Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 11
Geographic Variability and Pathogen-Specific Considerations in the Diagnosis and Management of Chronic Granulomatous Disease
Authors Prince BT, Thielen BK ![]() , Williams KW
, Williams KW ![]() , Kellner ES, Arnold DE, Cosme-Blanco W, Redmond MT, Hartog NL
, Kellner ES, Arnold DE, Cosme-Blanco W, Redmond MT, Hartog NL ![]() , Chong HJ, Holland SM
, Chong HJ, Holland SM ![]()
Received 21 April 2020
Accepted for publication 26 June 2020
Published 22 July 2020 Volume 2020:11 Pages 257—268
DOI https://doi.org/10.2147/PHMT.S254253
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Roosy Aulakh
Benjamin T Prince,1 Beth K Thielen,2 Kelli W Williams,3 Erinn S Kellner,4 Danielle E Arnold,5 Wilfredo Cosme-Blanco,6 Margaret T Redmond,1 Nicholas L Hartog,7 Hey J Chong,8 Steven M Holland9
1Division of Allergy and Immunology, Nationwide Children’s Hospital, The Ohio State University College of Medicine, Columbus, OH, USA; 2Division of Pediatric Infectious Diseases and Immunology, University of Minnesota, Minneapolis, MN, USA; 3Department of Pediatrics, Division of Pediatric Pulmonology, Allergy & Immunology, Medical University of South Carolina, Charleston, SC, USA; 4Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA; 5Division of Allergy and Immunology, Children’s Hospital of Philadelphia, Philadelphia, PA, USA; 6Department of Allergy and Immunology, Veteran Affairs Caribbean Healthcare System, San Juan, Puerto Rico; 7Department of Allergy and Immunology, Spectrum Health Helen DeVos Children’s Hospital, Michigan State University College of Human Medicine, Grand Rapids, MI, USA; 8Division of Allergy and Immunology, UPMC Children’s Hospital of Pittsburgh, Pittsburgh, PA, USA; 9National Institute of Allergy and Infectious Diseases, Bethesda, Maryland National Institutes of Health, Bethesda, MD, USA
Correspondence: Benjamin T Prince
Nationwide Children’s Hospital, 700 Children’s Drive, Columbus, OH 43205, USA
Tel +1 (614) 722-4404
Fax +1 (614) 722-4423
Email [email protected]
Abstract: Chronic granulomatous disease (CGD) is a rare but serious primary immunodeficiency with varying prevalence and rates of X-linked and autosomal recessive disease worldwide. Functional defects in the phagocyte nicotinamide adenine dinucleotide phosphate oxidase complex predispose patients to a relatively narrow spectrum of bacterial and fungal infections that are sometimes fastidious and often difficult to identify. When evaluating and treating patients with CGD, it is important to consider their native country of birth, climate, and living situation, which may predispose them to types of infections that are atypical to your routine practice. In addition to recurrent and often severe infections, patients with CGD and X-linked female carriers are also susceptible to developing many non-infectious complications including tissue granuloma formation and autoimmunity. The DHR-123 oxidation assay is the gold standard for making the diagnosis and it along with genetic testing can help predict the severity and prognosis in patients with CGD. Disease management focuses on prophylaxis with antibacterial, antifungal, and immunomodulatory medications, prompt identification and treatment of acute infections, and prevention of secondary granulomatous complications. While hematopoietic stem-cell transplantation is the only widely available curative treatment for patients with CGD, recent advances in gene therapy may provide a safer, more direct alternative.
Keywords: CGD, primary immunodeficiency diseases, nicotinamide adenine dinucleotide phosphate oxidase complex, DHR-123 oxidation assay
Introduction
Chronic granulomatous disease (CGD) is a rare primary immunodeficiency that is caused by defects in the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, which is critical for superoxide production. The disease was initially described in 1957 and appropriately named “fatal granulomatous disease of childhood” as all 4 patients died before 6 years of age.1 Although it was initially thought to solely be an X-linked disease, the first females were diagnosed in 1968, leading to the identification of autosomal recessive (AR) forms.2 Affected individuals develop recurrent and severe infections from a narrow range of characteristic bacteria and fungi and suffer from granulomatous inflammation that has become pathognomonic for the disease. Since its original description, improvements in antibacterial, antifungal, and immunomodulatory prophylaxis as well as hematopoietic stem-cell transplantion (HSCT) have allowed patients to live well into adulthood. Herein, we will review important concepts in the diagnosis and management of CGD as well as the genetic and geographic variability of the disease.
Epidemiology and Pathophysiology
CGD has a minimum estimated prevalence of 1 in 200,000–250,000 live births in the United States (US).3,4 Around the world, the prevalence is somewhat variable. While Japan appears to have a similar rate to the US, the prevalence of CGD has been reported to be as high as 3.43 in 100,000 live births in Jeju Island, Korea, due in part because of a common ancestor. In Israel, the prevalence of CGD is estimated to be 1.17 in 100,000 live births and is predominantly autosomal recessive.5 Even within Europe, however, the prevalence ranges from 0.1 in 100,000 in Italy to 0.85 in 100,000 live births in the United Kingdom and Ireland.6,7
The NADPH oxidase complex is comprised of 5 protein subunits and is responsible for generating the reactive oxygen species necessary for phagocyte killing of bacteria and fungi both directly and by the activation of intraphagosomal proteases.8,9 Pathogenic variants in any of the 5 genes that encode the 2 membrane-bound subunits (gp91phox encoded by CYBB and p22phox encoded by CYBA) or the 3 cytosolic subunits (p47phox encoded by NCF1, p67phox encoded by NCF2, and p40phox encoded by NCF4) of the complex lead to the clinical presentations that are CGD.10,11 Recently, biallelic, loss-of-function mutations in CYBC1 have also been found to cause decreased NADPH function resulting in CGD.12,13 While CYBC1 is not directly involved in the production and release of reactive oxygen species, it appears to be essential for gp91phox and p22phox dimerization and formation of the NADPH complex.12
Pathogenic variants in the CYBB gene result in the X-linked form of the disease. This is the most common form of CGD worldwide with most cases being hemizygous males.10 X-linked, female carriers of CYBB mutations are usually protected from typical CGD infections since they have two separate neutrophil populations secondary to random X chromosome lyonization – one with normal and the other with aberrant NADPH oxidase function.14,15 Some carriers, however, demonstrate skewed X chromosome lyonization, in which less than 20% of their phagocytes express the wild type CYBB gene. These patients can present with CGD-like manifestations often later in life, with carriers that have less than 10% normal phagocytes having the highest risk for severe infection.15–20
Biallelic pathogenic variants in NCF1, the most common AR cause of the disease, tend to be associated with a better prognosis compared to CYBB and NCF2, and account for approximately 25% of CGD cases in North America and Europe.3,11 Together, CYBB and NCF1 variants account for 75–90% of cases of CGD in North American, European, and Japanese populations.3,4,6,21 In contrast, areas with higher rates of consanguinity have predominantly AR disease.4,5,22,23 Cohorts have shown that in populations with >50% rates of consanguinity, 62–100% cases of CGD are secondary to autosomal recessive forms of the disease.5,22-24
Phenotype-Genotype Considerations
Phenotypic differences have been described between X-linked and AR disease. In X-linked disease, the mean age of symptom onset is between 4 and 14 months of age, and the mean age of diagnosis is 2.7–3.1 years.4,6,7,24 In comparison, AR disease often presents later, with a mean symptom onset at 30–36 months of age and mean age of diagnosis at 5.8 years-7.8 years.4,6 The difference in the age at diagnosis between X-linked and AR CGD persists even when controlling for gender.4 Patients with X-linked disease also have a more frequent and severe infections leading to a higher mortality compared to AR disease.4
In addition to phenotypic differences seen between X-linked and AR disease, there can be variability in presentation resulting from how the specific mutations affect gene function. Studies have shown the level of phagocyte-derived reactive oxygen intermediate (ROI) production is inversely proportional to survival and is the most important factor in predicting disease severity regardless of the gene affected.25 Generally, patients with nonsense, frameshift, and splice-site variants, or deletions are more likely to have decreased residual ROI production and have worse survival than patients with missense variants. The specific gene that is defective can also have a variable effect on ROI production. Individuals with defects in NCF4 and CYBC1 typically have more residual superoxide production and present with more mild infections compared to those with defects in the 4 other known genetic causes of CGD.26 Information regarding a patient’s ROI and specific genetic variant can be beneficial in guiding clinical decision-making and providing individualized patient care. These values can often be derived directly from the sequence data.
