Back to Journals » Eye and Brain » Volume 11
Giant cell arteritis: early diagnosis is key
Authors Baig IF, Pascoe AR, Kini A ![]() , Lee AG
, Lee AG
Received 14 August 2018
Accepted for publication 27 November 2018
Published 17 January 2019 Volume 2019:11 Pages 1—12
DOI https://doi.org/10.2147/EB.S170388
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Margaret Wong-Riley
Video abstract presented by Andrew G Lee
Views: 12285
Iyza F Baig,1 Alexis R Pascoe,1 Ashwini Kini,2 Andrew G Lee2–9
1McGovern Medical School, The University of Texas Health Science Center in Houston, Houston, TX, USA; 2Department of Ophthalmology, Blanton Eye Institute, Houston Methodist Hospital, Houston, TX, USA; 3Department of Ophthalmology, Baylor College of Medicine, Houston, TX, USA; 4Department of Ophthalmology, 5Department of Neurology, 6Department of Neurosurgery, Weill Cornell Medical College, Houston, TX, USA; 7The University of Texas Medical Branch, Galveston, TX, USA; 8The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 9Ophthalmology, The University of Iowa Hospitals and Clinics, Iowa City, IA, USA
Abstract: Giant cell arteritis (GCA) is an inflammatory vasculitis typically affecting elderly that can potentially cause vision loss. Studies have demonstrated that early recognition and initiation of treatment can improve visual prognosis in patients with GCA. This review addresses the benefits of early diagnosis and treatment, and discusses the available treatment options to manage the disease.
Keywords: giant cell arteritis, imaging in GCA, laboratory values in GCA, steroids in GCA, steroid-sparing agents in GCA
Introduction
Giant cell arteritis (GCA) is a granulomatous vasculitis that targets large- and medium-sized arteries, most commonly affecting the aorta, branches of the ophthalmic artery, and extracranial branches of the carotid arteries.1–5 Complications of the vasculitis including GCA typically result from ischemic injury, systemic inflammation, and aneurysm formation and rupture.6 From an ophthalmologic perspective, GCA is an urgent diagnosis because if not recognized and treated early, ischemic complications may result in permanent vision loss (up to 15%–25% of cases).7 In one study evaluating cases of delayed diagnosis of GCA resulting in permanent vision loss, 35% of patients had systemic symptoms for an average of 10.8 months before suffering permanent vision loss and 65% had transient visual symptoms for 8.5 days prior to diagnosis.1 In light of the research conducted in the past 20 years, it has become clear that the early diagnosis and initiation of treatment is essential to improve visual and systemic prognosis in patients with GCA.1,8,9 Numerous recent studies evaluating the implementation of fast-track clinics (FTCs) have found that the rate of permanent blindness decreased significantly with early initiation of corticosteroid treatment for the vasculitis.7–9 The purpose of this review is to highlight the early recognition and treatment of GCA.
Diagnostic approach
In 1990, the American College of Rheumatology (ACR) developed criteria for the diagnosis of GCA (Table 1).10 Although initially developed for research purposes, the ACR criteria (with a diagnosis threshold of 3 points) had a sensitivity of 93.5% and a specificity of 91.2% for the diagnosis of GCA.2,10 Since its development, the ACR criteria have been used to clinically diagnose suspected GCA patients, allowing for early detection and treatment without a temporal artery biopsy (TAB).4
 | Table 1 ACR diagnostic criteria for GCA Note: Data from Hunder et al.10 Abbreviations: ACR, American College of Rheumatology; ESR, erythrocyte sedimentation rate; GCA, giant cell arteritis; TAB, temporal artery biopsy. |
Despite the high sensitivity (93.5%) and specificity (91.2%) of the ACR criteria, a positive TAB, however, is not required for the diagnosis of GCA. El-Dairi et al set out to develop different diagnostic algorithms to increase the diagnostic yield of TAB by analyzing the laboratory, demographic, and clinical data from their study cohort. These authors proposed a seven-criteria scoring system including 1) evidence of anterior extracranial circulation ischemia (ie, arteritic anterior ischemic optic neuropathy [A-AION], posterior ischemic optic neuropathy [PION], ophthalmic artery occlusion, central retinal artery occlusion [CRAO], cilioretinal artery occlusion, or amaurosis fugax); 2) new-onset neck pain or headache; 3) abnormal erythrocyte sedimentation rate (ESR), platelets, or c-reactive protein (CRP) levels; 4) jaw claudication; 5) abnormal superficial temporal artery on exam (ie, nodularity, absence of pulse, local tenderness, beading); 6) constitutional symptoms (ie, fatigue, malaise, weight loss, fatigue); and 7) polymyalgia rheumatica (PMR).4 Each criterion confers one point, but a point is detracted when a criterion can be explained by an alternative chronic preexisting condition.
With a score of one point, the patient has a “very low” clinical suspicion for GCA, and an evaluation for another diagnosis is recommended. A score of 2 provides a moderate clinical suspicion (33%), and the authors recommended the initiation of oral prednisone (1 mg/kg/day) followed by TAB. If the TAB is negative with this “moderate” clinical suspicion, an alternative diagnosis other than GCA should be considered. In contrast, for patients with a high clinical suspicion (56%) (ie, score >2), it was recommended that empiric steroids (eg, intravenous [IV] methylprednisolone [1 g/day] or high-dose oral prednisone [1 mg/kg/day]) be started and a gold standard TAB be performed. Regardless of moderate or high clinical suspicion (ie, pretest probability of disease) in this scoring system, a positive TAB is considered a high post-test probability for GCA. If the clinical suspicion remains high despite a negative initial TAB (presumed false positive) then a contralateral TAB was recommended, and empiric steroids should be continued.4 With this new algorithm, a positive TAB was shown to have a sensitivity of 91.4%.
When comparing these proposed diagnostic criteria with the ACR criteria, 21% of TAB-negative patients were found to be false positives and thus would have been started on steroid therapy incorrectly.4 Considering the possible side effects of long-term corticosteroid treatment, incorrectly placing a patient on steroid treatment should be avoided. Furthermore, a study reports that 25.7% of their biopsy-proven GCA patients would not have met the ACR criteria.4 The lower specificity of the ACR criteria could, therefore, have dire consequences, potentially resulting in inadequate treatment.
