Back to Journals » Clinical Ophthalmology » Volume 14
Giant Cell Arteritis: The Experience of Two Collaborative Referral Centers and an Overview of Disease Pathogenesis and Therapeutic Advancements
Authors Dammacco R ![]() , Alessio G
, Alessio G ![]() , Giancipoli E
, Giancipoli E ![]() , Leone P
, Leone P ![]() , Cirulli A, Resta L
, Cirulli A, Resta L ![]() , Vacca A
, Vacca A ![]() , Dammacco F
, Dammacco F ![]()
Received 20 December 2019
Accepted for publication 29 January 2020
Published 11 March 2020 Volume 2020:14 Pages 775—793
DOI https://doi.org/10.2147/OPTH.S243203
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Rosanna Dammacco,1 Giovanni Alessio,1 Ermete Giancipoli,2 Patrizia Leone,3 Anna Cirulli,3 Leonardo Resta,4 Angelo Vacca,3 Franco Dammacco3
1Department of Ophthalmology and Neuroscience, University of Bari “Aldo Moro”, Medical School, Bari, Italy; 2Department of Biomedical Sciences, Ophthalmology Unit, University of Sassari, Sassari, Italy; 3Department of Biomedical Sciences and Human Oncology, University of Bari “Aldo Moro”, Medical School, Bari, Italy; 4Department of Emergency and Organ Transplantation, University of Bari “Aldo Moro”, Medical School, Bari, Italy
Correspondence: Franco Dammacco
Department of Biomedical Sciences and Human Oncology, University of Bari “Aldo Moro”, Medical School, Polyclinic, Piazza Giulio Cesare, 11, Bari 70124, Italy
Tel +39 080 5478 863
Fax +39 080 5478 820
Email [email protected]
Purpose: Giant cell arteritis (GCA), a chronic vasculitis of the large and medium-sized arteries, affects people > 50 years of age. This study assessed the prevalence of visual manifestations and other clinical features at presentation in an Italian cohort of GCA patients. Recent advances in the pathophysiology, diagnosis, and therapy of GCA are also reviewed.
Methods: This retrospective, single-center study conducted by the ophthalmology and internal medicine clinics of one university recruited 56 patients from 2005 to 2016 and followed them for 11– 54 months.
Results: Ocular involvement was diagnosed in 19 patients (33.9%), with permanent vision loss in 19.6% (7.1% of the cohort with bilateral vision loss). Arteritic anterior and posterior ischemic optic neuropathy were diagnosed in 11 patients (57.9%) and 1 patient (5.3%), respectively, cotton wool spots in 3 patients (15.8%), central retinal artery occlusion in 2 patients (10.5%), and anterior segment ischemia and multifocal choroidal ischemia in 1 patient each (5.3%). Polymyalgia rheumatica was associated with GCA in 44.6% of the patients. The most common extra-ocular manifestation was constitutional symptoms (82.1% of the patients). Large-vessel involvement, including of the ascending aorta, aortic arch, and left axillary artery, was diagnosed by magnetic resonance or computed tomography (CT) angiography and 18FDG positron emission/CT. Glucocorticoids (GCs) remain the standard-of-care worldwide, but methotrexate, provided as a steroid-sparing drug in 41% of the patients, resulted in earlier tapering, a lower cumulative dose of GCs, and a lower rate of relapse. Among the combinations of GCs and immunosuppressive drugs proposed to treat GCA, only tocilizumab has effectively induced and maintained disease remission.
Conclusion: According to our data and literature reports: a) GCA is a systemic disease; b) its diagnosis is expedited by the adjunct use of imaging techniques; c) insights into the pathogenesis of GCA may allow an improved, differentiated therapeutic approach.
Keywords: giant cell arteritis, ocular manifestations, diagnostic imaging, glucocorticoids, pathogenetic advances, tocilizumab
Introduction
Giant cell arteritis (GCA) is a medium- to large-vessel granulomatous vasculitis of typically acute onset that affects the elderly. Because irreversible visual loss is common, GCA should be considered an ophthalmic emergency requiring prompt diagnosis and immediate therapeutic measures.1–3 The inflammatory infiltrate in the arterial wall results in arterial stenosis of variable extent and in some cases even luminal occlusion, with consequent ischemic complications. Although GCA is recognized as a chronic systemic vasculitis, the superficial cranial branches of the external carotid artery, including the temporal and occipital arteries, and the ophthalmic branch of the internal carotid artery are more often involved, followed in terms of frequency by the vertebral, distal subclavian, and axillary arteries. When the thoracic aorta is involved, aneurysm formation and aortic dissection rather than occlusion may ensue.2,4,5 Nonetheless, it should be emphasized that, while GCA is principally a large-vessel vasculitis, arteries of any size may in fact be involved.6
The risk of developing ocular involvement has been studied in large cohorts of patients with biopsy-proven GCA, but the results have been largely variable. For example, visual manifestations were reported in 30.1% of 136 patients,7 in 72% of 127 patients,8 and in 13.4% in a large-scale nationwide study in Sweden.9 Visual difficulties may occur at the onset of the disease or appear at any time during its course. Permanent visual loss, a devastating consequence of GCA, has been reported in 8.2%,10 16%,11 up to 20%,12 and 32%13 of patients. The lower percentages are likely related to a better awareness of the disease and an earlier initiation of therapy. Consequently, even vague ophthalmologic symptoms should not be underestimated, as their timely and appropriate treatment may prevent potentially irreversible damage to the visual system.
Polymyalgia rheumatica (PMR), a chronic inflammatory disorder of unknown etiology, is found in 40–60% of patients with GCA and may become clinically apparent before, concomitant with, or after the diagnosis of GCA.4,5,14 Conversely, GCA is diagnosed in 16–21% of patients with PMR.15,16 Both GCA and PMR affect the elderly and the two diseases may well be closely related conditions, although the nature of their association is unclear.16,17
In a collaboration between two tertiary referral centers with specific experience in clinical immunology and ophthalmology respectively, the aims of our study were: 1) to determine the prevalence and major characteristics of the ocular involvement in a cohort of Italian GCA patients; 2) to underscore the changing concepts related to the nature of GCA and its pathogenesis; and 3) to assess recent therapeutic advancements.
Materials and Methods
This is a retrospective, observational, cohort study carried out on a series of 56 patients diagnosed with GCA from 2005 to 2016 and examined in a collaboration between the Ophthalmology Section, Department of Ophthalmology and Neuroscience, and the Internal Medicine Section, Department of Biomedical Sciences and Human Oncology, of the University of Bari, Italy. All procedures were carried out in accordance with the ethical standards of the University of Bari Medical School (that approved the study) and conformed to the tenets of the 1964 Helsinki Declaration and its later amendments. Given that this is a retrospective case record review, patients’ written informed consent to study enrollment was waived by the Ethics Committee of the hospital. In addition, all patient data was deidentified and patients in the figures provided written informed consent for the images to be published.
All patients underwent a thorough ophthalmologic examination, including visual acuity, pupillary reaction, ocular motility, visual field testing, external inspection, and direct ophthalmoscopy with fluorescein staining. Additional tests were performed as requested, both at the time of diagnosis and at variable intervals during follow-up, which ranged from 11 to 54 months.
GCA was diagnosed according to the American College of Rheumatology (ACR) classification criteria, which require the detection of three or more of the following items: 1) age ≥ 50 years; 2) headache of new onset or a new type of localized pain in the head; 3) temporal artery abnormalities, including tenderness to palpation and decreased pulsation; 4) an erythrocyte sedimentation rate (ESR) ≥ 50 mm/h; and 5) arterial biopsy showing a predominance of mononuclear cell infiltration or granulomatous inflammation of the vessel wall, usually but not necessarily accompanied by multinucleated giant cells. The reported sensitivity and specificity of the ACR criteria for the classification of GCA are 93.5% and 91.2%, respectively.18
The diagnosis of PMR in our patients was based on the following scoring algorithm developed by the European League Against Rheumatism (EULAR)/ACR classification criteria.19 Patients ≥ 50 years of age with bilateral shoulder pain and an abnormal C-reactive protein (CRP) concentration or ESR, plus at least four (without ultrasonography) or five (with ultrasonography) of the following: 1) morning stiffness for > 45 min (2 points); 2) hip pain or restricted range of hip motion (1 point); 3) absence of rheumatoid factor or anti-citrullinated protein antibodies (2 points); 4) absence of other joint involvement (1 point); 5) if ultrasonography is available, at least one shoulder with subdeltoid bursitis, biceps tenosynovitis, or gleno-humeral synovitis (either posterior or axillary) and at least one hip with synovitis or trochanteric bursitis (1 point); 6) if ultrasonography is available, both shoulders with subdeltoid bursitis, biceps tenosynovitis, or gleno-humeral synovitis (1 point). A score ≥4 was shown to have a 68% sensitivity and 78% specificity for discriminating PMR.19
Results
Demographic Features
In our cohort of 56 patients, 39 were females (69.6%) and 17 were males (30.3%). The mean age at disease onset was 69.3 ± 7.2 (mean ± SD) years. Occupational and socio-economic factors do not seem to be risk factors for GCA.20
Twenty-five of our 56 patients (44.6%) had PMR that was diagnosed at the same time as GCA, with the exception of two patients in whom PMR was recognized 3 and 5 months after the diagnosis of GCA. In none of our patients did the diagnosis of PMR precede that of GCA. A combined occurrence of GCA and PMR was more common in females (16/25: 64%).