Clinical Presentation
Infection continues to be the most common clinical feature of the disease and the leading cause of death in patients with CGD.4,27 The majority of patients present with characteristic infections of the skin, lymph nodes, lung, liver, and bone before 5 years of age. Depending on the genotype and residual ROI production, however, diagnosis may be delayed well until the third decade of life.4
Skin, soft tissue infections, and lymphadenitis are frequently the initial presenting sign of CGD.7,28 In Middle Eastern countries, lymphadenitis is the most common infection overall.4,29 While Staphylococcus aureus, Serratia marcescens, and Klebsiella spp are common causes of skin and lymph node infections, occasionally no organism is identified. In these cases, careful consideration must be taken to identify more unusual and fastidious organisms (see pathogen-specific considerations below). Pulmonary infections are generally the most common type of infection in CGD patients overall, primarily presenting as pneumonia, although lung abscesses and empyemas have also been reported.27 Recurrent pulmonary infections can lead to chronic complications such as fibrosis, pleural thickening, pulmonary hypertension, and bronchiectasis. Mulch pneumonitis, a severe inflammatory response to inhaled fungal elements in decayed organic matter, can also be a presenting sign of CGD.30,31 Aspergillus spp. are the most common causes of pneumonia in CGD, but other organisms, including Staphylococcus spp. and Burkholderia cepacia complex, may be identified. Liver abscesses, primarily due to S. aureus, are an important cause of morbidity in CGD, and often present with non-specific symptoms of fever and an elevated erythrocyte sedimentation rate.28 Similar to skin and lymph node infections, development of a liver abscess may lead to the diagnosis of CGD.
In addition to recurrent infections, patients with CGD have aberrant inflammation leading to the formation of granulomata and can rarely present with associated macrophage activation syndrome.32,33 While granuloma formation can occur in a variety of organs, it most commonly affects the gastrointestinal and genitourinary tracts.34,35 Granulomatous colitis is commonly seen in children with CGD and nearly half of the patients with the disease eventually develop inflammatory bowel disease (IBD).34 Gastrointestinal inflammation can present with broad symptoms of fever, abdominal pain, diarrhea, rectal bleeding, and weight loss or in some cases bowel obstruction.36 Within the genitourinary tract, granulomatous inflammation can lead to outlet obstruction and in rare cases eosinophilic cystitis.35,37 It is important to consider CGD in any patient presenting with granuloma formation as it can be an early clue to establishing a diagnosis of CGD.4
Patients with CGD and X-linked female carriers are both at increased risk for developing a wide range of autoimmune conditions. These manifestations do not seem to have a specific age or geographic predisposition with 5–15% of CGD patients being affected and up to two-thirds of X-linked carriers reporting symptoms in some studies.36,38 Commonly reported autoimmune conditions include the following: discoid and systemic lupus erythematosus, rheumatoid arthritis, immunoglobulin A (IgA) nephropathy, chorioretinitis, idiopathic thrombocytopenia (ITP), autoimmune hepatitis, Raynaud phenomenon, and stomatitis.36 It is important to note that ROI production does not seem to be related to the risk of autoimmune disease.15
Pathogen Variability Within the United States
Recent comprehensive data on the pathogens causing infection in patients with CGD in the US primarily come from two single-center cohorts.27,39 There are also data prior to 2000 that came from a national registry of patients with CGD.4 It is notable that both of the more recent single-center cohorts included patients who were diagnosed either because of affected siblings or IBD but had never had a serious infection (3–11%), which was not reported in the older registry data.
The pathogen profile in patients with CGD is small, but those few pathogens cause a high percentage of infections. Most bacteria causing infection in CGD are catalase positive, as are most pathogenic bacteria in general. Catalase negative bacteria can also cause infection however, and catalase is not necessary for pathogenicity in CGD model systems.40 Filamentous molds are the most common fungal pathogens in patients with CGD, but patients do not show an increased susceptibility to the dimorphic molds like coccidioidomycosis, histoplasmosis, or blastomycosis that are endemic in the United States.31
Aspergillus fumigatus is the most common fungal pathogen. Other Aspergillus species including A. nidulans and A. tanneri have also been isolated and have been associated with increased azole resistance.41 With the advent of fungal prophylaxis, several species of non-Aspergillus fungi have been found to cause serious infection in patients with CGD and also have increased azole resistance, including Phellinus spp., Paecilomyces variotti, Paecliomyces lilacinus, and Neosartorya udagawae.31,42 Fungal pathogens are associated with higher mortality than bacterial infections in patients with CGD. In the larger cohort study in the US, 55% of non-stem cell transplant-related deaths were caused by fungal infections.27
In CGD, the most common bacterial pathogens in the United States are S. aureus, Serratia marcescens, Burkholderia cepacia complex, Nocardia spp., and Klebsiella spp.27,39 Both cohort studies revealed a higher fatality rate with Burkholderia than other bacterial infections. Other bacteria found in soil and brackish water that uncommonly cause disease in healthy individuals but are more common pathogens in CGD are Granulibacter bethesdensis, Chromobacterium violaceum, and Francisella philomiragia.43–45 While most of the cases of Chromobacterium violaceum within the US have been primarily described in the southeastern United States, this facultatively anaerobic, Gram-negative bacillus can also be found in tropical and sub-tropical soil worldwide.44,46 Granulibacter bethesdensis, a fastidious Gram-negative organism that causes indolent lymphadenitis and splenitis in CGD occurs in the US and Europe, but its natural reservoir is unknown.45
Pathogen Variability Outside of the United States
The pathogen profile of CGD outside the United States varies greatly depending on vaccination history, antibiotic prophylaxis, country of origin, and likely other incompletely understood factors. When evaluating infectious symptoms in patients with CGD, it is important to consider their country of birth, climate, and living situation, which may predispose them to types of infections that may be atypical to your routine practice. While there does not appear to be an ethnic predisposition, it is also important to consider cultural practices as incidence rates may be higher in cultures where consanguinity is more common.4,5,22,23
Similar to patients with CGD who are born in the United States, Aspergillus spp. and/or Staphylococcus spp. were the most common microorganisms cultured in cohorts from Europe, Northern India, Israel, Iran, Latin America, and Turkey.7,22,23,47-51 Unlike the United States., Salmonella spp. had higher incidences in Europe and Israel and were found to be the most frequent causes of septicemia in both of these studies.47,49 Higher rates of Pseudomonas spp. were seen in CGD patients from Northern India and Israel.48,49 Also, dissimilar to CGD patients within the United States was the low rate of Burkholderia cepacia in the European CGD cohorts.7,47 The biggest difference in presentation of pathogens, however, is in Bacillus Calmette-Guerin (BCG)-related disease and tuberculosis, which is more frequently found outside of the United States.
