Back to Journals » Journal of Inflammation Research » Volume 11
Glucagon-like peptide-1 exerts anti-inflammatory effects on mouse colon smooth muscle cells through the cyclic adenosine monophosphate/nuclear factor-κB pathway in vitro
Authors Al-Dwairi A ![]() , Alqudah TE, Al-Shboul O, Alqudah M, Mustafa AG
, Alqudah TE, Al-Shboul O, Alqudah M, Mustafa AG ![]() , Alfaqih MA
, Alfaqih MA ![]()
Received 28 September 2017
Accepted for publication 22 January 2018
Published 20 March 2018 Volume 2018:11 Pages 95—109
DOI https://doi.org/10.2147/JIR.S152835
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Ahmed Al-Dwairi,1 Tamara E Alqudah,1 Othman Al-Shboul,1 Mohammad Alqudah,1 Ayman G Mustafa,2 Mahmoud A Alfaqih1
1Department of Physiology and Biochemistry, Faculty of Medicine, Jordan University of Science and Technology, Irbid, Jordan; 2Department of Anatomy, Faculty of Medicine, Jordan University of Science and Technology, Irbid, Jordan
Background: Intestinal smooth muscle cells (SMCs) undergo substantial morphological, phenotypic, and contractile changes during inflammatory bowel disease (IBD). SMCs act as a source and target for different inflammatory mediators, however their role in IBD pathogenesis is usually overlooked. Glucagon-like peptide-1 (GLP-1) is an incretin hormone reported to exert multiple anti-inflammatory effects in different tissues including the gastrointestinal tract through various mechanisms.
Aim: The aim of this research is to explore the effect of GLP-1 analog exendin-4 on the expression and secretion of inflammatory markers from mouse colon smooth muscle cells (CSMCs) after stimulation with lipopolysaccharide (LPS).
Materials and methods: Freshly isolated CSMCs from male BALB/c mice were cultured in DMEM and treated with vehicle, LPS (1 µg/mL), LPS+exendin-4 (50 nM), or LPS+exendin-4 (100 nM) for 24 h. Expression of inflammatory cytokines was then evaluated by antibody array membrane.
Results: CSMCs showed basal expression of several cytokines which was enhanced with the induction of inflammation by LPS. However, exendin-4 (50 and 100 nM) significantly (p<0.05) reduced the expression of multiple cytokines including tumor necrosis factor-α (TNF-α), interleukin-1α (IL-1α), T cell activation gene-3 (TCA-3), stromal cell-derived factor-1 (SDF-1), and macrophage colony stimulating factor (M-CSF). To confirm these results, expression of these cytokines was further assessed by enzyme-linked immunosorbent assay and real-time polymerase chain reaction and similar results were also observed. Moreover, secretion of TNF-α and IL1-α into the conditioned media was significantly downregulated by exendin-4 when compared to LPS-treated cells. Furthermore, LPS increased NF-κB phosphorylation, while exendin-4 significantly reduced levels of NF-κB phosphorylation.
Conclusion: These data indicate that GLP-1 analogs can exert significant anti-inflammatory effects on CSMCs and can potentially be used as an adjunct treatment for inflammatory bowel conditions.
Keywords: GLP-1, inflammation, inflammatory bowel disease, smooth muscle
Corrigendum for this paper has been published
Introduction
Inflammatory conditions of the gastrointestinal tract (GIT) are a growing problem in the industrialized nations, affecting the quality of the patient’s life and having significant financial burden.1,2 Inflammatory bowel disease (IBD) is a group of chronic incurable gastrointestinal inflammatory disorders with undetermined etiology. However, it is thought to result from dysregulated mucosal immune response to intestinal commensal flora in genetically predisposed individuals.1
Ulcerative colitis (UC) and Crohn’s disease (CD) are the most common forms of IBD affecting the colon and small intestine respectively and can involve the mucosa and submucosal layers. These inflammatory conditions impair the ability of affected gastrointestinal tissues to function properly, leading to various symptoms such as diarrhea, malabsorption, abdominal pain, cramping, rectal bleeding, and fatigue.1 Patients with IBD often alternate between periods of flares and remission. Untreated IBD can result in many intestinal and systemic complications such as intestinal perforations, toxic megacolon, abscesses, and colon cancer.3 Currently, no cure is available for IBD.4 Current treatment modalities are focused on reduction of the inflammatory process to alleviate the symptoms and to prevent future complications to improve the patient’s quality of life. Pharmaceutical treatment of IBD includes five major categories: anti-inflammatory drugs, immunosuppressants, biological agents, antibiotics, and drugs for symptomatic relief. However, these treatments are associated with significant side effects and are of limited success in some patients, highlighting the need for novel therapeutic agents with minimal side effects.5
At the histological level, the hallmark of IBD is recruitment of inflammatory cells, infiltration and activation within the intestinal mucosa and lamina propria, and enhanced production of pro-inflammatory mediators.4,6 Analysis of the mucosa from patients with IBD showed increased expression of several cytokines such as interleukin-1α (IL-1α), IL-6, IL-8, and tumor necrosis factor-α (TNF-α).7 Cytokines are mainly produced by monocytes and polymorphonuclear cells that have migrated to the inflammatory focus and to a lesser extent by the tissue macrophages and epithelial cells.6
The smooth muscle cells (SMCs) within the intestine are found in the longitudinal and circular muscle layers, as well as the muscularis mucosa layer. They are the main contractile nonproliferative apparatus needed for mixing and propelling the intraluminal contents, however they can exhibit phenotypic switching, hyperplasia, hypertrophy, and altered contractile behavior upon intestinal pathologies.8,9 Studies conducted using animal models have shown that mucosal inflammation in the gut leads to changes in the growth and contractile properties of SMCs. Other studies have shown that the altered intestinal motility observed in patients with CD is partially attributed to hypertrophy and alterations in the contractility of the intestinal SMCs.10
Glucagon-like peptide-1 (GLP-1) is an incretin hormone synthesized by the enteroendocrine L cells which are located mainly in the distal small intestine and colon. GLP-1 is secreted in response to nutrient ingestion leading to various effects on the body such as increased insulin secretion, decreased glucagon secretion, regulating proximal gut transit, and suppression of appetite signaling in the brain; these ultimately result in improved glucose homeostasis and weight loss.11 GLP-1 has an extremely short half-life (~1–2 min) due to rapid degradation by the enzyme dipeptidyl peptidase IV (DPP-4). DPP-4 is widely expressed by the gut and vascular endothelium. It cleaves GLP-1 from its N-terminal at position 2 occupied by alanine, resulting in the generation of inactive GLP-1(9–37) and GLP-1(9–36) amide.12 During the past decade, several GLP-1-based therapies became available for the treatment of type 2 diabetes mellitus and obesity.13,14 Recently, GLP-1 was found to exert several beneficial effects beyond glycemic control. It exerts anti-inflammatory and antioxidant effects on various tissues and cells including cardiomyocytes, endothelial cells, and vascular SMCs through various mechanisms via receptor-dependent and receptor-independent pathways.15 Although the primary function of GLP-1 is to serve as an anorexigenic (appetite-reducing) and incretin hormone, as well as a mediator of the “ileal brake” through inhibition of upper gastrointestinal function to prevent malabsorption and postprandial metabolic disturbances, the influence of GLP-1 on colon function was evaluated by a limited number of studies. Amato et al55 reported that GLP-1 receptor (GLP-1R) is expressed in the nitrergic neurons of the myenteric plexus in human colon circular muscular strips and, once activated by exogenous GLP-1, mediates an inhibitory effect on large intestinal motility through nitric oxide neural release resulting in reduction of the spontaneous contractile activity of the smooth muscles, which may contribute to delaying of intestinal propulsion to increase water and electrolyte absorption. Moreover, May16 recently reported that GLP-1R is expressed in pure mouse SMC cultures of the colon and intestine. They showed that organ bath studies identified a role of GLP-1R in the regulation of smooth muscle function by influencing the basal tone in colonic muscle strips and mediating relaxation of acetylcholine-induced phasic and tonic contractions in the colon. Furthermore, recent studies showed that GLP-1R agonists modulate enteric immune responses through the intestinal intraepithelial lymphocyte GLP-1R which may influence microbiome composition and intestinal inflammation; therefore, GLP-1R agonists may exert beneficial nonglycemic actions partially through modulation of intraepithelial lymphocyte GLP-1R.46 A recent study showed that GLP-1 nanomedicine is effective in treating intestinal inflammation in a mouse model of colitis, by preserving epithelial architecture and inhibition of cytokine production; however, the anti-inflammatory effects of GLP-1 on colon smooth muscle cells (CSMCs) have not yet been explored.17 Interestingly, GLP-1 levels are elevated in critically ill patients in different chronic inflammatory diseases including CD, UC, metabolic syndrome, coronary artery disease, and heart failure, and have been found to be increased in animal models of inflammatory conditions in response to bacterial stimuli, suggesting a correlation between inflammation and GLP-1 secretion.18–20 Interestingly, DPP4 inhibitors accelerated mucosal healing in an animal model of experimental colitis, and GLP-1 analog ROSE-010 reduced pain exacerbation in patients with irritable bowel syndrome.21–23
The aim of this research is to explore the anti-inflammatory effects of the GLP-1 analog exendin-4 on the expression and secretion of inflammatory markers from mouse colon smooth muscle under lipopolysaccharide (LPS)-induced inflammation in vitro. LPS is a bacterial endotoxin reported to increase cytokine secretion in different cell types.