Although the algorithm proposed by El-Dairi et al4 increased the diagnosed yield of a TAB, overall, the diagnosis of GCA should reside more on pretest (TAB) clinical suspicion than number of symptoms present. In one study, biopsy-proven GCA patients that had no systemic symptoms and only a single complaint of vision loss comprised 21.2% of patients with vision loss.1 If systemic manifestations are viewed as a primary component for GCA diagnosis, detection will not occur in time to save the patient’s sight. For instance, headache is the most common complaint for GCA patients4 but Hayreh et al found that the statistically significant difference (P-value: 0.084) of 55.7% of patients with positive TABs complaining of headache while 45.5% of patients with negative TABs complained of headache as well.1 Even though headache may be the most common symptom, headache alone is not a very specific symptom for GCA. When considering whether early treatment of corticosteroid treatment is appropriate, the predictive power of certain symptoms should be taken into account. For instance, jaw claudication is associated with a nine times greater risk for having a positive TAB.1
Pathophysiology
GCA is immune-mediated inflammation involving the medium- and large-size arteries. An unknown antigen is the presumed trigger for the immunologic cascade that begins with the dendritic cell processing the antigen and presenting it to T cells via the major histocompatibility complex II interaction with the T cell receptors. There is then a downstream activation and differentiation of T cells to TH1 and TH17 cells, which in turn express interferon γ, a potent macrophage activator. There is a proinflammatory cascade triggered by the macrophage activation with further release of chemokines including but not limited to IL-6 and tumor necrosis factor (TNF) alpha. There is recruitment of a large number of inflammatory cells with production of ROS and matrix metalloproteinases (MMPs), which then primarily attack the internal elastic lamina. This leads to damage of the vessel wall followed by abnormal vascular remodeling and ultimately occlusion the lumen of the vessel. Tissue expression of these proinflammatory cytokines including IL-6 has been found to co-relate to severity of disease activity and treatment response to steroids and IL-6 blockade (eg, tocilizumab [TCZ]).11,12
Histopathologic evaluation of TAB specimens (see Figures 1 and 2) have revealed that GCA may not be limited to just the internal elastic lamina and can involve the media, the adventitia, or the entire thickness of vessel wall. Recently, CD68, which is a cluster differentiating factor for macrophages found in TAB, was found to correlate with more steroid-resistant cases. It has been suggested that CD68 positivity could serve as a marker to triage patients earlier toward a steroid-sparing immunomodulatory therapy.13
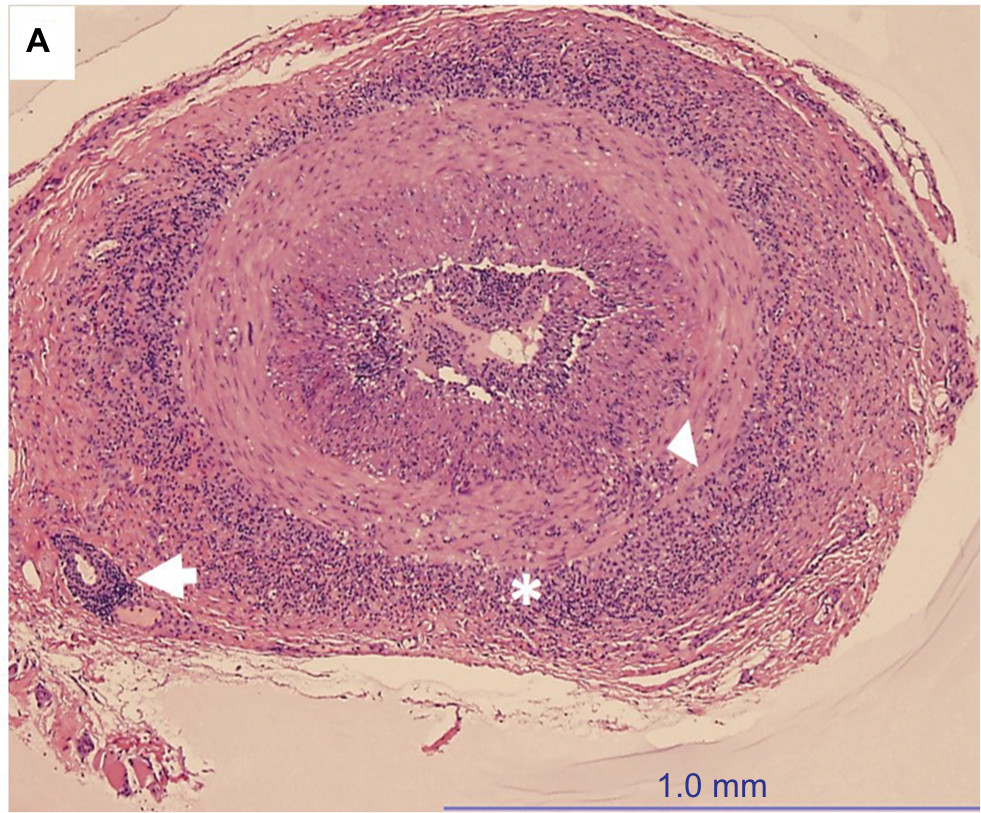 | Figure 1 H&E-stained, low-power temporal artery biopsy, showing the inflammatory infiltrate, mostly lymphocytes and macrophages, in multiple layers of the vessel: adventitia (asterisk), muscularis (arrow head), and even at the level of the vasa vasorum (arrow). Notes: There is irregular intimal hyperplasia and almost complete narrowing of the vascular lumen. Credit: Claudia ProsperoPonce, Department of Ocular-Pathology, Houston Methodist. |
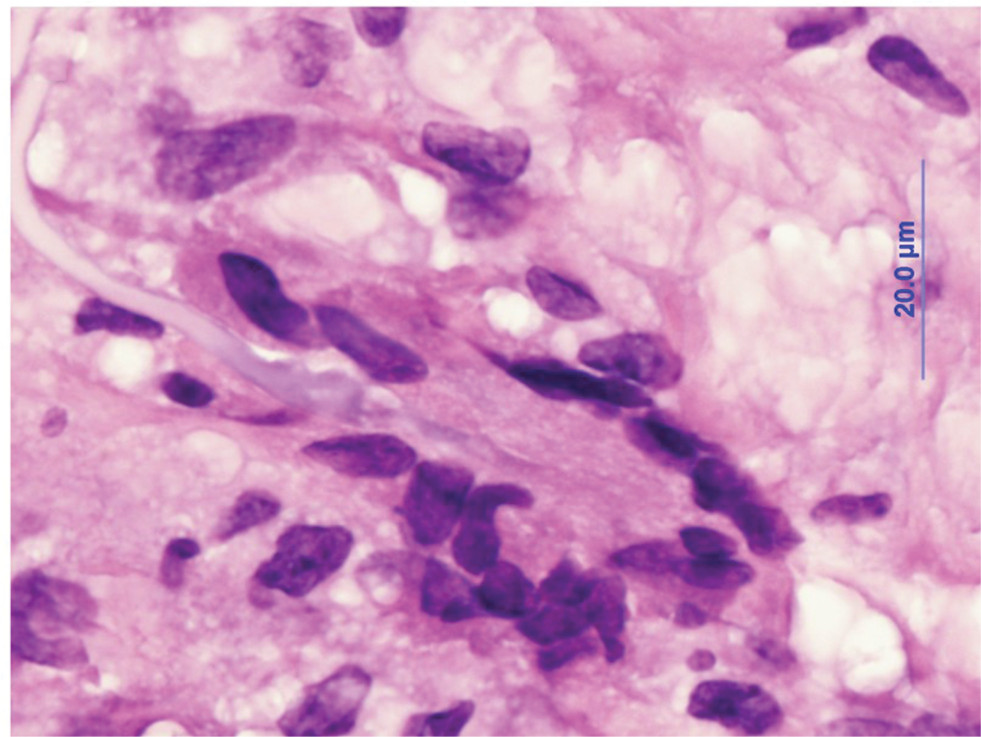 | Figure 2 H&E-stained, high-power temporal artery biopsy, showing a multinucleated giant cell attacking the elastic lamina. Note: Credit: Claudia ProsperoPonce, Department of Ocular-Pathology, Houston Methodist. |
Risk factors
GCA is a disease of elderly patients and patients are generally older than 50 years.1,3–5 The average age of presentation is 74–76 years with an increasing incidence as the patient ages, peaking at 80 years.3,5 While GCA can occur in both men and women, it is more common in women. One review revealed that women have an increased risk ranging from 2.3 to 2.6 times compared to the risk of men.1,3,15 Additionally, Caucasians are more commonly affected especially those of Scandinavian, Nordic, or Northern-European ancestry1,3,5,14,16 but the disease has been reported among many different ethnicities with variable incidence, and ethnicity alone should not be used to exclude consideration of GCA. There is evidence of GCA in southern European and Mediterranean countries, but incidence occurs at a lower rate.16 Lower incidence was also found in populations of African and Asian descent.3,16 Other independent risk factors include smoking, low body mass index, and early menopause.17
Symptoms
GCA causes both systemic and ocular symptoms. The systemic manifestations often precede the ocular manifestations, with new-onset headache being the most common systemic symptom.4,5 Systemic symptoms occur in about 50% of GCA patients and can include myalgias, headaches, scalp tenderness, tender temporal arteries, jaw claudication, and constitutional symptoms (eg, fever, anorexia, and weight loss).1,5 Although scalp tenderness is a symptom typically associated with GCA, it has been shown to be an unreliable symptom in the clinical diagnosis of GCA. Using a positive TAB as the definition of a GCA diagnosis, 18% of patients who complained of scalp tenderness had a positive TAB while 10.5% of patients with the same complaint had a negative TAB.1
Arm claudication may suggest subclavian vessel involvement.3 The subclavian and axillary arteries become narrowed due to inflammation and result in ischemia manifesting as arm pain with exertion.5
Jaw claudication occurs secondary to ischemia of the masseter muscle, which is supplied by maxillary artery. Therefore, exertional ischemia ensues while chewing or using the jaw. Jaw claudication occurs in up to 50% of cases and is considered as a modestly sensitive finding.4,5,18 Jaw claudication also is very specific and is most likely to be associated with a positive TAB.4,8 Hayreh et al1 found that jaw claudication increased the odds of having a ninefold positive TAB.
