Back to Journals » Journal of Blood Medicine » Volume 6
Glanzmann's thrombasthenia: pathogenesis, diagnosis, and current and emerging treatment options
Authors Solh T, Botsford A, Solh M
Received 8 March 2015
Accepted for publication 29 April 2015
Published 8 July 2015 Volume 2015:6 Pages 219—227
DOI https://doi.org/10.2147/JBM.S71319
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Tia Solh,1 Ashley Botsford,2 Melhem Solh3
1Department of Physician Assistant Studies, Philadelphia College of Osteopathic Medicine, Suwanee, GA, USA; 2College of Health Care Sciences, Nova Southeastern University, Orlando, FL, USA; 3The Blood and Marrow Transplant Group of Georgia, Northside Hospital, Atlanta, GA, USA
Abstract: Glanzmann’s thrombasthenia (GT) is a genetic platelet surface receptor disorder of GPIIb/IIIa (ITG αIIbß3), either qualitative or quantitative, which results in faulty platelet aggregation and diminished clot retraction. Spontaneous mucocutaneous bleeding is common and can lead to fatal bleeding episodes. Control and prevention of bleeding among patients with GT is imperative, and remains challenging. Local measures, including anti-fibrinolytic therapy, with or without platelet transfusions, used to be the mainstay of therapy. However, in recent years the use of recombinant factor VIIa (rFVIIa) has increased significantly, with excellent response rates in treating and preventing hemorrhage among GT patients. Gene therapy and stem cell transplantation offer a potential cure of this disease, but both are costly and remain experimental at this point. This manuscript offers a comprehensive review of our understanding of GT and the available treatment options.
Keywords: Glanzmann, thrombasthenia, treatment
Introduction
Platelets are a central component of many restorative physiological processes, including hemostasis. During hemostasis, damaged subendothelium releases adhesive proteins (ie, collagen and thromboplastin) and fibrinogen, which bind with aggregated platelets at the site of injury, forming a platelet plug. Platelets then provide a surface and phospholipid source for attachment of coagulation cofactors. Subsequent activation of the coagulation pathways prompts fibrin attachment to activated platelets, creating a thrombus. Any disruption in platelet function, whether acquired or inherited, will generate bleeding.
Acquired platelet disorders are more likely to be encountered in clinical practice than their inherited counterpart, and often result from the initiation of medications or underlying medical conditions. In contrast, inherited platelet disorders are rare, and until recently, have been under less scrutiny. One of the most recognized and clearly defined disorders of inherited abnormal hemostasis today is Glanzmann’s thrombasthenia (GT), a disease that has helped pave the way for key research findings in hematology.
Eduard Glanzmann was a Swiss pediatrician who first discovered the condition of thrombasthenia in 1918.4 Formerly known as “hereditary hemorrhagic thrombasthenia”, Glanzmann recognized a disorder that was not attributed to an abnormal number of platelets, but rather a faulty clot retraction, which paved the way for future work.4 According to a biographical account, Glanzmann’s encounter with a symptomatic 7-year-old girl led him to study the disease within families. Noting a familial pattern and symptoms manifesting in children, he considered a possible hereditary component. The disease was later defined as a heritable platelet disorder secondary to a dysfunction in GPIIb/IIIa complex.
Today GT receives much recognition, as it was one of the first disorders to define GPIIb/IIIa as a platelet receptor for adhesive molecules (such as VWF and fibrinogen). The disease also served as a template for understanding processes of platelet aggregation as well as targets for therapeutic measures.
Pathogenesis of GT
GT is an autosomal recessive platelet surface receptor disorder of GPIIb/IIIa (ITG αIIbβ3), either qualitative or quantitative, which results in faulty platelet aggregation and diminished clot retraction.
The GPIIb/IIIa, or ITG αIIbβ3, is a large heterodimeric cell transmembrane receptor comprised of a larger αIIb and a smaller β3 subunit. These subunits are non-covalently linked, allowing for duplex signaling between the cell membrane and extracellular matrix, while instituting intracellular signaling pathways. Electron microscope images of the heterodimer have shown an 8×12 nm nodular head and two 18 nm stalks.12 The stalks extend through the cell, and contain the cytoplasmic and transmembrane domains that serve as attachment points for intracellular signaling molecules and proteins, while the bent head contains the ligand-binding site.12 The β3 subunit consists of large, disulfide epidermal growth factor (EGF)-domains responsible for activation of ITG αIIbβ3 as a whole. The calcium binding sites involved in complex formation and platelet-platelet adherence are located on the β-propeller region of the αIIb subunit.6 The receptor head functionality – binding fibrinogen, VWF, vitronectin and fibronectin – is necessary for platelet aggregation. ITG αIIbβ3 controls cell-to-cell communications by regulation of cell migration, platelet aggregation and adhesion, and the formation of a thrombus.