Materials and methods
Solutions, drugs, and chemicals
Smooth muscle buffer (SMB) was prepared in house and contained 120 mM NaCl, 4 mM KCl, 2.6 mM KH2PO4, 2 mM CaCl2, 25 mM HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid), 14 mM glucose, and 2.1% essential amino acid mixture (pH was adjusted to 7.4). The digestion solution contained 0.5 mg collagenase and 0.33 mg trypsin inhibitor per milliliter of SMB. All of these chemicals were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Cell culture media used during the incubation process were prepared by adding 10% fetal bovine serum (FBS) (GE Healthcare UK Ltd, Little Chalfont, UK), 100 U/mL of penicillin, 100 µg/mL of streptomycin, and 2.5 µg/mL of amphotericin B to DMEM with L-glutamine (Capricorn Scientific GmbH, Ebsdorfergrund, Germany). The remaining reagents were purchased from Euroclone S.P.A. (Pero, Italy). Rabbit GLP-1R polyclonal antibody (H-55; sc-66911) was purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA) and anti-calponin antibodies were purchased from Abcam Inc. (Cambridge, UK). LPS was purchased from Sigma-Aldrich Co. GLP-1 analog exendin-4 was purchased from Abcam Inc. The polymerase chain reaction (PCR) primers were purchased from Integrated DNA Technologies, Inc. (Coralville, IA, USA) (Table 1). The protein assay kit, SYBR Green Master Mix, and cDNA synthesis kit were purchased from Bio-Rad Laboratories Inc. (Hercules, CA, USA). Mouse inflammation antibody array membrane (Cat. No. ab133999) was purchased from Abcam Inc. Specific enzyme-linked immunosorbent assay (ELISA) kits for mouse TNF-α, M-CSF, IL-1α, TCA-3, and SDF-1 were purchased from Sigma Aldrich Co. The cyclic adenosine monophosphate (cAMP) ELISA kit was purchased from R&D Systems Inc. (Minneapolis, MN, USA), and the nuclear factor (NF)-κB p65 pS536 ELISA kit was purchased from Abcam Inc. The nuclear protein extraction kit was purchased from Abcam Inc.. The RNA extraction kit was purchased from Zymo Research Corp. (Irvine, CA, USA). The 500-μm Nitex mesh was purchased from Sigma-Aldrich Co.
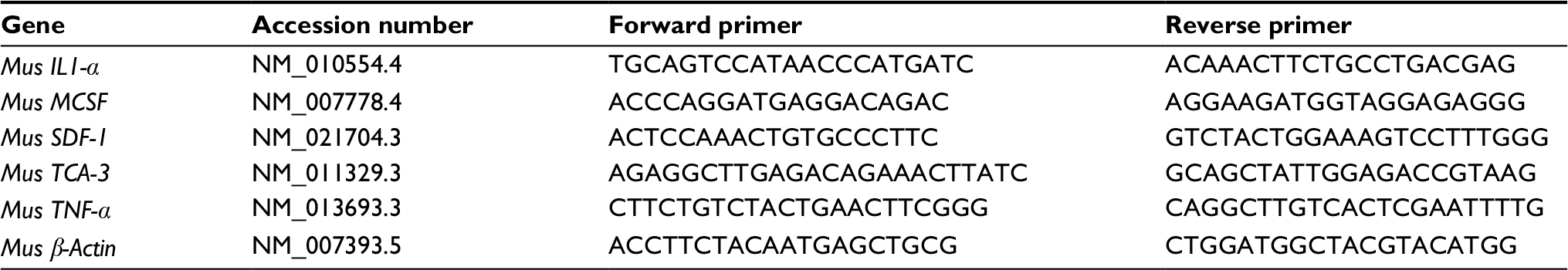 | Table 1 Primers used in real-time/reverse transcription polymerase chain reaction and their sequences |
Animals
Twenty young mature male BALB/c mice (~12 weeks of age, 26.5–30 g) were provided by the animal house of Jordan University of Science and Technology. Mice were euthanized by inhalation of CO2 and the colon was rapidly excised. The colon was cut into small pieces (2–3 cm in length) and then placed in cold SMB with the composition described previously. All procedures were approved and performed according to the guidelines of the Animal Care and Use Committee at Jordan University of Science and Technology.
Preparation of dispersed smooth muscle cells
CSMCs were isolated by sequential enzymatic digestion of muscle strips, filtration, and centrifugation. The mucosa was scraped off with fine scissors under a dissecting scope, and then the tissues were cut into thin slices (2 mm long; 2 mm thin) and incubated for 20 min in SMB containing 0.5 mg/mL collagenase and 0.33 mg/mL soybean trypsin inhibitor in a shaking water bath at 31 °C. The tissues were continuously gassed with 100% oxygen during the entire isolation procedure. The partly digested tissue was washed twice with 50 mL of collagenase-free SMB and the muscle cells were allowed to disperse spontaneously for 10 min in collagenase-free medium. Cells were harvested by filtration through 500-μm Nitex mesh and centrifuged twice at 1000 rpm for 10 min to eliminate broken cells and organelles. The process was repeated four or five times. The cells were counted in a hemocytometer, and it is estimated that 95% are viable using a trypan blue exclusion assay.
Identification of smooth muscle cells
Identification of mouse CSMCs was verified by immunohistochemical staining of paraffin-embedded dispersed colon muscle cells using anti-calponin antibody at 1:100 dilution.
Expression of inflammatory cytokines using antibody array membrane
Ten thousand dispersed mouse CSMCs (n=3/group) were seeded in a 6-well plate in DMEM containing 10% FBS, 100 U/mL of penicillin, 100 µg/mL streptomycin, and 2.5 µg/mL amphotericin B. The cells were incubated in a humidified incubator with 95% air and 5% CO2 at 37 °C for 24 h with either vehicle, LPS (1 μg/mL), LPS (1 μg/mL)+exendin-4 (50 nM), or LPS (1 μg/mL)+exendin-4 (100 nM). After completion of the incubation period (24 h) the treated samples were centrifuged at 1000 × g for 5 min at 4 °C. The conditioned media were stored at –20 °C for further analysis, and the cell pellets were immediately lysed.
Cell lysates were prepared using BashingBeads Lysis tubes (Zymo Research Corp.) and the cell lysis buffer containing protease inhibitor cocktail provided by the kit according to the manufacturer’s instructions. Then lysates were centrifuged for 10 min at 14000 × g at 4 °C and the supernatant collected for further analysis. Total protein concentration of the supernatants was measured using the DC protein assay kit. The protein concentration was then adjusted to 100 µg/mL in all samples.
To evaluate differential cytokine expression in the treated samples, antibody array membranes were used according to the manufacturer’s instructions. Briefly, the membranes were placed in the 8-well tray provided in the kit, and then 1 mL of each sample was incubated overnight into the designated well at 4 °C. Twenty-four hours later, the samples were aspirated and the membranes were washed with wash buffer and then incubated with 1 mL of biotin-conjugated anti-cytokines overnight at 4 °C. Then the biotin-conjugated anti-cytokines were aspirated and washing performed again as previously described. Finally, 2 mL of horseradish peroxidase-conjugated streptavidin was pipetted into each well and incubated overnight at 4 °C; then it was aspirated and the membranes were washed again by the previously described method. Detection buffer was used to develop a chemiluminescent signal and the C-DIGIT blot scanner (LI-COR Biotechnology, Lincoln, NE, USA) was used to detect the signal intensity. Measurements of the intensity of signals on the array membrane were performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Data were normalized to the positive control signals and the relative cytokine expression was determined.
CSMC viability assay
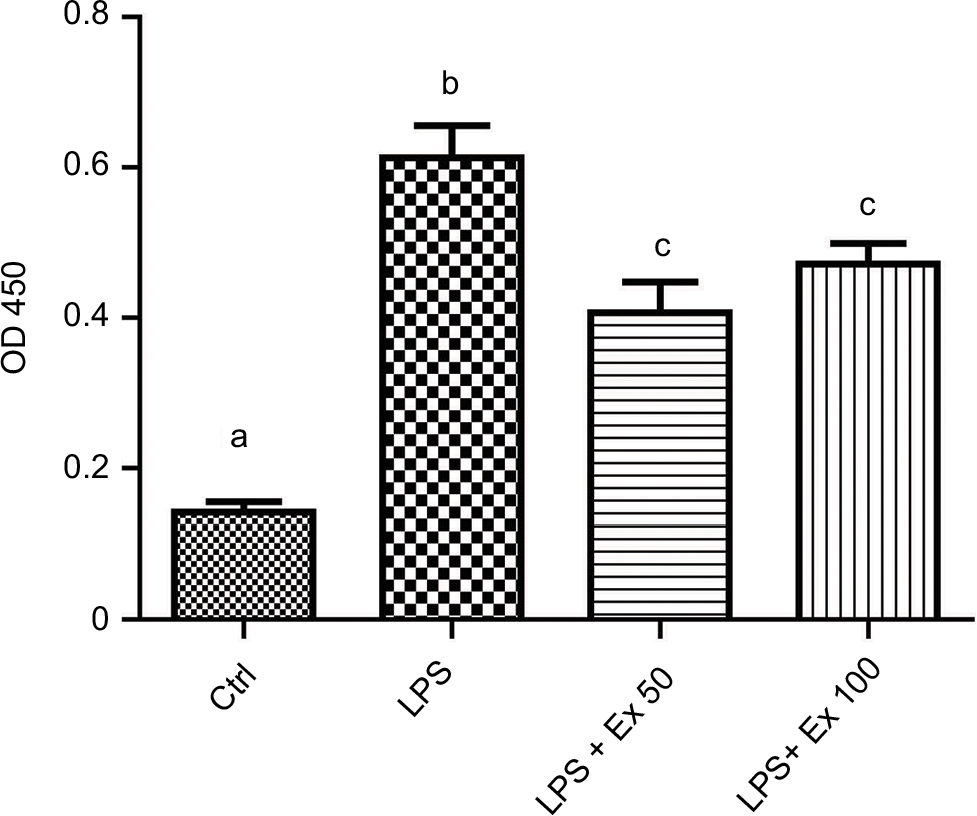
Freshly isolated CSMCs were cultured in 96-well plates at 1×105 cells/well. Cells were serum-deprived for 24 h before stimulation with vehicle, LPS (1 μg/mL), LPS (1 μg/mL)+exendin-4 (50 nM), or LPS (1 μg/mL)+exendin-4 (100 nM). Cell proliferation was assessed after 24 h using the MTT assay (Intron Biotechnology, Korea) according to the manufacturer’s instructions. Briefly, CSMCs were incubated with MTT reagent for 4 h at 37 °C. MTT reagent was converted to an insoluble formazan. The formazan is then solubilized, and the concentration determined by optical density at 570 nm.
Quantification of protein expression by ELISA
Protein levels of specific cytokines identified previously by the membrane array were further evaluated by ELISA. Specific ELISA kits for TNF-α, IL-1α, M-CSF, TCA-3, SDF-1, and NF-κB p65 (pS536) were used to measure the levels of these cytokines in the lysates and the conditioned media in the control and treated samples according to the manufacturer’s protocol.
Cellular cAMP determination assay
Freshly isolated CSMCs were cultured in DMEM supplemented with 10% FBS, 100 U/mL of penicillin, and 100 μg/mL of streptomycin at 37 °C in 5% CO2. Cells were seeded at a density of 10000 cells per well into a 96-well plate in the presence of vehicle, LPS (1 μg/mL), LPS (1 μg/mL)+exendin-4 (50 nM), or LPS (1 μg/mL)+exendin-4 (100 nM) for 10 min (n=3/group). Cells were then immediately lysed and assayed for cAMP accumulation using the cAMP determination kit according to the manufacturer’s protocol.