Baseline Clinical Features
Visual manifestations, which are the main object of the present study, are described in detail below, whereas the main clinical features of our GCA patients at the time of diagnosis are summarized, for the sake of brevity, in Figure 1 and Table 1. Fever, defined as a body temperature ≥ 38°C, was recorded in 41%, and constitutional symptoms including weakness, fatigue, and anorexia, were described by 82%, followed in terms of decreasing frequency by tenderness to palpation of the temporal artery (77%), headache of new onset localized in the temporal region or more rarely in the occipital area (75%), jaw claudication (55%), tongue pain during mastication (23%), and scalp tenderness (28%) that resulted in large eschars in one male patient (Figure 2). However, 3 of the 56 patients had either no or only vague general symptoms and were diagnosed with occult GCA21 based on an ophthalmologic examination, performed to determine the cause of visual disturbances, and a positive temporal artery biopsy (TAB).
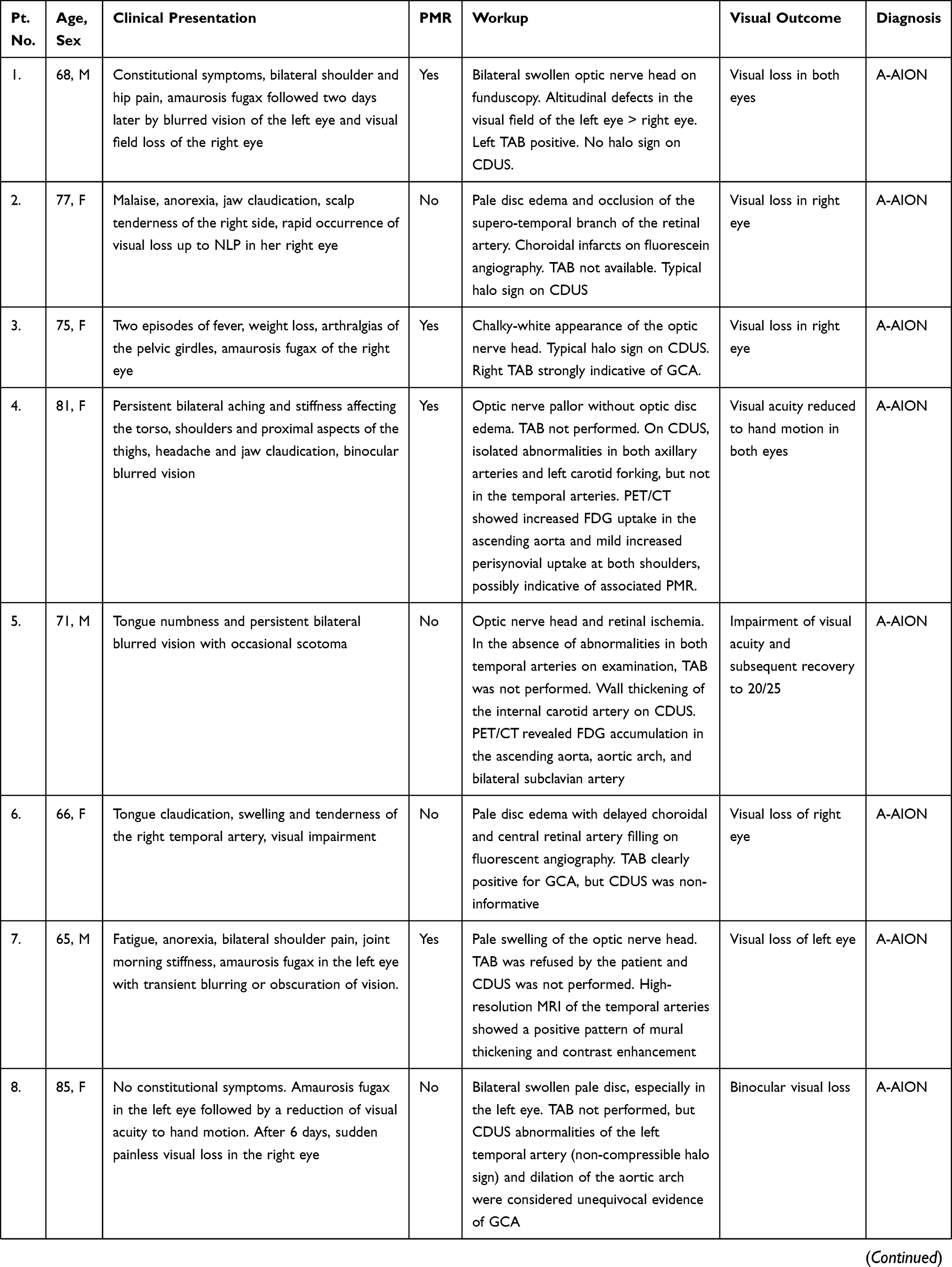 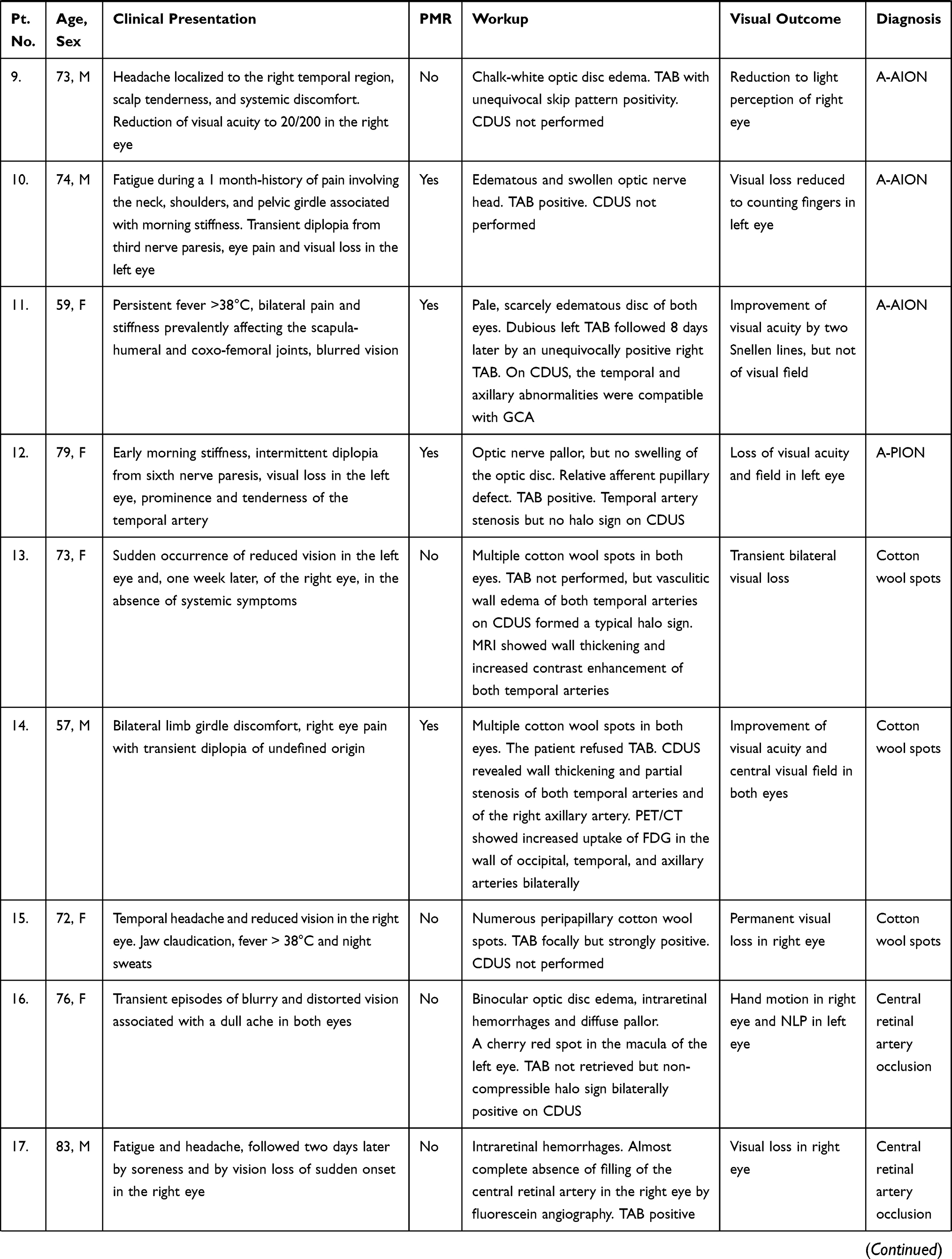 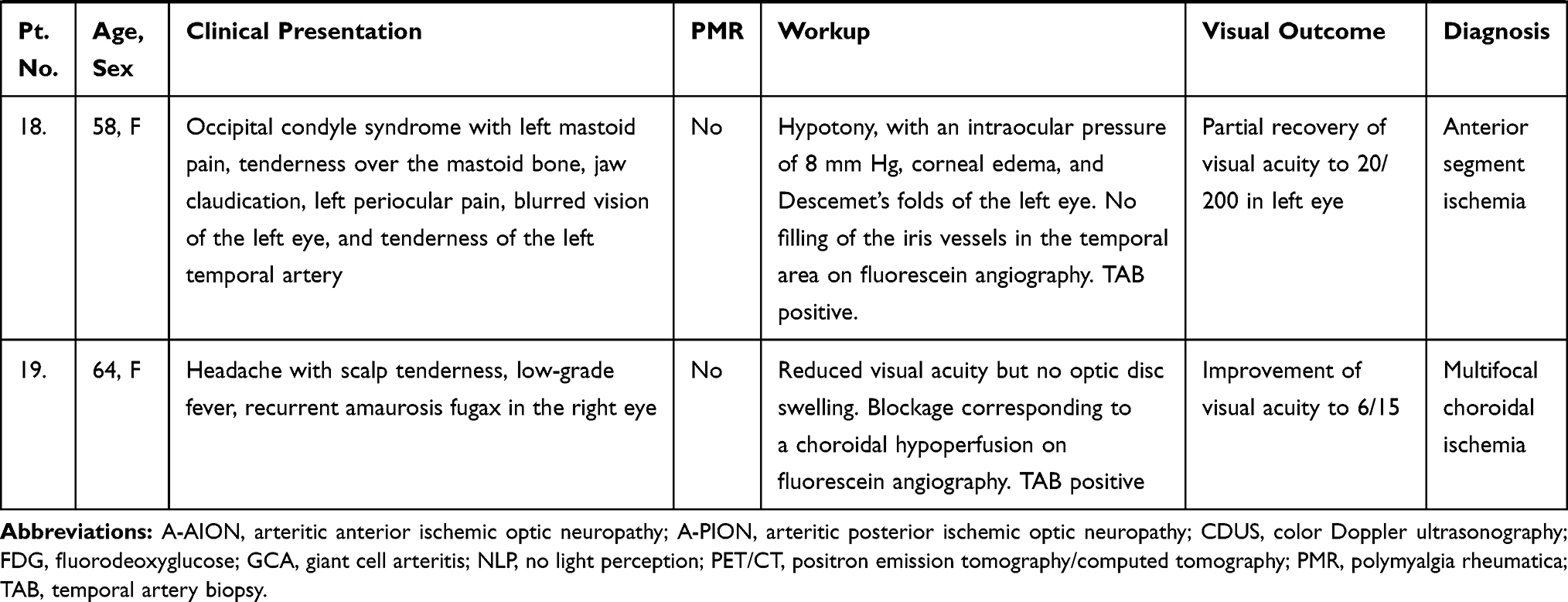 |
Table 1 Summary of the Main Characteristics in 19 Patients with Ocular Manifestations from Giant Cell Arteritis |
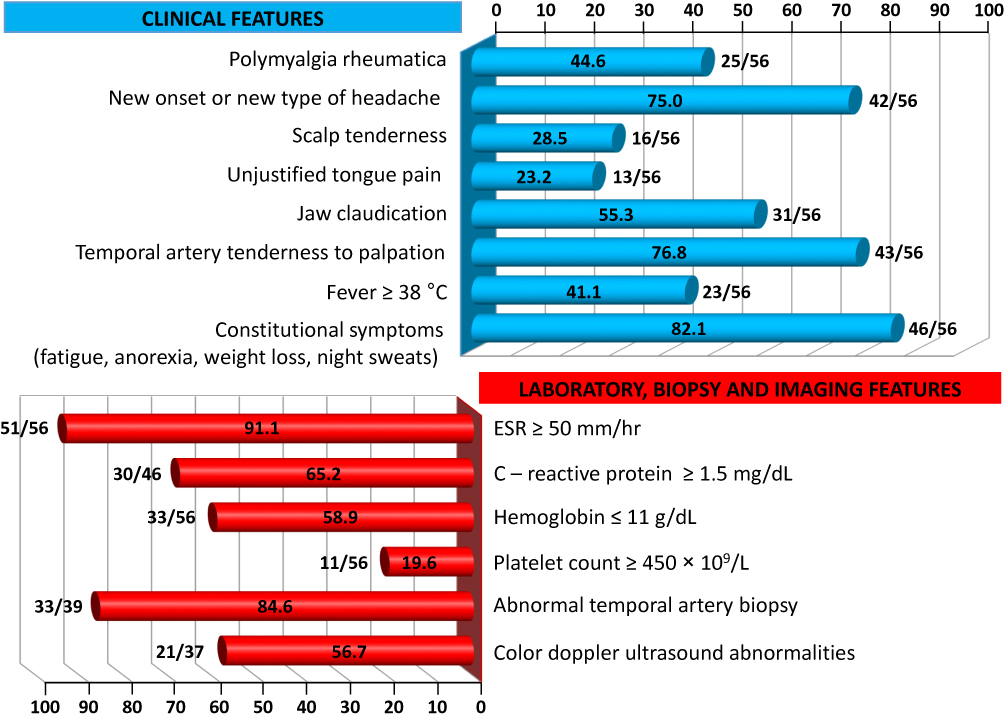 |
Figure 1 Frequency of clinical features and abnormalities of laboratory data, temporal artery biopsy findings and color Doppler ultrasound imaging in our cohort of GCA patients. |
 |
Figure 2 Giant cell arteritis with subatrophy of the right eyeball as a consequence of the ophthalmic artery occlusion (panels A and B). In addition to constitutional symptoms, the patient complained of persistent headaches and scalp tenderness. Three weeks later, one large and one smaller eschars appeared on the scalp that eventually resulted in cicatricial areas of alopecia (panels C and D). |
PMR was characterized by morning stiffness exceeding 45 mins in all but two patients (92%). Both shoulder and pelvic girdle pain, mostly bilateral, were found in 15 patients (60%) and aching in the neck, torso, shoulders and upper arms was largely prevalent in 6 patients (24%). Hip and pelvic girdle pain, always bilateral and conditioning a restricted range of motion, was the major complaint in the remaining 4 patients (16%). In addition, a variable combination of fatigue, weight loss and depression was recorded in 8 patients (32%).
Laboratory Data