Roughly 100,000 copies of the GPIIb/IIIa receptor are expressed along a platelet’s surface, which differs by two-fold between individuals.6,23 The gene ITGA2B, located on chromosome 17q21.31, codes for the platelet GPαIIb, while the gene ITGB3 encoding for the glycoprotein subunit IIIa lies on chromosome 17q21.32. Mutations have been found more commonly in the ITGA2B gene, possibly due to the voluminous number of exons when compared to the ITGB3 gene (30 compared to 15).6,10 Deletions, insertions, frameshifts, nonsense, and missense mutations have been frequently recognized. Missense mutations have been further studied, and display interruption in integrin maturation or subunit formation. Biogenesis of ITG αIIbβ3 arises from the hematopoietic stem cell. The subunit αIIb is arranged from a single peptide, and is closely linked to the megakaryocyte lineage, whereas β3 is linked to the vitronectin receptor (αvβ3) involved in transport processes, with distribution among multitudes of tissues.10 Both subunits are amassed from endoplasmic reticulum precursors, with further processing occurring in the Golgi apparatus. The αvβ3 receptor will be most abundant on platelets in patients with an ITGA2B mutation. Both αIIbβ3 and αvβ3 will be absent when a mutation prevents β3 synthesis, but a missense mutation in β3 can have varying effects. For example, mutations in β3 Leu262Pro and Ser162Leu have been shown to provide residual αIIbβ3 platelet complexes with the capacity to bind fibrin and retract clots, but, when stimulated, are incapable of adhering fibrinogen. In contrast, a mutation in β3 Leu196Pro is able to sustain partial clot retraction.2,6 A review by Nurden et al more closely examined the β-propeller ectodomain mutations of the αIIb subunit. Nurden et al concluded that a large series of mutations affecting the β-propeller domain interrupted calcium binding and had numerous deleterious effects on αIIbβ3 expression and function, giving rise to the different types of GT.5 Mutations in the αIIb subunit that allow for partial complex formation were found to be distant from the junction between αIIb and β3, inferring a variant form of GT. Different effects are noted between mutations occurring in either subunit of αIIbβ3, and between αIIbβ3 and αvβ3; however, αvβ3 is more resilient to change than αIIbβ3. Some mutations of αIIbβ3 do not lead to GT. For example, Kashiwagi et al recently described three gain of function mutations, ITGA2B p.Gly991Cys, ITGA2B p.Phe993del, and ITGB3 p.(Asp621_Glu660del), that led to a highly activated conformation of αIIbβ3 and spontaneous tyrosine phosphorylation of FAK in transfected cells. These mutations resulted in abnormalities in both platelet morphology and number, with impaired surface αIIbβ3 expression, but did not lead to GT.9
Homozygous or heterozygous mutations found at either gene locus determine the severity of abnormality seen in GT. Mutations can arrest subunit manufacturing, impede complex formation, and/or inhibit intracellular trafficking. When complex formation is hindered, residual subunits of αIIb or β3 degrade. Based on the expression and functionality of residual subunits, GT is classified into three types: <5% of residual αIIbβ3 signifies type I GT; 5%–20% of residual αIIbβ3 comprises type II GT; and rarely, >20% of residual αIIb β3, with dysfunctional properties, constitutes variant-type GT. Early work by George et al failed to correlate the subtype of GT with severity of bleeding.10 However, it has been noted by Fiore et al that phenotypic bleeding is more influenced by a mutation in the ITGB3 gene.6
Acquired GT is commonly the result of an autoantibody attack on platelet αIIbβ3, or isoantibodies inhibiting proper function. The production of autoantibodies has been associated with platelet transfusions, as well as numerous hematologic conditions, including immune thrombocytopenic purpura, non-Hodgkin’s lymphoma, multiple myeloma, myelodysplastic syndrome, hairy cell leukemia, and acute lymphoblastic leukemia. In a case report done by Blickstein et al, a patient with systemic lupus erythematosus presented in adulthood with degrees of mucocutaneous bleeding secondary to antibody production against GPIIb/IIIa.5 Also, some anti-thrombotic therapies use αIIbβ3 antagonists, such as abciximab, eptifibatide, and tirofiban, for treatment of acute coronary events, which can trigger a transient GT-like state.10
Incidence and prevalence
It is estimated that one in 1,000,000 individuals have GT, though the exact number is unknown.14 Studies have shown a slight predominance in women compared to men (58% versus 42%), consistent with its mode of inheritance.7 GT can be found in individuals across the globe, but is more common in ethnic groups that display higher incidences of consanguinity, such as Iraqi Jews, selective Arab populations (eg, Palestinian), and French Gypsies. According to one study, the disease has a high prevalence among the French Gypsy Manouche community, with approximately 150 cases of 300 in France from that origin. The inflated number among these population groups illustrates a founder effect.2