RNA isolation and quantitative real-time/reverse transcription polymerase chain reaction
To extract RNA from CSMCs, a Direct-Zol RNA MiniPrep kit from Zymo Research was used according to the manufacturer’s instructions. The Direct-Zol method assures efficient recovery of small and large RNAs. Dispersed mouse CSMCs were treated with either a vehicle, LPS (1 μg/mL), LPS (1 μg/mL)+exendin-4 (50 nM), or LPS (1 μg/mL)+exendin-4 (100 nM). The samples were incubated for 12 h in a humidified incubator with 95% air and 5% CO2 at 37 °C. The concentration and purity of RNA were determined using a NanoDrop 2000 spectrophotometer from Thermo Fisher Scientific (Waltham, MA, USA). One microgram of RNA was reverse-transcribed to obtain cDNA using the iScript cDNA synthesis kit (Bio-Rad Laboratories Inc.). Expression of target genes was assayed by quantitative real-time/reverse transcription (qRT)-PCR using Bio-Rad iTaq SYBR Green Supermix. Relative gene expression was performed using a standard curve-based method. Data for target genes were normalized to β-actin (Actb) mRNA levels. The SYBR Green Master Mix from Bio-Rad Laboratories Inc. contains antibody-mediated hot-start iTaq DNA polymerase, dNTPs, MgCl2, SYBR green I dye, and fluorescein. The 7500 Fast Real-Time PCR System from Applied Biosystems (Thermo Fisher Scientific) was used to run the reaction. The Master Mix was first activated at 95 °C for 3 min and then 35 cycles of amplification were performed each consisting of 15 sec at 95 °C and 60 sec at 60 °C. A melting point dissociation curve generated by the instrument was used to confirm that only a single product was present.
Immunohistochemistry
Dispersed mouse CSMCs were fixed in 10% neutral-buffered formalin (pH 7.4) overnight and were then embedded in paraffin matrix. Mid-colon tissue from male BALB/c mice was fixed in 10% neutral-buffered formalin (pH 7.4) overnight, and embedded in paraffin. Five-micrometer sections were prepared, and the sections were dewaxed and rehydrated through a graded alcohol series. Antigen unmasking was performed by boiling the sections in Citra Plus (BioGenex, San Ramon, CA, USA) in a microwave oven for 2 min at power 10 and then for 10 min at power 1, followed by cooling for 20 min. Sections were treated with 3% hydrogen peroxide to quench endogenous peroxidase activity and incubated in blocking solution containing goat IgG (Vectastain Elite ABC kit; Vector Laboratories, Inc., Burlingame, CA, USA) for 30 min. Sections were incubated overnight with rabbit GLP-1R polyclonal antibody (Santa Cruz Biotechnology) and anti-Calponin antibody (Abcam Inc.) followed by incubation with goat anti-rabbit secondary antibody (Vectastain Elite ABC kit; Vector Laboratories) for 30 min. Sections were stained with 3,3′-diaminobenzidine tetra-hydrochloride (Dako Denmark A/S, Glostrup, Denmark). Slides were dehydrated in an alcohol series and cleared in xylene. Images were obtained with 20× objective by an inverted Nikon TMS-F microscope (Nikon Instruments, Irving, TX, USA).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). One-way analysis of variance followed by Fisher’s post hoc analysis was used to examine significant differences between groups. A p-value less than 0.05 was considered significant. All data are shown as mean±standard error of the mean.
Results
Colonic smooth muscle cell identification
Freshly isolated mouse cells were viewed at 20× objective on the inverted Nikon TMS-F microscope and they appeared spindle-shaped with varying lengths (Figure 1A). The identity of mouse CSMCs was verified by immunohistostaining with anti-calponin antibody (Figure 1B). The results showed that more than 95% of the cells stained positive for calponin. Calponin is specific for differentiated SMCs.
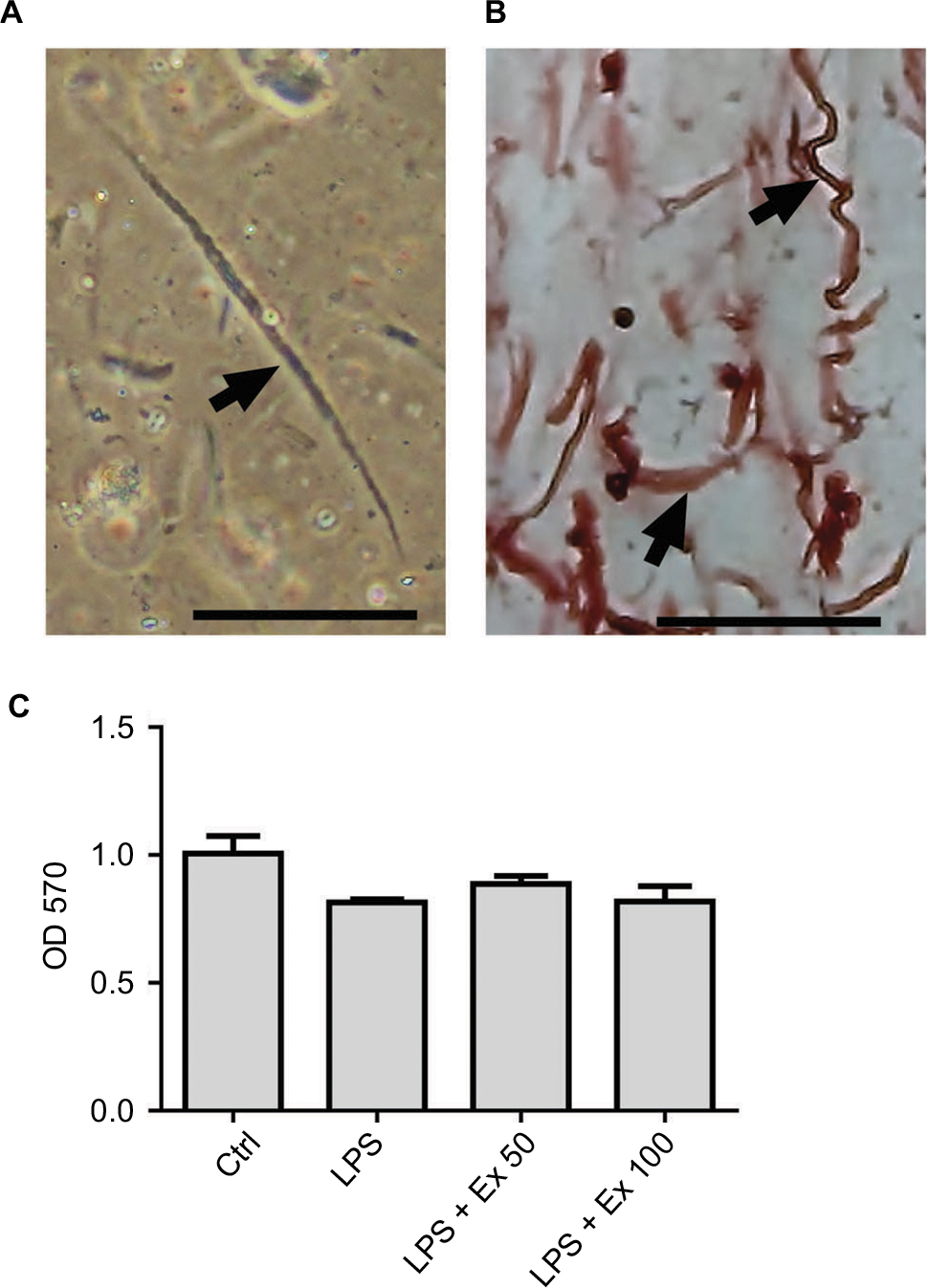 | Figure 1 Identification of mouse CSMCs. (A) Single CSMC appeared in spindle shape (arrow) under phase contrast microscopy. Cells were viewed via inverted Nikon TMS-F microscope, and the image captured with a Canon digital camera. (B) Immunohistochemical staining of paraffin-embedded mouse CSMCs (arrows) using anti-h1-calponin antibody. (C) Cell viability assay for cells treated with vehicle (Ctrl), LPS, LPS+exendin-4 (50 nM), or LPS+exendin-4 (100 nM). Pictures were viewed at 20× magnification, scale bar=50 µm. Abbreviations: CSMC, colon smooth muscle cell; Ex 50, exendin-4 (50 nM); Ex 100, exendin-4 (100 nM); LPS, lipopolysaccharide; OD, optical density; Ctrl, control. |
Inflammatory cytokine expression by antibody array membrane
Antibody array membrane analysis showed a basal expression for several cytokines by mouse CSMCs. Treatment with LPS caused a marked increase (~50–100%) in the expression of several cytokines. Co-treatment with exendin-4 significantly inhibited the overexpression of these cytokines by ~40–50% (Figure 2A and B). This initial screening step showed that five cytokines were especially affected by the LPS and exendin-4 treatment: TNF-α, IL-1α, macrophage colony stimulating factor (M-CSF), T cell activation gene-3 (TCA-3), and stromal cell-derived factor-1 (SDF-1). These data suggest that CSMCs are capable of producing several inflammatory mediators that can play an important role in the pathogenesis of inflammatory conditions of the gastrointestinal system; on the other hand, GLP-1 analog exendin-4 could modulate these inflammatory conditions by affecting smooth muscle’s cytokine and chemokine expression.
