Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 8
Glibenclamide population pharmacokinetic/pharmacodynamic modeling in South African type 2 diabetic subjects
Authors Rambiritch V, Naidoo P ![]() , Pillai G
, Pillai G
Received 17 December 2015
Accepted for publication 31 May 2016
Published 26 September 2016 Volume 2016:8 Pages 141—153
DOI https://doi.org/10.2147/CPAA.S102674
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Virendra Rambiritch,1 Poobalan Naidoo,2 Goonaseelan Pillai3
1Pharmacology Department, University of KwaZulu-Natal, Durban, 2Department of Internal Medicine, RK Khan Regional Hospital, Chatsworth, South Africa; 3Scientific Capability Development, Novartis Pharma AG, Basel, Switzerland
Aim: To determine the effective dose of glibenclamide by quantifying the dose–response relationship in South African type 2 diabetic patients.
Patients and methods: A total of 24 type 2 diabetic patients participated in a glibenclamide dose-escalation study during which glibenclamide, glucose, and insulin concentrations were quantified, while the dose of glibenclamide was progressively increased. All except four subjects contributed data on all dose-escalation steps; however, data from all 24 patients were included in the model-based analysis. Pharmacokinetic/pharmacodynamic (PKPD) relationships were modeled using the software Nonmem®. Six models were utilized to explore the effect of alternative glibenclamide dose and plasma concentration inputs on various metrics of glucose response.
Results: Six models adequately described the experimental data. The effective dose for a glucose-lowering effect suggested by PKPD modeling is less than 5 mg/day. Doses beyond 5 mg/day do not meaningfully add to glibenclamide effects on blood-glucose response.
Conclusion: The effective dose of glibenclamide, suggested by PKPD modeling, is less than 5 mg/day. Higher doses of glibenclamide, eg, 15 mg/day as originally recommended by the manufacturer, do not produce further decrease in the blood glucose level but may predispose the patients to adverse effects.
Keywords: type 2 diabetes, glibenclamide, pharmacokinetic/pharmacodynamic modeling, dose–response relationships, Nonmem
Introduction
Sulfonylureas (SUs) are oral antidiabetic agents used in clinical practice for decades.1 Despite the growing armamentarium of antidiabetic agents, SUs remain the commonly used second-line agents, in many countries, largely because of their low cost. The dose–response relationship of SUs remains contentious with continued conjecture as to optimal dosing.2
Our study was initially motivated by an audit of the prescribing of glibenclamide at selected public health facilities in KwaZulu-Natal, South Africa, which revealed that doses of 20 mg/day were frequently being used and that this was higher than the manufacturer’s recommended maximum dose of 15 mg/day.3 A further motivation for this analysis was the more general observation that the doses of marketed drugs were frequently reduced after achieving marketing authorization, highlighting the challenges of characterizing and understanding the dose response.4
Pharmacokinetic/pharmacodynamic (PKPD) modeling assists in characterizing and predicting the time course of drug effects (both intensity and duration) in healthy and diseased subjects.5 The PKPD model helps with dose interpolation. For example, in a clinical trial that utilizes conventional methodologies, comment can only be made on the doses tested. It is not possible to consider doses other than those actually employed in the study. However, with PKPD modeling, it is possible to interpolate between doses, and thus assists in determining optimal dose.
The aim of this study was to clarify the dose–response relationship of glibenclamide, a second-generation SU,6 by using PKPD modeling of data generated in a previously conducted clinical study.7
Patients and methods
Data for population PKPD modeling
A clinical study that has been previously published was considered in this study.7 Briefly, written informed consent was obtained from all the study participants, and the study was approved by The Biomedical Research Ethics Committee of the University of Durban- Westville. Twenty-four subjects with type 2 diabetes were entered into a dose-escalation study of increasing doses (0, 2.5, 5, 10, and 20 mg/day) of glibenclamide at 2-week intervals. Glibenclamide, glucose, and insulin levels were determined. All except two subjects contributed data on all dose-escalation steps. Subject 16 could not proceed to a dose escalation beyond 5 mg/day due to the presence of symptoms of hypoglycemia and subjects 20, 21, and 24 did not have complete data sets. However, within the modeling framework, all 24 study participants were included in the PKPD data set.
Bioanalytics
Glibenclamide levels were determined using rapid high-performance liquid chromatography based on the procedure reported by Hamid-Abdel et al.8 Blood glucose level was measured using a glucometer (Accutrend Alpha®; Hoffman-La Roche Ltd., Basel, Switzerland).
Nonlinear mixed-effects modeling
The population approach using nonlinear mixed-effects modeling as implemented in the software Nonmem® (Globomax LLC, Ellicott City, MD, USA and Nonmem Project Group, University of California, San Francisco, CA, USA) was used in this analysis. This model-based analysis was conducted to fully characterize the dose–exposure–response relationship as a monotonic function and thereby facilitate interpolation and prediction of response for doses and exposures not formally studied or observed.