Clinical presentation
GT may manifest itself shortly after birth, beginning with purpura in the neonate, and followed by episodes of mucocutaneous bleeding and spontaneous bruising. The majority of patients will be diagnosed before the age of 5, with recurring episodes of epistaxis and gingival bleeding being among the most common manifestations. In one Parisian study, 84% of patients with GT required red blood cell transfusion. The most common cause of epistaxis in any child is digital manipulation; however, in patients with GT, severe bleeding can occur in this highly vascularized area, which can be fatal. While fatal bleeding episodes can occur at any point in the lifespan of a patient with GT, the prevalence of severe bleeding does decrease with age.7
To date, there is little evidence to support any correlation between gene defect in GT and the severity of bleeding, as affected individuals demonstrate different bleeding tendencies, even within the same family or ethnic group.10 In some cases, disproportionately severe bleeding after minor trauma or surgery may be the introductory sign of the condition. Excessive bleeding during circumcision has been noted, and is often the reason for investigation and diagnosis of GT in males. In some cases, women have not been diagnosed until menarche, when severe blood loss due to menorrhagia has required transfusion, as reported in a study by George et al. Labor and delivery is of special concern, as women are at increased risk for severe or fatal hemorrhage during this time.7
Diagnosis
The diagnosis of GT is often overlooked, as the condition shares common clinical and laboratory features with other acquired platelet disorders. One must remember to include GT in the differential diagnosis, carefully analyzing the medical history (eg, asking about unprovoked bruising or bleeding episodes, or episodes of severe bleeding after minor trauma), family history (eg, inquiring about possible consanguinity), and clinical presentation (eg, examining for purpura and ecchymoses).
When considering a diagnosis of GT, selecting the appropriate laboratory evaluation is essential. For example, a normal platelet count on a routine blood smear does not rule out a diagnosis of GT, as patients with GT usually show no abnormalities in the platelet count. The complete blood count may be entirely normal, or may reveal iron deficiency. The prothrombin time and activated partial thromboplastin time will also be normal. However, the bleeding time will be prolonged, which warrants further investigation.
Light transmission aggregometry (LTA) is widely accepted as the gold standard diagnostic tool for assessing platelet function. Centrifuged platelet-rich plasma samples are monitored before and after the addition of an agonist (ADP, collagen, epinephrine, arachidonic acid, ristocetin, thrombin receptor activating peptide, and thromboxane A2 mimetic), assessing shape change, lag phase, percent of aggregation, slope of aggregation, and deaggregation. This test is highly specific for GT, as platelet aggregation fails to occur with any agonist, except ristocetin, where the reaction is preserved. Although this test yields specific data, LTA is very time consuming and personnel dependent, requiring the use of experienced laboratories. It is also more difficult to obtain platelet-rich plasma in patients with thrombocytopenia and in pediatric patients under the testing guidelines.
Platelet function analyzer (PFA) is a highly sensitive test for detecting GT. The PFA assay uses collagen + ADP- and collagen + epinephrine-embedded cartridges to mimic a damaged vessel endothelium. As citrated whole blood flows at a high shear stress rate through these cartridges, platelets bind, creating a platelet plug. The PFA assay is prolonged among patients with GT.6
Flow cytometry can be beneficial, as GT includes glycoprotein receptor deficiency and/or dysfunction. Flow cytometry will measure distinct platelet receptor densities using a variety of monoclonal antibodies to those receptors, singling out the deficient αIIbβ3. Under flow cytometric analysis, CD41 and CD61 levels are markedly decreased or absent, while normal levels of CD42, and identification of deficient and non-functional αIIbβ3 can be expressed, consistent with a diagnosis of G.10
The best way to diagnose GT in its entirety is through mutation analysis. Genomic DNA sequencing of the 45 exons comprising the αIIbβ3 unit, along with the splice sites of the ITGB3 and ITGA2B gene, should be investigated, and the established mutations should be confirmed with a second DNA sample analysis.