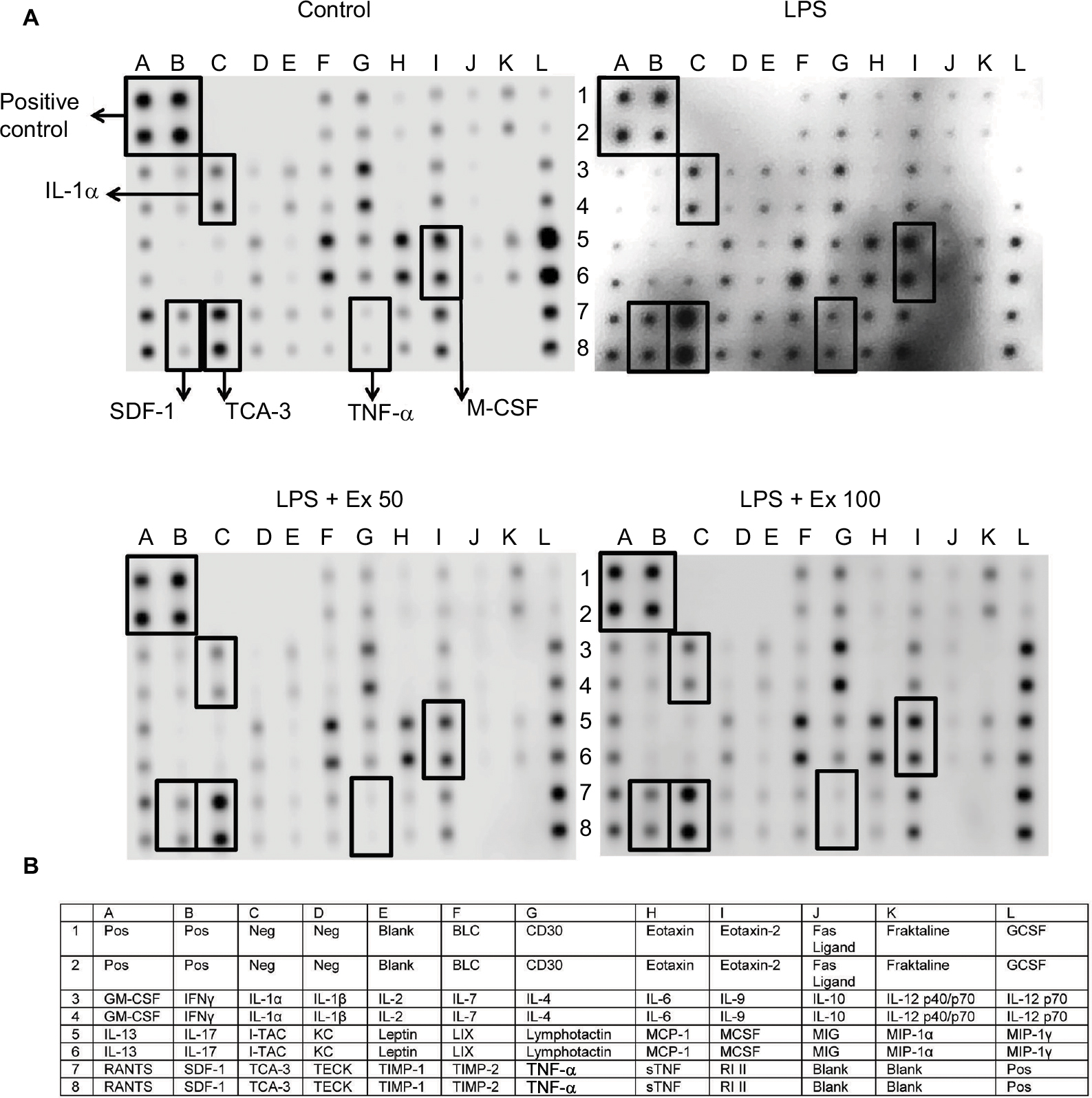 | Figure 2 Effect of exendin-4 treatment on the expression of inflammatory cytokines by mouse CSMCs using antibody array membrane. (A) Freshly isolated CSMCs from 20 male Balb/C mice were cultured in DMEM and divided into four treatment groups: control, LPS, LPS+exendin-4 (50 nM), and LPS+exendin-4 (100 nM). After 24 h of incubation the cells were collected, lysed, and incubated with the antibody array membrane. There was a basal expression of various cytokines by the normal CSMCs which was increased once inflammation was induced by LPS. Exendin-4 at both concentrations inhibited the overproduction of cytokines induced by inflammation. A visual comparison between the four arrays performed by two independent investigators and by ImageJ software (National Institutes of Health) revealed that five cytokines were especially affected by the treatment: tumor necrosis factor-α (TNF-α), interleukin-1α (IL-1α), T cell activation gene-3 (TCA-3), stromal cell-derived factor-1 (SDF-1), and macrophage colony stimulating factor (M-CSF). (B) List of cytokines and chemokines represented by the antibody array membrane (Cat. No. ab133999; Abcam Inc.). Abbreviations: CSMC, colon smooth muscle cell; Ex 50, exendin-4 (50 nM); Ex 100, exendin-4 (100 nM); GCSF, granulocyte colony stimulating factor; GM-CSF, granulocyte–macrophage colony stimulating factor; IFN, interferon; IL, interleukin; LPS, lipopolysaccharide; Pos, positive; Neg, negative. |
Colon smooth muscle cell viability assay
To ensure that CSMC viability is not affected by LPS or exendin-4 treatment, we performed an MTT assay on the treated cells. Our results indicate that cell viability is comparable between all treatment groups, indicating that neither LPS nor exendin-4 is affecting the growth of CSMCs within a 24-h period (Figure 1C).
Assessment of cytokine expression and secretion by ELISA
To further evaluate the levels of differentially expressed cytokines in the treated samples and control, specific ELISAs were used. Data showed that LPS treatment induced ~1–2-fold increase in the expression of TNF-α, IL1-α, M-CSF, TCA-3, and SDF-1 (p<0.05). However, co-treatment with exendin-4 (50 or 100 nM) significantly reduced their expression by ~20–60% compared to LPS alone (p<0.05) (Figure 3). Moreover, LPS treatment upregulated nuclear NF-κb p65 (pS536) protein levels, while exendin-4 reduced nuclear NF-κB p65 (pS536) levels, suggesting that GLP-1 analogs may modulate inflammatory cytokine expression by interfering with NF-κB p65 phosphorylation and translocation to the nucleus (Figure 7). To further evaluate whether altered cytokine expression may affect the cytokine secretion into the media, the levels of these cytokines were measured in the conditioned media of treated groups. Data showed that both TNF-α and IL-1α levels were increased with LPS treatment, while co-treatment with exendin-4 significantly reduced their secretion into the media (Figure 4). However, M-CSF, TCA-3, and SDF-1 secretion was not detected in the media. These data suggest that inflammatory stimuli can alter the behavior of gastrointestinal SMCs by enhancing NF-κB p65 phosphorylation and nuclear translocation, thereby increasing the expression of inflammatory cytokines. On the other hand, exendin-4 can inhibit cytokine production by reducing NF-κB p65 activation and translocation to the nucleus.
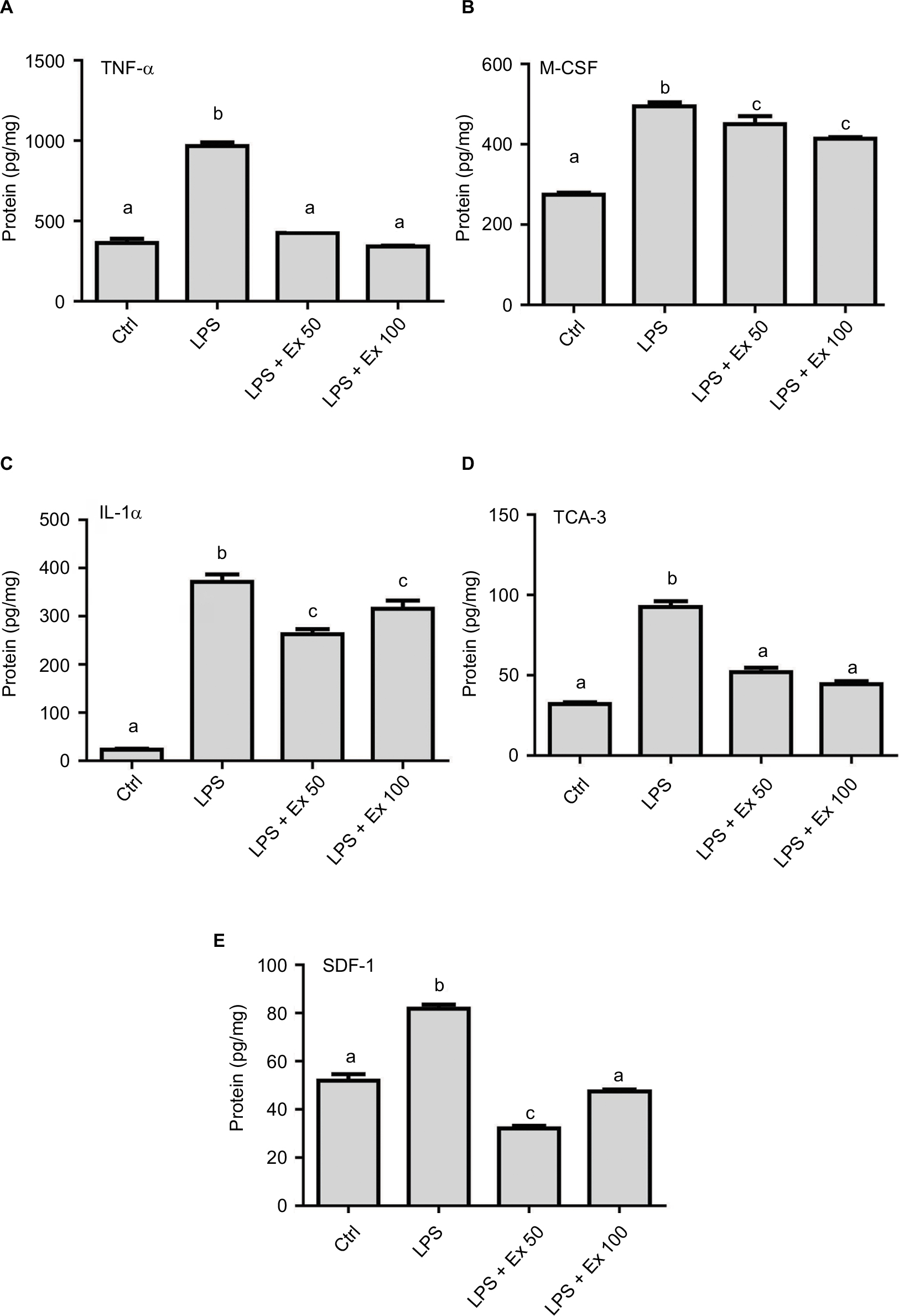 | Figure 3 Effect of exendin-4 treatment on the expression of inflammatory cytokines by mouse CSMCs using ELISA. Freshly isolated CSMCs from 20 male Balb/C mice were cultured in DMEM in 6-well plates and divided into four treatment groups (n=3/group): control (Ctrl), LPS, LPS+exendin-4 (50 nM), and LPS+exendin-4 (100 nM). After 24 h of incubation the cells were collected and lysed. Levels of different cytokines in the lysate were assayed using specific ELISA kits. Bar graphs represent levels of: (A) tumor necrosis factor-α (TNF-α), (B) macrophage colony stimulating factor (M-CSF), (C) interleukin-1α (IL-1α), (D) T cell activation gene-3 (TCA-3), and (E) stromal cell-derived factor-1 (SDF-1). Different lowercase letters indicate significant statistical difference between groups where p<0.05. Values shown are representative of three independent experiments performed in triplicates. Bars represent mean±standard error of the mean. Abbreviations: CSMC, colon smooth muscle cell; ELISA, enzyme-linked immunosorbent assay; Ex 50, exendin-4 (50 nM); Ex 100, exendin-4 (100 nM); LPS, lipopolysaccharide. |
