Glucocorticoids are widely used in clinical practice and it is estimated that >1.5 million (~1.65%) postmenopausal women and men over 50 years of age in the United States are treated with glucocorticoids with the vast majority being treated for prolonged periods (many weeks to months).3 Chronic and excessive use of the hormone can cause a large number of undesired side effects. These include increased appetite and weight gain with central obesity, fatty liver, glucose intolerance, muscle wasting, hypertension, dyslipidemia, and increased susceptibility to infections; other untoward effects include skin fragility, negative calcium balance from excessive bone loss leading to osteoporosis and skeletal fractures, and psychological abnormalities.4,5 Even short term (days) excessive use of the hormone can lead to multiple metabolic disturbances including resistance of peripheral tissues and liver to insulin action, hyperglycemia and hyperinsulinemia, and elevated plasma triglyceride levels. More prolonged use can lead to excessive deposition of lipids (predominantly triglycerides) in liver resulting in non-alcoholic fatty liver disease (NAFLD). Of note, the host of abnormalities listed above are commonly seen in patients with Cushing’s disease or syndrome who have chronically elevated levels of cortisol.4,5 Similar changes are also seen in patients who are treated with pharmacological doses of glucocorticoids for prolonged periods.6,7
In this manuscript, we will examine the pathways leading to increased fat deposition in the liver induced by excess glucocorticoids. The topic is of importance because increases in hepatic fat deposits over time can lead to inflammation, fibrosis and cirrhosis of the liver that can be a precursor to hepatocellular carcinoma in a subset of patients.8–12
Major Pathways Contributing to Hepatic Lipid Content
Hepatic lipid content reflects the contribution input and output pathways depicted in Figure 1. Input (entry) pathways leading to increased deposition of lipids (mostly triglycerides in lipid droplets) include (1) Food intake, (2) Glucose derived from gluconeogenesis, (3) Hyperglycemia, insulin resistance and hyperinsulinemia, (4) Increased de novo fatty acid synthesis (lipogenesis), and (5) Elevated circulating free fatty acid levels. Output (exit) pathways that can decrease hepatic lipid content include (6) Formation and secretion of very-low-density lipoprotein (VLDL) and (7) β-oxidation of fatty acids. Although some of the above pathways are inter-related, it is important to note that glucocorticoids independently regulate each of the steps enumerated above. Figure 1 also depicts the direct effects of glucocorticoids on other tissues that greatly impact hepatic lipid content.
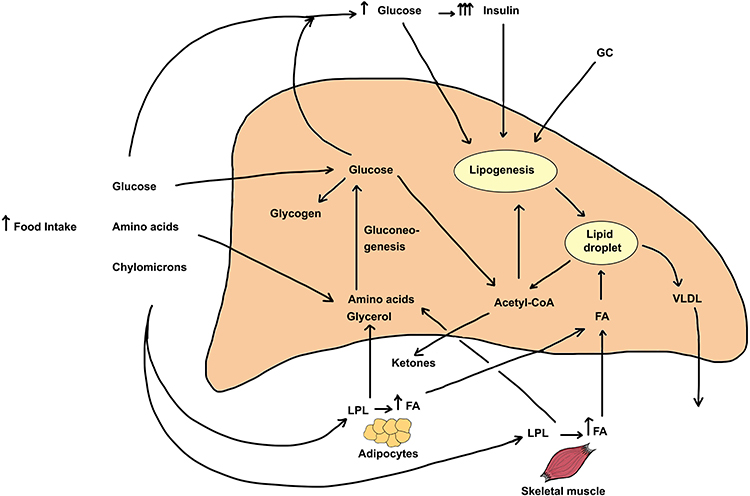 |
Figure 1 Metabolic pathways leading to excess triglyceride deposition in the liver and the effect of increased glucocorticoids on the pathways. The steady-state hepatic triglyceride content is determined by rates of TG formation and TG metabolism or secretion from hepatocytes. Several pathways and sources directly and indirectly serve to increase hepatic triglyceride content. These include 1) Increased appetite and food consumption. Following digestion of food, the bulk of the absorbed glucose is taken up by the liver (by GLUT2 transporters) and the rest is avidly taken up by the skeletal and cardiac muscle and adipose tissue by GLUT4 transporters that are stimulated by elevated insulin levels. The rise in blood glucose leads to increased secretion and blood levels of insulin, a response that is exaggerated by GC treatment. Amino acids are taken up mostly by liver and skeletal muscle. Chylomicrons release fatty acids (FA) and glycerol due to action of plasma LPL, and the released FA are taken up by adipose tissue, skeletal muscle as well as by the liver. GC excess stimulates tissue hormone-sensitive lipase and increases fatty acid and glycerol release from adipose tissue. 2) The uptake of FA by the liver represents the major pathway of lipid deposition in liver and this transport process is stimulated by GC. 3) Upon entry into the liver, amino acids and glycerol as well as glucose metabolites can form glucose through gluconeogenesis, a pathway that is stimulated by GC and inhibited by insulin. Finally, the elevated blood glucose and insulin both stimulate de novo fatty acid synthesis (lipogenesis), and excess GC in conjunction with elevated insulin synergistically stimulates this process. Two pathways mediate the decrease of triglycerides in intracellular stores. These include 1) the production and release of triglycerides as VLDL particles into the bloodstream, a process that is mildly stimulated by glucocorticoids and 2) β-oxidation of fatty acids, a metabolic pathway that is inhibited by glucocorticoids. All the major steps of TG metabolism in liver, adipose tissue, and skeletal muscle are affected by increased GC action. In sum, the net effect of excess glucocorticoids is to increase hepatic triglyceride stores leading to fatty liver. |
The contribution of the various sources leading to triglycerides (TG) deposition in the liver varies greatly depending on whether the individual is in the fasted or fed state. Based on several studies using a variety of techniques including stable isotopes in experimental animals and humans, there is general agreement that the major source of TG accretion in liver (~60% or more) is derived from plasma free (non-esterified) fatty acids (FFAs); release of fatty acids from adipose tissue accounts for the bulk of plasma FFAs.13–19 FFAs are predominately derived from TG deposited in adipose tissue. The contribution of de novo fatty acid synthesis varies from 1% to 5% in normal individuals after an overnight fast and can increase up to ~25% in persons in the fed state and in those with fatty liver.17,18 FFAs derived from breakdown of chylomicrons by plasma lipoprotein lipase (LPL) following a mixed meal account for 10–15% of hepatic uptake.17 In what follows, we will discuss mechanisms underlying glucocorticoid-induced fatty liver.
Glucocorticoid-Induced Hyperphagia and Disposition of Nutrients
Excess glucocorticoids increase food intake and cause central (visceral) obesity that is associated with hyperglycemia, hyperinsulinemia and hyperleptinemia.20 Leptin is an adipocyte-derived protein that regulates appetite by acting in the hypothalamus.21 Under normal conditions, leptin reduces appetite and leads to a decrease in body weight.22 However, studies in humans and experimental animals suggest that supra-physiological levels of glucocorticoids cause hyperphagia and obesity perhaps by reducing sensitivity to leptin.21,23,24 Leptin acts via the leptin receptor (OBRb) expressed in hypothalamic nuclei. Based on studies in vitro in a human hepatoma cell line and in vivo in rats, it has been shown that formation of leptin-OBRb complex leads to activation of tyrosine phosphorylation through JAK/STAT pathway.20 It is suggested that glucocorticoids inhibit leptin-induced JAK/STAT phosphorylation thereby leading to leptin resistance.20 Leptin resistance in turn leads to development of hyperphagia, obesity and metabolic disorders.21,22 It is probable that neuropeptide Y (NPY), an appetite-stimulating agent, also plays a role in the observed GC-induced hyperphagia despite elevated leptin levels.23,25
Following a normal meal, fatty acids derived from digestion of triglycerides are absorbed into enterocytes, resynthesized into triacylglycerol (TG) and secreted into lymphatic system as chylomicrons. Chylomicrons undergo lipolysis by plasma Lipoprotein Lipase (LPL) releasing FFAs and glycerol, and the released FFAs are taken up rapidly by muscle, adipose tissue and some by the liver, and are converted to TGs and are stored in lipid droplets; glycerol is taken up by the liver and kidneys and used for gluconeogenesis.26 Finally, the cholesterol-rich remnant chylomicrons are taken up by the liver through LDL-receptor-mediated mechanisms.