Laboratory data at the time of diagnosis are also summarized in Figure 1. An ESR ≥50 mm/h and a CRP level ≥1.5 mg/dL were recorded in 91% and 65% of our patients, respectively. In addition, anemia with hemoglobin levels ≤ 11 g/dL was detected in 59%, whereas a platelet count ≥ 450 × 109/L was an unusual finding, occurring in ~20%. Of note, seven patients with combined anemia and thrombocytosis experienced a relapse of ocular symptoms 2–5 months after achieving disease remission and while still receiving tapered doses of glucocorticoids (GCs).
Color Doppler Ultrasonography (CDUS)
Only 37 of our patients underwent CDUS, with abnormal findings detected in 21 of them (57%).Typical CDUS findings in GCA patients are a dark, hypoechoic concentric ring (halo sign) related to edematous wall thickening (Figure 3A and B) and areas of occlusion and/or stenosis (Figure 3C and D) that differ from the hyperechoic wall thickening that characterizes atherosclerosis. The reported sensitivity of the halo sign for TAB-positive GCA is 75% and the specificity 83%.22 The “compression sign”, a variant of the “halo” sign with higher sensitivity and specificity, indicates the persistence of a hypoechoic swelling despite the compression of the artery lumen with the ultrasound probe. Among the non-invasive imaging techniques used to diagnose GCA, CDUS has gained acceptance worldwide based on its ability to detect involvement of the temporal artery and supra-aortic branches. Although CDUS is equipment- and operator-dependent and does not readily allow visualization of the thoracic aorta, the procedure has several practical advantages, including easy repeatability, good resolution, and suitable sensitivity for the detection of cranial and extracranial disease.2 Compared with TAB, CDUS has better sensitivity but poorer specificity.23
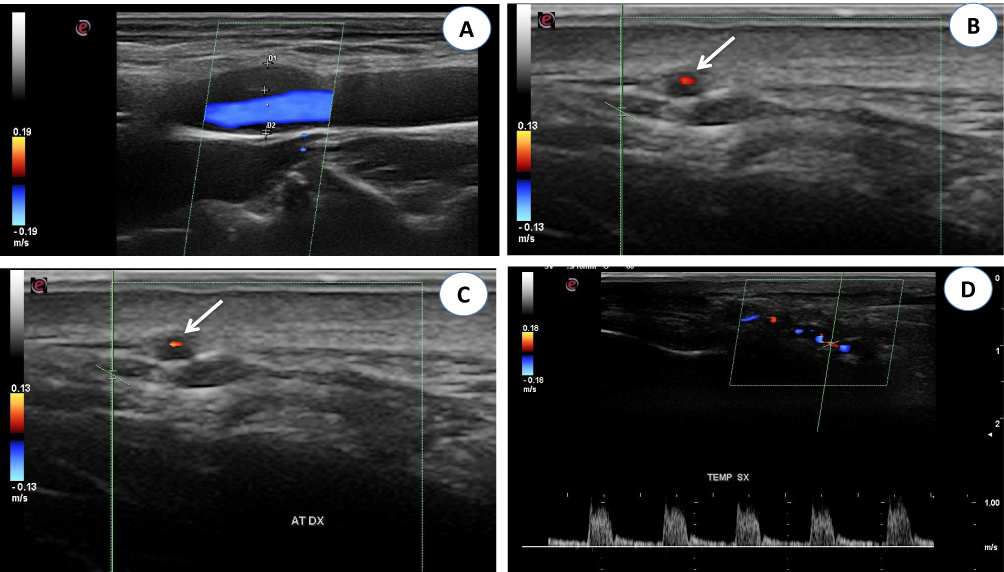 |
Figure 3 (A) Color Doppler ultrasonography (CDUS): longitudinal scan reveals a hypoechoic plaque in the proximal internal carotid artery. (B) CDUS of the right temporal artery shows a hypoechoic halo around the lumen in transverse view (arrow). (C) CDUS of the right temporal artery shows a hypoechoic halo around the lumen in transverse view (arrow). The halo sign corresponds to edema of the artery wall. (D) Longitudinal view of the right temporal artery by CDUS shows a hypoechoic halo of the temporal artery and the presence of turbulent and weak flow, suggesting the presence of stenosis. The peak systolic velocity is 1 m/s, that is double compared to the segment without stenosis. |
The optimal time to perform CDUS remains a matter of controversy related to the ability of GCs to induce the disappearance of the halo sign, which ranges from 2 days to 2 months.2 Nonetheless, CDUS should be performed as soon as possible in patients in whom a diagnosis of GCA is suspected.