PMR and GCA are closely associated, with ~50% of patients with PMR having biopsy-proven GCA.5,18 PMR is characterized by persistent pain for at least 1 month with episodes of aching and morning stiffness that lasts at least 30 minutes in the neck, shoulder, or pelvic girdle and an elevated ESR of at least 40 mm/h.5 One-third of patients with PMR complain of early morning stiffness, myalgias, fever, and anorexia, while 70%–95% complain of shoulder pain and 50%–70% of patients have hip and neck involvement.5 They may have issues with combing their hair or reaching to a shelf due to pain in their shoulders or getting out of a chair or climbing the stairs due to hip girdle pain (eg, the mnemonic, “hair, chair, stair” may be interesting).14 Patients also report bilateral involvement of the joints and proximal extremities with worsening of pain with movement of affected areas, which can negatively affect daily activities.5
The most concerning symptom for GCA is vision loss, with 50% of patients complaining of ocular involvement ranging from eye pain to amaurosis fugax.19 It has been observed that significantly older patients tended to experience ocular involvement more compared to younger patients, with no predilection for gender.19 Typically, ocular complaints include visual loss of varying severity, amaurosis fugax, diplopia, and eye pain. Visual loss of varying severity was present in 98% of patients with a positive TAB, while 31% complained of amaurosis fugax, 6% complained of diplopia, and 8.2% complained of eye pain.19
Vision loss secondary to GCA can be monocular or binocular. It can be permanent or more transient like amaurosis fugax, which has been shown to precede permanent vision loss in 44% of GCA patients.5 The most common cause of amaurosis fugax is transient ischemia to the optic nerve head.1 A precipitating factor in these patients is change of posture such as when a patient stands up, has orthostatic hypotension, or stoops down to reach something, inadvertently increasing intraocular pressure.1 These changes in positions can further compromise the already poor circulation in the optic nerve head causing an intermittent ischemia and vision loss.5 If the vision loss episodes are transient, it has been shown that permanent vision loss can occur about 8.5 days later.20
Vision loss can occur initially in one eye and then later involve the unaffected eye.1,5,19 Some patients may state that they experience simultaneous bilateral visual loss, but fundus exam would show that one eye would have older changes when compared to the other eye.19 In one instance, the patient was not aware of the loss until both eyes were involved.19 If left untreated, contralateral eye involvement commonly occurred between 1 and 14 days after initial onset with the longest interval being 9 months.5,19 If there is no further visual deterioration within the first week of adequate corticosteroid treatment, existing vision in affected eye and the vision in the unaffected eye will remain intact.19 When treated early and adequately, GCA-mediated blindness is preventable in majority of cases.19
Signs on physical examination
A thorough physical examination of a patient with suspected GCA is essential in informing clinical diagnosis. Palpation and inspection of the temporal artery may disclose erythema, tenderness, nodularity, or thickening.3 A clinically abnormal temporal artery may prompt a clinician to suspect GCA; however, a clinically abnormal temporal artery is not a statistically significant determinant for GCA. In one study, 19.8% of patients with a positive TAB were found to have a clinically abnormal temporal artery while 12.8% with a negative TAB were also found to have this clinical sign (P-value: 0.105).1 The affected temporal artery can be compared with the contralateral artery to determine if there is a decreased pulse. Auscultation can be performed over the carotid, subclavian, axillary, brachial, thoracic aorta, and abdominal aorta to assess for bruits and elucidate the underlying cause of the claudication symptoms previously discussed but the sensitivity is low.3,5
Visual acuity and visual field loss in GCA can be transient or constant; unilateral or bilateral; and can be variable in severity. A relative afferent pupillary defect will be present in unilateral or bilateral but asymmetric visual loss.1,19 Ophthalmoplegia is an uncommon but reported finding in GCA and acute transient or constant diplopia in an elderly patient should still prompt consideration for GCA.1 GCA can produce transient or permanent arteritic occlusion of the vascular supply for the extraocular muscles, ultimately resulting in ischemic myopathy.1
The most common ocular ischemic lesion in GCA is A-AION (81.2%).19 Acute pallid edema (pale and swollen optic disc) is a red flag for GCA (as opposed to typical NAION) and PION (retrobulbar optic neuropathy) (7.1%) in an elderly patient is especially concerning for GCA.19 In addition, other ocular ischemic events including nonembolic CRAO (14.1%), cilioretinal artery occlusion (21.8%), or ophthalmic artery occlusion can occur in GCA.19 The main blood supply for the optic nerve head are the posterior ciliary arteries, whose occlusion produces A-AION.1 Fluorescein angiography in A-AION, CRAO, cilioretinal artery occlusion, or PION may confirm choroidal perfusion loss consistent with GCA.1 A chalky white edematous optic disc is highly suggestive of A-AION.1
Other less common signs associated with GCA include cortical vision loss, retinal cotton wool spots, and choroidal ischemic lesions with or without A-AION.1,19 Retinal cotton wool spots may be observed on exam in the earlier stages of disease at the posterior pole.1,19 The choroidal ischemic lesions initially present mainly in the mid-periphery of the fundus as white triangular lesions, but as time passes, they may appear as chorioretinal degenerative lesions 2–3 weeks later.1
Ancillary diagnostic methods
Imaging
Ultrasound
Studies evaluating the ability of imaging to aid in early diagnosis and treatment of GCA have shown mixed results. In their study of color Doppler ultrasonography (CDUS) in GCA, Schmidt et al described a halo around the temporal artery in GCA patients21 and in 2018, Schmidt et al described four pathologic findings that can be detected by ultrasound in GCA: 1) hypoechoic wall thickening (termed the “halo sign”); 2) noncompressible arteries (“compression sign”); 3) stenosis; and 4) vessel occlusion.22 These authors reported a sensitivity of 77% and specificity of 96% for temporal artery ultrasound, compared to the clinical diagnosis likelihood ratios of 19 and 0.2, respectively, for positive and negative ultrasounds. In a study by Aranda-Valera et al examining 451 patients with suspected GCA, the ultrasound sensitivity and specificity was 91.6% and 95.8% compared to clinical diagnosis.23 In their review, Schmidt22 suggested that ultrasound may be a reliable technique to diagnose GCA without TAB and they also noted that the TAB is less sensitive overall than ultrasound, possibly because TAB assesses only a small segment of the temporal artery and can be susceptible to false negatives (eg, skip lesions). They further suggested that in cases when TA ultrasound does not provide a definitive diagnosis that a confirmatory TAB may be used. Hayreh et al, however, found that in patients with positive TAB, CDUS could be normal.1 The authors concluded that due to the possibility of false-negative results with CDUS, it should not replace the gold standard TAB in definitively diagnosing GCA.1 However, ultrasound has numerous advantages over TAB; it is a noninvasive, relatively inexpensive modality that provides results rapidly. In contrast, it may take up to 2 weeks to receive the results of a TAB.22 Currently, it is unclear what eventual role TA ultrasound will play in GCA both for diagnosis and disease activity monitoring. Serial ultrasound may in fact be better suited for a role in disease monitoring for response to treatment.