Glucose derived from carbohydrates ingested in food is rapidly taken up by the liver through action of the GLUT2 transporter, and by skeletal muscle, heart, and adipocytes in response to increased insulin levels and GLUT4 action.27 Glucose is deposited as glycogen in liver and skeletal muscle or is metabolized (see below).
Skeletal muscle and liver take up much of the amino acids derived from dietary proteins. Most amino acids can be converted to glucose through gluconeogenesis in liver (and to a lesser extent in kidneys) and are released into the circulation; this latter process plays a relatively minor role under fed conditions.28,29 However, under conditions of glucocorticoid excess, larger amounts of amino acids are released by the skeletal muscle and are converted to glucose in the liver.27,30 This is in keeping with the observation that prolonged use of excess glucocorticoids leads to significant muscle atrophy in humans and animals. In addition, some of the amino acids that are converted to glucose can be subsequently metabolized to fatty acids and deposited as triglycerides in adipocytes, especially in visceral adipose tissue.31 Of note, the cycles of excess food intake, excess lipid deposits in the liver and adipose tissue, and increased insulin resistance are self-re-enforcing.
It is worth mentioning that a subgroup (5–20%) of individuals in different countries with non-alcoholic fatty liver disease (NAFLD) are lean rather than being obese and have lesser degrees of insulin resistance and liver enzyme elevations.32 The role of potential alterations in glucocorticoid metabolism and cortisone to cortisol conversion in this subgroup requires investigation.
Glucocorticoid Stimulation of Gluconeogenesis and the Role of Glucagon
Glucocorticoids promote gluconeogenesis in the liver and kidney by stimulating the transcription of genes encoding gluconeogenic enzymes, namely phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase). Control of these genes is not unique to glucocorticoids, and other hormones including glucagon and catecholamines stimulate, and insulin inhibit their expression.30,33,34 Binding of GC receptor complex to the glucocorticoid response unit (GRU) located in the promoter region of the above genes, along with several accessory transcription factors lead to upregulation of PEPCK and G6Pase expression.27,35 PGC1α (nuclear receptor co-factor peroxisome proliferator-activated receptor γ co-activator 1 alpha) whose expression is stimulated by GC interacts with GRUs by increasing the binding of other transcription factors.27,35 PGC1α also induces the expression of G6Pase gene. G6Pase is an ER-resident enzyme that converts glucose-6-phosphate to glucose and phosphate. GLUT2 and phosphate transport proteins facilitate the movement of glucose and phosphate from the lumen of ER to cytosol and are subsequently released into the interstitial space.36–38
A recent study showed that glucocorticoids increase the expression of the transcription factor Krϋppel-like factor 9 (KLF9), which in turn augments the expression of PGC1α.39 Deletion of the KLF9 gene abolished the stimulatory effect of glucocorticoids on glucose output. Hence, GC-induced stimulation of gluconeogenesis involves stimulation of the cascade of KLF9-PGC1α-GRU-containing genes.40
Elevated glucagon levels are observed as a result of GC treatment.41,42 Increased glucagon in the presence of elevated insulin levels is often found in patients with type 2 diabetes (T2DM).42 High glucagon levels stimulate gluconeogenesis and glycogenolysis while inhibiting glycolysis and glycogen synthesis.27,42
The overall effect of glucocorticoid- and glucagon-induced stimulation of gluconeogenesis is to increase the delivery of glucose into the bloodstream for uptake and use by peripheral tissues, and importantly, the brain. Of course, some of the released glucose is taken up by the liver itself and can be converted to fatty acids and deposited as TGs in lipid-containing vesicles.
Glucocorticoid-Induced Insulin Resistance, Hyperglycemia, and Hyperinsulinemia
A hallmark of acute or chronic exposure to excess glucocorticoids is the development of hepatic and systemic insulin resistance (IR) and hyperinsulinemia.43–46 There are various mechanisms that can lead to GC-induced decrease in insulin sensitivity of tissues. While increase in visceral fat with chronic use of GCs contributes to systemic increase in IR, acute actions of GCs on muscle, liver, and other tissues play a vital role.47,48 GC inhibits a number of steps in the insulin signaling network. In rodent skeletal muscle, GC decreases the transcription of insulin receptor substrate-1 (IRS-1), while it increases the transcription of two proteins that counter insulin action, namely protein tyrosine phosphatase type 1B (PTP1B) and p38MAPK.49 GC also decreases IRS-1 and IRS-2 levels in adipose tissue50,51 and decreases IRS-1 phosphorylation in liver.50,52 Adiponectin secreted by adipocytes promotes insulin sensitivity in peripheral tissues; the expression of adiponectin is suppressed by GC treatment, thereby contributing to GC-induced increase in IR.53 Other mechanisms of GC-induced insulin resistance include accumulation of intra-myocellular lipids (droplets of TG) in skeletal muscle fibers,14,54-56 and suppression of osteocalcin synthesis and release from bone, which in turn contributes to IR and metabolic derangements as discussed further below.57
The elevation of glucose levels following exposure to excess GC reflects the suppression of insulin-stimulated glucose transport in insulin-responsive tissues due to GC-induced insulin resistance as well as the stimulation gluconeogenesis. A modest elevation of glucose levels is compensated by the much larger increment in insulin levels (Figure 1). The exact mechanism underlying the stimulation of insulin synthesis and secretion in response to generalized decrease in insulin sensitivity of tissues is not fully understood. Higher amounts of insulin were secreted by islets of dexamethasone-treated rats than by islets of control rats in response to equal concentration of extracellular glucose.58 In addition, central action of glucocorticoids may enhance vagal stimulation of insulin secretion.59 Surprisingly, in experiments using normal rat pancreatic islets in culture, addition of dexamethasone to the medium resulted in inhibition of insulin secretion.60–62 It is possible that the modest elevation of glucose levels following GC-induced insulin resistance leads to much larger increment in insulin secretion and hyperinsulinemia.
Effect of Cortisol, Glucose, and Insulin on de novo Fatty Acid Synthesis
Triglycerides (TG) are the major component of lipids that are contained in hepatic lipid droplets. Accretion of TGs is derived from esterification and re-esterification of fatty acids (FA) with glycerol or by de novo synthesis of fatty acids.63 The pathway of de novo fatty acid synthesis (referring to FA synthesis from acetyl-CoA) is stimulated by elevations of insulin and glucose levels, and by GC itself. The bulk of TG synthesis (~60%) results from esterification of fatty acids from food and FAs released from adipose tissue. Depending on the status of food intake, the percentage contribution of de novo FA synthesis can vary from small percentages with food deprivation to up to ~20% following food intake based on the availability of acetyl-CoA derived from glucose metabolism.17,18 Of note, the rate of triglyceride synthesis during fasting can be up to 5-fold higher in people who have high insulin-resistance (such as type 2 diabetes) with the associated increases in blood glucose and insulin.64–66
The process of de novo FA synthesis is under the control of specific transcription factors: sterol regulatory element-binding protein 1c (SREBP-1c) that is stimulated by insulin, and carbohydrate response element-binding protein (ChREBP) that is stimulated by elevated glucose.67,68 As noted above, in humans and experimental animals, both glucose and insulin levels become elevated in response to excess glucocorticoids.45,58,69 Activated SREBP-1c in conjunction with activated ChREBP stimulates the transcription of genes involved in de novo FA synthesis, including acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS).70 SREBP-1c is also involved in competitive inhibition of PPARγ coactivator 1α (PGC-1α), a co-regulator that activates PEPCK promotor activity and gluconeogenesis,71 this can help increase the supply of acetyl CoA for fatty acid synthesis. ChREBP acts in synergy with SREBP-1c to induce glycolytic and lipogenic gene expression.68,70,72 In addition, glucose that is phosphorylated by glucokinase augments the synergistic action of ChREBP and SREBP-1c on lipogenic gene expression.72 The net effect of these changes is that the rate of fatty acid synthesis is augmented in response to glucocorticoids.