Tuberculosis continues to be one of the top ten causes of death and the leading single infectious cause of death among all individuals worldwide.52 Incidence rates are low in the United States and new cases are more frequently seen in patients who are non-U.S.-born.53 The BCG vaccine is a live attenuated strain of M. bovis that was first used in humans in 1921 to help combat tuberculosis in children, and it continues to be the only vaccine currently used to protect against this disease.54 The World Health Organization recommends vaccination of infants at birth or as soon thereafter to protect against meningeal and miliary tuberculosis in endemic areas.55 Patients with CGD and other immune deficiencies are susceptible to adverse outcomes from the vaccine, ranging from regional disease like BCG lymphadenitis to disseminated BCGosis.56 Many European countries no longer give the BCG vaccine except to high-risk patients. Among reports from countries who still give BCG, the incidence of BCG adenitis ranged widely from 16.6% to 59.2% (Table 1).22,23,50,51,57,58 The BCG World Atlas is a helpful database to help determine BCG status of patients from other countries.59
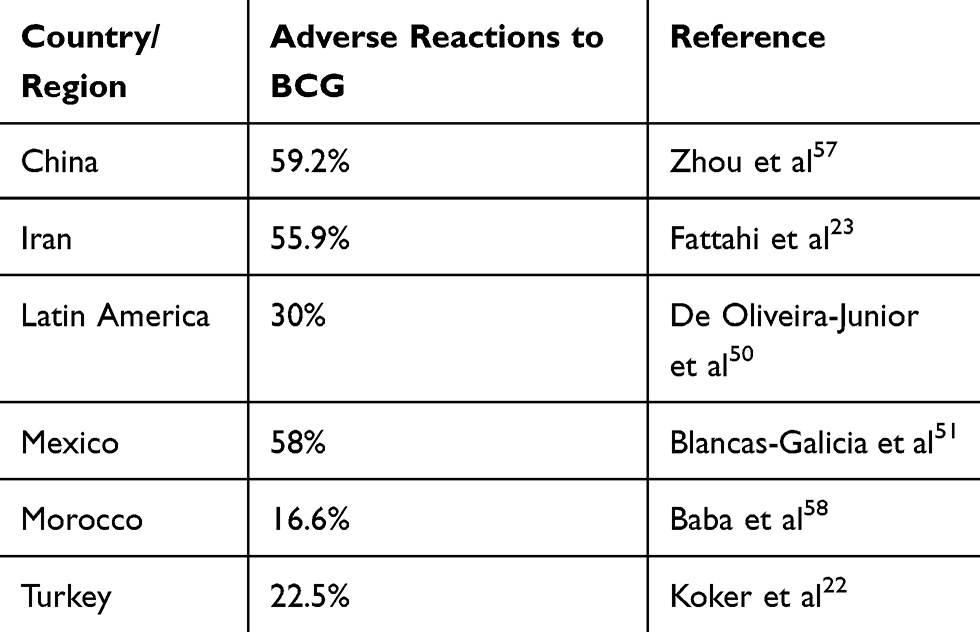 |
Table 1 CGD Patients with High Rates of Reactions to BCG in Countries with a National Vaccination Policy |
CGD patients living in areas endemic for tuberculosis have seen rates of infection that exceeds what is considered usual for those populations.51,60,61 In this population, pulmonary tuberculosis was the most common manifestation followed by peripheral lymph node involvement; however, a subset of patients with CGD can present with more disseminated, recurrent, and severe disease.51,61 In some of these endemic areas that are resource poor, the inability to distinguish BCG from tuberculosis has led to a possibly higher rate of mortality in CGD patients compared to what is seen in other countries.62
Pathogen Specific Considerations
Many of the common CGD pathogens discussed in preceding sections (Aspergillus spp. Staphylococcus aureus, Burkholderia cepacia, Serratia marcescens, Nocardia spp., Klebsiella spp.) can present with varied clinical manifestations with substantial overlap in disease manifestations across these organisms.4,7,27,39 Nevertheless, certain organisms are more commonly associated with particular disease manifestations. For example, Aspergillus most frequently causes pulmonary disease, ranging from asymptomatic imaging findings to locally invasive disease involving the chest wall.63 It is also among the top causes of osteomyelitis and brain abscesses.4,64 In contrast, Staphylococcus aureus and Serratia marsescens are more frequently isolated from skin abscesses, lymph nodes and liver abscesses.4,27 In addition to well-known pathogens, new pathogens are being identified with the advent of molecular diagnostics. For example, a syndrome of fever and lymphadenitis refractory to typical broad-spectrum antibacterials and prone to relapse was associated with Granulibacter bethesdensis infection.43,65
While some patients with CGD develop fungemia or bacteremia as a result of their infections, the majority of these infections are localized and require invasive sampling of involved tissues (eg, bronchoalveolar lavage, lung and bone biopsies, lumbar puncture for cerebrospinal fluid, and abscess aspirates) to establish a microbiological diagnosis and guide antimicrobial therapy. The most frequent pathogens associated with CGD can be grown on aerobic bacterial media and in fungal cultures. Acid-fast smears and cultures are also important as CGD patients may have infections with both tuberculous and non-tuberculous mycobacteria. However, despite invasive testing, a microbiological diagnosis is often elusive in half or more of cases.7,39 This is likely multifactorial, including infection due to fastidious organisms that are difficult to culture and/or inhibition of organism growth due to antimicrobial exposures prior to obtaining diagnostic specimens. With some organisms, it can also be difficult to discern whether they are truly pathogenic or a colonizer. For example, Actinomyces is a common colonizer of the gastrointestinal tract and Paecilomyces is a common environmental saprophyte, but both have also been described as pathogenic in CGD.40,66 Cytology and histopathology provide important clues to the presence of fastidious or non-viable organisms. For example, Granulibacter bethesdensis was initially detected by special Warthin-Starry stain and subsequently cultured using buffered charcoal yeast extract agar.43
More recently, additional culture-independent techniques have been utilized to increase the rates of microbiological diagnosis. For common pathogens, targeted testing for specific microbial products can be a helpful adjunct to traditional culture. For Aspergillus, detection of galactomannan antigen and Aspergillus by PCR in blood and bronchoalveolar lavage have high sensitivity and specificity for the diagnosis of invasive aspergillosis in BAL specimens.67
For detection of rare and novel pathogens, additional approaches are needed and becoming more readily available. One such technique involves amplification and sequencing of a conserved region of the bacterial 16S or fungal 28S ribosomal RNA. This can lead to the identification of a causative organism either from culture or directly from specimens.43,68 More recently, metagenomic sequencing of whole blood and bronchoalveolar lavage fluid has been used for the detection of pathogens with the potential to detect a broader range of pathogens and mixed pathogens.69,70 While these techniques hold promise for diagnosing challenging infections, they are also at risk of detecting colonizing and contaminating organisms and data on their practical clinical utility is still emerging.