2-[18F]-fluorodeoxyglucose (FDG) PET/CT
In two other studies, Hocevar et al8 and Diamantopoulos et al9 found that CDUS as well as positron emission tomography-computed tomography (PET/CT) aided in the early diagnosis of GCA and were associated with improved visual prognosis.
Due to its ability to detect increased glucose uptake from active inflammatory cells, FDG PET/CT can be a useful modality to detect inflammation in arterial walls. Numerous studies support FDG-PET/CT as a useful diagnostic imaging modality in GCA.24 In a meta-analysis of four studies, Soussan et al25 found that FDG PET/CT had a pooled sensitivity and specificity of 90% and 98% for GCA, respectively, leading to the conclusion that FDG PET/CT has good diagnostic accuracy for GCA. In a joint procedural recommendation article, Riemer et al24 provide recommendations to set a procedural standard for the use of FDG PET/CT in the imaging of large vessel vasculitis such as GCA. In general, their consensus recommends withdrawing or delaying corticosteroid therapy until after imaging, as corticosteroids can decrease vascular wall uptake of FDG and potentially cause a false-negative result.24 However, in the case of GCA, their consensus recommends not delaying therapy due to the risk of ischemic complications. Instead, they recommend FDG-PET within 3 days of starting corticosteroids as an alternative.
Few studies have examined FDG PET/CT as a potential imaging modality to monitor GCA disease activity in patients treated with corticosteroid-sparing therapies, such as anti-TNF and TCZ. In their retrospective study of 12 GCA patients treated with TCZ, Vitiello et al26 evaluated the value of FDG PET/CT in detecting and monitoring GCA disease activity. While patients were under low-dose corticosteroid therapy, a statistically significant reduction in FDG uptake was found, suggesting that FDG PET/CT may have the potential to monitor treatment response in TCZ-treated patients.26 Although the use of FDG PET/CT has promising implications, further prospective studies with larger sample sizes are required to determine the clinical significance of FDG PET/CT in the diagnosis and management of GCA.
“Fast-track” approaches
In recent years, studies have explored implementing “fast-track” methods that strive to quickly and efficiently recognize and treat GCA. In their study, Patil et al7 examined how a “fast-track pathway” (FTP) implemented in a secondary care rheumatology department affected rates of vision loss in patients with suspected GCA compared to those patients seen through a conventional referral route. In the FTP, general practitioners were educated regularly on the typical and atypical presentations of GCA, and suspected GCA referrals went through an expedited process to be reviewed by a rheumatologist within one business day. In this study, 79% of these FTP patients were seen within one business day compared to 64.6% of patients seen in the conventional referral route. Permanent visual deficits were less commonly observed in the FTP (9%) compared to conventional referral methods (37%). The study concluded that due to the reduced time between symptom onset and rheumatologist evaluation, the elimination of complex referral pathways, and increased general practitioner awareness, the FTP for GCA resulted in a reduced rate of permanent visual deficits.
Diamantopoulos et al9 also studied the implementation of an FTC in a Norwegian rheumatology clinic compared to conventionally evaluated GCA suspects. In the FTC approach, treatment for GCA was quickly started based on rapid clinical and CDUS assessment. Results showed that rapid CDUS assessment significantly reduced the relative risk (by 88%) of permanent visual deficits in patients examined in the FTC vs the conventional evaluation group. Furthermore, the use of ultrasound also significantly reduced the days of inpatient care needed (3.6 inpatient days in the conventionally evaluated group vs 0.6 days in the FTC). The study concluded that the FTC was not only associated with improved visual prognosis, but also was cost-effective by reducing the length of hospitalization needed to diagnose GCA.
Finally, Hocevar et al8 performed a prospective longitudinal study examining how prompt diagnosis and treatment in GCA affected the incidence of permanent vision loss. Through an early intervention clinic, rheumatologists performed CDUS and TAB within 1 day of the patient visit and obtained results of TAB within 3 hours. Although 35% of patients experienced visual symptoms, <6% of patients in this early intervention clinic experienced permanent deficits, as compared to the 15%–20% rate reported in the literature. The study also concluded that CDUS and PET/CT significantly aided in quickly recognizing and initiating corticosteroid therapy in patients with GCA.
These FTCs further provide evidence that early diagnosis and then treatment largely impact permanent vision loss,9 and warrant further investigation as models of GCA intervention.
Diagnostic value of laboratory studies
To increase clinical suspicion for GCA and diagnose the condition as early as possible, it is useful to interpret a patient’s signs and symptoms in light of certain laboratory studies. The diagnostic and therapeutic value of these laboratories will be discussed below.
Erythrocyte sedimentation rate
ESR measured with the Westergren method is a commonly used laboratory value that assists in the diagnosis of GCA.2 While ESR is a sensitive test, when interpreted in isolation, the test is nonspecific; therefore, it must be interpreted in light of the patient’s clinical picture. ESR can vary based on numerous factors, such as age and gender; ESR tends to increase with age, and women generally have higher ESR than men.27 Therefore, in order to estimate appropriate upper limits for what is considered a “normal” ESR, Miller et al provide a formula that accounts for how discrepancies in age and gender affect ESR. For men, the upper limit of normal ESR can be calculated as age divided by 2, while for women, this is calculated by the woman’s age plus 10, the sum of which is divided by 2.28 ESR may be elevated in numerous systemic conditions besides GCA, such as anemia, diabetes, malignancy, pregnancy, hypercholesterolemia, and infection.1,29
While a markedly elevated ESR level (>50 mm/h) has been included by ACR as one of the five criteria used to classify GCA, studies have shown that in 5%–30% of cases, patients may have a normal to low ESR.1,27 In their 27-year clinical study, Hayreh and Zimmerman1 report that initial ESR in those with TAB-proven GCA varied between 4 and 140 mm/h, while in healthy individuals, ESR was found to be between 1 and 59 mm/h. This overlap in ESR values between normal individuals and positive-TAB GCA patients suggests that GCA cannot be excluded based on a normal–to-low ESR.1 Salvarani et al27 also sought to evaluate the frequency of normal ESR in patients with biopsy-proven GCA. Results showed that when using the ACR definition of significant ESR (>50 mm/h), 10.8% of patients with biopsy-proven GCA had ESR <50 mm/h. These studies lead to the conclusion that a normal ESR should not delay the initiation of corticosteroid treatment if the other clinical criteria for GCA are met.27
Finally, the potential impracticality of obtaining a patient’s ESR can also limit its value in the early recognition of disease. Factors such as a low testing temperature, use of an inappropriately sized tube (<2 mm in diameter), and a delay of >3 hours between obtaining and measuring ESR can alter ESR results, possibly affecting clinical decision-making.2
C-reactive protein
CRP is an acute-phase plasma protein produced by the liver, and like ESR, is a sensitive but nonspecific test that when elevated has been associated with a host of other conditions besides GCA, such as tissue necrosis, infection, surgical tissue injury, transplantation, inflammation, and myocardial infarction.29 Gender also seems to slightly affect the diagnostic value of CRP. In males, the sensitivity and specificity are 100% and 83%, respectively, while for females, they are 100% and 79%.29 Still, the use of CRP has some advantages over ESR in the diagnosis of GCA. Unlike ESR, CRP is not affected by age or hematologic factors.2 CRP has been found to be a more sensitive and reproducible laboratory value in GCA, and, unlike ESR, is quick and easy to obtain. Hayreh et al29 demonstrated that compared to other laboratory values, CRP correlated the most with a positive TAB. Due to these advantages, CRP is considered a superior test to ESR and is a useful marker for diagnosis and disease monitoring in GCA patients.29 Studies have shown that having a CRP >2.45 mg/dL correlates with a 3.2–5.3 times higher chance of having GCA compared to CRP values <2.45 mg/dL.1,30 Finally, when monitoring response to treatment, CRP returns to normal more quickly than ESR.5
Interleukin 6
IL-6, a product of T-cells, B-cells, endothelial cells, fibroblasts, and macrophages, has been shown to be increased in inflamed arteries, potentiating the inflammatory responses of GCA.31 While Hayreh et al29 concluded that IL-6 was not a significantly more useful marker than CRP, Weyand et al6 found that compared to both ESR and CRP, IL-6 was a more sensitive biologic marker in predicting disease activity, found to be elevated (>6.1 pg/mL) in 92% of untreated patients.