Temporal Artery Biopsy
TAB, that has long been considered the gold standard in the diagnosis of GCA and is invariably unilateral, was performed in 39 of our 56 patients (70%), with abnormal findings determined in 33 of them (85%) (Figure 4). The histopathologic features were highly variable, both between patients and within the same patient, as the inflammatory infiltrate affects some arterial segments while sparing others.3 The usual findings consisted of inflammatory infiltrates with lymphocytes, macrophages, and giant cells affecting the three layers of the arterial wall (Figure 5). A variable number of giant cells forming granulomas were seen, mostly at the intima-media junction. Frequently, the elastic lamina was fragmented and the vascular smooth muscle cells (VSMCs) were destroyed.4
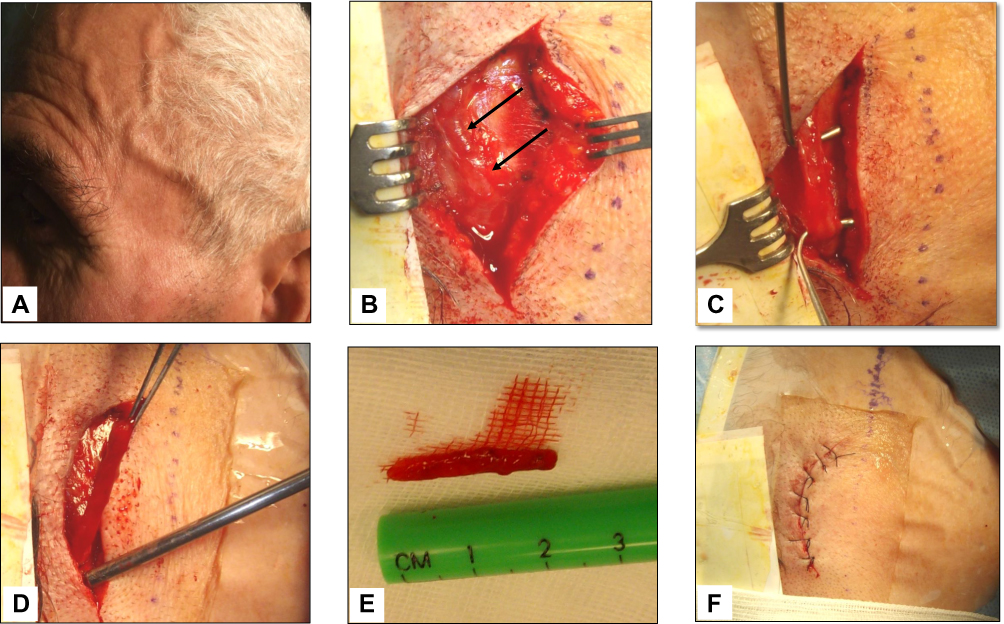 |
Figure 4 Giant cell arteritis of the left temporal artery. Panel A shows a prominent, tender and beaded artery, that was also hypo-pulsating on palpation. Panels B to F illustrate the various phases of the temporal artery biopsy. A 2 cm-long segment of the frontal branch of the superficial temporal artery was surgically removed. |
 |
Figure 5 (A) Histological findings in the wall of a giant cell arteritis. Wall thickening is mainly due to myofibroblastic proliferation of the intima, with a minimal exudate consisting of lymphocytes and plasma cells. The media is distorted by the granulomatous process, which extends into the adventitia. In the latter, a minimal fibrosis is present. (B) The media of the artery shows a layer of giant cells on its inner aspect, the site of the inner elastic membrane, and small fragments of elastic fibers in the cytoplasm of giant cells (arrows). Numerous other inflammatory cells are seen in the intima wall. The muscular fibers are degenerate, as evidenced by the cytoplasmic vacuoles. |
In the patients with a negative or no biopsy, the diagnosis of GCA was definitely supported by the occurrence of the other 3 or 4 items established by the ACR classification criteria as well as the results of imaging studies including ultrasonography, high-resolution magnetic resonance imaging (MRI) and 18F-fluorodeoxyglucose positron emission tomography/computed tomography (FDG-PET/CT).2,3,5
A question of practical interest is the maximum recommended delay for performing a TAB in patients started on GC therapy. Based on the findings of a small prospective study and several retrospective cohort studies, TAB should be performed within 4 weeks of GC initiation.24 The British Society for Rheumatology supports a more stringent approach and recommends that TAB be performed within 1 week of GC administration, although positivity may persist for 2–6 weeks.25 However, to prevent irreversible vision loss, in no case should TAB delay the initiation of GCs.
Visual Manifestations
Ocular involvement was diagnosed in 19 of our 56 GCA patients (34%), and in 11 of them (20%) it resulted in permanent visual loss, which was bilateral in four patients (7% of the whole cohort and 36% of those with permanent visual impairment). The interval between the involvement of the first and second eye was 6 days in two patients, and 7 and 10 days in the remaining two patients, respectively. The ocular manifestations observed in our patients are summarized in Figure 6 and the clinical presentation, PMR association, diagnostic workup, and visual outcome of these 19 patients are reported in Table 1.
 |
Figure 6 Distribution of ocular manifestations in 19 GCA patients of our cohort. The numbers between brackets indicate the relative percentages. |
Overall, the severity of visual outcome reflected both the impairment of central visual acuity and the loss of visual field. Visual acuity was affected to a variable extent in 16 of 19 patients (84%) within the first 7 to 10 days following the beginning of GC administration, whereas an improvement in visual acuity of two to five Snellen lines was recorded in 6 patients (31%) within the first 30 to 40 days from the start of therapy. However, this improvement was associated to a corresponding improvement in the central visual field in only two of them.
Arteritic Anterior Ischemic Optic Neuropathy (A-AION)
Consistent with the data reported in the literature,1,4,10,26 A-AION was the most frequent ocular manifestation in our cohort of patients (Figure 6). A-AION is the product of inflammation, thrombosis, and the consequent ischemia of the short posterior ciliary arteries (PCAs) originating from the ophthalmic artery. Most people have two PCAs, whereas some have only one and others as many as five.27 Fluorescein fundus angiography studies have shown that the medial PCA is more frequently occluded, which results in infarction of the optic nerve head.
As reported for the majority of the patients described in Table 1, in addition to the systemic symptoms typical of GCA but absent in patients with occult GCA, ocular manifestations include a rapidly developing, mostly unilateral loss of vision associated with severely reduced visual acuity and/or altitudinal visual field defects. Ophthalmoscopy reveals (Figure 7A and B) a pale, edematous, chalky-white optic disc that less commonly may show hyperemic swelling, an ophthalmoscopic picture quite similar to that of patients with non-A-AION. Flame hemorrhages adjacent to the disc and narrowing of the peripapillary retinal arterioles may also be seen. In less ominous situations, only a segment of the optic disc is affected, but this occurs in < 25–30% of patients.1
 |
Figure 7 Scanning laser ophthalmoscopy and fluorangiography patterns in a GCA patient with acute ischemia of the papilla. Pale colored edema and light swelling are evident on the optic disc (panels A and B). Four months later the pattern changed to optic nerve atrophy (panel C). |
Optical coherence tomography (OCT) angiography may help to identify microvascular defects and vessel density reduction in patients with acute optic disc edema, thus providing useful clues in assessing ischemia in the optic nerve head.28 In addition, three-dimensional high-resolution black-blood MRI enables a more reliable and earlier diagnosis of PCA involvement in these patients than achieved with funduscopy. This capability is particularly useful in the detection of “vision-at-risk” in patients with suspected GCA in whom the funduscopy findings are unremarkable.29
The obvious outcome of A-AION is the onset of optic atrophy (Figure 7C), a condition that requires 6–8 weeks to become fully established and may affect the whole disc or manifest segmentally. When optic atrophy simulates glaucomatous optic nerve cupping, OCT imaging may help differentiate the optic disc changes of GCA from those of glaucoma, as in the former the nasal and temporal retinal nerve fiber layers are thinner and macular thickness and volume are reduced.30
Arteritic Posterior Ischemic Optic Neuropathy (A-PION)
This insidious presentation of GCA accounts for ≤ 10% of all IONs and is to a large extent a diagnosis of exclusion. A-PION was diagnosed in only one of our patients, corresponding to 5% of our GCA patients with ocular involvement. As a consequence of the ischemia affecting the retrobulbar portion of the optic nerve, the patient complained of acute and painless reduction in visual acuity and color vision as well as visual field defects in the left eye associated with a relative afferent pupillary defect. No abnormalities of the anterior segment and intraocular pressure (IOP) were detected, and the optic nerve head was funduscopically normal. Although A-PION more frequently involves the PCAs, ischemia of other orbital arteries may also be responsible for its occurrence.27 Two months later, optic nerve atrophy with optic rim pallor was clearly evident.
These features allow A-PION-related post-ischemic optic disc cupping to be distinguished from glaucomatous cupping.31 Although diffusion-weighted MRI may aid in the diagnosis of acute-phase PION in patients with normal funduscopy findings,1,32 this procedure has not gained wide acceptance.
Cotton Wool Spots
Three of our GCA patients with ocular manifestations developed cotton wool spots (16%) that affected both eyes in two patients and were confined to the right eye in the third patient. Similar findings have been reported to occur in almost one-third of GCA patients suffering visual loss in the early phase of the disease.21 The spots are due to localized accumulations of axoplasmic debris as a result of focal inner retinal ischemia. Platelet microembolization and/or hypoperfusion of the terminal portions of the retinal vasculature are thought to play a role in their pathogenesis. Cotton wool spots may be an isolated finding or associated with other ocular features, such as optic disc edema. It should be emphasized that they can also be detected in patients with arterial hypertension, rheumatologic disease, HIV infection, and diabetic retinopathy.33
Central Retinal Artery Occlusion (CRAO)
Two of our patients with ophthalmic GCA were diagnosed with CRAO. In both, the PCAs were affected, a finding that can be explained by the fact that in the majority of people the central retinal artery and one or more of the PCAs derive from a common trunk of the ophthalmic artery.21 Vision loss of sudden onset was bilateral in one patients and restricted to the right eye in the other. The fundus examination showed diffuse intraretinal hemorrhages, a pallid retina, and optic disc edema of the affected eyes. A cherry red spot against a pale retina was also observed in the left eye of the first patient. As expected, the fluorescein column of the central retinal artery was almost completely absent by fluorescein angiography in the right eye of the second patient. Spectral domain OCT was not performed.