In the first step of de novo FA synthesis, acetyl-CoA is carboxylated to malonyl-CoA by ACC that is subsequently converted to palmitate (or other fatty acids) via multiple steps catalyzed by fatty acid synthase (FAS) enzyme complex.73 Of the two isoforms of ACC, ACC1 predominates and is the key regulatory step for FA synthesis. Insulin and glucocorticoids stimulate the transcription of ACC1 and thus play a critical role in lipid synthesis.74 Glucocorticoids also stimulate the expression of FAS leading to increased de novo hepatic fatty acid synthesis.75–78 Some studies in humans and experimental animals suggest that GC and insulin have a synergistic effect on stimulating ACC gene expression,74,79,80 while other studies show that insulin is required for the stimulation of ACC1 by GC.77,81,82 In addition, recent evidence shows that up-regulation of mitogen-activated protein kinase (MAPK) phosphatase-3 also plays a critical role in GC-induced hepatic lipid synthesis.83
In summary, glucocorticoids regulate several genes encoding enzymes involved in de novo FA synthesis (including ACC and FAS). Glucocorticoids and the hyperinsulinemia resulting from GC excess appear to have an additive or synergistic effect on the expression of enzymes mediating FA synthesis.
Glucocorticoids Increase Release of Fatty Acids from Adipose Tissue and Stimulate Their Uptake by the Liver
It has long been appreciated that plasma triglycerides and free fatty acid (FFA) levels become elevated in response to glucocorticoids.84,85 Moreover, using arterial and venous sampling of adipose tissue in humans under normal conditions, both FFA and glycerol levels rise during food deprivation and fall with feeding consistent with effects on lipolysis.85 These findings are largely attributable to the effect of insulin on hormone-sensitive lipase and in part due to activation of lipoprotein lipase.79,86 FFAs are a major substrate for energy production by tissue, but excessive and prolonged high FFA levels lead to increased fat deposition in liver and other tissues contributing to insulin resistance.14,16,27,87
Effects of glucocorticoids on adipocytes differ based on location of fat stores. GC stimulates lipolysis in subcutaneous adipose tissue, whereas it stimulates differentiation and hypertrophy of adipocytes in visceral adipose tissue, effects that are consistent with the phenotype of patients with Cushing’s disease.4,5,27 Using constant venous infusion of physiological concentrations of cortisol in normal human volunteers, it was shown that appearance of palmitate into the circulation is increased by 60%.88 Likewise, employing micro-dialysis techniques in femoral and abdominal adipose tissue in normal volunteers, cortisol infusion at physiological levels stimulated fatty acid release.89 Furthermore, in isolated rat hepatocytes in culture, addition of a glucocorticoid (dexamethasone) stimulated hormone-sensitive lipase (HSL) similar to effect seen upon addition of epinephrine, glucagon and growth hormone.90 However, unlike the other hormones mentioned, dexamethasone also stimulates the expression of the mRNA encoding HSL in experimental animals.90 While there is agreement that glucocorticoids significantly increase FFA release from subcutaneous adipose tissue,91 the steady-state circulating levels of FFAs is determined by their rate of entry into the circulation and rate of their uptake by tissues; FFA levels are only modestly elevated by glucocorticoids consistent with increased tissue uptake.
Tissues, especially heart, skeletal muscle, and liver avidly take up circulating FFAs.92 A host of fatty acid transporters mediate the transmembrane transport of FFAs by tissue.93–95 Similar to stimulation of glucose transport by insulin that is mediated by translocation of GLUT4 to the plasma membrane, fatty acid transporters are likewise translocated to plasma membranes to facilitate FFA uptake.94 The expression of CD36, a well-studied fatty acid transporter, is upregulated in liver of glucocorticoid-treated rats.96
Elevated insulin levels, in the absence of excess glucocorticoids strongly inhibit lipolysis in adipocytes. It is of interest that the glucocorticoid stimulation of FFA release from adipocytes and the rise of circulating FFA levels occur despite the presence of elevated plasma insulin levels that results from GC effect on increasing systemic insulin resistance.
In sum, GC-stimulation of lipolysis with elevated plasma levels of FFA and stimulation of FFA uptake by the liver represents the dominant path of hepatic lipid deposition.13,16,17,19,97
Effect of Glucocorticoids on Hepatic VLDL Synthesis and Release
Glucocorticoid excess in humans and experimental animals results in a significant increase in plasma triglyceride levels contained in very-low-density lipoprotein (VLDL) particles.76,98,99 In isolated rat hepatocytes, incubation with dexamethasone increased the secretion of triglycerides into the medium in particles with properties of VLDL.100,101 Similar increases in VLDL levels were observed in rats treated for 5 days with low doses of triamcinolone, with the effect being attributed to stimulation of hepatic fatty acid and VLDL synthesis.102 Glucocorticoids increase plasma free fatty acid levels by stimulating lipolysis in peripheral adipose tissue and stimulates FFA uptake by the liver where they are converted to TGs and deposited in lipid droplets.76,102 Triacylglycerol hydrolase (TGH) is an ER-localized enzyme that hydrolyzes TGs within hepatocyte. A portion of the released fatty acids can be re-esterified back to TGs.103 Some of the re-synthesized TGs bind to ApoB100 in the endoplasmic reticulum to form VLDL particles that are then concentrated in the Golgi apparatus and are secreted as VLDL.76,104,105 A larger fraction of the synthesized TGs, especially in fed conditions, are stored in lipid droplets.104,106 Through action of lipoprotein lipase, fatty acids and glycerol are released from VLDL and the lipoprotein is converted to cholesterol-rich LDL particles that are taken up via LDL receptors by the liver (and other tissues).
As noted in an earlier section, glucocorticoids stimulate hepatic fatty acid synthesis and lead to accumulation of TGs as well as to a small increase in VLDL secretion.75,77,98,104,107 Other mechanisms that contribute to accumulation of TGs in the liver by glucocorticoids are, reduction of apolipoprotein B degradation and decreased TGH expression and activity; the latter is primarily due to a decrease in TGH mRNA stability that leads to reduced lipolysis of TGs.76,101 The combination of increased TG synthesis and the relatively smaller increase in VLDL secretion by excess GC results in net hepatic TG accumulation.76,104
GC-induced elevation of plasma triglyceride and VLDL levels is mediated in part by increased secretion of VLDL by the liver. However, the stimulated rate of VLDL secretion does not fully explain the much larger increase in plasma TG levels, suggesting that additional mechanisms might be operative. It is generally agreed that glucocorticoids also inhibit plasma lipoprotein lipase which increases the accumulation of VLDL particles in the circulation.104,108
Glucocorticoid Inhibition of β-Oxidation of Fatty Acids
Fatty acids are activated by acyl-CoA synthetases to form fatty acyl-CoAs that are then transported into mitochondria via the carnitine shuttle. Fatty acyl-CoAs undergo beta-oxidation in the mitochondrial matrix and are converted into two carbon fragments of acetyl-CoA in a series of sequential reactions mediated by acyl-CoA-dehydrogenases and three other enzymatic steps.109 The bulk of acetyl-CoA enters the tricarboxylic acid cycle for energy production, while excess acetyl-CoA can be converted to “ketone bodies”, first into acetoacetate by reactions catalyzed by mitochondrial HMG-CoA synthase and lyase and then the generated acetoacetate can be reduced to β-hydroxybutyrate by β-hydroxybutyrate dehydrogenase.110
An important regulator of hepatic mitochondrial fatty acid oxidation is peroxisome proliferator-activated receptor α (PPARα).111,112 In conjunction with PGC-1, it induces the expression of mitochondrial acyl-CoA dehydrogenases and stimulates hepatic fatty acid oxidation.111,112 Glucocorticoids inhibit mitochondrial acyl-CoA dehydrogenases by inhibiting the transcriptional activity of PPARα resulting in decreased beta-oxidation thereby contributing to intracellular lipid accumulation.1,109
Glucagon levels are increased in response to glucocorticoids and can enhance fatty acid oxidation.41,46 Glucagon increases the expression of cyclic AMP-responsive element-binding (CREB) protein that in turn stimulates the transcription of carnitine acyl transferase 1 (CPT-1). CPT-1 catalyzes the conversion of fatty acids to acyl carnitines that are then transported into the mitochondria and the released acyl-CoAs undergo beta-oxidation.42,109 In addition, glucagon stimulates the expression of mitochondrial HMG-CoA synthase that catalyzes the formation of ketones.110 Hence, elevation of glucagon levels per se can stimulate fatty acid β-oxidation. In keeping with this, use of glucagon receptor antagonists can lead to elevated lipid deposits in the liver.42 However, the large increases in insulin levels that occur in conjunction with the glucocorticoid-induced insulin-resistant state greatly overshadows the stimulation of ketone body formation by glucagon.27,113 Part of the effect on ketone body formation is due to the significant repression of mitochondrial HMG-CoA synthase expression in rat hepatocytes in response to insulin.110,114,115
Taken together, the net effect of glucocorticoids on β-oxidation of fatty acids in the liver is one of the suppressions. The inhibition of one of the important pathways of decreasing hepatic lipid content serves to enhance the development of fatty liver that is commonly seen in the condition of excess glucocorticoids.