CGD Diagnosis
It is important to have a high index of suspicion of CGD in patients with recurrent or severe infections with any of the characteristic organisms described above, as prompt diagnosis and treatment are crucial to a better prognosis. The primary diagnostic tests used in CGD functionally assess the NADPH complex in stimulated neutrophils. The nitroblue tetrazolium (NBT) reduction test was historically used; however, it has been largely superseded by the dihydrorhodamine (DHR) assay as the gold standard for diagnosing CGD. While positive results in either of these tests are generally diagnostic, genetic sequencing confirms the diagnosis.71
DHR-123 Oxidation Assay
The DHR-123 oxidation assay is the gold-standard method to diagnose CGD. In this test, stimulated phagocytes normally oxidize DHR-123 to a fluorescent green signal, rhodamine, by the NADPH complex. A flow cytometer is then used to measure the oxidative burst activity. Phagocytes with a defective NADPH complex are unable to effectively oxidize DHR-123 and a change in fluorescence is not seen. The DHR-123 assay is widely commercially available, and it does not rely on operator subjective interpretation. Based on quantitative results, it can distinguish between X-linked and recessive forms of CGD, as well as carriers of pathogenic variants in gp91phox. This assay is highly sensitive and combined with the capacity to assess residual superoxide production, it can predict the severity and prognosis in patients with CGD.25 There are limitations to its use, however as this test can be abnormal in ehrlichiosis infection, myeloperoxidase deficiency, and the syndrome of synovitis, acne, pustulosis, hyperostosis, and osteitis (SAPHO).72,73
NBT Reduction Test
This is one of the oldest qualitative assays used to test neutrophil function. In this test, a drop of blood is placed on a slide and incubated with an activating agent and NBT. Normal granulocytes reduce the yellow NBT dye to blue formazan, a dark blue precipitate. Although it does not require sophisticated equipment, its use has been limited. While it can be used to identify gp91phox carriers, the interpretation of this test is subjective leading to increase false-negative results.74 It is also unable to differentiate between X-linked and autosomal recessive CGD.75
Genetic Sequencing
An abnormal neutrophil function test should be followed by confirmatory genetic sequencing. Knowledge about how a specific variant effects protein function as well as residual superoxide production have been predictive of disease severity and mortality risk.25 Both single-gene and multi-gene panels are available to sequence DNA from peripheral blood leukocytes. In patients suspected to have AR disease, it is also important to note that NCF1 is flanked by pseudogenes, which may complicate sequencing and interpretation.76
Management of Infectious and Granulomatous Complications
Given the risk for recurrent and/or severe infections in patients with CGD and the characteristic organisms that are known to commonly cause disease, the mainstay of management of this disease is targeted prophylaxis. Optimal prophylactic regimens should include a combination of antibacterial agents, antifungal agents, and immunomodulation with interferon-gamma (IFN-γ) therapy. In addition, the prompt evaluation and treatment of acute infections along with adjunctive anti-inflammatory medications are important to minimize infectious and granulomatous complications.
Daily oral trimethoprim-sulfamethoxazole (5mg/kg of the trimethoprim component with a maximum daily dose of 1 double-strength tablet) is recommended for antibacterial prophylaxis.77 Other antibacterial agents used include trimethoprim alone, 2nd or 3rd generation cephalosporins, and quinolones.78 Antifungal prophylaxis with itraconazole has been shown to significantly reduce the incidence of invasive fungal disease in patients with CGD.4,27,47 First-line antifungal prophylaxis is itraconazole (100mg for <13 years old or <50kg and 200mg daily for >13 years old or >50kg), but voriconazole and posaconazole are being increasingly used.77,79 Transaminitis has been associated with the azoles, so liver function tests should be periodically monitored. Due to variability in azole absorption, some clinicians assess drug levels, especially in those CGD patients with gastrointestinal granulomas. Voriconazole can lead to significant phototoxicity and, over long periods of time, there is a concern for fluoride toxicity.
The use of interferon gamma in patients with CGD allows for a more targeted therapeutic immunomodulatory approach. Interferon gamma has been shown to stimulate superoxide release and improve phagocyte-mediated bacterial killing in vitro.80–82 Studies have also shown a significant reduction in infection frequency, duration and hospitalization rates in patients with CGD.83,84 In the United States, recombinant interferon gamma is FDA approved and commonly used for infection prophylaxis in CGD. In Europe, interferon gamma therapy is often reserved for patients with persistent, recurrent infections despite appropriate antibacterial and antifungal prophylaxis.6 It may also be beneficial in CGD populations living in tuberculosis endemic areas. Interferon gamma is started at a dose of 50 μg/m2 (or 1.5 μg/kg if BSA is <0.5m2) and administered subcutaneously three times weekly. Common side effects include fevers, malaise, chills, fatigue and location injection site erythema or tenderness.
If and when there is a concern for acute infection, patients with CGD warrant a prompt and thorough physical and laboratory evaluation as there can be great variability in clinical presentation. Some patients, especially the younger children, with CGD may not present with classic infectious signs or symptoms of fever, fatigue, malaise, leukocytosis, or neutrophilia. In patients with CGD, an erythrocyte sedimentation rate and C-reactive protein can be very helpful in both assessing for possible infection and monitoring for response to treatment. There should be a low threshold for imaging the chest and/or abdomen (eg, liver) given the high risk for indolent bacterial and fungal infections. Early diagnosis of infections not only may keep the patient from being hospitalized, but also will reduce the risk for further organ damage and inflammation (eg, microgranuloma formation) at sites of infection.
Granulomatous complications in CGD can be quite profound and at times debilitating to patients. When treating acute infections in patients with CGD, an anti-inflammatory agent may be necessary in addition to antimicrobial therapy since granuloma formation can be seen at infection sites. Systemic corticosteroids have been used as adjunctive therapy to antibiotics in the treatment of persistent infections, such as staphylococcal liver abscesses. Staphylococcal liver abscesses can be very difficult to treat in CGD and may even require surgical intervention. Steroids are typically dosed at 1mg/kg/day for 2–3 weeks, followed by a taper over several months (on average 5 months).85,86 Corticosteroids have also been used to treat obstructive granulomatous lesions in CGD, such as bladder granulomas causing bladder outlet obstruction or gastrointestinal lesions causing obstruction.87 The use of systemic immunosuppressive medications, such as ustekinumab or vedolizumab, continues to be explored in patients with CGD colitis and has had varying effects to date.88–90 TNF-inhibitors should be avoided in patients with CGD, as they have been associated with serious bacterial and fungal infections and increased mortality.91
Long-Term Management of CGD
Allogeneic HSCT is the only widely available curative treatment for CGD with the potential for resolution of both infectious and inflammatory complications. Outcomes have improved significantly over the last few decades with reported overall survival rates now consistently >90% in pediatric patients less than 14 years of age regardless of donor source.92–99 Furthermore, pediatric patients who undergo HSCT have fewer infections, improved growth parameters, and higher quality of life measures compared to those treated conventionally.100,101
Patients with intractable infection or autoinflammation at the time of HSCT and adolescents or young adults have traditionally been difficult to transplant with historically high rates of graft-versus-host disease (GVHD) and transplant-related mortality.92,93,102,103 However, in 2014, Güngör et al published a large, multi-center HSCT trial that included 13 adult patients with impressive overall and event-free survival rates,104 and several institutions have subsequently reported excellent transplant outcomes in adolescent and young adult patients, including those with severe infection and significant autoinflammation at time of transplantation.105–107 Another multi-center study also recently demonstrated the presence of CGD-related colitis does not appear to negatively affect survival or significantly increase the risk of GVHD.108 The role of autoinflammation and organ dysfunction on transplant outcomes remains unclear; however, these recent publications indicate that HSCT for definitive cure may be considered for high-risk patients with reasonable outcomes.
Gene therapy is an appealing alternative to HSCT and a number of small trials have been conducted to treat gp91phox deficiency using γ-retroviral vectors and reduced intensity conditioning.109–112 All trials demonstrated initial engraftment of transduced neutrophils at 10% to 30% of circulating neutrophils, and gene therapy resulted in full or partial resolution of infection in most patients. However, cell engraftment progressively decreased with time, and several patients developed myelodysplastic syndrome (MDS) due to insertional activation of proto-oncogenes.112 In response to the high incidence of MDS seen with γ-retroviral vectors, gene therapy trials are currently underway using self-inactivating (SIN) lentiviral vectors. Encouragingly, studies using similar vectors for the treatment of Severe Combined Immunodeficiency and Wiskott Aldrich have showed good success.113 Long-term outcomes with gene therapy are unknown, and as with HSCT, it is unclear what level of oxidase-positive neutrophils is necessary for resolution of autoinflammation and sustained correction of the CGD phenotype.