Using IL-6 as a marker of GCA activity has several advantages over ESR. IL-6 is not affected by hematologic factors (ie, anemia, red cell morphology, plasma protein concentration), and is the specific inflammatory cytokine released during vascular insults.6 IL-6 also plays an important role in the production of downstream acute-phase reactants such as CRP; therefore, as IL-6 is more directly related to the mechanism of injury in GCA (vascular insult) and is upstream from markers like CRP, it is a more direct reflection of the disease activity in GCA.6
Thrombocytosis
Studies have suggested that elevated platelet counts may have predictive value for a positive TAB.30,32 Foroozan et al32 found that while an elevated platelet count (>400k) was a less sensitive marker than ESR, the presence of thrombocytosis may be a more specific marker than ESR with a specificity of 91%, far exceeding the specificity of ESR in their study. Furthermore, according to a study by Walvick et al,30 the odds of a positive TAB result were 1.5 times greater with an elevated ESR (47–107 mm/h), 5.3 times higher with elevated CRP (>2.45 mg/dL), and 4.2 times higher in patients with platelet counts >400,000/μL. The study concluded that the combination of an elevated CRP and platelet count was a better predictor of a positive biopsy than elevated ESR alone, and if all three lab values were elevated, then the odds of a positive TAB increased by eightfold.30 Therefore, it is recommended to obtain a complete blood count, ESR, and CRP in patients with suspected GCA.
Value of laboratories in monitoring treatment response
Compared to the clinical signs and symptoms of GCA, markers such as ESR and CRP are considered the more reliable and sensitive indicators of disease activity and relapses, often informing the course of corticosteroid treatment.1 The rate of corticosteroid taper and ideal maintenance dosing is, therefore, currently guided by the goal of achieving the lowest levels of ESR and CRP with the lowest dose of corticosteroids.1 While this is general practice, it should be mentioned that in their study, Kermani et al33 found that in 21% of patients, relapses were associated with normal ESR (defined as <20 mm/h) and normal CRP (defined as <5 mg/dL). Furthermore, studies suggest that even after initiation of corticosteroids, arterial inflammation often persists.6 As ESR often returns to normal levels soon after corticosteroid therapy has been started, ESR may not be the ideal sensitive marker for monitoring vascular inflammation.6 Therefore, using ESR and CRP to monitor disease activity and guide treatment decisions may not always be reliable. Furthermore, in their study assessing IL-6 as a biologic marker of disease activity, Weyand et al6 report that during disease flares, elevated ESR was only found in 58% of flares compared to an elevated IL-6 level in 89% of flares. This suggests that IL-6 may be a more sensitive alternative to ESR as an indicator of inadequate immunosuppression and disease relapse.
GCA patients with negative TAB
Although TAB is considered by many authors to be the “gold standard” for the diagnosis of GCA, a negative TAB does not completely rule out GCA as a diagnosis. According to a study by Bornstein et al,34 20.3% of those with a negative TAB were eventually diagnosed with GCA. Factors that contribute to the limitations of TAB include sampling error due to segmental nature, previous steroid use, and a phenotype not associated with cranial arteritis.34 It is of great importance to recognize these limitations as it has been estimated that 40% of GCA cases have a negative TAB result.34
Due to this limitation, according to Bornstein, diagnosis should be based more on clinical presentation, lab features, and response to high-dose corticosteroids.34 As stated previously, the ACR criteria should not be used as a sole diagnostic criteria, although the fulfillment of the criteria in combination with PMR-like symptoms increases the likelihood of GCA despite a negative TAB.34 In fact, it has been shown that 81.6% of GCA patients with a negative TAB fulfilled the ACR criteria.34 It is these TAB negative patients who might benefit most from concomitant TA ultrasound.
GCA patients with negative TABs have been studied by Bornstein et al to determine the best predictors for their diagnosis. These predictors were found to be fulfillment of the ACR criteria, clinical diagnosis of PMR, and thrombocytosis.34 The most common symptoms found in these patients at presentation were headache, constitutional symptoms, PMR, and anemia. It was also found that TAB negative patients tended to have higher rates of elevated ESR, platelet counts, liver function test levels, white blood cell counts, and jaw claudication.
Treatment of giant cell arteritis
Corticosteroids
Rapid and effective control of inflammation is of paramount importance. Since the 1950s, this has been achieved by the mainstay of GCA treatment: urgently administered high-dose corticosteroids.35 While corticosteroids are by no means a cure for GCA, ever since their introduction as a standard treatment for GCA, the incidence of blindness in patients with the disease has significantly decreased.6,36
In one study examining GCA patients with visual symptoms (ie, amaurosis fugax), 58% of patients whot were started on corticosteroids within 24 hours of visual symptom onset experienced improvement in those symptoms.37 However, in those patients with delayed corticosteroid initiation, only 6% experienced improvement in visual symptoms.37 Furthermore, in those patients who do not receive corticosteroids, up to 60% suffer vision loss in the contralateral eye, whereas if corticosteroids are given, that probability is decreased to 10%–20%.38 Prompt initiation of corticosteroids is, therefore, essential to improve the visual prognosis in patients with GCA.