Additional Critical Actions of Glucocorticoids in the Regulation of Metabolism
In addition to the above, topics of high relevance to GC’s effect of hepatic steatosis should be considered. These include the reversible intracellular conversion of cortisol to cortisone and the role this critical reaction has in controlling intracellular cortisol concentrations. The second is the role of osteocalcin, the bone-derived γ-carboxylated polypeptide, on insulin sensitivity, metabolism and glucose homeostasis.
Importance of Cortisol and Cortisone Interconversion to Glucocorticoid’s Actions on Metabolism
The availability and concentration of cortisol in the intracellular milieu is governed not only on its concentration in the circulation but also by its reversible conversion to cortisone, an inactive product. The conversion of inactive cortisone to active cortisol in cells is catalyzed by 11β-hydroxysteroid dehydrogenase type 1 or 11-ketoreductase (11β-HSD1).116,117 The enzyme is highly expressed in liver, adipose tissue, and skeletal muscle. The reverse inactivation reaction, namely conversion of cortisol to cortisone is catalyzed by 11β-dehydrogenase (type 2 11β-HSD; 11β-HSD2) that is predominantly expressed at high levels in kidneys, colon, and salivary glands.117 Through 11β-HSD2 activity, the concentration of cortisol is kept low in the latter tissues and serves to prevent the activation of mineralocorticoid receptors by cortisol.
Studies in transgenic mice have shown that over-expression of the 11β-HSD1 in adipose tissue results in insulin resistance, visceral adiposity, elevated blood FFA levels, and fatty liver.116,118 More specifically, overexpression of the enzyme in adipose tissue of transgenic mice on a high-fat diet resulted in hepatic insulin resistance and increased liver fat deposition.119 In keeping with the above, selective inhibition of 11β-HSD1 activity in knock-out (KO) mice increases insulin sensitivity in skeletal muscle and decreases the expression of genes involved in lipogenesis and lipolysis.117,120 Additionally, overexpression of 11β-HSD1 in the liver of transgenic mice stimulated the development of components of metabolic syndrome and its KO prevented this development.121,122
An enlightening detailed study of a patient with Cushing’s disease with increased blood levels of cortisol due to an ACTH-secreting pituitary tumor who exhibited none of the features of excess cortisol was reported; the patient had no central obesity, hirsutism, proximal myopathy, easy bruising, or hypertension.123 This led to mechanistic studies on the role of 11β-HSD1 in glucocorticoid action. Using 11β-HSD1 knockout mice, these novel studies showed that tissue regeneration of glucocorticoids instead of high circulating GC levels mediates the Cushing’s disease phenotype.117 They also found that the KO mice showed resistance to the effects of exogenously administered glucocorticoids. More importantly, mice with liver-specific KO of 11β-HSD1 developed the Cushing’s-like phenotype upon administration of excess GC. In contrast, adipose tissue-specific 11β-HSD1 KO mice were partially protected the untoward effects of the administered GC. It is probable that these mechanisms are, at least in part, operative in humans. Because of the potential importance of cortisol metabolism by 11β-HSD1 and 11β-HSD2 in human disease, the effect of inhibitors of 11β-HSD1 has been studied in patients with type 2 diabetes as well as in patients with fatty liver disease.124–126 However, the results of these studies on fatty liver and metabolic syndrome have been modest,127,128 and the use of the inhibitors is associated with some untoward effects.
It appears from the above that regulation of cortisol levels in specific tissue including liver, skeletal muscle and adipose tissue plays a contributing, but not a determining role in metabolism, obesity, insulin resistance, and glucose homeostasis. In addition, synthetic modulators of glucocorticoid receptor can attenuate obesity and inflammation and reverse fatty liver in experimental animals.129,130 Much further work is required for a better understanding of the role of the 11β-HSD1 and 11β-HSD2 in glucocorticoid action.
Role of Osteocalcin in Glucocorticoid Regulation of Glucose Homeostasis
Human osteocalcin is a 49 amino acid protein expressed in osteoblasts.131 The mature protein, first identified in 1980, contains three vitamin K-dependent Ɣ-carboxyglutamic acids.132 Osteocalcin stimulates bone formation and its lack leads to osteopenia.133 Once secreted into the bone lacunar space, it loses one or more of its carboxyglutamic acids. Both fully carboxylated and under-carboxylated forms of the protein appear in the circulation; however, only the under- or non-carboxylated forms mediate the metabolic actions of osteocalcin in peripheral tissues.131 Importantly, expression of osteocalcin is stimulated by insulin and is inhibited by glucocorticoids.46,57,134 Suppression of osteocalcin expression resulting from excess glucocorticoids leads to the commonly observed bone loss and increased bone fragility.
Studies performed in mice and rats concluded that osteocalcin has important effects outside of bone metabolism.46 It was first discovered that mice lacking osteocalcin exhibit decreased insulin sensitivity with glucose intolerance that is associated with decreased β-cell proliferation.135 These observations were extended by the finding that elevated serum under-carboxylated osteocalcin leads to increased insulin synthesis and secretion, elevated adiponectin levels, and increased insulin sensitivity in liver and muscle.131 The accumulated evidence suggests that the adverse effects of glucocorticoids on glucose homeostasis is mediated, at least in part, by inhibition of synthesis and secretion of osteocalcin.57
Conclusions and Future Directions
Ongoing research focused on the mechanisms underlying glucocorticoid stimulation of hepatic lipid accumulation continues to be a topic of great interest. The clinical importance of this field of investigation is the very large overlap and parallelism between glucocorticoid-induced fatty liver disease and the growing pandemic of NAFLD and NASH; the later conditions have become one of the leading causes of cirrhosis in the United States and other countries in the past decade. Elevated circulating glucocorticoid levels induce insulin resistance that is a hallmark of metabolic syndrome, type 2 diabetes, and fatty liver disease.12 GC-induced increase in hepatic deposition of lipids (mostly triglycerides) is mediated by multiple mechanisms including increased food intake, stimulated gluconeogenesis, stimulation of de novo fatty acid synthesis by high glucose, insulin, and GC levels, and increased release of FFAs from adipose tissue and their uptake and deposition in liver as TGs. While there is an increase in hepatic VLDL synthesis and secretion that in part accounts for the elevated circulating triglycerides, the effect of glucocorticoids on increasing plasma TG levels is largely mediated by inhibition of plasma lipoprotein lipase activity. In addition, glucocorticoid excess leads to inhibition of β-oxidation of fatty acids that can result in a further increase in liver TG levels. This set of changes helps explain the rapid development of fatty liver in experimental animals following a few days of treatment with excess glucocorticoids.16,79,96
There is also increasing evidence that much of the actions of cortisol is mediated by the cellular levels of the hormone; tissue cortisol levels are controlled by reversible interconversion of inactive cortisone to active cortisol mediated by β-hydroxysteroid dehydrogenases. Moreover, studies in experimental animals have also demonstrated a critical role for glucocorticoids in the regulation of osteocalcin synthesis and the effect of this bone-derived hormone in regulating the sensitivity of tissues to insulin. The extent that this later control mechanism is operative in humans is not known.