Anterior Segment Ischemia
Only one of our patients with ophthalmic GCA was diagnosed with anterior segment ischemia, that is a rare occurrence in larger cohorts as well.34,35 It is more commonly diagnosed in patients with general ocular ischemic syndrome with involvement of the anterior ciliary arteries and sometimes of the long branches of the PCAs.1 Corneal edema, anterior uveitis, and Descemet’s folds followed by the development of striate keratopathy were found in the initial phase, whereas an IOP of 12 mm Hg and rubeosis iridis were detected later.
Choroidal Ischemia
Here again, only one of our patients with ophthalmic GCA was diagnosed with choroidal ischemia, whose clinical signs appeared later in the course of the disease. Fundus examination of both eyes revealed pigment epithelial changes in the macular area and reduction of the choroidal vascular pattern, with choroidal filling defects. On fundus fluorescein angiography, the foveal avascular zone was enlarged, with delayed filling. No electrodiagnostic testing was performed to identify the site of ischemia.1
Pathogenesis of GCA
Although the patho-etiology of GCA remains poorly known and is possibly related to the interaction of genetic and environmental risk factors,1 recent developments in the pathogenesis of GCA have been the object of excellent reviews5,36,37 and will be summarized here, in that they are paving the way for targeted and more effective therapeutic measures.
The current hypothesis on the pathogenesis of GCA invokes interactions occurring between the innate and adaptive immune systems and the different compartments of the vessel wall, including endothelial cells (ECs) and VSMCs, such that vascular inflammation, remodeling, and occlusion may ensue. As depicted in Figure 8, unknown “danger signals” (bacterial or viral products?) are thought to trigger the maturation and activation of dendritic cells (DCs) resident in the adventitia of the artery via toll-like receptors (TLR).36 Two distinct cytokine networks are then thought to be involved in disease development: 1) In the first, mature DCs produce chemokines (eg, CCL19 and CCL21) that attract and retain additional DCs. 2) In the second, T cells (mostly CD4+ T cells) express the co-stimulatory molecules CD83 and CD86 that are responsible for T cell activation, resulting in the release of cytokines such as interleukin (IL)-1 β, IL-6, IL-23 and IL-21 or IL-12 and IL-18.
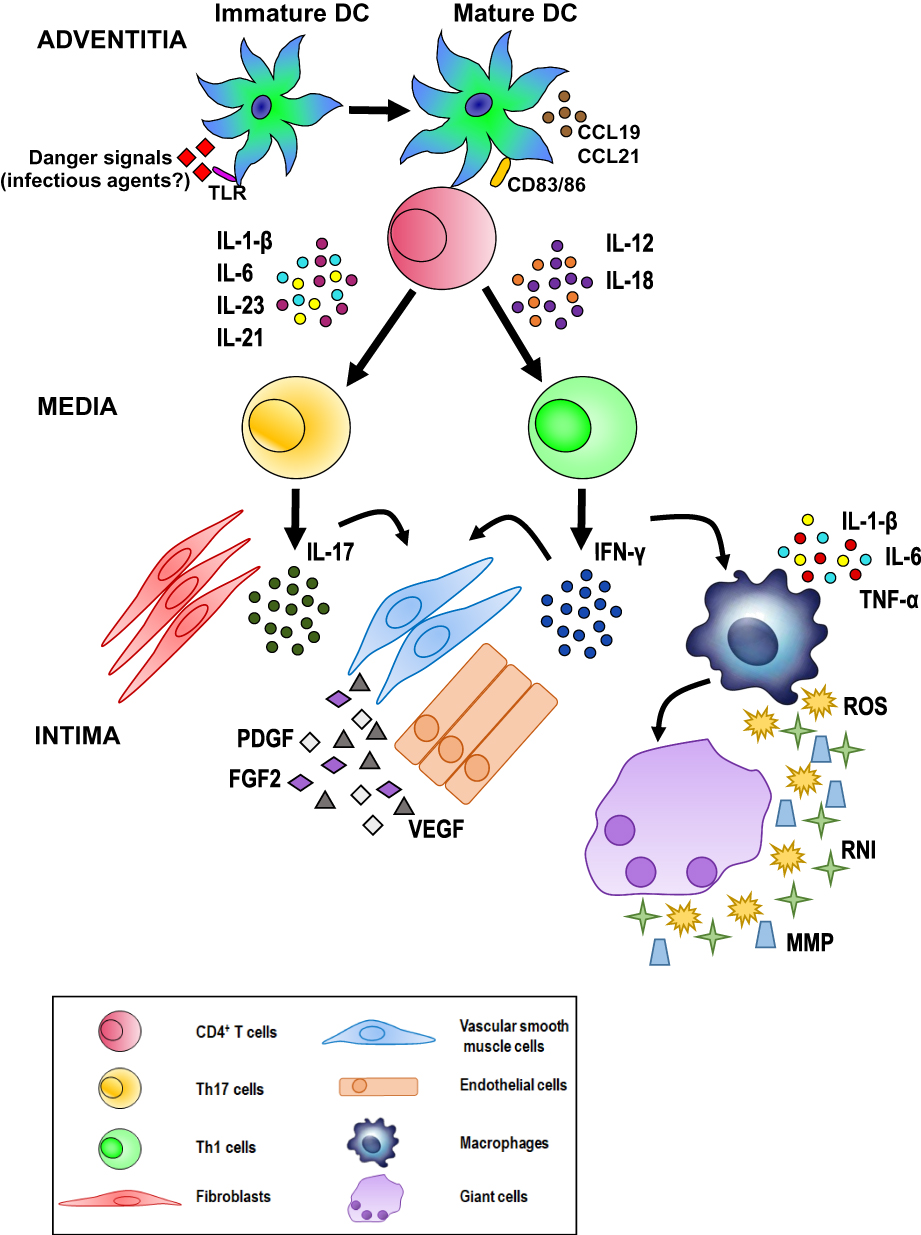 |
Figure 8 Possible pathogenetic algorithm of giant cell arteritis (GCA). After activation by danger signals, dendritic cells resident in the arterial adventitia mature, produce chemokines such as CCL19 and CCL21, and express the co-stimulatory molecules CD83 and CD86 required for their interaction with CD4+ T cells. Dendritic cells also release cytokines, such as IL-1 β, IL-6, IL-23 and IL-21 or IL-12 and IL-18 that trigger two distinct networks. The first network induces the differentiation of activated T cells into Th17 cells; the second drives Th1 cell formation. Both T cell lineages participate in the evolving granulomatous inflammation. See text for details. |
The clonal expansion and differentiation of T cells into Th17 cells is mediated by the first group (IL-1 β, IL-6, IL-23) while the second group (IL-12 and IL-18) induces Th1 cell formation.37 Th17 cells secrete IL-17, which modulates the function of ECs, VSMCs, fibroblasts, and bone marrow stromal cells. Th1 cells release interferon-γ (IFN-γ), which induces the activation of macrophages, ECs, VSMCs, and cytotoxic cells. Activated macrophages release pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), IL-1 β, and IL-6, thus amplifying the inflammatory response. Macrophage fusion results in the formation of giant cells, which contribute to the development of granulomas at the intima-media junction.
Giant cells and macrophages release reactive oxygen species (ROS), reactive nitrogen intermediates (RNI), and matrix metalloproteinases (MMP) to promote tissue injury and intimal hyperplasia.38,39 In addition, activated macrophages and injured VSMCs release platelet-derived growth factors (PDGF), vascular endothelial growth factor (VEGF), and fibroblast growth factor-2 (FGF2), which together trigger a vascular remodeling process that includes the proliferation and migration of medial myofibroblasts, the induction of neoangiogenesis, and the deposition of extracellular matrix proteins, resulting in luminal stenosis.40 Under the influence of the cytokines and growth factors produced by activated macrophages, ECs participate in the vascular remodeling process sustaining local inflammation, neoangiogenesis, and T cell recruitment.38–40
Treatment
Based on current understanding of disease pathogenesis, schematically summarized in Figure 8, two components play an important role in the onset of GCA: 1) a systemic extravascular inflammatory syndrome mainly sustained by the IL-6/IL-17 axis; 2) vascular damage with breaching of the artery wall, followed by ischemia largely conditioned by the IL-12/IFN-γ axis.3,4 These mechanisms have remarkable therapeutic implications, as the extravascular component is promptly responsive to GCs whereas the vascular component responds poorly or not at all, thus accounting for potential relapses characterized by visual loss and inflammatory lesions in patients treated with GCs for months, and especially during drug tapering.3
The worldwide standard-of-care for patients with GCA and/or PMR is GCs, which were in fact used to treat all of the patients in our study. However, since the cohort was collected over a time frame of 12 years, the initial optimal dose was not the same for all patients, including those with ophthalmic manifestations. No patient was given intravenous pulse methylprednisolone, whose better efficacy compared to oral therapy has not been unequivocally demonstrated.41 However, in patients with acute visual loss in one eye and signs of ischemia in the other, neuro-ophthalmologists usually start treatment with a 3-day course of intravenous pulse methylprednisolone (500–1000 mg daily!) that is then switched to 100–120 mg/day oral prednisone.4
In our experience, 15 patients with clinical and laboratory features of highly active disease were treated with 1 mg GCs/kg/day, whereas in 27 patients the fixed starting dose of GCs was 50 mg/day. The 11 patients in whom the diagnosis was initially uncertain were treated with 0.5 mg GCs/kg/day, but the dose was increased to 1 mg/kg/day 2–3 weeks later, when their TAB results confirmed the diagnosis. Finally, three patients with combined GCA and PMR but without ocular involvement were initially incorrectly diagnosed with rheumatoid arthritis, for which they were treated with 25 mg methotrexate (MTX)/week plus 200 mg hydroxychloroquine/day for 3 weeks. When these patients were determined to be TAB-positive, the hydroxychloroquine was discontinued and 50 mg GCs/day was added to the MTX regimen.