Typically, corticosteroid therapy is promptly initiated if the patient’s signs, symptoms, and/or laboratory studies (ESR/CRP) intimate that GCA is the likely diagnosis.37,39 Treatment should not be delayed awaiting the results of the TAB.1 Initial dosing depends on the patient’s symptoms: in those without visual or neurologic symptoms, initial steroid dosing of 40–60 mg (not <0.75 mg/kg) per day is appropriate.40 In patients presenting with visual or neurologic symptoms (ie, jaw claudication, amaurosis fugax, and so on), however, higher doses consisting of either 1–1.5 mg/kg of oral prednisone per day or IV methylprednisone (1 g daily for 3–5 days) should be started.1,40,41 Patients with GCA should experience significant symptomatic improvement within 1–2 days of starting corticosteroid treatment; if they do not, this suggests that GCA may not have been the diagnosis.41
The question of whether these steroids should be delivered to the patient orally or intravenously still remains unanswered. Multiple studies have been performed to elucidate whether the method of corticosteroid delivery impacts outcomes in patients with GCA. In their randomized, double-blinded, placebo-controlled clinical trial, Mazlumzadeh et al42 found that initially treating patients with IV steroid pulses was associated with faster weaning of oral corticosteroids, higher rates of sustained remission after treatment cessation, and lower cumulative corticosteroid dosing. Also, in favor of initial treatment with IV corticosteroids, Chan et al43 found improved visual outcomes in those treated with high-dose (1000 mg per day) IV corticosteroids for 3 days vs those solely treated with oral corticosteroids. However, Hayreh and Zimmerman39 found no benefit to high-dose IV dexamethasone (450 mg per day) for 3 days followed by oral prednisone vs oral prednisone alone. Although evidence is conflicting, considering the increased bioavailability and higher dosing potential of intravenously administered CS, general opinion leans in the favor of using IV corticosteroids as induction therapy for GCA, especially in patients at high risk for vision loss.2,36 In patients with visual symptoms, we recommend initial treatment with IV methylprednisolone (1000 mg/day) for 3 days, followed by 3–4 weeks of oral prednisone (80–100 mg/day).36 For patients who would benefit from IV corticosteroids, hospitalization to closely monitor visual changes and corticosteroid-associated adverse effects is recommended.3
Duration of treatment
The duration of treatment with corticosteroids may last months to years and is determined by both resolution of patients’ symptoms and normalization of inflammatory markers (ESR/CRP).36 CRP typically stabilizes earlier than ESR.1,37 A gradual tapering of high-dose corticosteroids can be started once ESR and CRP are stabilized, eventually culminating in either complete weaning off of corticosteroids or finding a stable maintenance dose. Slower tapering is typically less likely to result in relapse of disease.3 The tapering regimen typically consists of a 10 mg decrease in dosage every 2 weeks until dose is 20 mg; dose is decreased by 2.5 mg every 2–4 weeks until reaching 10 mg. Afterwards, dose is decreased by 1 mg every 1–2 months.40 After inflammation has been appropriately suppressed, the goal of treatment is to maintain low levels of inflammatory markers with the lowest dose of prednisone.1 However, with increases in inflammatory markers, dosage is also immediately increased. Due to the individual variability of optimal corticosteroid dosage and time to reach lowest possible prednisone dose, maintenance therapy of GCA is customized for each patient.1 Recurrence of GCA is not uncommon during corticosteroid taper, with relapses occurring at least once in up to 50% of patients.1,31,44 For some patients, corticosteroid therapy may be discontinued within 1–2 years; however, due to the chronic relapsing nature of GCA, the duration of corticosteroid therapy in most GCA patients is indefinite.1,44 Therefore, patients with GCA in whom corticosteroid has been completely discontinued, periodic monitoring for relapses should still be performed.36
Unfortunately, long-term corticosteroid therapy is commonly associated with significant comorbidities related to the age of the patient and cumulative corticosteroid dosage.44 These complications include steroid-induced diabetes, arterial hypertension, osteoporosis, cataracts, infection, and psychosis.31,38 In fact, in the age group of GCA, 86% of patients treated with long-term corticosteroid therapy suffered from these corticosteroid-related complications within 10 years.38 Due to the need to consistently monitor for these significant adverse effects and flares, it is essential to consult with the patient’s primary care provider and/or rheumatologist.
In their article, Buttgereit et al45 review risk management for some of the most worrisome complications of corticosteroid therapy. It is essential to monitor and manage the risks of these adverse effects. For osteoporosis, risk should be assessed based on history and fracture risk assessment tool, and bone mineral densities and vitamin D levels may be included in risk monitoring. Lifestyle interventions such as physical exercise (specifically weight bearing), smoking cessation, limited alcohol, and increased dietary calcium should be encouraged. Unless contraindicated, all patients on high-dose corticosteroids should be referred for consideration for bone-protective therapies. The risk of steroid-induced osteoporosis may be managed with calcium and vitamin D supplementation, and possible preventative therapy with bisphosphonates. To best manage the risk of hyperglycemia, patients on corticosteroids should be encouraged to reduce their weight, consume a healthy diet, and exercise regularly. Monitoring by the primary care physicians or rheumatologists should include regular blood and urine testing. For the cardiovascular complications that may result from corticosteroid use, risk should be assessed per national guidelines, and patients should be encouraged to consume a healthy diet, exercise, restrict sodium intake, and quit smoking. For patients with high risk of cardiovascular complications, it is suggested to regularly monitor blood pressure and serum lipid panels before and after starting corticosteroids. The frequent complications of corticosteroid treatment highlight the need to develop steroid-sparing maintenance therapies. Therefore, several randomized, controlled trials have been performed to evaluate effective steroid-sparing regimens for the treatment of GCA.
Steroid-sparing agents
Tocilizumab
IL-6, a product of B-cells, T-cells, endothelial cells, macrophages, and fibroblasts, has been demonstrated to be elevated in the inflamed arteries affected by GCA.31 TCZ binds to both soluble and membrane-bound IL-6R and inhibits IL-6-mediated differentiation of naive TH cells to TH17 cells.1,46 The level of disease activity in GCA has been shown to correlate with the serum levels of IL-6, and in patients who were both treated and not treated with corticosteroids, IL-6 has been shown to be a more sensitive biologic marker for disease activity than ESR.6 On this basis, many studies, including a phase 2 trial, were performed to analyze the efficacy of the IL-6 receptor inhibitor, TCZ, and showed promise.47,48
Recently, the GiACTA trial confirmed these results via a multicenter, randomized, controlled trial.49 In this 1-year trial, 251 patients with GCA were randomly assigned to one of the treatment arms: a combination of TCZ weekly with 26-week prednisone taper, a combination of TCZ biweekly with 26-week prednisone taper, placebo with a 26-week prednisone taper, and placebo with 52-week prednisone taper.31 Disease remission was determined by normalization of CRP (<1 mg/dL) and absence of flare, which was defined as ESR >30 mm/h or relapse of the clinical signs and symptoms of GCA, as well as the necessity to increase prednisone dose. Sustained remission was defined by consistent remission between week 12 and 52 while adhering to prednisone taper. Outcomes measured at 52 weeks showed that 56% and 53% of patients receiving TCZ weekly and biweekly, respectively, achieved and sustained remission, compared to 14% and 18% of those on placebo and 26-week and 52-week prednisone taper, respectively. The TCZ treatment arms also had decreased rates of flare (23% of those on weekly TCZ, 26% on biweekly TCZ) compared to placebo (68% of those on 26-week taper, 49% on 52-week taper). Furthermore, in arms receiving TCZ, rate of adverse events was lower than those in the placebo group. Some criticisms have arisen since the publication of the GiACTA trial.50 While follow-up studies are needed to evaluate safety and efficacy of TCZ beyond 52 weeks, TCZ has been proven by randomized trial to be superior to placebo and prednisone regimens in maintaining remission and was associated with reduced cumulative corticosteroid dosage over a 52-week period.49
Anti-TNF agents
TNF alpha is a product of the activated macrophages and plays a role by promoting expression of various adhesion molecules and promoting leucocyte infiltration. It also upregulates MMP activity that can directly cause endothelial damage. Hence, blocking the TNF alpha inhibitors can play an important role limiting the inflammation-mediated damage in GCA.51,52
Numerous randomized, controlled trials have shown that anti-TNF agents are not effective in maintaining or inducing remission in patients with GCA. In their prospective double-blind placebo-controlled trial, Martínez-Tabaoada et al53 demonstrated that after 12 months, 50% of patients receiving etanercept were able to successfully wean off corticosteroids compared to 22.2% of patients in the placebo group; however, the P-value was not significant. Seror et al54 also conducted a multicenter, randomized, controlled trial to examine adalimumab as a potential steroid-sparing agent in the treatment of GCA. Results of this study showed that among the 70 patients enrolled, adding a 10-week course of adalimumab did not improve remission rates in patients on <0.1 mg/kg of corticosteroids at 6 months.54 The results of a randomized, controlled trial showed that the anti-TNF infliximab was also found not to decrease relapse rates, nor did it decrease relapse rates in patients tapered to 10 mg/day of corticosteroids.55
Methotrexate
Three prospective randomized, controlled trials have been performed to assess the efficacy of methotrexate in the treatment of GCA. Two of these studies yielded similar conclusions: during corticosteroid taper, no significant difference was found in cumulative dose or duration of corticosteroid therapy in patients assigned to the methotrexate group vs the placebo group.56,57 In the third study, however, it was found that compared to a regimen of corticosteroid and placebo, the treatment of patients with a combination of corticosteroid and methotrexate yielded lower rates of relapse (45% vs 84.2%).58 Finally, in a meta-analysis by Mahr et al59 of 161 patients, use of low-dose methotrexate as an adjunctive therapy was found to result in a reduction in cumulative corticosteroid dose and higher rates of maintaining steroid-free remission.