Currently, there are no standard HSCT guidelines and no consensus surrounding transplant protocols for CGD patients. Large, multi-center studies are currently underway to further investigate long-term transplant outcomes, identify optimal conditioning regimens, and determine the level of donor chimerism necessary for resolution of autoinflammation and sustained protection against new-onset inflammatory or autoimmune disease manifestations. Biomarkers predictive of disease course and transplant outcomes are also needed to help guide treatment decisions. With the new SIN lentiviral vectors and optimization of conditioning regimens, gene therapy may also become a viable alternative to HSCT in the future.
Conclusion
CGD is a rare but serious primary immunodeficiency with varying prevalence and rates of X-linked and AR disease worldwide. Functional defects in the phagocyte NADPH oxidase complex predispose patients to a relatively narrow spectrum of bacterial and fungal infections that are sometimes fastidious and often difficult to identify. When evaluating and treating patients with CGD, it is important to consider their native country of birth, climate, and living situation, which may predispose them to types of infections that are atypical to your routine practice. In addition to recurrent and often severe infections, patients with CGD and X-linked female carriers are also susceptible to developing many non-infectious complications including tissue granuloma formation and autoimmunity. The DHR-123 oxidation assay is the gold standard for making the diagnosis and it along with genetic testing can help predict the severity and prognosis in patients with CGD. Disease management focuses on prophylaxis with antibacterial, antifungal, and immunomodulatory medications, prompt identification and treatment of acute infections, and prevention of secondary granulomatous complications. While HSCT is the only widely available curative treatment for patients with CGD, recent advances in gene therapy may provide a safer, more direct alternative.
Abbreviations
CGD, chronic granulomatous disease; NADPH, nicotinamide adenine dinucleotide phosphate; AR, autosomal recessive; BCG, Bacillus Calmette-Guerin.
Disclosure
Dr Kelli W Williams is co-investigator in randomized, double blind placebo controlled study to investigate safety and efficacy of dupilimab in severe eczema in pediatric patients for Icon Clinical Research (Regeneron), principal Investigator in a Phase 3, randomized 3-part study to investigate safety and efficacy of dupilimab in eosinophilic esophagitis for Icon Clinical Research (Regeneron), co-investigator for research study on Food Allergy Symptom Stress-management with Technology in caregivers and reports grants from NIH/National Institute of Nursing Research, participates in Advisory Board Round table event and reports personal fees from Horizon Therapeutics, outside the submitted work. Dr Nicholas L Hartog reports personal fees from Horizon Pharmaceuticals, Takeda, Orchard Therapeutics, and Pharming Healthcare, outside the submitted work. Dr Hey J Chong reports personal fees from Actimmune, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Bridges RA, Berendes H, Good RA. A fatal granulomatous disease of childhood; the clinical, pathological, and laboratory features of a new syndrome. AMA J Dis Child. 1959;97(4):387–408. doi:10.1001/archpedi.1959.02070010389004
2. Azimi PH, Bodenbender JG, Hintz RL, Kontras SB. Chronic granulomatous disease in three female siblings. JAMA. 1968;206(13):2865–2870. doi:10.1001/jama.1968.03150130023004
3. Rider NL, Jameson MB, Creech CB. Chronic granulomatous disease: epidemiology, pathophysiology, and genetic basis of disease. J Pediatric Infect Dis Soc. 2018;7(suppl_1):S2–S5. doi:10.1093/jpids/piy008
4. Winkelstein JA, Marino MC, Johnston RB, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore). 2000;79(3):155–169. doi:10.1097/00005792-200005000-00003
5. Wolach B, Gavrieli R, de Boer M, et al. Chronic granulomatous disease: clinical, functional, molecular, and genetic studies. The Israeli experience with 84 patients. Am J Hematol. 2017;92(1):28–36. doi:10.1002/ajh.24573
6. Martire B, Rondelli R, Soresina A, et al. Clinical features, long-term follow-up and outcome of a large cohort of patients with chronic granulomatous disease: an Italian multicenter study. Clin Immunol. 2008;126(2):155–164. doi:10.1016/j.clim.2007.09.008
7. Jones LBKR, McGrogan P, Flood TJ, et al. Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry. Clin Exp Immunol. 2008;152(2):211–218. doi:10.1111/j.1365-2249.2008.03644.x
8. Tkalcevic J, Novelli M, Phylactides M, Iredale JP, Segal AW, Roes J. Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity. 2000;12(2):201–210. doi:10.1016/S1074-7613(00)80173-9
9. Reeves EP, Lu H, Jacobs HL, Messina C. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416(6878):291–297. doi:10.1038/416291a
10. Roos D, Kuhns DB, Maddalena A, et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol Dis. 2010;45(3):246–265. doi:10.1016/j.bcmd.2010.07.012
11. Roos D, Kuhns DB, Maddalena A, et al. Hematologically important mutations: the autosomal recessive forms of chronic granulomatous disease (second update). Blood Cells Mol Dis. 2010;44(4):291–299. doi:10.1016/j.bcmd.2010.01.009
12. Arnadottir GA, Norddahl GL, Gudmundsdottir S, et al. A homozygous loss-of-function mutation leading to CYBC1 deficiency causes chronic granulomatous disease. Nat Commun. 2018;9(1):4447. doi:10.1038/s41467-018-06964-x
13. Thomas DC, Charbonnier L-M, Schejtman A, et al. EROS/CYBC1 mutations: decreased NADPH oxidase function and chronic granulomatous disease. J Allergy Clin Immunol. 2019;143(2):782–785.e1. doi:10.1016/j.jaci.2018.09.019
14. Repine JE, Clawson CC, White JG, Holmes B. Spectrum of function of neutrophils from carriers of sex-linked chronic granulomatous disease. J Pediatr. 1975;87(6 Pt 1):901–907. doi:10.1016/S0022-3476(75)80902-4
15. Marciano BE, Zerbe CS, Falcone EL, et al. X-linked carriers of chronic granulomatous disease: illness, lyonization, and stability. J Allergy Clin Immunol. 2017. doi:10.1016/j.jaci.2017.04.035
16. Lewis EM, Singla M, Sergeant S, Koty PP, McPhail LC. X-linked chronic granulomatous disease secondary to skewed X chromosome inactivation in a female with a novel CYBB mutation and late presentation. Clin Immunol. 2008;129(2):372–380. doi:10.1016/j.clim.2008.07.022
17. Wolach B, Scharf Y, Gavrieli R, de Boer M, Roos D. Unusual late presentation of X-linked chronic granulomatous disease in an adult female with a somatic mosaic for a novel mutation in CYBB. Blood. 2005;105(1):61–66. doi:10.1182/blood-2004-02-0675
18. Anderson-Cohen M, Holland SM, Kuhns DB, et al. Severe phenotype of chronic granulomatous disease presenting in a female with a de novo mutation in gp91-phox and a non familial, extremely skewed X chromosome inactivation. Clin Immunol. 2003;109(3):308–317. doi:10.1016/j.clim.2003.08.002