Overall, in our patients GC therapy was started from 5 days to 3 weeks after the appearance of clinical symptoms. In 11 patients, not even a partial recovery of visual acuity was achieved. These observations highlight the urgency of early treatment to prevent the involvement of the contralateral eye and possibly to impede further worsening of visual acuity in the affected eye. However, GCs are of poor (if any) efficacy in appreciably improving the vision of patients who have already developed ophthalmic GCA, especially in terms of visual field recovery.4
GCs were slowly tapered on an individual basis, taking into account the mitigation of signs and symptoms and the reduction or normalization of laboratory features, including ESR, the CRP level, and the platelet count. The length of GC administration ranged from 18 to 36 months. Nonetheless, during GC tapering 9 out of 56 patients suffered disease relapse (16%), including 4 out of 19 with ophthalmic GCA (21%). In addition, four patients had disease recurrence (none in patients with ophthalmic GCA) 3–6 months following GC withdrawal. Patients with relapse or recurrence were retreated with medium to high doses of GCs that were gradually tapered. Thereafter, these patients were placed on a maintenance regime of indefinite low-dose GCs.
As expected given the length of GCs administration and because GCA is a disease of the elderly, these drugs are frequently responsible for the occurrence of adverse events.4,42,43 One or more treatment-associated events were in fact diagnosed in 18 of our patients (32%), including diabetes mellitus (5 patients), posterior subcapsular cataract (6 patients), arterial hypertension (7 patients), infection (4 patients), bleeding ulcer (1 patient), and femoral neck fracture (1 patient). Two patients died 11 and 19 months after starting steroid therapy: the first (a 79-year-old woman with hypertension) as a consequence of stroke and the second (a 77-year-old man with renal failure) due to Pneumocystis jirovecii pneumonia.
As mentioned above, three patients with combined GCA and PMR were initially treated with MTX plus hydroxychloroquine, based on incorrectly diagnosed rheumatoid arthritis, but 3 weeks later received MTX plus GC. MTX (15–25 mg/week) was administered together with GCs from the very beginning, as a steroid-sparing drug, in 23 patients (41%) of our cohort. These patients, diagnosed from 2011 onward, included 10 with ophthalmic GCA. All 23 patients underwent an earlier tapering of GCs, resulting in a lower cumulative dose of GCs at 18 months. Only two of the patients had a relapse (after 11 and 14 months of treatment, respectively), thus confirming that adjunctive MTX treatment lowers the risk of relapse and is a steroid-sparing agent.44–46
The last patient, enrolled in the study in October 2016 (patient number 19 of Table 1), was initially treated with the combination of GCs plus MTX, but the latter drug was discontinued because of unacceptable liver toxicity. However, after the occurrence of two relapses separated by a period of 3 months under GCs alone, tocilizumab (TCZ), an anti–IL-6 receptor humanized monoclonal antibody, was added to the drug regimen. At the time, TCZ was allowed in Italy only for the treatment of patients with refractory and persistently active rheumatoid arthritis. After HBV and HCV infection had been excluded and both a quantiFERON-TB-Plus test and a chest X-ray had ruled out tuberculosis, the patient was given four intravenous infusions of TCZ every 4 weeks at a standard dose of 8 mg/kg.47,48 After TCZ discontinuation, the patient experienced no disease flares during a follow-up of 11-months. Her abnormal laboratory features slowly subsided and her visual acuity improved considerably.
Discussion
The extensive medical literature and our own experience strongly indicate that GCA, which typically affects the medium and large arteries of people > 50 years of age, remains the most common type of granulomatous vasculitis. In addition, the more frequent adoption of imaging studies have clearly shown that the disease, rather than simply being a “temporal arteritis” (a misleading misnomer in use for many years), is in fact a systemic disease in which the large vessels, such as the aorta and its major branches, are progressively affected in step with repeated recurrences.1,4,49 Visual manifestations of lesser or greater severity are among the presenting symptoms or appear shortly after the diagnosis of GCA in approximately one-third of patients.4 Permanent visual loss, although remarkably reduced in the last 10–15 years as a consequence of a heightened medical awareness of the disease and prompt treatment, still affects up to 16% of GCA patients.11
The remarkable clinical heterogeneity of GCA should be emphasized, ranging from a silent variant, in which the typical cranial manifestations are absent and the systemic symptoms more prominent, to the so-called occult variant, in which systemic signs and symptoms are absent and the diagnosis is accordingly delayed. Occult GCA is diagnosed in approximately 20% of patients with ocular involvement and biopsy-confirmed GCA.1,21,50,51 In these patients, premonitory signs such as amaurosis fugax and visual loss should raise strong diagnostic suspicion of GCA. The ophthalmologist should then immediately start the patient on GCs, before or at the very early stage of the development of acute ocular ischemic lesions.21
Before GCs became the standard of care, 30–60% of patients with GCA experienced vision loss (according to Hoffman et al).52 Following the introduction of GCs therapy, the incidence of visual manifestations decreased, with reported rates of 29% of 239 patients53 and 16% of 200 patients.54 In a nationwide study in Sweden based on 12,048 patients with GCA (3737 males and 8311 females) evaluated between 2002 and 2010, a total number of 1,618 patients (13.4%) developed ocular manifestations.9 Thus, despite an earlier diagnosis and the prompt administration of GCs, permanent visual loss of variable degree still affects around 15% of patients4,50 and in our cohort was diagnosed in 20%.
It has been submitted that the clinical features and pathogenesis of GCA resemble those of Takayasu arteritis, another granulomatous large-vessel panarteritis that is sometimes considered in the differential diagnosis.16,17,39 However, Takayasu arteritis preferentially affects young women during their reproductive years and its leading presenting symptoms are claudication in one or both arms, malaise, fever and weight loss, a difference in blood pressure between arms, dizziness and headache, renal artery hypertension, and visual impairment.16,55 A disorder more closely related to GCA is PMR, which as noted in the Introduction occurs in 40–60% of patients with GCA.4,5,14 PMR was diagnosed in 45% of our cohort. In the two diseases, the onset occurs in older individuals, and similar genetic risk factors and pathogenic pathways have been identified. In fact, PMR may be an early subclinical, albeit predominantly peripheral, vasculitis.5,16,56 The similarities between GCA, PMR, and Takayasu arteritis have led to the suggestion that they are components of a spectrum of the same underlying disease.16,17,57
Histologic analysis with TAB still remains the gold-standard diagnostic procedure in many rheumatologic centers. However, although it will be positive in the large majority of these patients, it is an invasive examination and a negative biopsy result does not exclude a diagnosis of GCA, given that the inflammatory process may spare some arteries or segments thereof. In addition, the TAB may sometimes be contraindicated for the risk of bleeding consequent to the frequent assumption of anti-platelet drugs by these elderly patients or to the occurrence of other coagulation disorders. Also, the TAB is obviously unable to allow the assessment and the extent of arterial involvement that may sometimes include large vessels, such as the aortic arch and its major branches.
CDUS provides reliable information as regards superficial arteries such as the temporal artery and to a lower extent the subclavian and axillary arteries. Because ultrasonographic data of temporal arteries do not correlate with eye complications, OCT or CDUS of the orbital (retrobulbar) vessels are of critical importance in order to quickly differentiate the mechanism of eye involvement (arteritic, versus non-arteritic). The arteritic conditions should be treated promptly to prevent further visual loss of the fellow eye. Although CDUS of orbital vessels is a faster method compared with OCT, it is much less reliable when extra-cranial deep vessels are suspected to be affected. In these situations, MRI, CT and FDG-PET/CT are being used with increasing frequency in the diagnosis and monitoring of cranial and extracranial arteries, including the aorta. Compared to TAB, these imaging modalities offer the advantage of being less invasive, more sensitive and readily available.2,3,5
However, although these imaging techniques have questioned the diagnostic role of TAB, it is still debatable in the clinical practice which imaging tool is able to provide the most reliable assessments of disease extent and activity. The task force of the European League Against Rheumatism (EULAR) has recommended that an early imaging test be performed in patients with suspected GCA, with ultrasound being the first choice and CT or FDG-PET/CT being possible alternatives. When clinical examination and imaging are still unable to allow a definite diagnosis, temporal artery biopsy and/or additional imaging may be necessary. Furthermore, in case of a suspected flare, disease activity can be better assessed with the use of imaging procedures, but the frequency and choice of imaging modalities in the follow-up of the patient are left to the individual referral center decision.58
The treatment of GCA and PMR must take into account the disease course and the associated comorbidities.59 As noted above, MTX is frequently prescribed in conjunction with GCs, not only as a steroid-sparing therapy but also because of its demonstrated ability to reduce the risk of relapses.4,44,52 It seems, therefore, safe to state that the association of MTX to GCs is justified in both GCA and PMR patients with early relapse while tapering GCs, in those requiring long-term or more than 7.5–10 mg/day of maintenance prednisone equivalents after at least 3 months of sustained GC therapy, and in those experiencing unbearable GC-related side-effects.