Abatacept
In a recent multicenter, randomized, withdrawal-design trial, Langford et al35 analyzed the efficacy of the fusion protein, abatacept, in the treatment of GCA. Abatacept consists of the extracellular-ligand-binding domain of CTLA-4 and a modified Fc region of IgG1. By binding CD80 and CD86 with its CTLA-4 component, abatacept prevents CD28-mediated T-cell co-stimulation.35 While it has been approved by the United States Food and Drug Administration in the treatment of rheumatoid arthritis and juvenile idiopathic arthritis, until 2017, no randomized, controlled trials had been performed to assess the efficacy of the fusion protein in the treatment of GCA. In the study, 41 GCA patients who attained remission on standardized prednisone taper (down to 20 mg/day) and abatacept by week 12 were randomized to 2 groups: 1 continued to receive abatacept, while the other received placebo. Prednisone was then discontinued at week 28. Results revealed that in the abatacept arm, the relapse-free rate at 12 months was 48% vs 31% of those receiving placebo. Furthermore, a significantly longer median period of remission was associated with the abatacept (9.9 months) vs the placebo group (3.9 months). Finally, no difference was found between abatacept arm and placebo arm in rate or severity of adverse events (ie, infection).
Conclusion
In summary, clinicians should consider GCA in the differential diagnosis of elderly patients with acute pain in the distribution of the external carotid artery (eg, headache, scalp tenderness); PMR; or acute/transient visual loss or diplopia. Prompt laboratory evaluation (eg, ESR, CRP, platelet count) followed by empiric high-dose corticosteroid therapy is warranted in patients suspected of having GCA. Although ultrasound techniques are improving for the diagnosis of GCA, TAB remains the current best confirmatory test for GCA. TA ultrasound, however, may have higher sensitivity and may play a role in TAB negative patients or in monitoring of clinical response to treatment or for GCA relapse. Patients with GCA often require long durations of steroid therapy and steroid-related complications are common. The management of these side effects may require multidisciplinary care and the need of steroid-sparing regimens.
Disclosure
The authors report no conflicts of interest in this work.
References
Hayreh SS, Zimmerman B. Management of giant cell arteritis. Ophthalmologica. 2003;217(4):239–259. | ||
Rahman W, Rahman FZ, Cell G. Giant cell (temporal) arteritis: an overview and update. Surv Ophthalmol. 2005;50(5):415–428. | ||
Hoffman GS, Arteritis GC. Giant cell arteritis. Ann Intern Med. 2016;165(9):ITC65. | ||
El-Dairi MA, Chang L, Proia AD, Cummings TJ, Stinnett SS, Bhatti MT. Diagnostic algorithm for patients with suspected giant cell arteritis. J Neuroophthalmol. 2015;35(3):246–253. | ||
Salvarani C, Cantini F, Boiardi L, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med. 2002;347(4):261–271. | ||
Weyand CM, Fulbright JW, Hunder GG, Evans JM, Goronzy JJ. Treatment of giant cell arteritis: interleukin-6 as a biologic marker of disease activity. Arthritis Rheum. 2000;43(5):1041. | ||
Patil P, Williams M, Maw WW, et al. Fast track pathway reduces sight loss in giant cell arteritis: results of a longitudinal observational cohort study. Clin Exp Rheumatol. 2015;33(2 Suppl 89):S-103-6. | ||
Hocevar A, Rotar Z, Jese R, et al. Do early diagnosis and glucocorticoid treatment decrease the risk of permanent visual loss and early relapses in giant cell arteritis: a prospective longitudinal study. Medicine. 2016;95(14):e3210. | ||
Diamantopoulos AP, Haugeberg G, Lindland A, Myklebust G. The fast-track ultrasound clinic for early diagnosis of giant cell arteritis significantly reduces permanent visual impairment: towards a more effective strategy to improve clinical outcome in giant cell arteritis? Rheumatology. 2016;55(1):66–70. | ||
Hunder GG, Bloch DA, Michel BA, et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990;33(8):1122–1128. | ||
Cid MC. 3. Pathogenesis of giant cell arteritis. Rheumatology. 2014;53(suppl 2):i2–i3. https://doi.org/. | ||
Hernández-Rodríguez J, Segarra M, Vilardell C, et al. Tissue production of pro-inflammatory cytokines (IL-1beta, TNFalpha and IL-6) correlates with the intensity of the systemic inflammatory response and with corticosteroid requirements in giant-cell arteritis. Rheumatology. 2004;43(3):294–301. | ||
Sultan H, Smith SV, Lee AG, Chévez-Barrios P. Pathologic markers determining prognosis in patients with treated or healing giant cell arteritis. Am J Ophthalmol. 2018;193:45–53. | ||
Borchers AT, Gershwin ME. Giant cell arteritis: a review of classification, pathophysiology, geoepidemiology and treatment. Autoimmun Rev. 2012;11(6–7):A544–A554. | ||
Watts RA. 2. Epidemiology of giant cell arteritis: a critical review. Rheumatology. 2014;53(suppl 2):i1–i2. | ||
Gonzalez-Gay MA, Vazquez-Rodriguez TR, Lopez-Diaz MJ, et al. Epidemiology of giant cell arteritis and polymyalgia rheumatica. Arthritis Rheum. 2009;61(10):1454–1461. | ||
Larsson K, Mellström D, Nordborg E, Nordborg C, Odén A, Nordborg E. Early menopause, low body mass index, and smoking are independent risk factors for developing giant cell arteritis. Ann Rheum Dis. 2006;65(4):529–532. | ||
Gonzalez-Gay MA, Barros S, Lopez-Diaz MJ, Garcia-Porrua C, Sanchez-Andrade A, Llorca J. Giant cell arteritis: disease patterns of clinical presentation in a series of 240 patients. Medicine. 2005;84(5):269–276. | ||
Hayreh SS, Podhajsky PA, Zimmerman B. Ocular manifestations of giant cell arteritis. Am J Ophthalmol. 1998;125(4):509–520. | ||
Kawasaki A, Purvin V. Giant cell arteritis: an updated review. Acta Ophthalmol. 2009;87(1):13–32. | ||
Schmidt WA, Kraft HE, Vorpahl K, Völker L, Gromnica-Ihle EJ. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med. 1997;337(19):1336–1342. | ||
Schmidt WA. Ultrasound in the diagnosis and management of giant cell arteritis. Rheumatology. 2018;57(suppl_2):ii22–ii31. | ||