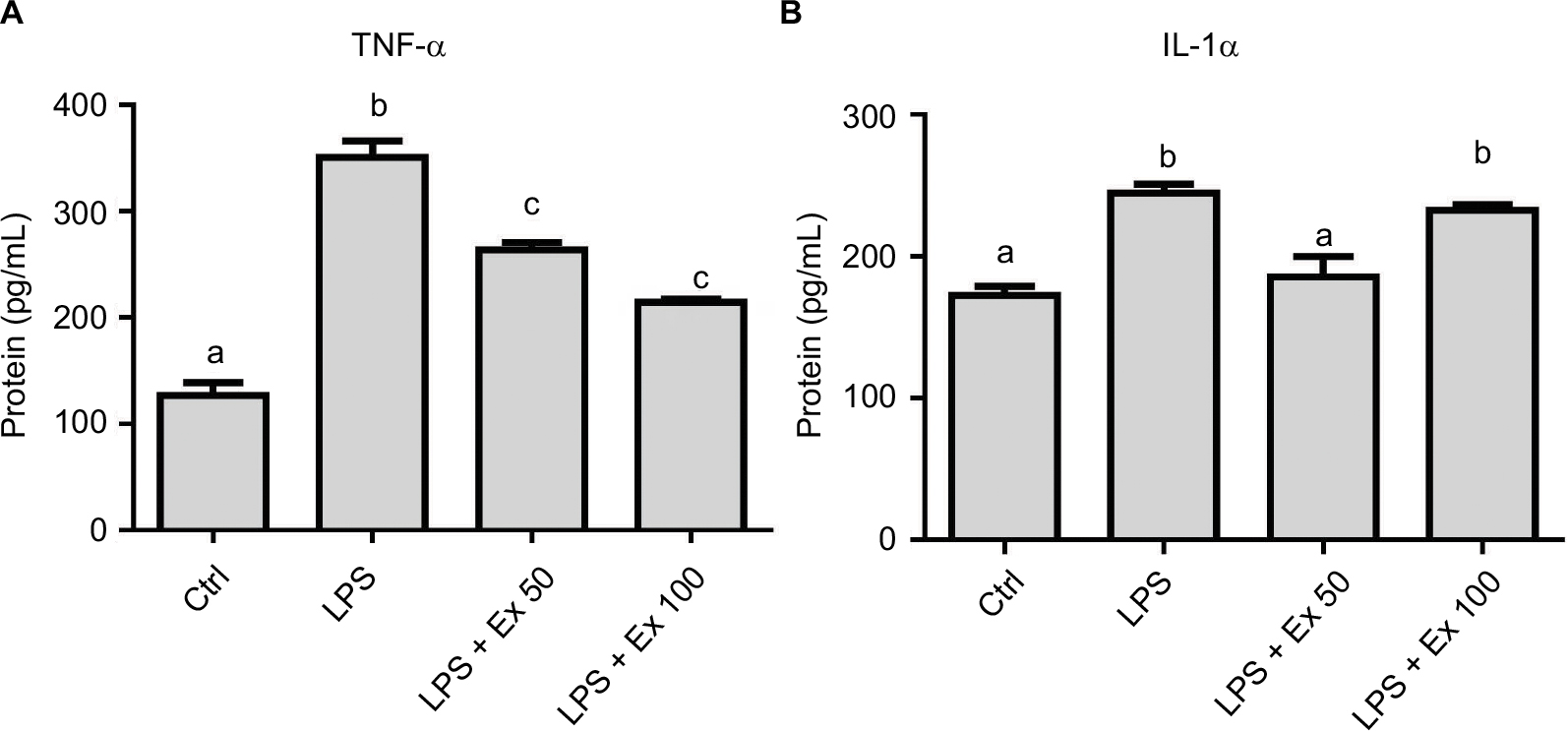 | Figure 4 Effect of exendin-4 treatment on the secretion of inflammatory cytokines by mouse CSMCs into the conditioned media using ELISA. Freshly isolated CSMCs from 20 male Balb/C mice were cultured in DMEM in 6-well plates and divided into four treatment groups (n=3/group): control (Ctrl), LPS, LPS+exendin-4 (50 nM), and LPS+exendin-4 (100 nM). After 24 h of incubation the cells were collected and lysed as mentioned previously, and the conditioned media were stored for later analysis at –20 °C. Levels of different cytokines in the conditioned media were assayed using specific ELISA kits. Bar graphs represent levels of: (A) tumor necrosis factor-α (TNF-α) and (B) interleukin-1α (IL-1α). Different lowercase letters indicate significant statistical difference between groups where p<0.05. Values shown are representative of three independent experiments performed in triplicates. Bars represent mean±standard error of the mean. Abbreviations: CSMC, colon smooth muscle cell; ELISA, enzyme-linked immunosorbent assay; Ex 50, exendin-4 (50 nM); Ex 100, exendin-4 (100 nM); LPS, lipopolysaccharide. |
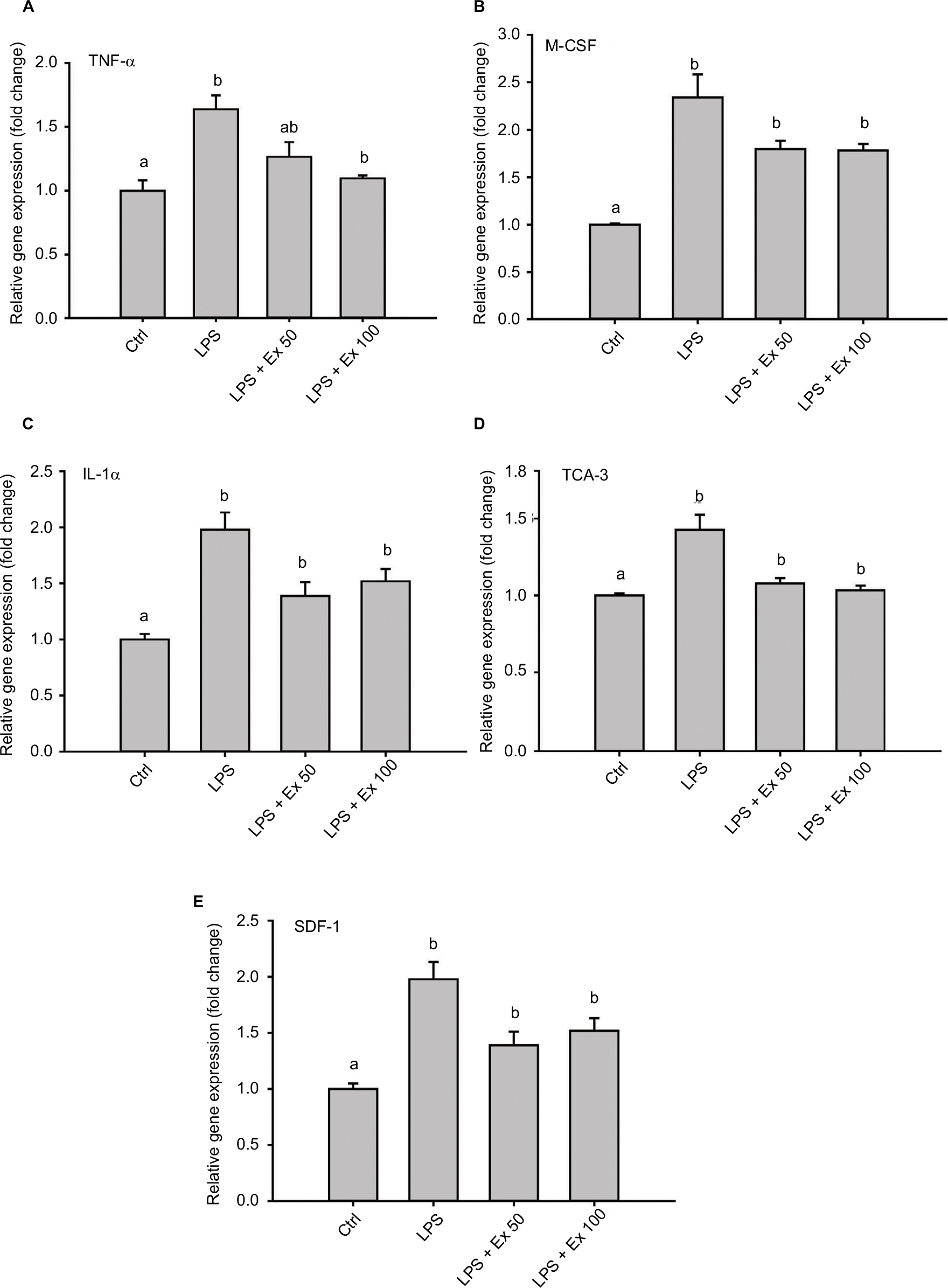 | Figure 5 Effect of exendin-4 treatment on mRNA abundance of inflammatory cytokines in mouse CSMCs using RT-PCR. Freshly isolated CSMCs from 20 male Balb/C mice were cultured in DMEM in 6-well plates and divided into four treatment groups (n=3/group): control (Ctrl), LPS, LPS+exendin-4 (50 nM), and LPS+exendin-4 (100 nM). After 12 h of incubation the cells were collected and RNA was extracted as mentioned previously. Relative gene expression of different cytokines in the treated cell was assayed using RT-PCR. Bar graphs represent gene expression levels of: (A) tumor necrosis factor-α (TNF-α), (B) macrophage colony stimulating factor (M-CSF), (C) interleukin-1α (IL-1α), (D) T cell activation gene-3 (TCA-3), and (E) stromal cell-derived factor-1 (SDF-1). Different lowercase letters indicate significant statistical difference between groups where p<0.05. Values shown are representative of two independent experiments performed in triplicates. Bars represent mean±standard error of the mean. Abbreviations: CSMC, colon smooth muscle cell; Ex 50, exendin-4 (50 nM); Ex 100, exendin-4 (100 nM); LPS, lipopolysaccharide; RT-PCR, real-time/reverse transcription polymerase chain reaction. |
 | Figure 6 Expression of GLP-1 receptor on mouse CSMCs. (A) Micrograph depicting immunohistochemical staining of GLP-1R in mouse isolated single CSMCs, scale bar=40 µM. Arrows show cytoplasmic and membranous staining indicative of GLP-1R expression. (B) Micrograph depicting immunohistochemical staining of GLP-1R in mouse mid-colon section, scale bar=100 µM. Arrows show GLP-1R staining in colon muscularis layer, indicating expression of GLP-1R in CSMCs. (C) cAMP levels in CSMCs treated with: control (Ctrl), LPS, LPS+exendin-4 (50 nM), and LPS+exendin-4 (100 nM) for 10 min. Different lowercase letters indicate significant statistical difference between groups where p<0.05. Abbreviations: cAMP, cyclic adenosine monophosphate; CSMC, colon smooth muscle cell; Ex 50, exendin-4 (50 nM); Ex 100, exendin-4 (100 nM); LPS, lipopolysaccharide. |
 | Figure 7 Exendin-4 reduces LPS-induced nuclear NF-κB phosphorylation. Freshly isolated CSMCs from 20 male Balb/C mice were cultured in DMEM in 6-well plates and divided into four treatment groups (n=3/group): control (Ctrl), LPS, LPS+exendin-4 (50 nM), and LPS+exendin-4 (100 nM). After 24 h of incubation the cells were collected and lysed. Total nuclear proteins were extracted using specific nuclear extraction kit. Nuclear NF-κB p65 pS536 levels were assayed in the lysate using a specific ELISA kit. Different lowercase letters indicate significant statistical difference between groups where p<0.05. Values shown are representative of two independent experiments performed in triplicates. Bars represent mean±standard error of the mean. Abbreviations: CSMC, colon smooth muscle cell; Ex 50, exendin-4 (50 nM); Ex 100, exendin-4 (100 nM); LPS, lipopolysaccharide; NF, nuclear factor; OD, optical density. |
Inflammatory cytokine expression by real-time PCR
To confirm the results obtained from the antibody array membrane and ELISA at the level of mRNA expression, real-time PCR was performed on mouse CSMCs treated with vehicle, LPS (1 μg/mL), LPS (1 μg/mL)+exendin-4 (50 nM), or LPS (1 μg/mL)+exendin-4 (100 nM). This experiment proved that LPS treatment induced a significant increase (~40–100%, p<0.05) in the level of expression of TNF-α, IL-1α, TCA-3, SDF-1, and M-CSF. On the other hand, exendin-4 co-treatment significantly (p<0.05) inhibited the overexpression of these cytokines by the CSMCs (Figure 5).