PKPD models
In modeling the PKPD relationships, two broad categories of models were examined – models with glibenclamide dose as the driving force for the pharmacodynamic (PD) response variable and models with steady-state glibenclamide concentration (Cpss) as the driving force. In this manner, the role of pharmacokinetic (PK) variability on the overall variability in response was examined. The PD response metric that was tested included fasting blood glucose (FBG), mean glucose concentration, and the full glucose concentration profile. Thus, during PKPD modeling, the following six models were tested:
- Dose as driving force on FBG
- Cpss as driving force on FBG
- Dose as driving force on mean glucose concentration
- Cpss as driving force on mean glucose concentration
- Dose as driving force on full glucose profile
- Cpss as driving force on full glucose profile
Model 1: Dose as driving force on FBG concentration
Model description
The effect of glibenclamide on glucose response was modeled with an inhibitory Emax model, as shown in Equation 1:
Effect = E0 × (1 – [Dose × Emax]/[Dose + ED50]) (1)
where E0 is the baseline glucose concentration, ED50 is the dose that leads to 50% inhibition of the glucose concentration, Emax is the maximum response to glibenclamide, and Effect is the FBG concentration.
This Emax model has properties that account for the hyperbolic shape of the dose–response relationship. The parameters are maximum response (Emax) and potency (ED50). The model itself is limited in explaining differences between regimens (ie, different PK inputs) or understanding the sources of variability. Differences in ED50 could be due to differences in drug exposure (PK), and/or differences in PD sensitivity. Hence, to elucidate these questions regarding regimen and the underlying variability, PK and PD data need to be modeled.
Model 2: Cpss as driving force on FBG concentration
Model description
This Emax model (Equation 2) has properties that account for the hyperbolic shape of the concentration–response relationships rather than the dose–response relationship. In contrast to the previous dose–response model, this model differentiates between PK variability (in drug clearance [CL/f]) and PD variability:
Effect = E0 × (1 – [Cpss × Emax]/[Cpss + EC50])(2)
where Cpss is the average steady-state plasma concentration and is calculated with Equation 3 using the CL/f estimated from a previously reported population PK model:9
Cpss = Dose/(CL/f × 24) (3)
Model 3: Dose as driving force on mean glucose concentration
Model description
The model is as described in Equation 1. However, Effect (response) is now the mean glucose concentration rather than FBG. The mean glucose concentration is calculated as the area under the glucose concentration versus time curve divided by the time over which the glucose concentrations were measured. The limitations of this model in accounting for PK variability are similar to those mentioned for Model 1.
Model 4: Cpss as driving force on mean glucose concentration
Model description
The model is as described in Equation 2. However, Effect (response) is now the mean glucose concentration calculated as the area under the glucose concentration versus time curve divided by the time over which the glucose concentrations were measured.
Models with full glucose profile as PD end point
Model description
Graphical exploration of the glucose time-course data during the placebo phase of the study revealed a consistent harmonic pattern. These data were modeled using a combination of sine and cosine functions, as shown in Equation 4. This placebo model was used to describe the full glucose versus time profile. The placebo model response was used to ensure that changes in glucose profiles were due to the drug and not to glucose homeostasis:
E0 = A0 + A1 × cos(2 × π × Time/Period)
+ B1 × sin(2 × π × Time/Period)
+ A2 × cos(4 × π × Time/Period)
+ B2 × sin(4 × π × Time/Period)(4)
where A0 represents the baseline glucose concentration (measurement at time 0), A1, A2, B1, and B2 are the coefficients for the harmonic function. Period was 8 hours, ie, the time during which glucose concentrations were measured.
Unexplained inter-subject variability in the baseline glucose concentration (A0) was estimated using the following model (Equation 5) with the random effect ηj:
A0j = TVP × exp(ηj) (5)
where TVP is the typical value of A0 in the population, A0j is the individual value for A0 in the jth individual, and ηj is a random variable with mean of zero and variance ωA02. This model assumes an exponential distribution for the A0j values so as to constrain the estimate of the baseline glucose concentration to positive values.
On the other hand, the coefficients of the harmonic function could take on both positive and negative values and were therefore modeled with an additive error distribution, as shown in Equation 6:
Pnj = TVPn+ ηj (6)
where TVPn is the typical value for the coefficient of interest (A1, A2, B1, and B2) in the population, Pnj is the individual value for the relevant coefficient in the jth individual, and ηj is a random variable with mean of zero and variance ωPn2. The omega matrix for all the coefficients was constrained to the same value, ie, the η values were drawn from the same distribution. The glucose concentration data were log transformed prior to fitting. The residual error model of this log-transformed data comprised of an additive model, as shown in Equation 7:
Cij = C × ij (1 + εij) (7)
where Cij is the ith glucose concentration measured at time ti in the jth individual, C × ij is the respective model-predicted concentration, and εij is a normally distributed error term with mean of zero and variances σ2. Examples of potential sources of residual variability include assay error, deviations from the model specification, and intra-subject variability.