Aranda-Valera IC, García Carazo S, Monjo Henry I, de Miguel Mendieta E. Diagnostic validity of Doppler ultrasound in giant cell arteritis. Clin Exp Rheumatol. 2017;35 Suppl 103(1):123–127. | ||
Slart RHJA, Members of Committees, SNMMI Cardiovascular, EANM Committee Coordinator, et al. FDGPET/CT(A) imaging in large vessel vasculitis and polymyalgia rheumatica: joint procedural recommendation of the EANM, SNMMI, and the PET Interest Group (PIG), and endorsed by the ASNC. Eur J Nucl Med Mol Imaging. 2018;45:1250–1269. | ||
Soussan M, Nicolas P, Schramm C, et al. Management of large-vessel vasculitis with FDG-PET: a systematic literature review and meta-analysis. Medicine. 2015;94(14):e622. | ||
Vitiello G, Orsi Battaglini C, Carli G, et al. Tocilizumab in giant cell arteritis: a real-life retrospective study. Angiology. 2018;69(9):763–769. | ||
Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurrence in a population-based study. Arthritis Rheum. 2001;45(2):140–145. | ||
Miller A, Green M, Robinson D. Simple rule for calculating normal erythrocyte sedimentation rate. BMJ. 1983;286(6361):266. | ||
Hayreh SS, Podhajsky PA, Raman R, Zimmerman B. Giant cell arteritis: validity and reliability of various diagnostic criteria. Am J Ophthalmol. 1997;123(3):285–296. | ||
Walvick MD, Walvick MP. Giant cell arteritis: laboratory predictors of a positive temporal artery biopsy. Ophthalmology. 2011;118(6):1201–1204. | ||
Unizony SH, Dasgupta B, Fisheleva E, et al. Design of the tocilizumab in giant cell arteritis trial. Int J Rheumatol. 2013;2013:1–10. | ||
Foroozan R, Danesh-Meyer H, Savino PJ, Gamble G, Mekari-Sabbagh ON, Sergott RC. Thrombocytosis in patients with biopsy-proven giant cell arteritis. Ophthalmology. 2002;109(7):1267–1271. | ||
Kermani TA, Warrington KJ, Cuthbertson D, et al. Disease relapses among patients with giant cell arteritis: a prospective, longitudinal cohort study. J Rheumatol. 2015;42(7):1213–1217. | ||
Bornstein G, Barshack I, Koren-Morag N, Ben-Zvi I, Furie N, Grossman C. Negative temporal artery biopsy: predictive factors for giant cell arteritis diagnosis and alternate diagnoses of patients without arteritis. Clin Rheumatol. 2018;37(10):2819–2824. | ||
Langford CA, Cuthbertson D, Ytterberg SR, et al. A randomized, double-blind trial of abatacept (CTLA-4Ig) for the treatment of giant cell arteritis. Arthritis Rheumatol. 2017;69(4):837–845. | ||
Almarzouqi SJ, Morgan ML, Lee AG. Treatment of giant cell arteritis. Curr Opin Ophthalmol. 2015;26(6):469–475. | ||
Eberhardt RT, Dhadly M. Giant cell arteritis: diagnosis, management, and cardiovascular implications. Cardiol Rev. 2007;15(2):55–61. | ||
Ness T, Bley TA, Schmidt WA, Lamprecht P. The diagnosis and treatment of giant cell arteritis. Dtsch Arztebl Int. 2013;110(21):376. | ||
Hayreh SS, Biousse V. Treatment of acute visual loss in giant cell arteritis: should we prescribe high-dose intravenous steroids or just oral steroids? J Neuroophthalmol. 2012;32(3):278–287. | ||
Caylor TL, Perkins A. Recognition and management of polymyalgia rheumatica and giant cell arteritis. Am Fam Physician. 2013;88(10):676–684. | ||
Chatterjee S, Flamm SD, Tan CD, Rodriguez ER. Clinical diagnosis and management of large vessel vasculitis: giant cell arteritis. Curr Cardiol Rep. 2014;16(7):498. | ||
Mazlumzadeh M, Hunder GG, Easley KA, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: a double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum. 2006;54(10):3310–3318. | ||
Chan CC, Paine M, O’Day J. Steroid management in giant cell arteritis. Br J Ophthalmol. 2001;85(9):1061–1064. | ||
Ferfar Y, Mirault T, Desbois AC, et al. Biotherapies in large vessel vasculitis. Autoimmun Rev. 2016;15(6):544–551. | ||
Buttgereit F, Matteson EL, Dejaco C, Dasgupta B. Prevention of glucocorticoid morbidity in giant cell arteritis. Rheumatology. 2018;57(suppl_2): ii11–ii21. | ||
Fujimoto M, Serada S, Mihara M, et al. Interleukin-6 blockade suppresses autoimmune arthritis in mice by the inhibition of inflammatory Th17 responses. Arthritis Rheum. 2008;58(12):3710–3719. | ||
Villiger PM, Adler S, Kuchen S, et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387(10031):1921–1927. | ||
Loricera J, Blanco R, Hernández JL, et al. Tocilizumab in giant cell arteritis: Multicenter open-label study of 22 patients. Semin Arthritis Rheum. 2015;44(6):717–723. | ||
Stone JH, Tuckwell K, Dimonaco S, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med Overseas Ed. 2017;377(4):317–328. | ||
Walker UA. Trial of tocilizumab in giant-cell arteritis. N Engl J Med. 2017;377(15):1493–1495. | ||
Jarrot PA, Kaplanski G. Anti-TNF-alpha therapy and systemic vasculitis. Mediators Inflamm. 2014;2014:1–9. | ||
Henderson CF, Seo P. Biologic agents in systemic vasculitis. Int J Clin Rheumtol. 2011;6(4):453–462. | ||
Martínez-Taboada VM, Rodríguez-Valverde V, Carreño L, et al. A double-blind placebo controlled trial of etanercept in patients with giant cell arteritis and corticosteroid side effects. Ann Rheum Dis. 2008;67(5):625–630. | ||
Seror R, Baron G, Hachulla E, et al. Adalimumab for steroid sparing in patients with giant-cell arteritis: results of a multicentre randomised controlled trial. Ann Rheum Dis. 2014;73(12):2074–2081. | ||
Hoffman GS, Cid MC, Rendt-Zagar KE, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med. 2007;146(9):621. | ||
Spiera RF, Mitnick HJ, Kupersmith M, et al. A prospective, double-blind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol. 2001;19(5):495. | ||
Hoffman GS, Cid MC, Hellmann DB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum. 2002;46(5):1309–1318. | ||
Jover JA, Hernández-García C, Morado IC, Vargas E, Bañares A, Fernández-Gutiérrez B. Combined treatment of giant-cell arteritis with methotrexate and prednisone. Ann Intern Med. 2001;134(2):106. | ||
Mahr AD, Jover JA, Spiera RF, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum. 2007;56(8):2789–2797. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.