In sum, there is overwhelming evidence that exposure to excess glucocorticoids leads to development of fatty liver, insulin resistance, hyperinsulinemia, hyperglycemia and altered glucose homeostasis. In a meta-analysis of normal healthy individuals treated with glucocorticoids, it was reported that the treatment resulted in increased fasting hyperglycemia, insulin levels, and insulin resistance.43 Another study reported that 32% of people develop GC-induced hyperglycemia and 19% develop T2DM after ≥4 weeks of treatment with glucocorticoids.44 Even a 24-hour treatment with glucocorticoids results in significant alterations in glucose homeostasis.45,136 Given that this class of medications are frequently used at pharmacologic doses and for prolonged durations for the treatment of a host of clinical disorders,44 it is imperative that the dosage and duration of exposure to GC that can lead to the development of fatty liver be closely examined. Moreover, the degree of reversibility of GC-induced fatty liver and the time course of such reversal upon discontinuation of glucocorticoids are largely unknown. Studies are required to answer these medically important questions.
Acknowledgments
We are indebted to Ms. Tara Haghighi for her help in producing Figure 1.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Faramarz Ismail-Beigi is a consultant for Sanofi and COVANCE. He has grants from the NIH, and has previously received grants from Novo Nordisk; he has shares in Thermalin Insulin. The authors report no other conflicts of interest in this work.
References
1. Marino JS, Stechschulte LA, Stec DE, Nestor-Kalinoski A, Coleman S, Hinds TD
2. Der Laan SV, Onno C, Meijer OC. Pharmacology of glucocorticoids: beyond receptors. Eur J Pharmcol. 2008;585:3483–3491.
3. Overman RA, Yeh JY, Deal CL. Prevalence of oral glucocorticoid usage in the United States: a general population perspective. Arthritis Care Res (Hoboken). 2013;65(2):294–298. doi:10.1002/acr.21796
4. Shibli-Rahhal A, Van Beek M, Schlechte JA. Cushing’s syndrome. Clin Dermatol. 2006;24(4):260–265.
5. Chanson P, Salenave S. Metabolic syndrome in Cushing’s syndrome. Neuroendocrinology. 2010;92(Suppl.1):96–101. doi:10.1159/000314272
6. Pijl H, Meinders AE. Bodyweight change as an adverse effect of drug treatment. Mechanisms and management. Drug Saf. 1996;14(5):329–342. doi:10.2165/00002018-199614050-00005
7. Schäcke H, Döcke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96(1):23–43. doi:10.1016/S0163-7258(02)00297-8
8. Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346(16):1221–1231. doi:10.1056/NEJMra011775
9. Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013;10(11):656–665. doi:10.1038/nrgastro.2013.183
10. Younossi ZM, Blissett D, Blissett R, et al. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology. 2016;64(5):1577–1586. doi:10.1002/hep.28785
11. Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313(22):2263–2273. doi:10.1001/jama.2015.5370
12. Gharaibeh NE, Rahhal MN, Rahimi L, Ismail-Beigi F. SGLT-2 inhibitors as promising therapeutics for non-alcoholic fatty liver disease: pathophysiology, clinical outcomes, and future directions. Diabetes Metab Syndr Obes. 2019;12:1001–1012. doi:10.2147/DMSO.S212715
13. Hellerstein MK, Christiansen M, Kaempfer S, et al. Measurement of de novo hepatic lipogenesis in humans using stable isotopes. J Clin Invest. 1991;87(5):1841–1852. doi:10.1172/JCI115206
14. Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unraveling the mechanism. Lancet. 2010;375(9733):2267–2277. doi:10.1016/S0140-6736(10)60408-4
15. Rui L. Energy metabolism in the liver. Compr Physiol. 2014;4(1):177–197.
16. Samuel VT, Shulman GI. Non-alcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 2018;27(1):22–41. doi:10.1016/j.cmet.2017.08.002
17. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–1351. doi:10.1172/JCI23621
18. Parks EJ. Dietary carbohydrate’s effects on lipogenesis and the relationship of lipogenesis to blood insulin and glucose concentrations. Br J Nutr. 2002;87(Suppl. 2):S247–S253. doi:10.1016/j.clindermatol.2006.04.012
19. Timlin MT, Parks EJ. Temporal pattern of de novo lipogenesis in the postprandial state in healthy men. Am J Clin Nutr. 2005;81(1):35–42. doi:10.1093/ajcn/81.1.35
20. Ishida-Takahashi R, Uotani S, Abe T, et al. Rapid inhibition of leptin signaling by glucocorticoids in vitro and in vivo. J Biol Chem. 2004;279(19):19658–19664. doi:10.1074/jbc.M310864200
21. Uddén J, Björntorp P, Arner P, Barkeling B, Meurling L, Rossner S. Effects of glucocorticoids on leptin levels and eating behavior in women. J Intern Med. 2003;253(2):225–231. doi:10.1046/j.1365-2796.2003.01099.x
22. Gruzdeva O, Borodkina D, Uchasova E, Dyleva Y, Barbarash O. Leptin resistance: underlying mechanisms and diagnosis. Diabetes Metab Syndr Obes. 2019;12:191–198. doi:10.2147/DMSO.S182406
23. Solano JM, Jacobson L. Glucocorticoids reverse leptin effects on food intake and body fat in mice without increasing NPY mRNA. Am J Physiol. 1999;277(4):E708–16. doi:10.1152/ajpendo.1999.277.4.E708
24. Perry RJ, Resch JM, Douglass AM, et al. Leptin’s hunger-suppressing effects are mediated by the hypothalamic–pituitary–adrenocortical axis in rodents. Proc Natl Acad Sci U S A. 2019;116(27):13670–13679. doi:10.1073/pnas.1901795116
25. Erickson JC, Hollopeter G, Palmiter RD. Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science. 1996;274(5293):1704–1707. doi:10.1126/science.274.5293.1704
26. Feingold K, Grunfeld C. Endotext [Internet]: Introduction to Lipids and Lipoproteins. South Darthmouth (MA): MDText.com, Inc; 2000. Available from: https://www.ncbi.nlm.nih.gov/books/NBK305896/.
27. Vegiopoulos A, Herzig S. Glucocorticoids, metabolism and metabolic diseases. Molecular Cellular Endocrinol. 2007;275(1–2):43–61. doi:10.1016/j.mce.2007.05.015
28. Fromentin C, Tomé D, Nau F, et al. Dietary proteins contribute little to glucose production, even under optimal gluconeogenic conditions in healthy humans. Diabetes. 2013;62(5):1435–1442. doi:10.2337/db12-1208
29. Adeva-Andany MM, Funcasta-Calderón R, Fernández-Fernández C, Castro-Quintela E, Carneiro-Freire N. Metabolic effects of glucagon in humans. J Clin Transl Endocrinol. 2018;15:45–53. doi:10.1016/j.jcte.2018.12.005
30. Zhang X, Yang S, Chen J, Su Z. Unraveling the regulation of hepatic gluconeogenesis. Front Endocrinol (Lausanne). 2018;9:802. doi:10.3389/fendo.2018.00802
31. Peckett AJ, Wright DC, Riddell MC. The effects of glucocorticoids on adipose tissue lipid metabolism. Metabolism. 2011;60(11):1500–1510. doi:10.1016/j.metabol.2011.06.012
32. Kumar R, Mohan S. Non-alcoholic fatty liver disease in lean subjects: characteristics and implications. J Clin Transl Hepatol. 2017;5(3):216–223. doi:10.14218/JCTH.2016.00068
33. Hanson RW, Reshef L. Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Annu Rev Biochem. 1997;66:581–611. doi:10.1146/annurev.biochem.66.1.581
34. Lochhead PA, Salt IP, Walker KS, Hardie DG, Sutherland C. 5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes. 2000;49(6):896–903.