Model 5: Dose as driving force on full glucose profile
Model description
The model is as described in Equation 1. However, Effect (response) is now the full glucose concentration versus time profile modeled using the placebo model, as described in Equations 4–7.
Model 6: Cpss as driving force on full glucose profile
Model description
The model is as described in Equation 2. However, response is now the full glucose concentration versus time profile modeled using the placebo model as described in Equations 4–7.
Results
In evaluating among the six alternative models presented in this study, various graphical and numerical model evaluation methods were used. For purposes of brevity and given their intuitive interpretation, only plots of observed glucose concentrations, population model predictions, and individual model predictions will be shown.
Models 1 and 2: Dose or Cpss as driving force on fasting glucose
Figure 1 shows the individual plots of observed glucose concentrations (open circles), population model predictions (dotted line), and individual model predictions (solid line) from the dose–fasting glucose model. A good fit of the model to the data can be inferred when the observed glucose concentrations (open circles) are close or directly on the model-predicted line. Examples where the observed and model prediction profiles are almost superimposable for the individual model predictions (solid line) include subjects 3 and 15 in Figure 1. This suggests that the model is adequately describing the observed data.
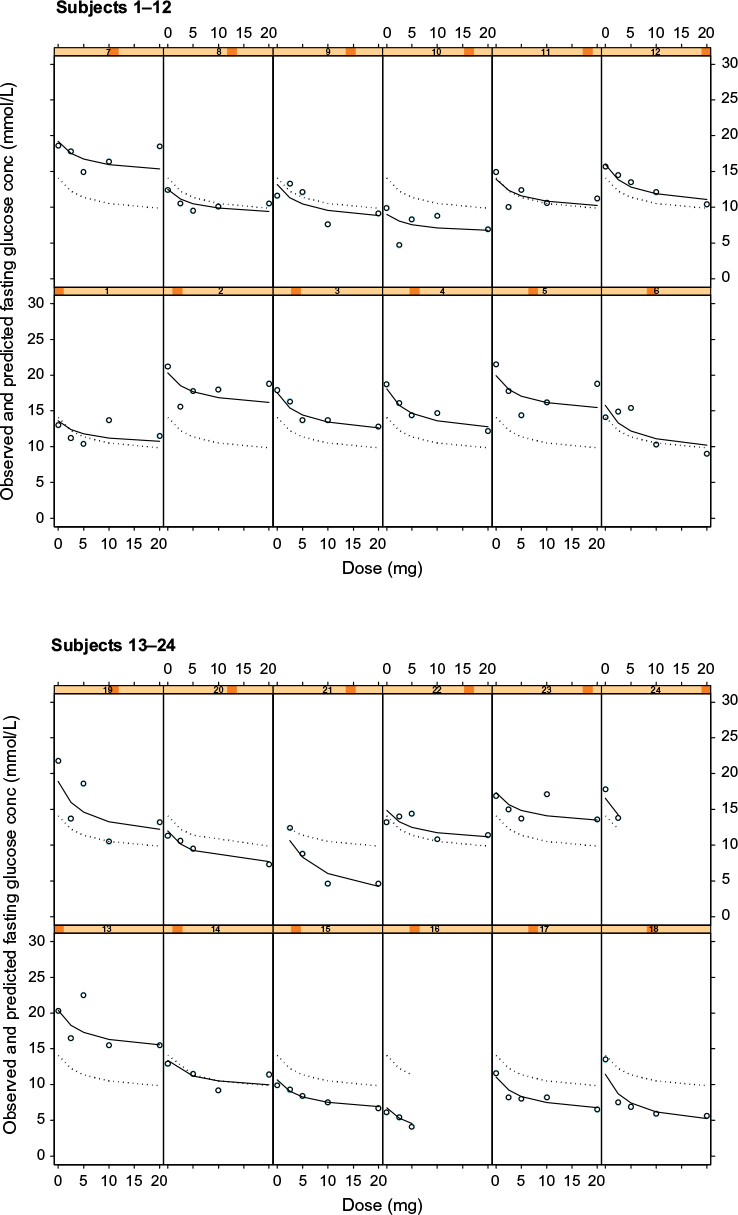 | Figure 1 Plots of observed glucose concentrations (open circles), population model predictions (dotted line), and individual model predictions (solid line) from the dose–fasting glucose concentration PKPD model for glucose response to glibenclamide. Notes: Each cell represents the data for an individual subject shown as a dose–response plot, ie, y-axis shows fasting glucose concentration in mmol/L and x-axis shows dose in mg. Abbreviations: PKPD, pharmacokinetic/pharmacodynamic. |
While this graphical representation suggests that Model 1 is suitable in describing the effect of dose on fasting glucose concentrations, it does neither consider the changing glucose profile throughout the 8-hour study period nor does it take into consideration the PKs of glibenclamide.
In Figure 2, the observed versus model-predicted FBG concentrations are shown for the model that now includes PK variability. This model using Cpss as the driving force for the PD response incorporates the individual subject’s clearance in determining the FBG concentrations, ie, it accounts for the individual PK variation which might contribute to the overall PD response.