Overall, the diagnosis of GT includes the presence of normal platelet count (typically on the lower end of normal), prolonged bleeding time, and prolonged PFA time. Platelets fail to aggregate under the conditions utilized in LTA, which is uniquely indicative of GT.
Treatment
All patients with GT require management by a specialist, and should be registered with a 24-hour center capable of handling diagnosis and treatment.5,6 Patients with GT do not need therapy on a regular basis, but will always require treatment during surgical procedures, controlling bleeding after injury, and during spontaneous bleeding episodes.
Patient education
As patients will likely receive transfusions of blood products at least once in their lifetime, all patients should receive immunization against hepatitis B. Patients should also be advised to avoid contact sports as well as the use of aspirin and non-steroidal anti-inflammatory drugs.6 In order to mitigate the risk of gingival bleeding, patients should maintain excellent oral hygiene and participate in regular dental visits. Women in particular, due to the consequences of menorrhagia, need monitoring for iron deficiency, and should be placed on iron supplementation if necessary. Patients should be HLA typed at diagnosis, and monitored for HLA and anti-platelet antibodies regularly.6 Patients should also be counseled that bleeding from a minor wound, the gingiva, or the nose may be able to be stopped by applying pressure, and/or by taking an oral anti-fibrinolytic, but to seek medical attention if bleeding persists.
General treatment concepts
The current standard of treatment of bleeding episodes in patients with GT is the use of local measures alone or in conjunction with anti-fibrinolytic therapy first, followed by platelet transfusion, and rFVIIa if bleeding persists.7 Local measures include the use of compression, fibrin sealants, and topical thrombin.3 When possible, platelet concentrates should be single-donor derived and HLA-matched due to the risk of developing alloantibodies against the platelet glycoproteins, αIIbβ3, or αIIbβ3, and/or the HLA antigens with repeated transfusions. If HLA-matched platelets are not available, patients should receive leukocyte-reduced platelets, as this has been shown to reduce the rate of HLA immunization. Patients with severe bleeding episodes should continue to receive platelet transfusions for 48 hours after the cessation of bleeding, and until wound healing has occurred in patients undergoing surgery. The PFA assay time will normalize with adequate transfusions.2
Management of minor to moderate bleeding
For mild to moderate bleeding episodes, local measures, and/or anti-fibrinolytic drugs may resolve bleeding (Figure 1). Anti-fibrinolytic drugs work particularly well for mucocutaneous bleeding, and have been shown to successfully decrease bleeding during dental procedures, in combination with local measures. Local measures include compression, gelatin sponges, fibrin sealants, and topical thrombin.6 Anti-fibrinolytic agents include tranexamic acid and epsilon aminocaproic acid. These agents, used alone or as adjunctive therapy with rFVIIa, have proven to be useful and safe agents in minimizing or resolving bleeding in patients with GT in multiple settings. As both agents can be given orally or intravenously, they have been used successfully for the treatment of epistaxis, gingival bleeding, and menorrhagia, and also as prophylaxis prior to dental extractions and other minor surgical procedures. The use of anti-fibrinolytics in cases of hematuria should be avoided due to the risk of clots in the urinary tract, and should be used with caution in patients undergoing procedures carrying a high risk of thrombosis.6
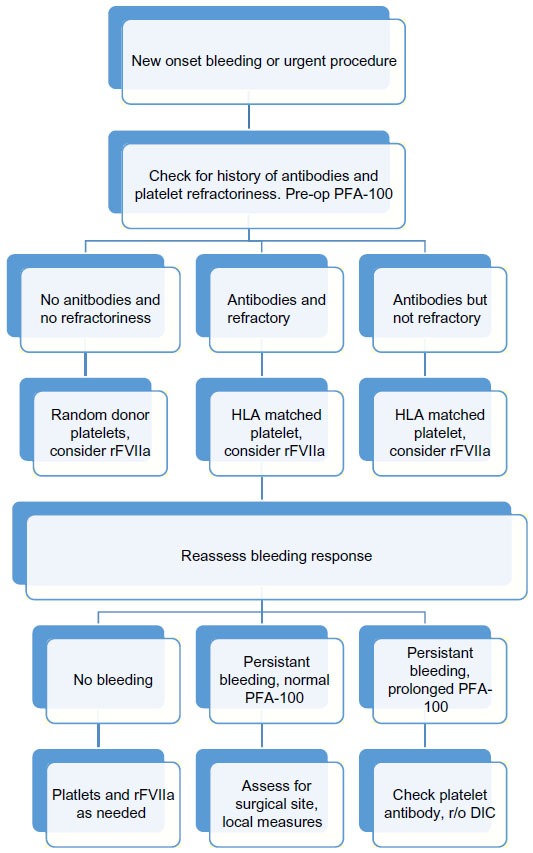 | Figure 1 Management algorithm for major bleeding episode or urgent invasive procedure. |
Management of epistaxis
Epistaxis is one of the most common bleeding manifestations in patients with GT.1,2,5 Patients may respond to compression, topical thrombin, anti-fibrinolytics, nasal packing, or a combination of these, but these methods are not always effective.12 A retrospective study comparing different methods used to control local nasal hemorrhage in children with GT found that the use of anterior and posterior packing only, without adjunctive prothrombotic agents, was at least as effective as the same procedure with the use of a prothrombotic agent. If conventional methods fail to control bleeding, further treatment with platelet transfusion and/or rFVIIa is necessary.12 Interestingly, nasal packing with salt pork strips has been used to successfully control life threatening nasal hemorrhage in a hemodynamically unstable child with GT, after anti-fibrinolytic agents, rFVIIa, nasal cautery, and nasal packing with synthetic materials had failed to control the bleeding.11