35. Patel R, Williams-Dautovich J, Cummins CL. Minireview: new molecular mediators of glucocorticoid receptor activity in metabolic tissues. Mol Endocrinol. 2014;28(7):999–1011.
36. Burchell A. Endoplasmic reticulum phosphate transport. Kidney Int. 1996;49(4):953–958. doi:10.1038/ki.1996.134
37. Hah JS, Ryu J, Lee W, Jung CY, Lachaal M. The hepatocyte glucose-6-phosphatase subcomponent T3: its relationship to GLUT2. Biochim Biophys Acta. 2002;1564(1):198–206. doi:10.1016/S0005-2736(02)00450-9
38. Adeva-Andany MM, Perez-Fepete N, Fernandez-Fernansez C, Donapetry-Garcia C, Pazos-Garcia C. Liver glucose metabolism in humans. Biosci Rep. 2016;36(6):e00416. doi:10.1042/BSR20160385
39. Cui A, Fan H, Zhang Y, et al. Dexamethasone-induced Krüppel-like factor 9 expression promotes hepatic gluconeogenesis and hyperglycemia. J Clin Invest. 2019;129(6):2266–2278. doi:10.1172/JCI66062
40. Sweet DR, Fan L, Jain MK. Taking KLF9 to “Cort” for crimes against metabolism. J Clin Invest. 2019;129(6):2178–2180. doi:10.1172/JCI128481
41. Rafacho A, Goncaalves-Neto LM, Santo-Silva JC, et al. Pancreatic alpha cells dysfunction contributes to the disruption of glucose homeostasis and compensatory insulin hypersecretion in glucocorticoid-treated rats. PLoS One. 2014;9(4):e93531. doi:10.1371/journal.pone.0093531
42. Galsgaard KD, Pedersen J, Knop FK, Holst JJ, Albrechtsen NJ. Glucagon receptor signaling and lipid metabolism. Front Physiol. 2019;10:413. doi:10.3389/fphys.2019.00413
43. Zhou PZ, Zhu YM, Zou GH, et al. Relationship between glucocorticoids and insulin resistance in healthy individuals. Med Sci Monit. 2016;22:1887–1894. doi:10.12659/MSM.895251
44. Liu XX, Zhu XM, Miao Q, Ye HY, Zhang ZY, Li YM. Hyperglycemia induced by glucocorticoids in non-diabetic patients: a meta-analysis. Ann Nutr Metab. 2014;65(4):324–332. doi:10.1159/000365892
45. Abdelmannan D, Tahboub R, Genuth S, Ismail-Beigi F. Effect of dexamethasone on oral glucose tolerance in normal individuals. Endocr Pract. 2010;16(5):770–777. doi:10.4158/EP09373.OR
46. Rafacho A, Ortsäter H, Nadal A, Quesada I. Glucocorticoid treatment and endocrine pancreas function: implications for glucose homeostasis, insulin resistance and diabetes. J Endocrinol. 2014;223(3):R49–R62. doi:10.1530/JOE-14-0373
47. Stojanovska L, Rosella G, Proietto J. Evolution of dexamethasone-induced insulin resistance in rats. Am J Physiol. 1990;258(5 Pt 1):E748–E756. doi:10.1152/ajpendo.1990.258.5.E748
48. Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol. 2011;12(11):722–734. doi:10.1038/nrm3198
49. Almon RR, Dubois DC, Jin JY, Jusko WJ. Temporal profiling of the transcriptional basis for the development of corticosteroid-induced insulin resistance in rat muscle. J Endocrinol. 2005;184(1):219–232. doi:10.1677/joe.1.05953
50. Saad MJ, Folli F, Kahn JA, Kahn CR. Modulation of insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of dexamethasone-treated rats. J Clin Invest. 1993;92(4):2065–2072. doi:10.1172/JCI116803
51. Buren J, Lai YC, Lundgren M, Eriksson JW, Jensen J. Insulin action and signaling in fat and muscle from dexamethasone-treated rats. Arch Biochem Biophys. 2008;474(1):91–101. doi:10.1016/j.abb.2008.02.034
52. Bazuine M, Carlotti F, Jahangir Tafrechi RS, Hoeben RC, Antonie Maassen J. Mitogen-activated protein kinase (MAPK) phosphatase-1 and −4 attenuate p38 MAPK during dexamethasone-induced insulin resistance in 3T3-L1 adipocytes. Mol Endocrinol. 2004;18(7):1697–1707. doi:10.1210/me.2003-0213
53. Mazziotti G, Gazzaruso C, Giustina A. Diabetes in Cushing syndrome: basic and clinical aspects. Trends Endocrinol Metab. 2011;22(12):499–506. doi:10.1016/j.tem.2011.09.001
54. Lim S, Son KR, Song IC, et al. Fat in liver/muscle correlates more strongly with insulin sensitivity in rats than abdominal fat. Obesity (Silver Spring). 2009;17(1):188–195. doi:10.1038/oby.2008.486
55. Greco AV, Mingrone G, Giancaterini A, et al. Insulin resistance in morbid obesity: reversal with intramyocellular fat depletion. Diabetes. 2002;51(1):144–151. doi:10.2337/diabetes.51.1.144
56. Coen PM, Goodpaster BH. Role of intramyocelluar lipids in human health. Trends Endocrinol Metab. 2012;23(8):391–398. doi:10.1016/j.tem.2012.05.009
57. Brennan-Speranza TC, Henneicke H, Gasparini SJ, et al. Osteoblasts mediate the adverse effects of glucocorticoids on fuel metabolism. J Clin Invest. 2012;122(11):4172–4189. doi:10.1172/JCI63377
58. Sood A, Ismail-Beigi F. Effect of dexamethasone on insulin secretion: examination of underlying mechanisms. Endocr Pract. 2010;16(5):763–769. doi:10.4158/EP09372.OR
59. Stubbs M, York DA. Central glucocorticoid regulation of parasympathetic drive to pancreatic B-cells in the obese fa/fa rat. Int J Obes. 1991;15(8):547–553.