19. Hauck F, Koletzko S, Walz C, et al. Diagnostic and treatment options for severe IBD in female X-CGD carriers with non-random X-inactivation. J Crohns Colitis. 2016;10(1):112–115. doi:10.1093/ecco-jcc/jjv186
20. Rösen-Wolff A, Soldan W, Heyne K, Bickhardt J, Gahr M, Roesler J. Increased susceptibility of a carrier of X-linked chronic granulomatous disease (CGD) to Aspergillus fumigatus infection associated with age-related skewing of lyonization. Ann Hematol. 2001;80(2):113–115. doi:10.1007/s002770000230
21. Kobayashi S, Murayama S, Takanashi S, et al. Clinical features and prognoses of 23 patients with chronic granulomatous disease followed for 21 years by a single hospital in Japan. Eur J Pediatr. 2008;167(12):1389–1394. doi:10.1007/s00431-008-0680-7
22. Köker MY, Camcıoğlu Y, van Leeuwen K, et al. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J Allergy Clin Immunol. 2013;132(5):1156–1163.e5. doi:10.1016/j.jaci.2013.05.039
23. Fattahi F, Badalzadeh M, Sedighipour L, et al. Inheritance pattern and clinical aspects of 93 Iranian patients with chronic granulomatous disease. J Clin Immunol. 2011;31(5):792–801. doi:10.1007/s10875-011-9567-x
24. Movahedi M, Aghamohammadi A, Rezaei N, et al. Chronic granulomatous disease: a clinical survey of 41 patients from the Iranian primary immunodeficiency registry. Int Arch Allergy Immunol. 2004;134(3):253–259. doi:10.1159/000078774
25. Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363(27):2600–2610. doi:10.1056/NEJMoa1007097
26. Dinauer MC. Inflammatory consequences of inherited disorders affecting neutrophil function. Blood. 2019;133(20):2130–2139. doi:10.1182/blood-2018-11-844563
27. Marciano BE, Spalding C, Fitzgerald A, et al. Common severe infections in chronic granulomatous disease. Clin Infect Dis. 2015;60(8):1176–1183. doi:10.1093/cid/ciu1154
28. Bennett N, Maglione PJ, Wright BL, Zerbe C. Infectious complications in patients with chronic granulomatous disease. J Pediatric Infect Dis Soc. 2018;7(suppl_1):S12–S17. doi:10.1093/jpids/piy013
29. Mortaz E, Azempour E, Mansouri D, et al. Common infections and target organs associated with chronic granulomatous disease in Iran. Int Arch Allergy Immunol. 2019;179(1):62–73. doi:10.1159/000496181
30. Siddiqui S, Anderson VL, Hilligoss DM, et al. Fulminant mulch pneumonitis: an emergency presentation of chronic granulomatous disease. Clin Infect Dis. 2007;45(6):673–681. doi:10.1086/520985
31. Holland SM. Chronic granulomatous disease. Hematol Oncol Clin North Am. 2013;27(1):
32. Álvarez-Cardona A, Rodríguez-Lozano AL, Blancas-Galicia L, Rivas-Larrauri FE, Yamazaki-Nakashimada MA. Intravenous immunoglobulin treatment for macrophage activation syndrome complicating chronic granulomatous disease. J Clin Immunol. 2012;32(2):207–211. doi:10.1007/s10875-011-9616-5
33. Akagi K, Kawai T, Watanabe N, et al. A case of macrophage activation syndrome developing in a patient with chronic granulomatous disease-associated colitis. J Pediatr Hematol Oncol. 2014;36(3):e169–e172. doi:10.1097/MPH.0b013e31828e5dae
34. Marciano BE, Rosenzweig SD, Kleiner DE, et al. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. 2004;114(2):462–468. doi:10.1542/peds.114.2.462
35. Walther MM, Malech H, Berman A, et al. The urological manifestations of chronic granulomatous disease. J Urol. 1992;147(5):1314–1318. doi:10.1016/S0022-5347(17)37552-3
36. Henrickson SE, Jongco AM, Thomsen KF, Garabedian EK, Thomsen IP. Noninfectious manifestations and complications of chronic granulomatous disease. J Pediatric Infect Dis Soc. 2018;7(suppl_1):S18–S24. doi:10.1093/jpids/piy014
37. Barese CN, Podestá M, Litvak E, Villa M, Rivas EM. Recurrent eosinophilic cystitis in a child with chronic granulomatous disease. J Pediatr Hematol Oncol. 2004;26(3):209–212. doi:10.1097/00043426-200403000-00014
38. Battersby AC, Braggins H, Pearce MS, et al. Inflammatory and autoimmune manifestations in X-linked carriers of chronic granulomatous disease in the United Kingdom. J Allergy Clin Immunol. 2017;140(2):628–630.e6. doi:10.1016/j.jaci.2017.02.029
39. Bortoletto P, Lyman K, Camacho A, Fricchione M, Khanolkar A, Katz BZ. Chronic granulomatous disease: a large, single-center US experience. Pediatr Infect Dis J. 2015;34(10):1110–1114. doi:10.1097/INF.0000000000000840
40. Reichenbach J, Lopatin U, Mahlaoui N, et al. Actinomyces in chronic granulomatous disease: an emerging and unanticipated pathogen. Clin Infect Dis. 2009;49(11):1703–1710. doi:10.1086/647945
41. Sugui JA, Peterson SW, Clark LP, et al. Aspergillus tanneri sp. nov., a new pathogen that causes invasive disease refractory to antifungal therapy. J Clin Microbiol. 2012;50(10):3309–3317. doi:10.1128/JCM.01509-12
42. Haidar G, Zerbe CS, Cheng M, Zelazny AM, Holland SM, Sheridan KR. Phellinus species: an emerging cause of refractory fungal infections in patients with X-linked chronic granulomatous disease. Mycoses. 2017;60(3):155–160. doi:10.1111/myc.12573
43. Greenberg DE, Ding L, Zelazny AM, et al. A novel bacterium associated with lymphadenitis in a patient with chronic granulomatous disease. PLoS Pathog. 2006;2(4):e28. doi:10.1371/journal.ppat.0020028
44. Meher-Homji Z, Mangalore RP, Johnson PD, Chua KY. Chromobacterium violaceum infection in chronic granulomatous disease: a case report and review of the literature. JMM Case Rep. 2017;4(1):e005084. doi:10.1099/jmmcr.0.005084
45. Relich RF, Humphries RM, Mattison HR, et al. Francisella philomiragia bacteremia in a patient with acute respiratory insufficiency and acute-on-chronic kidney disease. J Clin Microbiol. 2015;53(12):3947–3950. doi:10.1128/JCM.01762-15
46. Macher AM, Casale TB, Fauci AS. Chronic granulomatous disease of childhood and Chromobacterium violaceum infections in the southeastern United States. Ann Intern Med. 1982;97(1):51–55. doi:10.7326/0003-4819-97-1-51
47. van den Berg JM, van Koppen E, Åhlin A, et al. Chronic granulomatous disease: the European experience. PLoS One. 2009;4(4):e5234. doi:10.1371/journal.pone.0005234
48. Rawat A, Vignesh P, Sharma A, et al. Infection profile in chronic granulomatous disease: a 23-year experience from a tertiary care center in North India. J Clin Immunol. 2017;37(3):319–328. doi:10.1007/s10875-017-0382-x
49. Wolach B, Gavrieli R, de Boer M, et al. Chronic granulomatous disease in Israel: clinical, functional and molecular studies of 38 patients. Clin Immunol. 2008;129(1):103–114. doi:10.1016/j.clim.2008.06.012
50. de Oliveira-junior EB, Zurro NB, Prando C, et al. Clinical and genotypic spectrum of chronic granulomatous disease in 71 Latin American patients: first report from the LASID registry. Pediatr Blood Cancer. 2015;62(12):2101–2107. doi:10.1002/pbc.25674
51. Blancas-Galicia L, Santos-Chávez E, Deswarte C, et al. Genetic, immunological, and clinical features of the first mexican cohort of patients with chronic granulomatous disease. J Clin Immunol. 2020;40(3):475–493. doi:10.1007/s10875-020-00750-5