As mentioned before, the IL-6/IL-17 axis that is mainly responsible for the extravascular inflammatory syndrome of GCA is readily responsive to GCs. Conversely, the IL-12/IFN-γ axis is largely involved in the vascular damage and is poorly responsive to these drugs.5 To overcome this discrepancy, adjunctive therapies such as aspirin/anticoagulants, hydroxyl-3-methylglutaryl coenzyme-A reductase inhibitors also known as statins, and angiotensin II receptor blockers commonly defined as sartans have been proposed. However, in the absence of randomized clinical trials, the evidence supporting their usefulness is circumstantial and overall disappointing.2,16
With the aim of targeting the immune abnormalities underlying both the extravascular and vasculogenic components of GCA, many different therapeutic agents have been proposed, including immunosuppressive drugs (azathioprine, cyclophosphamide, leflunomide, and mycophenolate mofetil) and TNF-α inhibitors (infliximab, adalimumab, etanercept, abatacept, and rituximab). A survey of the results achieved with their administration, mostly in patients with difficult-to-treat GCA and in small series, can be found in recent reviews.16,60,61
While, overall, the results of treatment have been disappointing, an encouraging exception is biologic therapy using TCZ, shown in a trial setting to be effective in the induction and maintenance of remission in GCA patients.62 In a randomized, double-blind, placebo-controlled, Phase 3 trial (GiACTA trial), TCZ combined with a 26-week prednisone taper was shown to be significantly superior to either 26-week or 52-week prednisone tapering plus placebo, in terms of sustained GCs-free remission.63 Furthermore, when TCZ was included as add-on therapy to GCs during the first 3 months of treatment, at week 26 the primary endpoint of remaining in remission with ≤0.1 mg prednisone/kg/day was met by 75% of the patients, although half experienced relapse during the 9 months following TCZ discontinuation.47 Finally, in an observational, retrospective, multicenter study carried out on patients with GCA refractory to conventional therapy, 94% achieved prompt and persistent clinical improvement one month after TCZ introduction. Infections were the most common adverse event.48
The strengths of our study are: 1) the homogeneous collection of data, made possible through a carefully implemented collaboration between tertiary eye-care and clinical immunology centers of the same university hospital; 2) the length of follow-up, which exceeded 4 years in 39% of the study patients; and 3) the clinical and ophthalmological assessments made by the same internists and the same ophthalmologists, which avoided or at least reduced the risk of unwanted variability.
Nonetheless, the following potential shortcomings should be mentioned: 1) the retrospective nature of the study; 2) the relatively small cohort of 56 patients, compared to the much larger number of patients enrolled in polycenter or retrospective population-based cohort studies;7,10,48,63,64 3) the use of CDUS but the lack of imaging studies such as MRI and/or CT angiography, and/or FDG-PET/CT in the first 15 patients enrolled from 2005 to 2008, such that an involvement of the aortic arch and its branches or of other deep vessels may have remained undiagnosed in some cases.
The worldwide morbidity burden from GCA that has been projected by 2050 is a cause for concern.65 Given the expected increase in the prevalence of GCA as a consequence of aging populations, it is estimated that by 2050 over 3 million people in Europe, North America, and Oceania will be diagnosed with GCA. It is reasonable to predict that in some 500,000 of these patients the disease will result in visual impairments, with an estimated cost exceeding US$ 76 billion for the USA alone. In addition, the cost of in-patient care for patients with active GCA will be about US$ 1 billion, in addition to the estimated US$ 6 billion needed by 2050 to cope with GC-related adverse events, especially bone fractures.65 Both further insights into the pathogenesis of GCA and advancements in the treatment of this disease are therefore urgently needed if this alarming scenario is to be avoided.
Abbreviations
A-AION, arteritic anterior ischemic optic neuropathy; ACR, American College of Rheumatology; A-PION, arteritic posterior ischemic optic neuropathy; CDUS, color Doppler ultrasonography; CRAO, central retinal artery occlusion; CRP, C-reactive protein; DCs, dendritic cells; ECs, endothelial cells; ESR, erythrocyte sedimentation rate; EULAR, European League Against Rheumatism; FDG-PET/CT, 18F-fluorodeoxyglucose positron emission tomography/computed tomography; FGF2, fibroblast growth factor-2; GCA, giant cell arteritis; GCs, glucocorticoids; HBV, hepatitis B virus; HCV, hepatitis C virus; IFN-γ, interferon-γ; IL, interleukin; IOP, intraocular pressure; MMP, matrix metalloproteinases; MRI, magnetic resonance imaging; MTX, methotrexate; OCT, optical coherence tomography; PCAs, posterior ciliary arteries; PDGF, platelet-derived growth factors; PMR, polymyalgia rheumatica; RNI, reactive nitrogen intermediates; ROS, reactive oxygen species; TAB, temporal artery biopsy; TCZ, tocilizumab; TLR, toll-like receptors; TNF-α, tumor necrosis factor-α; VEGF, vascular endothelial growth factor; VSMCs, vascular smooth muscle cells.
Ethics Approval
For this retrospective study, patient consent was not required. No animal studies were included in the study.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Fondo di Sviluppo e Coesione 2007-2013 – APQ Ricerca Regione Puglia “Programma regionale a sostegno della specializzazione intelligente e della sostenibilità sociale ed ambientale - FutureInResearch”. The sponsors of this study are public or nonprofit organizations that support science in general; they had no role in gathering, analyzing, or interpreting the data.
Disclosure
The authors report no conflicts of interest in this work.
References
1. De Smit E, O’Sullivan E, Mackey DA, Hewitt AW. Giant cell arteritis: ophthalmic manifestations of a systemic disease. Graefes Arch Clin Exp Ophthalmol. 2016;254(12):2291–2306. doi:10.1007/s00417-016-3434-7
2. Ninan JV, Lester S, Hill CL. Giant cell arteritis: beyond temporal artery biopsy and steroids. Intern Med J. 2017;47(11):1228–1240. doi:10.1111/imj.2017.47.issue-11
3. Sammel AM, Fraser CL. Update on giant cell arteritis. Curr Opin Ophthalmol. 2018;29(6):520–527. doi:10.1097/ICU.0000000000000528
4. Vodopivec I, Rizzo JF
5. Weyand CM, Goronzy JJ. Clinical practice. Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med. 2014;371:50–57. doi:10.1056/NEJMcp1214825
6. Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised international Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013;65(1):1–11. doi:10.1002/art.37715
7. Salvarani C, Cimino L, Macchioni P, et al. Risk factors for visual loss in an Italian population-based cohort of patients with giant cell arteritis. Arthritis Rheum. 2005;53(2):293–297. doi:10.1002/(ISSN)1529-0131
8. Figus M, Talarico R, Posarelli C. d’Ascanio A, Elefante E, Bombardieri S. Ocular involvement in giant cell arteritis. Clin Exp Rheumatol. 2013;31(1 Suppl 75):S96.
9. Ji J, Dimitrijevic I, Sundquist J, Sundquist K, Zöller B. Risk of ocular manifestations in patients with giant cell arteritis: a nationwide study in Sweden. Scand J Rheumatol. 2017;46(6):484–489. doi:10.1080/03009742.2016.1266030
10. Chen JJ, Leavitt JA, Fang C, Crowson CS, Matteson EL, Warrington KJ. Evaluating the incidence of arteritic ischemic optic neuropathy and other causes of vision loss from giant cell arteritis. Ophthalmology. 2016;123(9):1999–2003. doi:10.1016/j.ophtha.2016.05.008
11. Liozon E, Dalmay F, Lalloue F, et al. Risk factors for permanent visual loss in biopsy-proven giant cell arteritis: a study of 339 patients. J Rheumatol. 2016;43(7):1393–1399. doi:10.3899/jrheum.151135
12. Soriano A, Muratore F, Pipitone N, Boiardi L, Cimino L, Salvarani C. Visual loss and other cranial ischaemic complications in giant cell arteritis. Nat Rev Rheumatol. 2017;13(8):476–484. doi:10.1038/nrrheum.2017.98
13. Berger CT, Wolbers M, Meyer P, Daikeler T, Hess C. High incidence of severe ischaemic complications in patients with giant cell arteritis irrespective of platelet count and size, and platelet inhibition. Rheumatology (Oxford). 2009;48(3):258–261. doi:10.1093/rheumatology/ken480
14. Salvarani C, Gabriel SE, O’Fallon WM, Hunder GG. Epidemiology of polymyalgia rheumatica in Olmsted County, Minnesota, 1970–1991. Arthritis Rheum. 1995;38(3):369–373. doi:10.1002/(ISSN)1529-0131
15. Gonzalez-gay MA. Giant cell arteritis and polymyalgia rheumatica: two different but often overlapping conditions. Semin Arthritis Rheum. 2004;33:289–293. doi:10.1016/j.semarthrit.2003.09.007
16. Frohman L, Wong AB, Matheos K, Leon-alvarado LG, Danesh-meyer HV. New developments in giant cell arteritis. Surv Ophthalmol. 2016;61(4):400–421. doi:10.1016/j.survophthal.2016.01.001
17. Cantini F, Niccoli L, Storri L, et al. Are polymyalgia rheumatica and giant cell arteritis the same disease? Semin Arthritis Rheum. 2004;33(5):294–301. doi:10.1016/j.semarthrit.2003.09.008
18. Hunder GG, Bloch DA, Michel BA, et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990;33(8):1122–1128. doi:10.1002/art.1780330810
19. Dasgupta B, Cimmino MA, Kremers HM, et al. 2012 provisional classification criteria for polymyalgia rheumatica: a European League Against Rheumatism/American College of Rheumatology collaborative initiative. Arthritis Rheum. 2012;64(4):943–954. doi:10.1002/art.34356
20. Zöller B, Li X, Sundquist J, Sundquist K. Occupational and socio-economic risk factors for giant cell arteritis: a nationwide study based on hospitalizations in Sweden. Scand J Rheumatol. 2013;42(6):487–497. doi:10.3109/03009742.2013.793777
21. Hayreh SS, Podhajsky PA, Zimmerman B. Occult giant cell arteritis: ocular manifestations. Am J Ophthalmol. 1998;125(4):521–526. doi:10.1016/S0002-9394(99)80193-7
22. Luqmani R, Lee E, Singh S, et al. The role of ultrasound compared to biopsy of temporal arteries in the diagnosis and treatment of giant cell arteritis (TABUL): a diagnostic accuracy and cost-effectiveness study. Health Technol Assess. 2016;20(90):1–238. doi:10.3310/hta20900
23. Ball EL, Walsh SR, Tang TY, Gohil R, Clarke JM. Role of ultrasonography in the diagnosis of temporal arteritis. Br J Surg. 2010;97(12):1765–1771. doi:10.1002/bjs.v97:12
24. Daily B, Dassow P, Haynes J, Nashelsky J. Giant cell arteritis: biopsy after corticosteroid initiation. Am Fam Physician. 2017;95(2):116–117.