Management of menorrhagia
Anti-fibrinolytic agents are first-line therapy to control menorrhagia. If these agents fail, hormone supplementation with either progesterone alone or progesterone with estrogen should be used. A continuous regimen of an estrogen-progestin oral contraceptive agent or intramuscular depot medroxyprogesterone acetate given once every 3 months has been used successfully in women with GT. Hormonal intrauterine devices have also been used to reduce bleeding, with or without the use of an anti-fibrinolytic agent.3
Severe menorrhagia, which plagues many women with GT and can be seen with menarche, can be treated with high-dose conjugated estrogen intravenously for 24–48 hours, followed by high doses of an oral combination of estrogen–progestin. However, intensive menstrual bleeding does not always respond to typical therapy. rFVIIa has been utilized with anecdotal success in GT when anti-fibrinolytics and platelet transfusions did not control excessive menorrhagia.4 Surgical therapy such as hysterectomy or endometrial ablation are also therapeutic options for severe menorrhagia, but should be reserved for women who no longer desire pregnancy.
Management of postpartum hemorrhage
Pregnancy in patients with GT is associated with increased risk of primary and secondary postpartum hemorrhage, as well as increased risk of fetal bleeding.5 Guidelines have advocated for the use of either platelet transfusions, or rFVIIa in combination with an anti-fibrinolytic, as prophylaxis for vaginal delivery. A systematic review of management and outcomes of 35 pregnant women with GT revealed that hemorrhage during or after delivery is common and severe, and occurred up to 20 days postpartum. Patients who were not given any platelet transfusions as prophylaxis were more likely to experience postpartum hemorrhage than patients who were given platelet transfusion as prophylaxis (63% versus 38%). The use of rFVIIa as prophylaxis was documented in three pregnancies, either alone or in combination with platelets, and did not prevent hemorrhage in those cases.5 However, successful use of rFVIIa for persistent postpartum hemorrhage has been reported elsewhere in the literature.6 Maternal platelet alloantibodies were documented in 16 pregnancies, and plasma exchange successfully reduced the alloantibody titer in one case. Four of the 16 cases resulted in neonatal deaths, three of which resulted from intracranial hemorrhage between 24 and 31 weeks gestation. All women with GT should be monitored for platelet alloantibodies throughout pregnancy.5
Management with desmopressin
Desmopressin (DDAVP) causes the release of VWF, FVIII, and tissue plasminogen activator into the plasma. DDAVP has no direct effect on platelets, and samples from patients treated with DDAVP do not show a significant effect on platelet deposition on collagen ex vivo.1 While DDAVP is successful in treating other platelet disorders, there is currently little data to support its use in patients with GT.6 In one small case series, DDAVP as a single agent successfully treated one out of nine patients with GT.1
Role of transfusions in the management of GT
Platelet transfusion allows the partial correction of the functional defect in patients with GT and is considered standard therapy for patients when local measures and/or anti-fibrinolytics fail to control bleeding.4 It is also the standard prophylaxis in GT patients undergoing major surgery (Figure 2).3 It is not uncommon for patients with severe bleeding after trauma or delivery to require multiple platelet and/or RBC transfusions. Repeated platelet transfusions are associated with the formation of isoantibodies to GPIIb/IIIa isotopes and/or HLA isotopes, and it has been estimated that between 30% and 70% of patients will develop alloantibodies after platelet transfusion.2,3 A study by Santoro et al followed 17 GT patients over 30 years, 16 of whom were transfused at least once with platelets and/or RBCs. Alloantibodies developed in 25% of the patients transfused; two patients were positive for anti-HLA antibodies, one patient was positive for anti-ITG αIIbβ3, and