52. World Health Organization. Global Tuberculosis Report 2018. 2018.
53. Stewart RJ, Tsang CA, Pratt RH, Price SF, Langer AJ. Tuberculosis - United States, 2017. MMWR Morb Mortal Wkly Rep. 2018;67(11):317–323. doi:10.15585/mmwr.mm6711a2
54. Calmette A. Preventive vaccination against tuberculosis with BCG. Proc R Soc Med. 1931;24(11):1481–1490.
55. BCG vaccines. WHO position paper – february 2018. Wkly Epidemiol Rec. 2018;93(8):73–96.
56. Norouzi S, Aghamohammadi A, Mamishi S, Rosenzweig SD, Rezaei N. Bacillus calmette-guérin (BCG) complications associated with primary immunodeficiency diseases. J Infect. 2012;64(6):543–554. doi:10.1016/j.jinf.2012.03.012
57. Zhou Q, Hui X, Ying W, et al. A cohort of 169 chronic granulomatous disease patients exposed to BCG vaccination: a retrospective study from a single center in Shanghai, China (2004–2017). J Clin Immunol. 2018;38(3):260–272. doi:10.1007/s10875-018-0486-y
58. Baba LA, Ailal F, El Hafidi N, et al. Chronic granulomatous disease in Morocco: genetic, immunological, and clinical features of 12 patients from 10 kindreds. J Clin Immunol. 2014;34(4):452–458. doi:10.1007/s10875-014-9997-3
59. Zwerling A, Behr MA, Verma A, Brewer TF, Menzies D, Pai M. The BCG world atlas: a database of global BCG vaccination policies and practices. PLoS Med. 2011;8(3):e1001012. doi:10.1371/journal.pmed.1001012
60. Lee PPW, Chan K-W, Jiang L, et al. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease: a review of 17 patients living in a region endemic for tuberculosis. Pediatr Infect Dis J. 2008;27(3):224–230. doi:10.1097/INF.0b013e31815b494c
61. Conti F, Lugo-Reyes SO, Blancas-Galicia L, et al. Mycobacterial disease in patients with chronic granulomatous disease: a retrospective analysis of 71 cases. J Allergy Clin Immunol. 2016;138(1):241–248.e243. doi:10.1016/j.jaci.2015.11.041
62. Fernando SJA, Faiz NM, Handunnetti SM, et al. Preliminary study on chronic granulomatous disease in Sri Lanka. Allergy Asthma Clin Immunol. 2018;14(1):37. doi:10.1186/s13223-018-0264-7
63. Khanna G, Kao SC, Kirby P, Sato Y. Imaging of chronic granulomatous disease in children. Radiographics. 2005;25(5):1183–1195. doi:10.1148/rg.255055011
64. Henriet S, Verweij PE, Holland SM, Warris A. Invasive fungal infections in patients with chronic granulomatous disease. Adv Exp Med Biol. 2013;764:27–55. doi:10.1007/978-1-4614-4726-9_3
65. Greenberg DE, Shoffner AR, Zelazny AM, et al. Recurrent granulibacter bethesdensis infections and chronic granulomatous disease. Emerg Infect Dis. 2010;16(9):1341–1348. doi:10.3201/eid1609.091800
66. Silliman CC, Lawellin DW, Lohr JA, Rodgers BM, Donowitz LG. Paecilomyces lilacinus infection in a child with chronic granulomatous disease. J Infect. 1992;24(2):191–195. doi:10.1016/0163-4453(92)92980-W
67. White PL, Wingard JR, Bretagne S, et al. Aspergillus polymerase chain reaction: systematic review of evidence for clinical use in comparison with antigen testing. Clin Infect Dis. 2015;61(8):1293–1303. doi:10.1093/cid/civ507
68. Wang Y, Marciano BE, Shen D, et al. Molecular identification of bacterial DNA in the chorioretinal scars of chronic granulomatous disease. J Clin Immunol. 2013;33(5):917–924. doi:10.1007/s10875-013-9899-9
69. Hong DK, Blauwkamp TA, Kertesz M, Bercovici S, Truong C, Banaei N. Liquid biopsy for infectious diseases: sequencing of cell-free plasma to detect pathogen DNA in patients with invasive fungal disease. Diagn Microbiol Infect Dis. 2018;92(3):210–213. doi:10.1016/j.diagmicrobio.2018.06.009
70. Schlaberg R, Chiu CY, Miller S, et al. Validation of metagenomic next-generation sequencing tests for universal pathogen detection. Arch Pathol Lab Med. 2017;141(6):776–786. doi:10.5858/arpa.2016-0539-RA
71. Yu JE, Azar AE, Chong HJ, Jongco AM, Prince BT. Considerations in the diagnosis of chronic granulomatous disease. J Pediatric Infect Dis Soc. 2018;7(suppl_1):S6–S11. doi:10.1093/jpids/piy007
72. Banerjee R, Anguita J, Roos D, Fikrig E. Cutting edge: infection by the agent of human granulocytic ehrlichiosis prevents the respiratory burst by down-regulating gp91phox. J Immunol. 2000;164(8):3946–3949. doi:10.4049/jimmunol.164.8.3946
73. Mauch L, Lun A, O’Gorman MRG, et al. Chronic granulomatous disease (CGD) and complete myeloperoxidase deficiency both yield strongly reduced dihydrorhodamine 123 test signals but can be easily discerned in routine testing for CGD. Clin Chem. 2007;53(5):890–896. doi:10.1373/clinchem.2006.083444
74. Ochs HD, Igo RP. The NBT slide test: a simple screening method for detecting chronic granulomatous disease and female carriers. J Pediatr. 1973;83(1):77–82. doi:10.1016/S0022-3476(73)80316-6
75. Roos D, de Boer M. Molecular diagnosis of chronic granulomatous disease. Clin Exp Immunol. 2014;175(2):139–149. doi:10.1111/cei.12202
76. Görlach A, Lee PL, Roesler J, et al. A p47-phox pseudogene carries the most common mutation causing p47-phox- deficient chronic granulomatous disease. J Clin Invest. 1997;100(8):1907–1918. doi:10.1172/JCI119721
77. Thomsen IP, Smith MA, Holland SM, Creech CB. A comprehensive approach to the management of children and adults with chronic granulomatous disease. J Allergy Clin Immunol Pract. 2016;4(6):1082–1088. doi:10.1016/j.jaip.2016.03.021
78. Kuruvilla M, La MorenaDe MT. Antibiotic prophylaxis in primary immune deficiency disorders. J Allergy Clin Immunol Pract. 2013;1(6):573–582. doi:10.1016/j.jaip.2013.09.013
79. Slack MA, Thomsen IP. Prevention of infectious complications in patients with chronic granulomatous disease. J Pediatric Infect Dis Soc. 2018;7(suppl_1):S25–S30. doi:10.1093/jpids/piy016
80. Ezekowitz RA, Orkin SH, Newburger PE. Recombinant interferon gamma augments phagocyte superoxide production and X-chronic granulomatous disease gene expression in X-linked variant chronic granulomatous disease. J Clin Invest. 1987;80(4):1009–1016. doi:10.1172/JCI113153
81. Condino-Neto A, Newburger PE. Interferon-gamma improves splicing efficiency of CYBB gene transcripts in an interferon-responsive variant of chronic granulomatous disease due to a splice site consensus region mutation. Blood. 2000;95(11):3548–3554. doi:10.1182/blood.V95.11.3548
82. Sechler JG. Recombinant Human Interferon-y Reconstitutes Defective Phagocyte Function in Patients with Chronic Granulomatous Disease Ofchildhood. 2005 January. 1–5.