GLP-1R expression in CSMCs
GLP-1R is a G protein-coupled receptor that is widely expressed in many tissues including the pancreatic islets, GIT, lung, kidneys, heart, and some regions of the central and peripheral nervous system. GLP-1R is also widely expressed by multiple immune cell populations, particularly by the intestinal intraepithelial lymphocytes. In the pancreatic islets, activation of GLP-1R results in activation of adenylyl cyclase which leads to generation of cAMP, which in turn activates various downstream effectors through activation of protein kinase A. Expression of GLP-1R in the CSMCs has not been explored previously, therefore we sought to determine its expression in mouse CSMCs by immunohistochemistry. Our results indicate that GLP-1R is expressed in isolated single CSMCs (Figure 6A). Furthermore, immunohistochemistry of whole colon sections showed that GLP-1R is expressed in the muscular wall of the colon as well as the epithelial cells (Figure 6B). Moreover, we evaluated the effect of GLP-1R activation by exendin-4 through measuring intracellular cAMP levels in CSMCs. Our results indicate that exendin-4 increased CSMC cAMP levels in a dose-dependent fashion, indicating that GLP-1R mediates its effects in CSMCs through activation of adenylyl cyclase, leading in turn to cAMP-dependent activation of various second messenger pathways (Figure 6C).
Discussion
The intestinal epithelial barrier is usually the first site to be involved in the inflammatory conditions affecting the gut,24 where microorganisms or allergens are first encountered, leading to activation of the local immune cells and recruitment of new immune cells which alter the local environment resulting in the major hallmarks of inflammation;25 however, emerging evidence supports the notion that intestinal SMCs are affected functionally and structurally by most inflammatory conditions involving the intestine leading to altered morphology and contractile behavior, and secretion of various inflammatory mediators.26,27 Some studies have shown that the intestinal SMCs are capable of producing several cytokines such as IL-6 and TNF-α under different pathological conditions. A study showed that TNF-α binds to receptors on the SMCs, leading to activation of NF-κB and expression of a number of chemotactic genes such as MCP-1, IL-8, and intercellular adhesion molecule-1.28 Moreover, human CSMCs can express IL-1α, IL-6, IL-8, cyclooxygenase-2, and regulated on activation, normal T cell expressed and secreted (RANTES) in response to exposure to TNF-α, IL-1α, and interferon-γ.29
The aim of this study was to evaluate the expression of inflammatory mediators by normal and inflamed CSMCs, and to evaluate the effects of GLP-1 analog exendin-4 on the expression of inflammatory mediators by CSMCs after induction of inflammation by LPS.
GLP-1 is an incretin hormone synthesized and secreted by the intestinal L cells in response to the presence of nutrients within the gut. It improves glucose homeostasis through various mechanisms including activation of insulin secretion and inhibition of glucagon secretion, delaying gastric emptying, reducing hepatic glucose production, and decreasing food intake.30 GLP-1 has been shown to exert various anti-inflammatory actions on different tissues and can potentially be used for the treatment of several chronic inflammatory conditions like atherosclerosis, neurodegenerative disorders, diabetic nephropathy, asthma, psoriasis, and nonalcoholic steatohepatitis.15,31 Moreover, GLP-1 has been shown to improve the inflammatory milieu, vascular endothelial function, lipid profile, and cholesterol in diabetic patients.31 Several synthetic forms of GLP-1 are now available for treatment of diabetes mellitus such as exenatide.32 Treatment with GLP-1 analogs can suppress multiple inflammation parameters in the vascular and bronchial SMCs including a reduction in the production of certain cytokines.33
In our study, we used an antibody array membrane as a screening tool to assess the effect of inflammation and GLP-1 co-treatment on the expression of cytokines and chemokines in mouse CSMCs. After careful analysis and comparison between the four treatment groups, we found that normal CSMCs are capable of producing several cytokines and chemokines at the basal level, while inducing inflammation by LPS stimulation caused a marked increase in the expression of TNF-α, IL-1α, M-CSF, TCA-3, and SDF-1. On the other hand, co-treatment with GLP-1 analog exendin-4 caused a significant reduction in the expression of these cytokines. We further evaluated these results by ELISA and RT-PCR and evaluated their secretion into the media. Our results indicate that exendin-4 is effective in suppressing secretion of TNF-α and IL-1α from CSMCs.
TNF-α is a potent pro-inflammatory cytokine primarily secreted from activated macrophages; however, many other cell types are capable of producing it. Dysregulation of TNF-α production has been implicated in a variety of human diseases. It has several biological activities like initiating systemic inflammatory responses to infection and sepsis, exerting an antitumor and antiviral activity, and playing a crucial role in the host response to some pathogens like Mycobacterium tuberculosis.34,35 TNF-α has been implicated in the pathogenesis of several autoimmune diseases like rheumatoid arthritis and IBD. Indeed, the role of TNF-α in the pathogenesis of IBD is well established and is highlighted by the effectiveness of anti-TNF-α treatment in the management of IBD.34 In our study, normal CSMCs were capable of producing TNF-α, while LPS enhanced this capability and GLP-1 co-treatment significantly inhibited its expression and secretion, suggesting an important role for CSMC-driven TNF-α in the pathogenesis of gastrointestinal inflammatory diseases through recruitment and activation of different inflammatory cells.
IL-1α is a central mediator of innate immunity and inflammation;7 in experimental colitis, it is proven to be a key mediator and an early marker of inflammation and its tissue levels correlate with the severity of inflammation. High levels of IL-1α have been reported in freshly homogenized colonic biopsies taken from IBD patients.36 Moreover, specific blockade of IL-1α receptors reduces the inflammatory responses associated with experimental colitis.37 Some of the effects of TNF-α are mediated through IL-1α. The IL-1α precursor is constitutively expressed by the epithelial layer of the entire GIT and several other cell types outside the GIT including the VSMCs, however its expression is greatly enhanced during inflammatory conditions. If cell death occurs by necrosis, this active precursor is released, thereby initiating a cascade of inflammatory cytokines and chemokines, hence accounting for the early phases of sterile inflammation. Moreover, increased expression and secretion of both IL-1α and TNF-α together are considered an alarm signal that initiates inflammatory responses by inducing a cascade of other pro-inflammatory genes.38
M-CSF is a cytokine produced by several types of cells in the body. It controls survival, proliferation, and differentiation of macrophages. It is proven that both M-CSF and macrophages are increased in the GIT in the presence of IBD, hence it is postulated that M-CSF may play a significant role in the pathogenesis of IBD.39,40 Expression of many cytokines is dependent on M-CSF. Marshall et al39 found that blockade of M-CSF with a neutralizing anti-M-CSF antibody inhibits dextran sulfate sodium-induced colitis in a mouse model. Our experiment has clearly shown that exendin-4 significantly inhibited M-CSF expression by the inflamed CSMCs.
Chemokines are chemotactic cytokines that play an essential role in the inflammatory response through recruitment and activation of various types of leukocytes. Several studies support the notion that chemokines play an essential role in the pathogenesis of IBD; rectal biopsies taken from patients during the active phase of IBD show high levels of IL-8 (CXCL8) and RANTES (CCL5).41–43 Moreover, MCP-1 (CCL2) mRNA expression was found to increase during the active disease state in endothelial cells and SMCs.44 In our study, expression of two chemokines, TCA-3 (CCL1) and SDF-1 (CXCL12), increased after induction of inflammation, while exendin-4 co-treatment significantly inhibited the production of these chemokines, suggesting that GLP-1 analogs may interfere in attraction and recruitment of inflammatory cells by inhibition of CSMC-derived chemokines.