one developed both types of alloantibodies.5 In the international survey conducted by Poon et al, a similar prevalence of anti-HLA alloantibodies was reported, and 17 out of the 23 patients who were platelet refractory in the survey had anti-platelet alloantibodies.7 Platelet alloimmunization can lead to relative or absolute platelet refractoriness, resulting in accelerated destruction of platelets and therapeutic failure of platelet transfusions. Therefore, HLA-matched platelets are recommended to avoid possible alloimmunization.5 Before the widespread use of rFVIIa, absolute refractoriness due to platelet alloantibodies was traditionally managed by plasmapheresis or IVIG, followed by platelet transfusion.2 rFVIIa is now recommended for use in patients who do not respond to platelet transfusions, with or without the presence of platelet alloantibodies.6
 | Figure 2 Management algorithm for elective surgical procedures in Glanzmann’s thrombasthenia. |
Platelet alloantibodies have been successfully removed prior to surgery by immunoadsorption on protein A Sepharose.4,5 In a series of three type I GT patients with alloantibodies, immunoadsorption on protein A Sepharose successfully cleared anti-GPIIb/IIIa antibodies on five different occasions, including episodes with life threatening hemorrhaging. However, the effect is transient; the response to platelet transfusion lasted 5 to 13 days in the series. The widespread use of the procedure is limited by the requirement of large vascular access and the need for the procedure to be performed in dedicated centers.4
rFVIIa
rFVIIa was first used successfully for severe, uncontrolled hemorrhaging in a 2-year-old boy with GT in 1996.1,2 Since then, its use has continued throughout the world, and while it successfully manages bleeding in most patients with GT, it is not effective in all patients.
NovoSeven RT is a recombinant activated human FVII commercial product that is available in North America, Europe, Australia, and Japan. It was first approved for use in the US as a hemostatic agent in patients with acquired hemophilia A and B, antibodies to factors VIII or IX, and those with congenital deficiency of FVII. In 2014, the US Food and Drug Administration (FDA) additionally approved NovoSeven RT for the treatment of bleeding episodes and perioperative management of patients with GT who are refractory to platelet transfusions, with or without the presence of anti-platelet alloantibodies.4
Mechanism of action of rFVIIa
The mechanism by which rFVIIa controls bleeding in some GT patients but not others has not been fully delineated. Through a tissue factor-independent mechanism, it is thought to weakly bind to the surface of platelets and increase activation of factor IX and X, thereby increasing thrombin generation. Doses of 90 mcg/kg and above have been shown to significantly increase thrombin generation. The increased amount of thrombin enhances platelet adhesion and promotes platelet aggregation, including those lacking GPIIb/IIIa.3 This has been demonstrated in perfusion models, in which rFVIIa increased thrombin generation and fibrin deposition, and partially restored platelet aggregation.7
Use of rFVIIa in GT
While not 100% efficacious, the high rates of success and relatively low risks associated with using rFVIIa as treatment or prevention of bleeding in patients with GT has been well documented, especially in those who are refractory to platelet transfusions or have antiplatelet antibodies.1,5,7,13 The use of HLA-matched platelets has historically been used and recommended as prophylaxis for major surgical procedures, including cesarean section. rFVIIa can be used to avoid platelet transfusion altogether, which would mitigate the risk of platelet alloimmunization, in cases of non-life threatening hemorrhage when local measures and anti-fibrinolytics have failed.3 This is of special concern in women of child-bearing age who may transfer platelet alloantibodies into the fetal circulation, causing fetal thrombocytopenia, which has been associated with intrauterine death and intracranial hemorrhage.7
The optimal dosage for use in GT patients has not been established, and currently there is no means of determining the optimal dose or dosing regimen. However, the recommended dose is bolus injections of 90 mcg/kg intravenously every 2 hours for three doses or until bleeding stops, followed by one or more maintenance doses.3,6,7 Repeat doses are necessary due to the short half-life of rFVIIa.