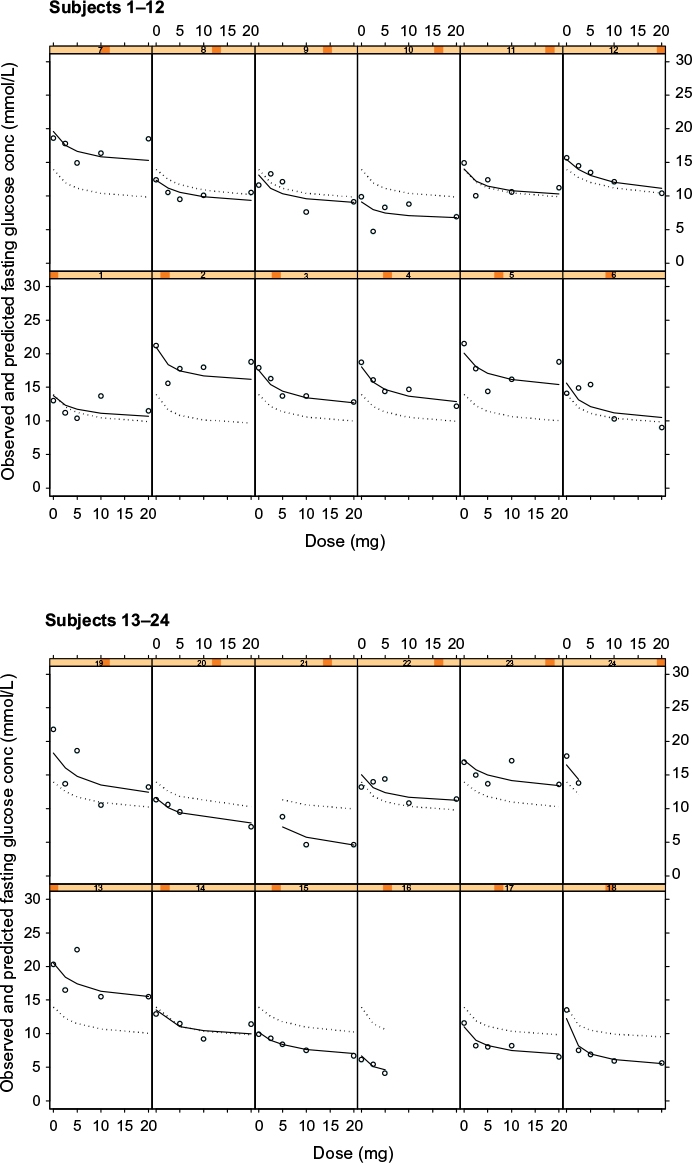 | Figure 2 Plots of observed glucose concentrations (open circles), population model predictions (dotted line), and individual model predictions (solid line) for the steady state concentration (CPss) fasting glucose concentration PKPD model for glucose response to glibenclamide. Notes: Each cell represents the data for an individual subject shown as a dose–response plot, ie, y-axis shows fasting glucose concentration in mmol/L and x-axis shows dose in mg. Abbreviation: PKPD, pharmacokinetic/pharmacodynamic. |
There does not appear to be any striking differences in the observed versus predicted or individual subject fits of the model to the data when compared to the model with dose as driving force on FBG.
The parameters from the dose and the Cpss models for FBG as the PD response are compared in Table 1. Comparing dose versus FBG or Cpss versus FBG as the driving force for the model, the parameters are estimated with better precision, ie, lower relative standard error (RSE 6.35% vs 6.04%) for the model with Cpss as the driving force. Apart from this, there is little difference in the estimated model parameters. There is a low variability in the estimate of potency for dose–fasting glucose model (ED50 =4.56 mg) as compared to Cpss–fasting glucose model (ED50 derived =4.41 mg).
 | Table 1 Population pharmacokinetic/pharmacodynamic parameters for Models 1 and 2: fasting blood glucose as the pharmacodynamic response Note: aDerived ED50 = EC50 × Cl/f × 24, where EC50 =85.20 mg/L and drug clearance (CL/f)9 =2.16 L/h. Abbreviations: E0, baseline glucose concentration; Emax, maximum inhibition of glucose concentration; EC50, glibenclamide concentration producing 50% inhibition of glucose concentration; ED50, glibenclamide dose producing 50% inhibition of glucose concentration; RSE, relative standard error of the estimate; BSV, between-subject variability; CV, coefficient of variation; Cpss, steady-state glibenclamide concentration. |
The point estimates for the fixed-effects model parameters are essentially the same for the two models; however, the parameters are estimated with better precision, ie, lower RSE with the Cpss model. The estimates for between-subject variability (BSV) are different for all the parameters – essentially larger for the Cpss model. In addition, there is remarkably low variability in the estimate of potency with both models.