TCA-3 is a β-chemokine produced from activated T lymphocytes at the site of inflammation; it functions as a chemotactic factor for neutrophils and macrophages. According to a study conducted by Scheerens et al,45 mRNA levels encoding TCA-3 and several other chemokines were significantly upregulated in the chronically inflamed colons of IL-10–/– mice when compared with wild-type mice. Furthermore, reversal of colitis in these mice was accompanied by inhibition in the expression of TCA-3.
SDF-1 is a chemokine secreted by various types of cells; it serves as a chemotactic factor for lymphocytes and monocytes. Several studies linked SDF-1 expression to IBD pathogenesis. SDF-1 is expressed by normal intestinal epithelial cells and its expression is upregulated in intestinal epithelial cells in IBD.41 In a mouse model of colitis, using an antagonist for SDF-1 receptor resulted in a reduction in colonic inflammation manifested as a reduction in the production of pro-inflammatory cytokines and an improvement in colonic pathology.46
Although CSMCs are important in the pathogenesis of IBD, their role is rarely explored compared to the role of inflammatory cells. Their secretory functions are also overlooked since most studies focus on the morphological changes and altered contractility that occur during colonic inflammation.
GLP-1 exerts its various functions by interacting with GLP-1R, which is a G protein-coupled receptor that is widely distributed in the pancreatic islets, GIT, lungs, kidneys, heart, and some regions of the central and peripheral nervous system. GLP-1R is also widely expressed by multiple immune cell populations, particularly by the intestinal intraepithelial lymphocytes.47 In our study, immunohistochemical staining revealed that GLP-1R is expressed in mouse CSMCs, suggesting that GLP-1 can directly influence CSMC function. In the pancreatic islets, activation of GLP-1R results in activation of adenylyl cyclase and generation of cAMP, activating various downstream mediators through protein kinase A. GLP-1R activation is also coupled to increased intracellular calcium concentration, inhibition of voltage-dependent potassium currents, and activation of gene expression through effects on Erk1/2, protein kinase C, and phosphatidylinositol 3-kinase.48 In the current study, treatment with GLP-1R agonist exendin-4 significantly increased cAMP levels, suggesting that mouse CSMCs utilize the protein kinase A signaling pathway to mediate GLP-1 effects.
It has been shown that agents that increase intracellular cAMP can exert multiple anti-inflammatory effects through reduction of release of cytokines and lysosomal enzymes from neutrophils, histamine, and leukotrienes from mast cells, and other inflammatory cell functions.49 It has been shown that inhibitors of phosphodiesterase type 4 can elevate cAMP intracellular levels, and can downregulate the release of pro-inflammatory cytokines in the mucosa of IBD patients.50
The exact mechanism through which GLP-1 was able to affect the production of cytokines in the CSMCs was beyond the scope of this study. We speculate that increased intracellular cAMP levels after binding of GLP-1 on GLP-1R on the surface of CSMCs results in downregulation of NF-κB phosphorylation and nuclear translocation which could ultimately result in a reduction in the expression of cytokines and chemokines (Figure 8). NF-κB is a transcription factor that controls the transcription of DNA, cytokine production, and cell survival, and has been shown to be suppressed by cAMP.51 Several studies have proved the inhibitory effect of GLP-1 on NF-κB. Dai et al52 found that incubating human umbilical vein endothelial cells with long-acting GLP-1 analog liraglutide was associated with an inhibition of NF-κB phosphorylation and translocation from the cytoplasm to the nucleus. In another study performed on a rat model of diabetes, it was found that exendin-4 treatment significantly inhibited the phosphorylation of NF-κB in the kidneys of diabetic rats.53 Chaudhuri et al32 found that 12-week treatment with exenatide for diabetic patients suppressed the generation of reactive oxygen species and the intranuclear NF-κB binding by mononuclear cells.
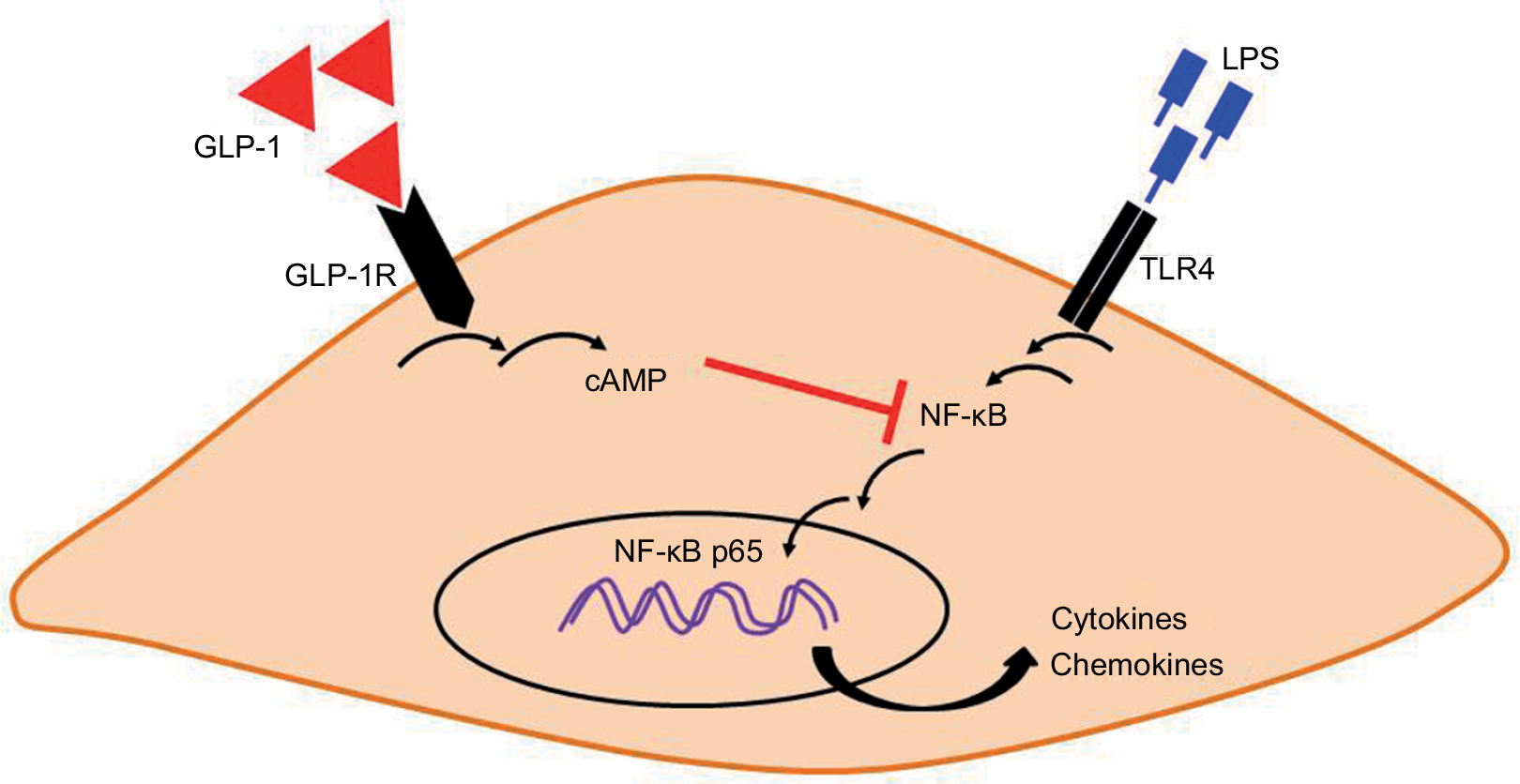 | Figure 8 A proposed integrative model for how GLP-1 analog exendin-4 may suppress LPS-induced inflammatory gene expression in mouse CSMCs. Binding of exendin-4 to cell surface GLP-1R may activate adenylyl cyclase and increase the formation of intracellular second messenger cAMP. cAMP can interfere with LPS-induced NF-κB activation, phosphorylation, and translocation to the nucleus, which in turn suppress inflammatory cytokine and chemokine expression. Abbreviations: cAMP, cyclic adenosine monophosphate; CSMC, colon smooth muscle cell; GLP-1, glucagon-like peptide-1; GLP-1R, GLP-1 receptor; LPS, lipopolysaccharide; NF, nuclear factor; TLR, toll-like receptor. |
Gut inflammation is associated with alterations in intestinal motility; this might result from changes in the SMC function and/or the enteric nervous system. It was observed through various studies that intestinal inflammation is associated with either an increase or a decrease in SMC contractility; this most probably reflects a difference in the cytokine profiles depending on the inflammatory stimuli.9,10 Vermillion et al54 found that the contraction of the smooth muscle tissue taken from the inflamed small intestine of patients with CD is abnormal compared with noninflamed tissues from a control group. In another study, IL-1α was found to be significantly elevated in the human sigmoid circular smooth muscle layer taken from patients with UC and that the circular smooth muscle from these samples does not contract normally.10
In an experiment performed on human colon circular muscular strips taken from normal subjects, it was found that GLP-1R is expressed in the human colon and when activated by exogenous GLP-1 it results in an inhibitory effect on the large intestine motility through nitric oxide neural release.55 If it proves to be beneficial in reversing the inflammation-induced effects on SMC contraction, GLP-1 can prove to be of value as a potential therapeutic option in inflammatory conditions involving the colon.
Conclusion
GLP-1 analog exendin-4 was successful in inhibiting the overexpression of cytokines associated with the induction of inflammation in CSMCs. Several studies are needed to evaluate the true value of GLP-1 analogs and the possibility of using them as adjunct therapy in the treatment of inflammatory bowel conditions.
Acknowledgments
This study was funded by the deanship of research, Jordan University of Science and Technology, Irbid, Jordan (grant number 20160138). We thank Princess Haya Biotechnology Center at Jordan University of Science and Technology for their assistance in performing RT-PCR assays.