No randomized controlled trials examining the use of rFVIIa in patients with GT currently exist. However, the safety and efficacy of rFVIIa in GT patients has been well documented. To date, the single largest published investigation into the efficacy of rFVIIa in patients with GT was reported by Poon et al through an international survey. The survey consisted of 59 GT patients who were given rFVIIa as either treatment of hemorrhage or for surgical prophylaxis. The overall success rate was 64% in bleeding episodes, and 94% during surgical procedures. The success rate was higher in those patients who received boluses of 80 mcg/kg or higher of rFVIIa, given intravenously, no more than 2.5 hours apart. Two of the 59 patients were given a high dose of rFVIIa as a continuous infusion, in combination with an anti-fibrinolytic agent, during surgical procedures and experienced thromboembolic events: pulmonary embolism with deep venous thrombosis, and ureteric clot, respectively.7 In patients given boluses of rFVIIa, there were no adverse or thromboembolic events reported.1,2 An open-label study with 28 GT patients found that 93% of patients with bleeding episodes responded to rFVIIa.4 In addition, a total of 40 published case reports in the literature showed 69% of patients during bleeding episodes and 96% of patient undergoing surgery responded to rFVIIa treatment. The incidence of thrombotic events in GT patients treated with rFVIIa is unknown; controlled clinical trials are needed to further assess the risk.
rFVIIa should be administered as early in bleeding episodes as possible, as a UK study has shown that rFVIIa was successful in 71% of patients treated within 12 hours of onset, but only 18% of patients responded to rFVIIa after 12 hours had passed.1–3 Minor surgeries in GT patients have been successfully performed without the need for platelet transfusion when rFVIIa is administered for prophylaxis, and its use is recommended by the United Kingdom Haemophilia Centre Doctors’ Organisation for minor surgical prophylaxis including dental extractions.5
Rituximab
Rituximab (anti-CD20) is a human-mouse chimeric monoclonal antibody that targets the B cell CD20 antigen, and has been used in the treatment of acquired immune cytopenias as well as in antibody-mediated hemostatic disorders.
The use of systemic corticosteroids, chemotherapy, plasma exchange, protein A Sepharose immunoadsorption, IVIG, rFVIIa, and rituximab have all been reported as successful therapy for patients with acquired GT.4,5,8 Multiple case reports have demonstrated the efficacy of rituximab in patients with acquired GT and bleeding manifestations or ecchymoses who were otherwise refractory to therapy with systemic steroids, cyclophosphamide, azathioprine, plasmapheresis, and IVIG. Administration of rituximab at 375 mg/m2/week for 4 weeks resulted in normal platelet aggregation and cessation of symptoms in these patients.8
Bevacizumab
Bevacizumab (Avastin) is an anti-VEGF antibody that has been used in various cancers in conjunction with chemotherapy.2 The use of bevacizumab as a potential treatment option for management of severe gastrointestinal bleeding in patients with refractory GT needs to be explored further. A single case report in the literature documented success using bevacizumab in a patient with type I GT, who was experiencing severe, recurrent GI bleeding due to angiodysplasia. The patient was refractory to platelet transfusions, tranexamic acid, and embolization, but responded to bevacizumab. A 5 mg/kg infusion of bevacizumab was first given every 2 weeks as rescue therapy, followed by use every 4 weeks as maintenance therapy, and resulted in cessation of bleeding.2
Hematopoietic stem cell transplantation
Hematopoietic stem cell transplantation (HSCT) offers a cure for patients with severe, recurrent bleeding episodes, and/or platelet alloantibodies who are refractory to platelet transfusions.4–6 There currently is no clearly defined algorithm for transplantation in GT, and HSCT for GT is rare. The first successful bone marrow transplant in GT occurred in 1985 in a 4-year-old boy with anti-GPIIb/IIIa antibodies.6 To date, at least 19 reported patients with severe GT have received successful stem cell transplantations from umbilical cord blood, HLA-identical siblings, matched unrelated donors, or matched family donors.3,4 Most patients received stem cells from a sibling with busulfan and cyclophosphamide conditioning. Patients have undergone both conventional and reduced-intensity conditioning with sustained engraftment.4,5
Future therapy
Our understanding of the pathogenesis of GT and the function of αIIbβ3 has improved substantially since Glanzmann first described the disease; however, the treatment of GT remains unsatisfactory. The quality of life of patients with GT remains impaired by multiple, spontaneous mucocutaneous bleeding episodes, and the high risk of hemorrhaging with surgery or any trauma. The available therapies, aside from the costly and risky allogeneic stem cell transplantation, are focused on treating bleeding episodes, and none of them provide a cure.