Models 3 and 4: Dose or Cpss as driving force on mean glucose concentration
There was minimal difference in the graphical outputs from Models 3 and 4: dose versus mean glucose concentration and the Cpss versus mean glucose concentration, and hence only plots of the latter (ie, Model 4) are shown in Figure 3. Inspection of Figure 3 shows that the model describes the individual observed glucose, individual- and population-predicted glucose concentrations for most subjects. It equally underestimates and overestimates the remaining subjects; however, the model adequately describes the observed mean glucose and individual-predicted mean glucose concentrations.
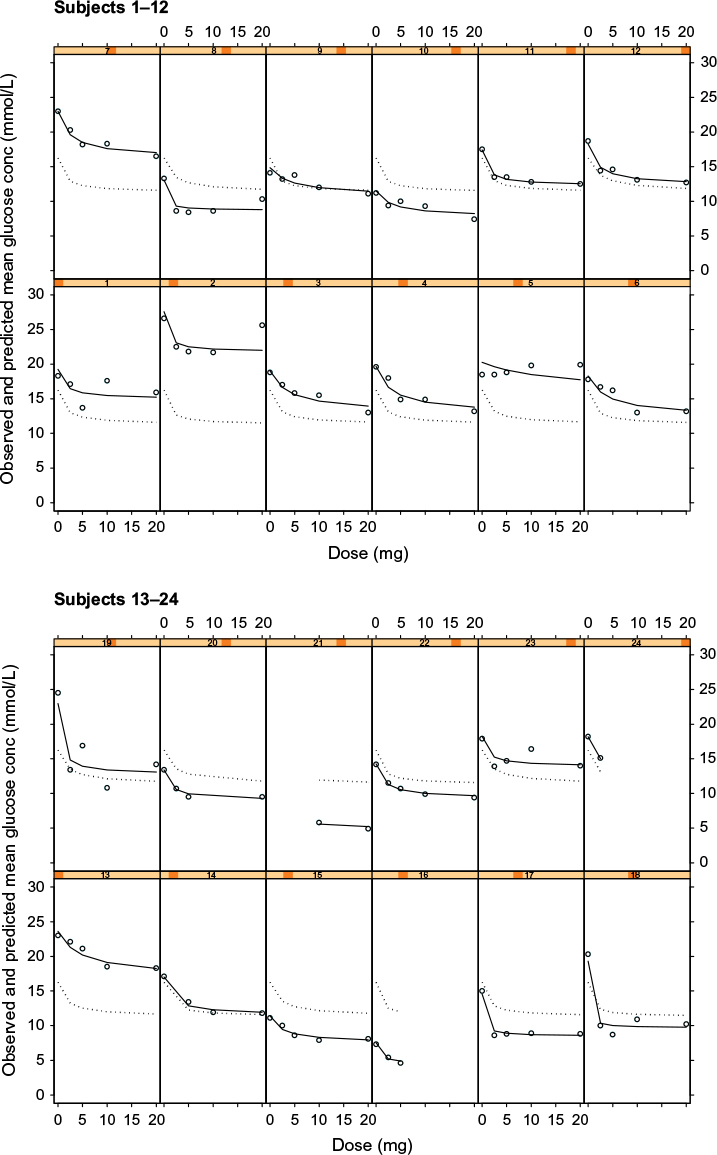 | Figure 3 Plots of observed glucose concentrations (open circles), population model predictions (dotted line), and individual model predictions (solid line) for the steady state concentration (Cpss) -mean glucose concentration PKPD model for glucose response to glibenclamide. Notes: Each cell represents the data for an individual subject shown as a dose–response plot, ie, y-axis shows fasting glucose concentration in mmol/L and x-axis shows dose in mg. Abbreviation: PKPD, pharmacokinetic/pharmacodynamic. |
Table 2 shows virtually no difference in the fixed-effect parameter estimates from the dose or the Cpss model when mean glucose is the PD response. The random-effects parameters are also similar – with the exception of the estimate of potency where a decrease in BSV is noted, ie, inclusion of PK variability in the model by using Cpss as the driving force resulted in a decrease in the BSV from 120% CV for ED50 to 108% CV for EC50.
 | Table 2 Population pharmacokinetic/pharmacodynamic parameters for Models 3 and 4: mean blood glucose as the pharmacodynamic response Note: aDerived ED50 = EC50 × Cl/f × 24, where EC50 = 36.00 mg/L and drug clearance (CL/f)9 = 2.16 L/h. Abbreviations: E0, baseline glucose concentration; Emax, maximum inhibition of glucose concentration; EC50, glibenclamide concentration producing 50% inhibition of glucose concentration; ED50, glibenclamide dose producing 50% inhibition of glucose concentration; RSE, relative standard error of the estimate; BSV, between-subject variability; CV, coefficient of variation; Cpss, steady-state glibenclamide concentration. |
While this model provides a good description of the data and is considered adequate for the purpose of this analysis, one further attempt at building a model for the glucose-lowering PD effect was considered, namely modeling the full glucose concentration versus time profile.