Author contributions
AA: conception and design of study, acquisition of data, analysis and interpretation of data, drafting the manuscript, revising the manuscript critically for important intellectual content. TEA: acquisition of data (RT-PCR and ELISA), analysis and interpretation of data, drafting the manuscript, revising the manuscript critically for important intellectual content. OA, AGM, MAA, and MA: analysis and interpretation of data, revising the manuscript critically for important intellectual content. All authors agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361(21):2066–2078. | ||
Click B, Binion DG, Anderson AM. Predicting costs of care for patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2017;15(3):393–395. | ||
Ott C, Schölmerich J. Extraintestinal manifestations and complications in IBD. Nat Rev Gastroenterol Hepatol. 2013;10(10):585–595. | ||
Fakhoury M, Negrulj R, Mooranian A, Al-Salami H. Inflammatory bowel disease: clinical aspects and treatments. J Inflamm Res. 2014;7:113–120. | ||
Ananthakrishnan AN, Donaldson T, Lasch K, Yajnik V. Management of inflammatory bowel disease in the elderly patient: challenges and opportunities. Inflamm Bowel Dis. 2017;23(6):882–893. | ||
Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448(7152):427–434. | ||
Papadakis KA, Targan SR. Role of cytokines in the pathogenesis of inflammatory bowel disease. Annu Rev Med. 2000;51(1):289–298. | ||
Nair DG, Miller KG, Lourenssen SR, Blennerhassett MG. Inflammatory cytokines promote growth of intestinal smooth muscle cells by induced expression of PDGF-Rβ. J Cell Mol Med. 2014;18(3):444–454. | ||
Nair DG, Han TY, Lourenssen S, Blennerhassett MG. Proliferation modulates intestinal smooth muscle phenotype in vitro and in colitis in vivo. Am J Physiol Gastrointest Liver Physiol. 2011;300(5): G903–G913. | ||
Vrees MD, Pricolo VE, Potenti FM, Cao W. Abnormal motility in patients with ulcerative colitis: the role of inflammatory cytokines. Arch Surg. 2002;137(4):439–445; discussion 445–446. | ||
Meier JJ. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8(12):728–742. | ||
Ervinna N, Mita T, Yasunari E, et al. Anagliptin, a DPP-4 inhibitor, suppresses proliferation of vascular smooth muscles and monocyte inflammatory reaction and attenuates atherosclerosis in male apo E-deficient mice. Endocrinology. 2013;154(3):1260–1270. | ||
Derosa G, Cicero AF, Franzetti IG, et al. Effects of exenatide and metformin in combination on some adipocytokine levels: a comparison with metformin monotherapy. Can J Physiol Pharmacol. 2013;91(9):724–732. | ||
Christensen M, Knop FK. Once-weekly GLP-1 agonists: how do they differ from exenatide and liraglutide? Curr Diab Rep. 2010;10(2):124–132. | ||
Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation. 2008;117(18):2340–2350. | ||
May AT. Identification of Expression and Function of the Glucagon-like Peptide-1 Receptor in Gastrointestinal Smooth Muscle [master’s thesis]. Richmond, VA: VCU University Archives, Virginia Commonwealth University; 2017. | ||
Anbazhagan AN, Thaqi M, Priyamvada S, et al. GLP-1 nanomedicine alleviates gut inflammation. Nanomedicine. 2017;13(2):659–665. | ||
Kahles F, Meyer C, Möllmann J, et al. GLP-1 secretion is increased by inflammatory stimuli in an IL-6-dependent manner, leading to hyperinsulinemia and blood glucose lowering. Diabetes. 2014;63(10):3221–3229. | ||
Lebherz C, Schlieper G, Möllmann J, et al. GLP-1 levels predict mortality in patients with critical illness as well as end-stage renal disease. Am J Med. 2017;130(7):833–841.e3. | ||
Lebherz C, Kahles F, Piotrowski K, et al. Interleukin-6 predicts inflammation-induced increase of Glucagon-like peptide-1 in humans in response to cardiac surgery with association to parameters of glucose metabolism. Cardiovasc Diabetol. 2016;15(1):21. | ||
Mimura S, Ando T, Ishiguro K, et al. Dipeptidyl peptidase-4 inhibitor anagliptin facilitates restoration of dextran sulfate sodium-induced colitis. Scand J Gastroenterol. 2013;48(10):1152–1159. | ||
Sakanaka T, Inoue T, Yorifuji N, et al. The effects of a TGR5 agonist and a dipeptidyl peptidase IV inhibitor on dextran sulfate sodium-induced colitis in mice. J Gastroenterol Hepatol. 2015;30 (Suppl 1):60–65. | ||
Camilleri M, Vazquez-Roque M, Iturrino J, et al. Effect of a glucagon-like peptide 1 analog, ROSE-010, on GI motor functions in female patients with constipation-predominant irritable bowel syndrome. Am J Physiol Gastrointest Liver Physiol. 2012;303(1):G120–G128. | ||
Clayburgh DR, Shen L, Turner JR. A porous defense: the leaky epithelial barrier in intestinal disease. Lab Invest. 2004;84(3):282–291. | ||
Berkes J, Viswanathan VK, Savkovic SD, Hecht G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut. 2003;52(3):439–451. | ||
Snape W Jr, Williams R, Hyman PE. Defect in colonic smooth muscle contraction in patients with ulcerative colitis. Am J Physiol. 1991;261(6 Pt 1): G987–G991. | ||
Severi C, Sferra R, Scirocco A, et al. Contribution of Intestinal Smooth Muscle to Crohn’s Disease Fibrogenesis. Eur J Histochem. 2014;58(4):2457. | ||
Shi XZ, Sarna SK. Transcriptional regulation of inflammatory mediators secreted by human colonic circular smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. 2005;289(2):G274–G284. | ||
Salinthone S, Singer CA, Gerthoffer WT. Inflammatory gene expression by human colonic smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. 2004;287(3):G627–G637. | ||
Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132(6):2131–2157. | ||
Lee YS, Jun HS. Anti-inflammatory effects of GLP-1-based therapies beyond glucose control. Mediators Inflamm. 2016;2016:3094642. | ||
Chaudhuri A, Ghanim H, Vora M, et al. Exenatide exerts a potent antiinflammatory effect. J Clin Endocrinol Metab. 2012;97(1):198–207. | ||
Zhu T, Wu XL, Zhang W, Xiao M. Glucagon like peptide-1 (GLP-1) modulates OVA-induced airway inflammation and mucus secretion involving a protein kinase A (PKA)-dependent nuclear factor-κB (NF-κB) signaling pathway in mice. Int J Mol Sci. 2015;16(9):20195–20211. | ||
Rizzo G, Pugliese D, Armuzzi A, Coco C. Anti-TNF alpha in the treatment of ulcerative colitis: a valid approach for organ-sparing or an expensive option to delay surgery? World J Gastroenterol. 2014;20(17):4839–4845. | ||
Bradley JR. TNF-mediated inflammatory disease. J Pathol. 2008;214(2):149–160. | ||
Dionne S, D’Agata ID, Hiscott J, Vanounou T, Seidman EG. Colonic explant production of IL-1 and its receptor antagonist is imbalanced in inflammatory bowel disease (IBD). Clin Exp Immunol. 1998;112(3):435–442. | ||
Cominelli F, Pizarro TT. Interleukin-1 and interleukin-1 receptor antagonist in inflammatory bowel disease. Aliment PharmacolTher. 1996;10 Suppl 2:49–53; discussion 54. | ||
Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14(5):329–342. | ||
Marshall D, Cameron J, Lightwood D, Lawson AD. Blockade of colony stimulating factor-1 (CSF-I) leads to inhibition of DSS-induced colitis. Inflamm Bowel Dis. 2007;13(2):219–224. | ||
Ghia JE, Galeazzi F, Ford DC, Hogaboam CM, Vallance BA, Collins S. Role of M-CSF-dependent macrophages in colitis is driven by the nature of the inflammatory stimulus. Am J Physiol Gastrointest Liver Physiol. 2008;294(3):G770–G777. | ||
Werner L, Guzner-Gur H, Dotan I. Involvement of CXCR4/CXCR7/CXCL12 interactions in inflammatory bowel disease. Theranostics. 2013;3(1):40–46. | ||
Danese S, Gasbarrini A. Chemokines in inflammatory bowel disease. J Clin Pathol. 2005;58(10):1025–1027. | ||
Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta. 2014;1843(11):2563–2582. | ||
Mazzucchelli L, Hauser C, Zgraggen K, et al. Differential in situ expression of the genes encoding the chemokines MCP-1 and RANTES in human inflammatory bowel disease. J Pathol. 1996;178(2):201–206. | ||
Scheerens H, Hessel E, de Waal-Malefyt R, Leach MW, Rennick D. Characterization of chemokines and chemokine receptors in two murine models of inflammatory bowel disease: IL-10-/- mice and Rag-2-/- mice reconstituted with CD4+CD45RBhigh T cells. Eur J Immunol. 2001;31(5):1465–1474. | ||
Xia XM, Wang FY, Zhou J, Hu KF, Li SW, Zou BB. CXCR4 antagonist AMD3100 modulates claudin expression and intestinal barrier function in experimental colitis. PLoS One. 2011;6(11):e27282. | ||
Yusta B, Baggio LL, Koehler J, et al. GLP-1R agonists modulate enteric immune responses through the intestinal intraepithelial lymphocyte GLP-1R. Diabetes. 2015;64(7):2537–2549. | ||
Nagayama K, Kyotani Y, Zhao J, et al. Exendin-4 prevents vascular smooth muscle cell proliferation and migration by angiotensin II via the inhibition of ERK1/2 and JNK signaling pathways. PloS One. 2015;10(9):e0137960. | ||
Moore A, Willoughby D. The role of cAMP regulation in controlling inflammation. Clin Exp Immunol. 1995;101(3):387–389. | ||
Spadaccini M, D’Alessio S, Peyrin-Biroulet L, Danese S. PDE4 inhibition and inflammatory bowel disease: a novel therapeutic avenue. Int J Mol Sci. 2017;18(6). pii: E1276. | ||
Minguet S, Huber M, Rosenkranz L, Schamel WW, Reth M, Brummer T. Adenosine and cAMP are potent inhibitors of the NF-kappa B pathway downstream of immunoreceptors. Eur J Immunol. 2005;35(1):31–41. | ||
Dai Y, Mehta JL, Chen M. Glucagon-like peptide-1 receptor agonist liraglutide inhibits endothelin-1 in endothelial cell by repressing nuclear factor-kappa B activation. Cardiovasc Drugs Ther. 2013;27(5):371–380. | ||
Kodera R, Shikata K, Kataoka HU, et al. Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia. 2011;54(4):965–978. | ||
Vermillion DL, Huizinga JD, Riddell RH, Collins SM. Altered small intestinal smooth muscle function in Crohn’s disease. Gastroenterology. 1993;104(6):1692–1699. | ||
Amato A, Baldassano S, Liotta R, Serio R, Mulè F. Exogenous glucagon-like peptide 1 reduces contractions in human colon circular muscle. J Endocrinol. 2014;221(1):29–37. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.