Gene therapy has gained significant attention in the last decade, and the concept for it was provided in animal models.11 The use of αIIb promoter-driven murine leukemia retrovirus vectors for gene transfer of CD34+ cells to target transgene expression in developing megakaryocytes was performed by Wilcox et al.11 Successful transduction of CD34+ cells was demonstrated in vitro from two patients with GT using murine leukemia retrovirus vectors with expression of αIIbβ3 on transduced megakaryocytes. This resulted in correction of GT. In an αIIb deficient canine model for GT, Fang et al showed improvement in hemostasis and modest restoration of platelet surface αIIbβ3 by lentivirus transduction of mobilized CD34+ stem cells.14 In a recent study, Sullivan et al developed a strategy to achieve a high level of protein expression in human megakaryocytes. Hemizygous insertion of Gp1ba promotor-driven human αIIb complementary DNA into the AAVS1 locus of induced pluripotent stem cells led to a high αIIb messenger RNA and protein expression, and resulted in the correction of αIIbβ3 in megakaryocytes.5 These studies offer promise for patients with GT as curative treatment, and will help restore the quality of life that most patients lack with this disease.
Disclosure
The authors have no conflicts of interest related to this work.
References
Diz-Kucukkaya R. Inherited platelet disorders including Glanzmann thrombasthenia and Bernard-Soulier syndrome. Hematology Am Soc Hematol Educ Program. 2013;2013:268–275. | |
Stevens RF, Meyer S. Fanconi and Glanzmann: the men and their works. Br J Haematol. 2002;119(4):901–904. | |
Bennett JS. Structure and function of the platelet integrin alphaIIbbeta3. J Clin Invest. 2005;115(12):3363–3369. | |
Nurden AT, Pillois X, Wilcox DA. Glanzmann thrombasthenia: state of the art and future directions. Semin Thromb Hemost. 2013;39(6):642–655. | |
Nurden AT, Fiore M, Nurden P, Pillois X. Glanzmann thrombasthenia: a review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood. 2011;118(23):5996–6005. | |
Fiore M, Nurden AT, Nurden P, Seligsohn U. Clinical utility gene card for: Glanzmann thrombasthenia. Eur J Hum Genet. 2012;20(10). | |
Nurden AT, Ruan J, Pasquet KM, et al. A novel 196Leu to Pro substitution in the beta3 subunit of the alphaIIbbeta3 integrin in a patient with a variant form of Glanzmann thrombasthenia. Platelets. 2002;13(2):101–111. | |
Morel-Kopp MC, Melchior C, Chen P, et al. A naturally occurring point mutation in the beta3 integrin MIDAS-like domain affects differently alphavbeta3 and alphaIIIbbeta3 receptor function. Thromb Haemost. 2001;86(6):1425–1434. | |
Kashiwagi H, Kunishima S, Kiyomizu K, et al. Demonstration of novel gain-of-function mutations of alphaIIbbeta3: association with macrothrombocytopenia and glanzmann thrombasthenia-like phenotype. Mol Genet Genomic Med. 2013;1(2):77–86. | |
George JN, Caen JP, Nurden AT. Glanzmann’s thrombasthenia: the spectrum of clinical disease. Blood. 1990;75(7):1383–1395. | |
Blickstein D, Dardik R, Rosenthal E, et al. Acquired thrombasthenia due to inhibitory effect of glycoprotein IIbIIIa autoantibodies. Isr Med Assoc J. 2014;16(5):307–310. | |
Levy JM, Mayer G, Sacrez R, et al. [Glanzmann-Naegeli thrombasthenia. Study of a strongly endogamous ethnic group]. Ann Pediatr (Paris). 1971;18(2):129–137. French. | |
Cattaneo M, Cerletti C, Harrison P, et al. Recommendations for the Standardization of Light Transmission Aggregometry: A Consensus of the Working Party from the Platelet Physiology Subcommittee of SSC/ISTH. J Thromb Haemost. Epub April 10, 2013. | |
Rosas RR, Kurth MH, Sidman J. Treatment and outcomes for epistaxis in children with Glanzmann’s thrombasthenia. Laryngoscope. 2010; 120(12):2374–2377. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.