Model for full glucose profile (placebo response)
The fluctuations in glucose concentrations over the observation period were due primarily to the response to food (Figure 4, column 1 [zero dose]). In this study, subjects were given breakfast, followed by lunch ~4 hours later. The initial peak in glucose corresponds to ingestion of breakfast and the subsequent peak 4 hours later to lunch. Sampling to characterize these two events showed two blood glucose peaks at ~2 and 6 hours, temporally related to the ingestion of food. The model selected to describe this data is empirical and has no physiological meaning – the data could also have been fit with a series of spline functions or a polynomial to the placebo data.
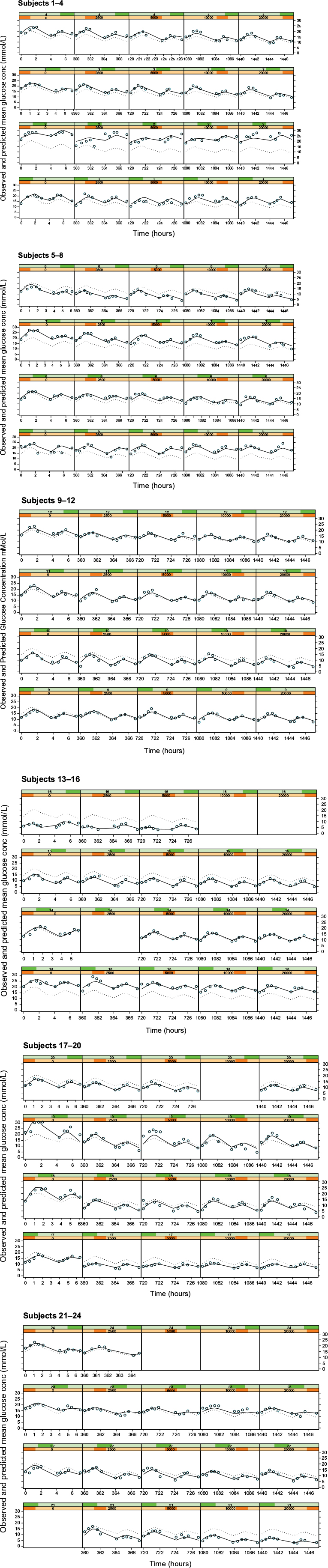 | Figure 4 Plots of observed glucose concentrations (open circles), population model predictions (dotted line), and individual model predictions (solid line) for the Cpss–full glucose profile PKPD model. Notes: Each row represents the data for an individual subject and each column represents a different dose level, with placebo on the extreme left and increasing doses of 2.5, 5, 10, and 20 mg in the subsequent columns. Abbreviations: PKPD, pharmacokinetic/pharmacodynamic; Cpss, steady-state glibenclamide concentration. |
The empirical placebo model was used to describe the time course of glucose concentration data over the 8-hour study observation period. It satisfactorily described the data save for a small number of subjects (Figure 4, column 1). In all other subjects, the observed, individual- and population-predicted curves were all almost superimposable, strengthening the validity of the model.
Models 5 and 6: Dose or Cpss as driving force on full glucose profile
As with Models 3 and 4, there was no noteworthy difference between the dose or Cpss as the driving force and hence only the individual subject fits for the Cpss–full glucose profile model are shown in Figure 4. In these plots, each row corresponds to the data for an individual subject, while each column shows a new dose level within an individual subject. The dose–response relationship is shown by the shift in the entire glucose concentration versus time profile downward as one progresses from left to right within a row, ie, from placebo on the left to the highest dose administered on the right.
The parameters from this complex full glucose profile model are shown in Table 3.The point estimates of the fixed-effects PD model parameters – baseline glucose concentration (A0), potency (EC50 and ED50), and Emax – are essentially not different in the dose and the Cpss models. However, there is a difference in the random-effects parameters, in particular, the Fourier coefficients have higher variability while the primary PD model parameters have lower variability in the dose model compared to the Cpss model. A likely explanation for this is that PK variability is being confounded with PD variability – the dose model can only allocate the PK variability into the Fourier coefficients. The variability in potency is also much larger in this model than that seen with the model where the PD response was the mean glucose concentration (221% CV vs 108% CV).
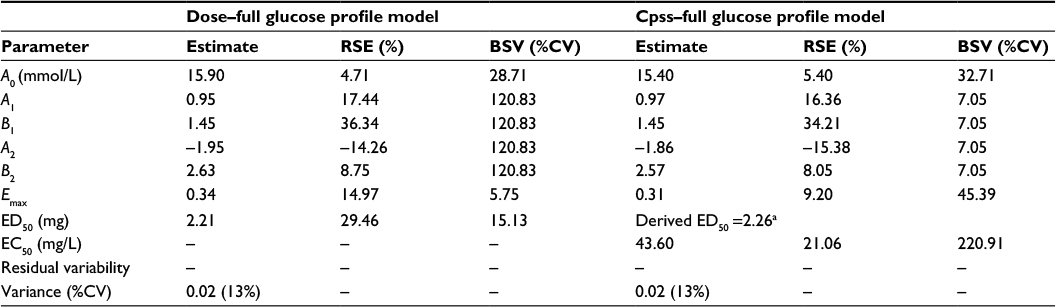 | Table 3 Population pharmacokinetic/pharmacodynamic parameters for the Models 5 and 6: full glucose profile as the pharmacodynamic response Note: aDerived ED50 = EC50 × Cl/f × 24 where EC50 = 43.60 mg/L and drug clearance (CL/f)9 =2.16 L/h. Abbreviations: A0, baseline glucose concentration; A1, B1, A2, B2, coefficients of the harmonic function; Emax, maximum inhibition of glucose concentration; ED50, glibenclamide dose producing 50% inhibition of glucose concentration; EC50, glibenclamide concentration producing 50% inhibition of glucose concentration; RSE, relative standard error of the estimate; BSV, between-subject variability; CV, coefficient of variation; Cpss, steady-state glibenclamide concentration. |
Thus, despite the very good fit of the model to the data, this is likely to be due to the large amount of flexibility that is allowed by the large number of model parameters.