25. Dasgupta B, Borg FA, Hassan N, et al. BSR and BHPR guidelines for the management of giant cell arteritis. Rheumatology (Oxford). 2010;49(8):1594–1597. doi:10.1093/rheumatology/keq039a
26. Antonio-santos AA, Murad-kejbou SJ, Foroozan R, Yedavally S, Kaufman DI, Eggenberger ER. Preserved visual acuity in anterior ischemic optic neuropathy secondary to giant cell (temporal) arteritis. J Vasc Interv Neurol. 2016;8(5):17–21.
27. Hayreh SS. Posterior ischaemic optic neuropathy: clinical features, pathogenesis, and management. Eye (Lond). 2004;18(11):1188–1206. doi:10.1038/sj.eye.6701562
28. Balducci N, Morara M, Veronese C, et al. Optical coherence tomography angiography in acute arteritic and non-arteritic anterior ischemic optic neuropathy. Graefes Arch Clin Exp Ophthalmol. 2017;255(11):2255–2261. doi:10.1007/s00417-017-3774-y
29. Sommer NN, Treitl KM, Coppenrath E, et al. Three-dimensional high-resolution black-blood magnetic resonance imaging for detection of arteritic anterior ischemic optic neuropathy in patients with giant cell arteritis. Invest Radiol. 2018;53(11):698–704. doi:10.1097/RLI.0000000000000500
30. Gupta PK, Asrani S, Freedman SF, El-dairi M, Bhatti MT. Differentiating glaucomatous from non-glaucomatous optic nerve cupping by optical coherence tomography. Open Neurol J. 2011;5:1–7. doi:10.2174/1874205X01105010001
31. Hayreh SS. Ischemic optic neuropathy. Prog Retin Eye Res. 2009;28(1):34–62. doi:10.1016/j.preteyeres.2008.11.002
32. Srinivasan S, Moorthy S, Sreekumar K, Kulkarni C. Diffusion-weighted MRI in acute posterior ischemic optic neuropathy. Indian J Radiol Imaging. 2012;22(2):106–107. doi:10.4103/0971-3026.101082
33. De Smit E, O’sullivan E. Cotton-wool spots in giant cell arteritis. CMAJ. 2013;185(9):796. doi:10.1503/cmaj.120540
34. Hamed LM, Guy JR, Moster ML, Bosley T. Giant cell arteritis in the ocular ischemic syndrome. Am J Ophthalmol. 1992;113(6):702–705. doi:10.1016/S0002-9394(14)74798-1
35. McKillop E, Tejwani D, Weir C, Jay J. Anterior segment ischaemia with giant cell arteritis. Can J Ophthalmol. 2006;41(2):201–203. doi:10.1139/I06-009
36. Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol. 2013;9:73140. doi:10.1038/nrrheum.2013.161
37. Terrades-garcia N, Cid MC. Pathogenesis of giant-cell arteritis: how targeted therapies are influencing our understanding of the mechanisms involved. Rheumatology (Oxford). 2018;57(suppl_2):ii51–ii62. doi:10.1093/rheumatology/kex423
38. Rittner HL, Kaiser M, Brack A, Szweda LI, Goronzy JJ, Weyand CM. Tissue-destructive macrophages in giant cell arteritis. Circ Res. 1999;84(9):1050–1058. doi:10.1161/01.RES.84.9.1050
39. Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med. 2003;349(2):160–169. doi:10.1056/NEJMra022694
40. Kaiser M, Weyand CM, Björnsson J, Goronzy JJ. Platelet-derived growth factor, intimal hyperplasia, and ischemic complications in giant cell arteritis. Arthritis Rheum. 1998;41(4):623–633. doi:10.1002/1529-0131(199804)41:4<623::AID-ART9>3.0.CO;2-6
41. Hayreh SS, Zimmerman B. Visual deterioration in giant cell arteritis patients while on high doses of corticosteroid therapy. Ophthalmology. 2003;110(6):1204–1215. doi:10.1016/S0161-6420(03)00228-8
42. Nesher G, Sonnenblick M, Friedlander Y. Analysis of steroid related complications and mortality in temporal arteritis: a 15-year survey of 43 patients. J Rheumatol. 1994;21(7):1283–1286.
43. Proven A, Gabriel SE, Orces C, O’Fallon WM, Hunder GG. Glucocorticoid therapy in giant cell arteritis: duration and adverse outcomes. Arthritis Rheum. 2003;49(5):703–708. doi:10.1002/(ISSN)1529-0131
44. Mahr AD, Jover JA, Spiera RF, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum. 2007;56(8):2789–2797. doi:10.1002/(ISSN)1529-0131
45. González-Gay MA, Pina T, Prieto-peña D, Calderon-goercke M, Blanco R, Castañeda S. Drug therapies for polymyalgia rheumatica: a pharmacotherapeutic update. Expert Opin Pharmacother. 2018;19(11):1235–1244. doi:10.1080/14656566.2018.1501360
46. Koster MJ, Yeruva K, Crowson CS, Muratore F, Labarca C, Warrington KJ. Efficacy of methotrexate in real-world management of giant cell arteritis: a case-control study. J Rheumatol. 2019;46(5):501–508. doi:10.3899/jrheum.180429
47. Samson M, Devilliers H, Ly KH, et al. Tocilizumab as an add-on therapy to glucocorticoids during the first 3 months of treatment of Giant cell arteritis: a prospective study. Eur J Intern Med. 2018;57:96–104. doi:10.1016/j.ejim.2018.06.008
48. Calderón-goercke M, Loricera J, Aldasoro V, et al. Tocilizumab in giant cell arteritis. Observational, open-label multicenter study of 134 patients in clinical practice. Semin Arthritis Rheum. 2019;49(1):126–135. doi:10.1016/j.semarthrit.2019.01.003
49. Prieto-gonzález S, Arguis P, García-martínez A, et al. Large vessel involvement in biopsy-proven giant cell arteritis: prospective study in 40 newly diagnosed patients using CT angiography. Ann Rheum Dis. 2012;71(7):1170–1176. doi:10.1136/annrheumdis-2011-200865
50. Paraskevas KI, Boumpas DT, Vrentzos GE, Mikhailidis DP. Oral and ocular/orbital manifestations of temporal arteritis: a disease with deceptive clinical symptoms and devastating consequences. Clin Rheumatol. 2007;26(7):1044–1048. doi:10.1007/s10067-006-0493-x
51. de Boysson H, Liozon E, Ly KH, Dumont A, Delmas C, Aouba A. The different clinical patterns of giant cell arteritis. Clin Exp Rheumatol. 2019;37 Suppl 117(2):57–60.
52. Hoffman GS, Cid MC, Hellmann DB, et al. International Network for the Study of Systemic Vasculitides. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum. 2002;46(5):1309–1318. doi:10.1002/art.v46:5
53. González-gay MA, Blanco R, Rodríguez-valverde V, et al. Permanent visual loss and cerebrovascular accidents in giant cell arteritis: predictors and response to treatment. Arthritis Rheum. 1998;41(8):1497–1504. doi:10.1002/(ISSN)1529-0131
54. Cid MC, Font C, Oristrell J, et al. Association between strong inflammatory response and low risk of developing visual loss and other cranial ischemic complications in giant cell (temporal) arteritis. Arthritis Rheum. 1998;41(1):26–32. doi:10.1002/(ISSN)1529-0131
55. Schmidt J, Kermani TA, Bacani AK, et al. Diagnostic features, treatment, and outcomes of Takayasu arteritis in a US cohort of 126 patients. Mayo Clin Proc. 2013;88(8):822–830. doi:10.1016/j.mayocp.2013.04.025
56. Buttgereit F, Dejaco C, Matteson EL, Dasgupta B. Polymyalgia rheumatica and giant cell arteritis: a systematic review. JAMA. 2016;315(22):2442–2458. doi:10.1001/jama.2016.5444
57. Kermani TA. Takayasu arteritis and giant cell arteritis: are they a spectrum of the same disease? Int J Rheum Dis. 2019;22 Suppl 1:41–48. doi:10.1111/1756-185X.13288
58. Dejaco C, Ramiro S, Duftner C, et al. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice. Ann Rheum Dis. 2018;77(5):636–643. doi:10.1136/annrheumdis-2017-212649
59. Pfeil A, Oelzner P, Hellmann P. The treatment of giant cell arteritis in different clinical settings. Front Immunol. 2019;24(9):3129. doi:10.3389/fimmu.2018.03129
60. Ninan J, Lester S, Hill C. Giant cell arteritis. Best Pract Res Clin Rheumatol. 2016;30(1):169–188. doi:10.1016/j.berh.2016.05.001
61. Low C, Conway R. Current advances in the treatment of giant cell arteritis: the role of biologics. Ther Adv Musculoskelet Dis. 2019. doi:10.1177/1759720X19827222
62. Villiger PM, Adler S, Kuchen S, et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a Phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387(10031):1921–1927. doi:10.1016/S0140-6736(16)00560-2
63. Stone JH, Tuckwell K, Dimonaco S, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med. 2017;377(4):317–328. doi:10.1056/NEJMoa1613849
64. Achkar AA, Lie JT, Hunder GG, O’fallon WM, Gabriel SE. How does previous corticosteroid treatment affect the biopsy findings in giant cell (temporal) arteritis? Ann Intern Med. 1994;120(12):987–992. doi:10.7326/0003-4819-120-12-199406150-00003
65. De Smit E, Palmer AJ, Hewitt AW. Projected worldwide disease burden from giant cell arteritis by 2050. J Rheumatol. 2015;42(1):119–125. doi:10.3899/jrheum.140318
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.