60. Jeong IK, Oh SH, Kim BJ, et al. The effects of dexamethasone on insulin release and biosynthesis are dependent on the dose and duration of treatment. Diabetes Res Clin Pract. 2001;51(3):163–171. doi:10.1016/S0168-8227(00)00229-1
61. Lambillotte C, Gilon P, Henquin JC. Direct glucocorticoid inhibition of insulin secretion: an in vitro study of dexamethasone effects in mouse islets. J Clin Invest. 1997;99(3):414–423. doi:10.1172/JCI119175
62. Martínez B, Pereira A, Muzetti J, Telles F, Mundim F, Teixeira M. Experimental model of glucocorticoid-induced insulin resistance. Acta Cir Bras. 2016;31(10):645–649. doi:10.1590/S0102-865020160100000001
63. Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol. 2013;48(4):434–441. doi:10.1007/s00535-013-0758-5
64. Wilke MS, French MA, Goh YK, Ryan EA, Jones PJ, Clandinin MT. Synthesis of specific fatty acids contributes to VLDL-triacylglycerol composition in humans with and without type 2 diabetes. Diabetologia. 2009;52(8):1628–1637. doi:10.1007/s00125-009-1405-9
65. Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118(3):829–838. doi:10.1172/JCI34275
66. Santoleri D, Titchenell PM. Resolving the paradox of hepatic insulin resistance. Cell Mol Gastroenterol Hepatol. 2019;7(2):447–456. doi:10.1016/j.jcmgh.2018.10.016
67. Ferré P, Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP‐1c. Diabetes Obes Metab. 2010;12(suppl2):83–92. doi:10.1111/j.1463-1326.2010.01275.x
68. Filhoulaud G, Guilmeau S, Dentin R, Girard J, Postic C. Novel insights into ChREBP regulation and function. Trends Endocrinol Metab. 2013;24(5):257–268. doi:10.1016/j.tem.2013.01.003
69. Schwarz JM, Linfoot P, Dare D, Aghajanian K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am J Clin Nutr. 2003;77(1):43–50. doi:10.1093/ajcn/77.1.43
70. Weickert MO, Pfeiffer AF. Signalling mechanisms linking hepatic glucose and lipid metabolism. Diabetologia. 2006;49(8):1732–1741. doi:10.1007/s00125-006-0295-3
71. Puigserver P, Rhee J, Donovan J, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423(6939):550–555. doi:10.1038/nature01667
72. Dentin R, Pégorier JP, Benhamed F, et al. Hepatic glucokinase Is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem. 2004;279(19):20314–20326. doi:10.1074/jbc.M312475200
73. Ruderman NB, Saha AK, Kraegen EW. Minireview: malonyl CoA, AMP-activated protein kinase, and adiposity. Endocrinology. 2003;144(12):5166–5171. doi:10.1210/en.2003-0849
74. Zhao LF, Iwasaki Y, Zhe W, et al. Hormonal regulation of Acetyl-CoA carboxylase isoenzyme gene transcription. Endocr J. 2010;57(4):317–324. doi:10.1507/endocrj.K09E-298
75. Reshef L, Olswang Y, Cassuto H, et al. Glyceroneogenesis and the triglyceride/fatty acid cycle. J Biol Chem. 2003;278(33):30413–30416. doi:10.1074/jbc.R300017200
76. Dolinsky VW, Douglas DN, Lehner R, Vance DE. Regulation of the enzymes of hepatic microsomal triacylglycerol lipolysis and re-esterification by the glucocorticoid dexamethasone. Biochem J. 2004;378(Pt 3):967–974. doi:10.1042/bj20031320
77. Diamant S, Shafrir E. Modulation of the activity of insulin-dependent enzymes of lipogenesis by glucocorticoids. Eur J Biochem. 1975;53(2):541–546. doi:10.1111/j.1432-1033.1975.tb04097.x
78. Smith GI, Shankaran M, Yoshino M, et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest. 2020;130(3):1453–1460. doi:10.1172/JCI134165
79. Wang JC, Gray NE, Kuo T, Harris CA. Regulation of triglyceride metabolism by glucocorticoid receptor. Cell Biosci. 2012;2(1):19. doi:10.1186/2045-3701-2-19
80. Altman KL, Miller LL, Bly CG. The synergistic effect of cortisone and insulin on lipogenesis in the perfused rat liver as studied with alpha-C14-acetate. Arch Biochem Biophys. 1951;31(2):329–331. doi:10.1016/0003-9861(51)90225-1
81. Wang Y, Jones VB, Urs S, et al. The human fatty acid synthase gene and de novo lipogenesis are coordinately regulated in human adipose tissue. J Nutr. 2004;134(5):1032–1038. doi:10.1093/jn/134.5.1032
82. Amatruda JM, Danahy SA, Chang CL. The effects of glucocorticoids on insulin-stimulated lipogenesis in primary cultures of rat hepatocytes. Biochem J. 1983;212(1):135–141. doi:10.1042/bj2120135
83. Feng B, He Q, Xu H. FOXO-1dependent up-regulation of MAP kinase phosphatase e (MKP-3) mediates glucocorticoid-induced hepatic lipid accumulation in mice. Mol Cell Endocrinol. 2014;393(1–2):46–55. doi:10.1016/j.mce.2014.06.001
84. Skanse B, Von Studnitz W, Skoogs N. The effect of corticotropin and cortisone on serum lipids and lipoproteins. Acta Endocrinol (Copenh). 1959;31(3):442–450. doi:10.1530/acta.0.XXXI0442
85. Frayn KN, Coppack SW, Fielding BA, Humphreys SM. Coordinated regulation of hormone-sensitive lipase and lipoprotein lipase in human adipose tissue in vivo: implications for the control of fat storage and fat mobilization. Adv Enzyme Regul. 1995;35:163–178. doi:10.1016/0065-2571(94)00011-Q
86. Macfarlane DP, Forbes S, Walker BR. Glucocorticoids and fatty acid metabolism in humans: fueling fat redistribution in the metabolic syndrome. J Endocrinol. 2008;197(2):189–204. doi:10.1677/JOE-08-0054
87. Saltiel AR, Kahn CR. Insulin signaling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799–806. doi:10.1038/414799a
88. Divertie GD, Jensen MD, Miles JM. Stimulation of lipolysis in humans by physiological hypercortisolemia. Diabetes. 1991;40(10):1228–1232. doi:10.2337/diab.40.10.1228
89. Djurhuus CB, Gravholt CH, Nielsen S, et al. Effects of cortisol on lipolysis and regional interstitial glycerol levels in humans. Am J Physiol Endocrinol Metab. 2002;283(1):E172–7.
90. Slavin BG, Ong JM, Kern PA. Hormonal regulation of hormone-sensitive lipase activity and mRNA levels in isolated rat adipocytes. J Lipid Res. 1994;35(9):1535–1541.
91. DjorDjevic A, Veličković N, Bursać B, Teofilović A, MaTić G. The role of glucocorticoid hormones in diet-induced metabolic diseases. Diabe Biologia Serbica. 2017;39(1):16–25.
92. Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J Lipid Res. 2009;50:S86–S90. doi:10.1194/jlr.R800085-JLR200
93. Alves-Bezerra M, Cohen DE. Triglyceride metabolism in the liver. Compr Physiol. 2017;8(1):1–8. doi:10.1002/cphy.c170012
94. Bonen A, Campbell SE, Benton CR, et al. Regulation of fatty acid transport by fatty acid translocase/CD36. Proc Nutr Soc. 2004;63(2):245–249. doi:10.1079/PNS2004331
95. Schwenk RW, Holloway GP, Luiken JJ, Bonen A, Glatz JF. Fatty acid transport across the cell membrane: regulation by fatty acid transporters. Prostaglandins Leukot Essent Fatty Acids. 2010;82(4–6):149–154. doi:10.1016/j.plefa.2010.02.029
96. D’souza AM, Beaudry JL, Szigiato AA, et al. Consumption of a high-fat diet rapidly exacerbates the development of fatty liver disease that occurs with chronically elevated glucocorticoids. Am J Physiol Gastrointest Liver Physiol. 2012;302(8):G850–63. doi:10.1152/ajpgi.00378.2011
97. Parks EJ, Krauss RN, Christiansen MP, Neese RA, Hellerstein MK. Effects of a low-fat, high-carbohydrate diet on VLDL-triglyceride assembly, production, and clearance. J Clin Invest. 1999;104(8):1087–1096. doi:10.1172/JCI6572
98. Brindley DN. Regulation of hepatic triacylglycerol synthesis and lipoprotein metabolism by glucocorticoids. Clin Sci. 1981;61(2):129–133. doi:10.1042/cs0610129
99. Choi SH, Ginsberg HN. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol Metab. 2011;22(9):353–363. doi:10.1016/j.tem.2011.04.007
100. Mangiapane EH, Brindley DN. Effects of dexamethasone and insulin on the synthesis of triacylglycerols and phosphatidylcholine and the secretion of very-low-density lipoproteins and lysophosphatidylcholine by monolayer cultures of rat hepatocytes. Biochem J. 1986;233(1):151–160. doi:10.1042/bj2330151
101. Wang C-N, Mcleod R, Yao Z, Brindley DN. Effects of dexamethasone on the synthesis, degradation, and secretion of apolipoprotein B in cultured rat hepatocytes. Arterioscler Thromb Vasc Biol. 1995;15(9):1481–1491. doi:10.1161/01.ATV.15.9.1481
102. Krausz Y, Bar-On H, Shafrir E. Origin and pattern of glucocorticoid-induced lipidemia in rats. Dose-dependent bimodal changes in serum lipids and lipoproteins in relation to hepatic lipogenesis and tissue lipoprotein lipase activity. Biochem Biophys Acta. 1981;663(1):69–82. doi:10.1016/0005-2760(81)90195-8
103. Liu M, Chung S, Shelness GS, Parks JS. Hepatic ABCA1 and VLDL triglyceride production. Biochim Biophys Acta. 2012;1821(5):770–777. doi:10.1016/j.bbalip.2011.09.020
104. Cole TG, Wilcox HG, Heimberg M. Effects of adrenalectomy and dexamethasone on hepatic lipid metabolism. J Lipid Res. 1982;23(1):81–91.