Consequently, the models where FBG and mean glucose concentrations are the PD responses were considered as being adequate for this data.
Discussion
While it is possible to mathematically describe the full glibenclamide–insulin–glucose system, the glibenclamide–glucose system was modeled in this analysis, in keeping with the work of other researchers.10,11 The clinical effectiveness of antidiabetic treatment, and hence decisions on management of diabetes, is determined by fasting and/or postprandial blood glucose levels. Hence, this study utilized the glibenclamide–glucose system for PKPD analysis.
In this dose-escalation study, a zero dose (absence of glibenclamide) was employed to characterize the insulin and glucose profiles. Since insulin and glucose are interdependent, we selected the glucose response, and modeled this using a mathematical basis and an algorithm described by Krzyzanski et al12 for application of Fourier analysis to quantify variable, but biorhythmic, physiologic substances to generate input functions for use in PD response models. This method does not require trial and error nonlinear regression analysis to identify the optimal number of harmonics that describe the response pattern.12
Graphical exploration of the within-subject drug response data for glucose, insulin, and glibenclamide profiles with increasing doses of glibenclamide shows that there is no direct relationship between drug administration and glucose or insulin response, ie, there is no acute effect of drug on these parameters. The lack of acute antihyperglycemic effect may be due to the requirement for formation of active metabolites. Rydberg et al10 observed that at a “given concentration of glibenclamide, a more intense effect is observed at the later sample time due to additional hypoglycemic effect of in vivo formed metabolites.” Consequently, we modeled dose or the average drug concentration (Cpss) as the driving force for the drug effect rather than a full glibenclamide plasma concentration versus time profile.
In comparing dose versus FBG or Cpss versus FBG as the driving force for the model, the parameters are estimated with better precision, ie, lower RSE (6.35% vs 6.04%) for the model with Cpss as the driving force. Apart from this, there is little difference in the estimated model parameters. There is a low variability in the estimate of potency for dose–fasting glucose model (ED50 =4.56 mg) as compared to Cpss–fasting glucose model (ED50 derived =4.41mg).
There is no difference in the parameter estimates from the dose or the Cpss models, where the PD response is mean glucose concentration. As expected, there is a higher estimate for the modeled baseline glucose concentration for the dose versus mean glucose concentration model as compared to the dose versus FBG model (16.70 vs 14.10 mmol/L). The ED50 (1.85 mg) and the derived ED50 (1.87 mg) for the dose versus mean glucose concentration model are almost identical with a BSV of 108 – 120%.
In addition, a much larger variability is noted in the potency parameters relative to the model where the PD response was FBG. By calculating a mean glucose concentration, the variability in concentrations during the course of the observation period is being brought into the model. Intuitively, one would expect a fasting measurement, ie, one taken when the biological system is not being subjected to the known factors that can influence glucose response (eg, food), to be subject to lower variability. FBG is also the PD marker directly influenced by the SUs. This accounts for the differences in the estimates of random-effects parameter estimates. The maximum glucose-lowering effect seen with both groups of models is also very similar, ie, between 31% and 37% relative to baseline.
The point estimates of the primary PD model parameters for the dose versus full glucose profile and the Cpss versus full glucose profile are not essentially different, ie, baseline glucose concentration, A0 (15.9 and 15.4, respectively), potency (ED50 =2.21 and ED50-derived =2.26, respectively), and Emax (0.34 and 0.31, respectively). However, there is a difference in the random elements, in particular, the Fourier coefficients have higher variability while the primary PD model parameters have lower variability in the dose model compared to the Cpss model. It seems therefore that the greater freedom allowed to the model with the large number of extra parameters results in “inappropriate” apportioning of relative contributions of variability. An attempt was made to resolve this by estimating the Fourier coefficients from the placebo data alone and then subsequently fixing these in the estimation step for the full data set. However, the model with the full data set failed to converge.
The Emax for the six models evaluated varies from 0.37 for the dose versus FBG to 0.31 for the Cpss versus full glucose profile models. This means that the maximal benefit of glibeclamide therapy in this population lies in a decrease of 31%–37% from baseline. This translates to a reduction of the FBG from 15.4 to 10.6 mmol/L and 9.7 mmol/L, respectively.