83. International Chronic Granulomatous Disease Cooperative Study Group. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. The international chronic granulomatous disease cooperative study group. N Engl J Med. 1991;324(8):509–516. doi:10.1056/NEJM199102213240801
84. Marciano BE, Wesley R, De Carlo ES, et al. Long-term interferon-gamma therapy for patients with chronic granulomatous disease. Clin Infect Dis. 2004;39(5):692–699. doi:10.1086/422993
85. Leiding JW, Freeman AF, Marciano BE, et al. Corticosteroid therapy for liver abscess in chronic granulomatous disease. Clin Infect Dis. 2012;54(5):694–700. doi:10.1093/cid/cir896
86. Straughan DM, McLoughlin KC, Mullinax JE, et al. The changing paradigm of management of liver abscesses in chronic granulomatous disease. Clin Infect Dis. 2018;66(9):1427–1434. doi:10.1093/cid/cix1012
87. Chin TW, Stiehm ER, Falloon J, Gallin JI. Corticosteroids in treatment of obstructive lesions of chronic granulomatous disease. J Pediatr. 1987;111(3):349–352. doi:10.1016/s0022-3476(87)80452-3
88. Hahn KJ, Ho N, Yockey L, et al. Treatment with anakinra, a recombinant IL-1 receptor antagonist, unlikely to induce lasting remission in patients with CGD colitis. Am J Gastroenterol. 2015;110(6):938–939. doi:10.1038/ajg.2015.135
89. Butte MJ, Park KT, Lewis DB. Treatment of CGD-associated colitis with the IL-23 blocker ustekinumab. J Clin Immunol. 2016;36(7):619–620. doi:10.1007/s10875-016-0318-x
90. Campbell N, Chapdelaine H. Treatment of chronic granulomatous disease-associated fistulising colitis with vedolizumab. J Allergy Clin Immunol Pract. 2017;5(6):1748–1749. doi:10.1016/j.jaip.2017.07.035
91. Uzel G, Orange JS, Poliak N, Marciano BE, Heller T, Holland SM. Complications of tumor necrosis factor-α blockade in chronic granulomatous disease-related colitis. Clin Infect Dis. 2010;51(12):1429–1434. doi:10.1086/657308
92. Soncini E, Slatter MA, Jones LBKR, et al. Unrelated donor and HLA-identical sibling haematopoietic stem cell transplantation cure chronic granulomatous disease with good long-term outcome and growth. Br J Haematol. 2009;145(1):73–83. doi:10.1111/j.1365-2141.2009.07614.x
93. Schuetz C, Hoenig M, Gatz S, et al. Hematopoietic stem cell transplantation from matched unrelated donors in chronic granulomatous disease. Immunol Res. 2009;44(1–3):35–41. doi:10.1007/s12026-008-8068-3
94. Goździk J, Pituch-Noworolska A, Skoczeń S, et al. Allogeneic haematopoietic stem cell transplantation as therapy for chronic granulomatous disease–single centre experience. J Clin Immunol. 2011;31(3):332–337. doi:10.1007/s10875-011-9513-y
95. Martinez CA, Shah S, Shearer WT, et al. Excellent survival after sibling or unrelated donor stem cell transplantation for chronic granulomatous disease. J Allergy Clin Immunol. 2012;129(1):176–183. doi:10.1016/j.jaci.2011.10.005
96. Tewari P, Martin PL, Mendizabal A, et al. Myeloablative transplantation using either cord blood or bone marrow leads to immune recovery, high long-term donor chimerism and excellent survival in chronic granulomatous disease. Biol Blood Marrow Transplant. 2012;18(9):1368–1377. doi:10.1016/j.bbmt.2012.02.002
97. Morillo-Gutierrez B, Beier R, Rao K, et al. Treosulfan-based conditioning for allogeneic HSCT in children with chronic granulomatous disease: a multicenter experience. Blood. 2016;128(3):440–448. doi:10.1182/blood-2016-03-704015
98. Mehta B, Mahadeo K, Kapoor N, Abdel-Azim H. Low-dose total-body irradiation and alemtuzumab-based reduced-intensity conditioning regimen results in durable engraftment and correction of clinical disease among children with chronic granulomatous disease. Pediatr Transplant. 2015;19(4):408–412. doi:10.1111/petr.12471
99. Lum SH, Flood T, Hambleton S, et al. Two decades of excellent transplant survival for chronic granulomatous disease: a supraregional immunology transplant center report. Blood. 2019;133(23):2546–2549. doi:10.1182/blood.2019000021
100. Cole T, Pearce MS, Cant AJ, Cale CM, Goldblatt D, Gennery AR. Clinical outcome in children with chronic granulomatous disease managed conservatively or with hematopoietic stem cell transplantation. J Allergy Clin Immunol. 2013;132(5):1150–1155. doi:10.1016/j.jaci.2013.05.031
101. Cole T, McKendrick F, Titman P, et al. Health related quality of life and emotional health in children with chronic granulomatous disease: a comparison of those managed conservatively with those that have undergone haematopoietic stem cell transplant. J Clin Immunol. 2013;33(1):8–13. doi:10.1007/s10875-012-9758-0
102. Seger RA, Güngör T, Belohradsky BH, et al. Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: a survey of the European experience, 1985–2000. Blood. 2002;100(13):4344–4350. doi:10.1182/blood-2002-02-0583
103. Horwitz ME, Barrett AJ, Brown MR, et al. Treatment of chronic granulomatous disease with nonmyeloablative conditioning and a T-cell-depleted hematopoietic allograft. N Engl J Med. 2001;344(12):881–888. doi:10.1056/NEJM200103223441203
104. Güngör T, Teira P, Slatter M, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. 2014;383(9915):436–448. doi:10.1016/S0140-6736(13)62069-3
105. Parta M, Kelly C, Kwatemaa N, et al. Allogeneic reduced-intensity hematopoietic stem cell transplantation for chronic granulomatous disease: a single-center prospective trial. J Clin Immunol. 2017;37(6):548–558. doi:10.1007/s10875-017-0422-6
106. Fox TA, Chakraverty R, Burns S, et al. Successful outcome following allogeneic hematopoietic stem cell transplantation in adults with primary immunodeficiency. Blood. 2018;131(8):917–931. doi:10.1182/blood-2017-09-807487
107. Arnold DE, Seif AE, Jyonouchi S, Sullivan KE, Bunin NJ, Heimall JR. Allogeneic hematopoietic stem cell transplantation in adolescent patients with chronic granulomatous disease. J Allergy Clin Immunol Pract. 2019;7(3):1052–1054.e1052. doi:10.1016/j.jaip.2018.10.034
108. Marsh RA, Leiding JW, Logan BR, et al. Chronic granulomatous disease-associated ibd resolves and does not adversely impact survival following allogeneic HCT. J Clin Immunol. 2019;39(7):653–667. doi:10.1007/s10875-019-00659-8
109. Ott MG, Schmidt M, Schwarzwaelder K, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12(4):401–409. doi:10.1038/nm1393
110. Bianchi M, Hakkim A, Brinkmann V, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009;114(13):2619–2622. doi:10.1182/blood-2009-05-221606
111. Kang HJ, Bartholomae CC, Paruzynski A, et al. Retroviral gene therapy for X-linked chronic granulomatous disease: results from Phase I/II trial. Mol Ther. 2011;19(11):2092–2101. doi:10.1038/mt.2011.166
112. Stein S, Ott MG, Schultze-Strasser S, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16(2):198–204. doi:10.1038/nm.2088
113. Keller MD, Notarangelo LD, Malech HL. Future of care for patients with chronic granulomatous disease: gene therapy and targeted molecular medicine. J Pediatric Infect Dis Soc. 2018;7(suppl_1):S40–S44. doi:10.1093/jpids/piy011
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.