105. Reaven EP, Kolterman OR, Reaven GM. Ultrastructural and physiological evidence for corticosteroid-induced alterations in hepatic production of very low density lipoprotein particles. J Lipid Res. 1974;15(1):74–83.
106. Onal G, Kutlu O, Gozuacik D, Dokmecei Emre S. Lipid droplets in health and disease. Lipids Health Dis. 2017;16(1):128. doi:10.1186/s12944-017-0521-7
107. Berg AL, Nilsson-Ehle P. Direct effects of corticotropin on plasma lipoprotein metabolism in man – studies in vivo and in vitro. Metabolism. 1994;43(1):90–97. doi:10.1016/0026-0495(94)90162-7
108. Bagdade JD, Yee E, Albers J, Pykalisto OJ. Glucocorticoids and triglyceride transport: effects on triglyceride secretion rates, lipoprotein lipase, and plasma lipoproteins in the rat. Metabolism. 1976;25(5):533–542. doi:10.1016/0026-0495(76)90007-X
109. Lettéron P, Brahimi-Bourouina N, Robin MA, Moreau A, Feldmann G, Pessayre D. Glucocorticoids inhibit mitochondrial matrix acyl-CoA dehydrogenases and fatty acid beta-oxidation. Am J Physiol. 1997;272(5 Pt 1):G1141–50. doi:10.1152/ajpgi.1997.272.5.G1141
110. Hegardt FG. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: a control enzyme in ketogenesis. Biochem J. 1999;338(Pt 3):569–582. doi:10.1042/bj3380569
111. Leone TC, Lehman JJ, Finck BN, et al. PGC-1α deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3(4):e101. doi:10.1371/journal.pbio.0030101
112. Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20(5):1868–1876. doi:10.1128/MCB.20.5.1868-1876.2000
113. Muoio DM, Newgard CB. Molecular and metabolic mechanisms of insulin resistance and β-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(3):193–205. doi:10.1038/nrm2327
114. Gil-Gómez G, Ayté J, Hegardt FG. The rat mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme-A-synthase gene contains elements that mediate its multihormonal regulation and tissue specificity. Eur J Biochem. 1993;213(2):773–779. doi:10.1111/j.1432-1033.1993.tb17819.x
115. Yoo-Warren H, Monahan JE, Short J, et al. Isolation and characterization of the gene coding for cytosolic phosphoenolpyruvate carboxykinase (GTP) from the rat. Proc Natl Acad Sci U S A. 1983;80(12):3656–3660. doi:10.1073/pnas.80.12.3656
116. Geer EB, Islam J, Buettner C. Mechanisms of glucocorticoid-induced insulin resistance: focus on adipose tissue function and lipid metabolism. Endocrinol Metab Clin North Am. 2014;43(1):75–102. doi:10.1016/j.ecl.2013.10.005
117. Morgan SA, McCabe EL, Gathercole LL, et al. 11β-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc Natl Acad Sci USA. 2014;111(24):E2482–91. doi:10.1073/pnas.1323681111
118. Woods CP, Hazlehurst JM, Tomlinson JW. Glucocorticoids and non-alcoholic fatty liver disease. J Steroid Biochem Mol Biol. 2015;154:94–103. doi:10.1016/j.jsbmb.2015.07.020
119. Abulizi A, Camporez JP, Zhang D, Samuel VT, Shulman GI, Vatner DF. Ectopic lipid deposition mediates insulin resistance in adipose specific 11β-hydroxysteroid dehydrogenase type 1 transgenic mice. Metabolism. 2019;93:1–9. doi:10.1016/j.metabol.2018.12.003
120. Morgan SA, Sherlock M, Gathercole LL, et al. 11beta-hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes. 2009;58(11):2506–2515. doi:10.2337/db09-0525
121. Paterson JM, Morton NM, Fievet C, et al. Metabolic syndrome without obesity: hepatic overexpression of 11beta-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc Natl Acad Sci U S A. 2004;101(18):7088–7093. doi:10.1073/pnas.0305524101
122. Harno E, Cottrell EC, Keevil BG, et al. 11-Dehydrocorticosterone causes metabolic syndrome, which is prevented when 11β-HSD1 is knocked out in livers of male mice. Endocrinology. 2013;154(10):3599–3609. doi:10.1210/en.2013-1362
123. Tomlinson JW, Draper N, Mackie J, et al. Absence of Cushingoid phenotype in a patient with Cushing’s disease due to defective cortisone to cortisol conversion. J Clin Endocrinol Metab. 2002;87(1):57–62. doi:10.1210/jcem.87.1.8189
124. Feig PU, Shah S, Hermanowski-Vosatka A, et al. Effects of an 11β-hydroxysteroid dehydrogenase type 1 inhibitor, MK-0916, in patients with type 2 diabetes mellitus and metabolic syndrome. Diabetes Obes Metab. 2011;13(6):498–504. doi:10.1111/j.1463-1326.2011.01375.x
125. Rosenstock J, Banarer S, Fonseca VA, et al. The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care. 2010;33(7):1516–1522. doi:10.2337/dc09-2315
126. Stefan N, Ramsauer M, Jordan P, et al. Inhibition of 11β-HSD1 with RO5093151 for non-alcoholic fatty liver disease: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2(5):406–416. doi:10.1016/S2213-8587(13)70170-0
127. Pereira CD, Azevedo I, Monteiro R, Martins MJ. 11β-Hydroxysteroid dehydrogenase type 1: relevance of its modulation in the pathophysiology of obesity, the metabolic syndrome and type 2 diabetes mellitus. Diabetes Obes Metab. 2012;14(10):869–881. doi:10.1111/j.1463-1326.2012.01582.x
128. Dammann C, Stapelfeld C, Maser E. Expression and activity of the cortisol-activating enzyme 11β-hydroxysteroid dehydrogenase type 1 is tissue and species-specific. Chem Biol Interact. 2019;303:57–61. doi:10.1016/j.cbi.2019.02.018
129. Van den Heuvel JK, Boon MR, van Hengel I, et al. Identification of a selective glucocorticoid receptor modulator that prevents both diet-induced obesity and inflammation. Br J Pharmacol. 2016;173(11):1793–1804. doi:10.1111/bph.13477
130. Kooneef LL, van den Heuvel JK, Kroon J, et al. Selective glucocorticoid receptor modulation prevents and reverses nonalcoholic fatty liver disease in male mice. Endocrinology. 2018;159(12):3925–3936. doi:10.1210/en.2018-00671
131. Ferron M, Lacombe J. Regulation of energy metabolism by the skeleton: osteocalcin and beyond. Arch Biochem Biophys. 2014;561:137–146. doi:10.1016/j.abb.2014.05.022
132. Price PA, Nishimoto SK. Radioimmunoassay for the vitamin K-dependent protein of bone and its discovery in plasma. Proc Natl Acad Sci U S A. 1980;77(4):2234–2238. doi:10.1073/pnas.77.4.2234
133. Prummel MF, Wiersinga WM, Lips P, Sanders GT, Sauerwein HP. The course of biochemical parameters of bone turnover during treatment with corticosteroids. J Clin Endocrinol Metab. 1991;72(2):382–386. doi:10.1210/jcem-72-2-382
134. Ferris HA, Kahn CR. New mechanisms of glucocorticoid-induced insulin resistance: make no bones about it. J Clin Invest. 2012;122(11):3854–3857. doi:10.1172/JCI66180
135. Lee NK, Sowa H, Hinoi E, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130(3):456–469. doi:10.1016/j.cell.2007.05.047
136. Van Raalte DH, Nofrate V, Bunck MC, et al. Acute and 2-week exposure to prednisolone impair different aspects of β-cell function in healthy men. Eur J Endocrinol. 2010;162(4):729–735. doi:10.1530/EJE-09-1034
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.