The ED50 and the derived ED50 for the six models described are 4.56, 4.41, 1.85, 1.87, 2.21, and 2.26 mg, respectively. When this low estimate of potency is assessed together with the maximum drug effect of under 37%, 5 mg brings only marginal benefit. In addition, an evaluation
of glycemic control in individual patients showed that only seven (30%) and four (17%) subjects achieved acceptable and optimal control, respectively, at doses greater than 5 mg/day. Thus, both the observed and the modeled data suggest a maximal dose not exceeding 5 mg in the study population. This conclusion is supported by the findings of Rydberg et al10 and Groop et al13 that “the maximum effect of glibenclamide would be obtained by 5 mg or less.” Further increase in glibenclamide dose is not likely to produce significant reductions in blood glucose levels. High-dose glibenclamide is associated with various side effects,12 including hypoglycemia, increased cardiovascular risk, and masking of the severity of a myocardial infarction.14,15
Conclusion
The six PKPD models adequately described the dose–response relationship of glibenclamide. The models are congruent in supporting a conclusion that doses of glibenclamide of less than 5 mg/day are effective in lowering blood glucose level with no appreciable increase in effect with higher doses. The higher doses of glibenclamide that have previously been used (up to 20 mg/day) should be avoided because they do not have greater glucose-lowering effect and exposure to high doses are likely to predispose patients to adverse events.
Disclosure
G Pillai has stocks in Novartis. The other authors report no conflicts of interest in this work.
References
Naidoo P, Rambiritch V, Butkow N, Saman S. Optimal utilisation of sulphonylureas in resource-constrained settings. Cardiovasc J Afr. 2014;25(2):83–85. | ||
Rambiritch V, Naidoo P, Butkow N. Dose–response relationships of sulfonylureas: will doubling the dose double the response? South Med J. 2007;100(11):1132–1136. | ||
Rambiritch V, Pillai G, Maharaj B, Robertson LI. Glibenclamide – what dose? S Afr Med J. 2007;97(7):474. | ||
Cross J, Lee H, Westelinck A, Nelson J, Grudzinskas C, Peck C. Postmarketing drug dosage changes of 499 FDA-approved new molecular entities, 1980–1999. Pharmacoepidemiol Drug Saf. 2002;11:439–446. | ||
Breimer DD, Danhof M. Relevance of the application of pharmacokinetic/pharmacodynamic modeling concepts in drug development. The ‘wooden shoe’ paradigm. Clin Pharmacokinet. 1997;32:259–267. | ||
Groop L, Groop P-H, Stenman S, et al. Comparison of pharmacokinetics, metabolic effects and mechanisms of action of glyburide and glipizide during long-term treatment. Diabetes Care. 1987;10(6):671–678. | ||
Rambiritch V, Maharaj B, Naidoo P. Glibenclamide in patients with poorly controlled type 2 diabetes: a 12-week, prospective, single-center, open-label, dose-escalation study. Clin Pharmacol. 2014;6: 63–69. | ||
Hamid-Abdel ME, Suleiman MS, el-Sayed YM, Najib NM, Hasan MM. A rapid high performance liquid chromatography assay of glibenclamide in serum. J Clin Pharm Ther. 1989;14(3):181–188. | ||
Rambiritch V, Maharaj B, Naidoo P, Pillai G. Population pharmacokinetic modeling of glibenclamide in South African type 2 diabetic subjects. Clin Pharmacol. 2016;8:83–92. | ||
Rydberg T, Jonsson A, Karlsson MO, Melander A. Concentration–effect relations of glibenclamide and its active metabolites in man: modelling of pharmacokinetics and pharmacodynamics. Br J Clin Pharmacol. 1997;43(4):373–381. | ||
Tracewell WG, Stalker DJ, Maloley PA, Gallagher TF, Gwift PR. Population pharmacokinetics of glyburide in patients with well controlled diabetes. Pharmacotherapy. 1998;18(1):51–56. | ||
Krzyzanski W, Chakraborty A, Jusko WJ. Algorithm for application of Fourier analysis for biorhythmic baselines of pharmacodynamic indirect response models. Chronobiol Int. 2000;17(1):77–93. | ||
Groop LC, Barzilai N, Ratheiser K, et al. Dose-dependent effects of glyburide on insulin secretion and glucose uptake in humans. Diabetes Care. 1991;14(8):724–727. | ||
Feldman JM. Glyburide: a second-generation sulfonylurea hypoglycemic agent. History, chemistry, metabolism, pharmacokinetics, clinical use and adverse effects. Pharmacotherapy. 1985;5(2): 43–62. | ||
Huizar JF, Gonzalez LA, Alderman J, Smith HS. Sulfonylureas attenuate electrocardiographic ST-segment elevation during an acute myocardial infarction in diabetics. J Am Coll Cardiol. 2003;42(6):1017–1021. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.