Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 8
Genomic profiling toward precision medicine in non-small cell lung cancer: getting beyond EGFR
Authors Richer A, Friel J, Carson V, Inge L, Whitsett TG
Received 2 October 2014
Accepted for publication 13 November 2014
Published 20 February 2015 Volume 2015:8 Pages 63—79
DOI https://doi.org/10.2147/PGPM.S52845
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin Bluth
Amanda L Richer,1 Jacqueline M Friel,1 Vashti M Carson,2 Landon J Inge,1 Timothy G Whitsett2
1Norton Thoracic Institute, St Joseph’s Hospital and Medical Center, 2Cancer and Cell Biology Division, Translational Genomics Research Institute, Phoenix, AZ, USA
Abstract: Lung cancer remains the leading cause of cancer-related mortality worldwide. The application of next-generation genomic technologies has offered a more comprehensive look at the mutational landscape across the different subtypes of non-small cell lung cancer (NSCLC). A number of recurrent mutations such as TP53, KRAS, and epidermal growth factor receptor (EGFR) have been identified in NSCLC. While targeted therapeutic successes have been demonstrated in the therapeutic targeting of EGFR and ALK, the majority of NSCLC tumors do not harbor these genomic events. This review looks at the current treatment paradigms for lung adenocarcinomas and squamous cell carcinomas, examining genomic aberrations that dictate therapy selection, as well as novel therapeutic strategies for tumors harboring mutations in KRAS, TP53, and LKB1 which, to date, have been considered “undruggable”. A more thorough understanding of the molecular alterations that govern NSCLC tumorigenesis, aided by next-generation sequencing, will lead to targeted therapeutic options expected to dramatically reduce the high mortality rate observed in lung cancer.
Keywords: non-small cell lung cancer, precision medicine, epidermal growth factor receptor, Kirsten rat sarcoma viral oncogene homolog, serine/threonine kinase 11, tumor protein p53
Introduction
Lung cancer remains the leading cause of cancer-related mortality worldwide.1 In the United States alone, lung cancer is expected to affect more than 224,000 people in 2014, representing 13.5% of all new cancer cases with a 5-year survivorship of 16.8% (http://seer.cancer.gov/csr/1975_2011/) and will be responsible for an estimated 160,000 deaths.2 The high mortality associated with lung cancer is due to the frequent presence of regional and distant metastasis at diagnosis (78% of diagnoses), that carries 5-year survival rates of 25% (regional) and 4% (distant),3 as well as increased incidence in relapse following treatment and resistance to standard therapeutics. These challenges necessitate a thorough understanding of the molecular biology of lung cancer toward the discovery and development of novel therapeutic approaches. The advent of genomic technologies and, more recently, next-generation sequencing (NGS), allow for a more comprehensive look at lung cancer, with the promise of therapeutically actionable discoveries.
Lung cancer is separated into two major histological categories: small cell lung cancer and non-small cell lung cancer (NSCLC). Of the two, NSCLC accounts for the vast majority of lung cancer cases.4 This review will focus on the molecular drivers and genomics-enabled treatment strategies in NSCLC. NSCLC is further divided into histological subtypes: lung adenocarcinomas (LAC) that arise in cells that line the alveoli; squamous cell carcinomas (SCC); and large-cell carcinomas. Adenocarcinomas and SCC account for the majority of NSCLC cases. In addition to distinct histological features, adenocarcinomas and SCC differ in terms of their molecular drivers, pathogenesis, and disease progression, and they require differential treatment strategies.
Genomics-enabled precision medicine
The molecular landscape of many tumor types is currently being explored by NGS. A more thorough understanding of the molecular alterations in tumors offers opportunities to not only discover driver events, but also to predict therapeutic strategies that might benefit patients based on the individual tumor biology (precision medicine). Genomics-based therapeutic selection has become the standard-of-care for LAC patients, with mutant epidermal growth factor receptor (EGFR) and anaplastic lymphoma receptor tyrosine kinase (ALK) gene rearrangements dictating therapies (gefitinib/erlotinib and crizotinib, respectively) with improved response rates over conventional chemotherapy.5,6 While these have been hailed as therapeutic successes, the majority of lung tumors (>75%) do not harbor these molecular alterations. The discovery of recurrent genomic alterations has also paved the way for multiplexed biomarker tests. Platforms such as Sequenom7 or SNaPShot8 are now available to identify therapeutically actionable molecular alterations in lung cancer.9 In addition, the application of genomic technologies may also enable more the accurate identification of NSCLC histological subtypes from limited tissue samples, an area of current clinical need. With the cost of NGS rapidly diminishing, and time-to-results getting shorter, the genome-wide characterization of an individual tumor toward therapy selection is now an imminent possibility. Importantly, the discovery of therapeutic options in prevalent, previously “undruggable” genes such as Kirsten rat sarcoma viral oncogene homolog (KRAS), serine/threonine kinase 11 (STK11/LKB1), and tumor protein p53 (TP53), as well as the prediction of combinational therapies suggested by tumor alterations, will forward precision medicine toward the reduction of lung cancer mortality.
Molecular landscape of NSCLC
Lung adenocarcinoma
LAC are the most common histological subtype of lung cancer, and are characterized by abnormal growth of peripheral glandular epithelial tissue. LACs are highly heterogeneous, demonstrating high rates of somatic mutations and genomic rearrangements.10 Comprehensive molecular profiling of 230 LAC tumors and matched normal tissue by The Cancer Genome Atlas (TCGA) Research Network11 identified mutations to several oncogenes (KRAS [33%], EGFR [14%], BRAF [10%], MET [7%], and RIT1 [2%]) and tumor suppressors (TP53 [46%], STK11 [17%], KEAP1 [17%], NF1 [11%], RB1 [4%], and CDKN2A [4%]). Chromatin-modifying genes, SETD2 (9%), ARID1A (7%), and SMARCA4 (6%), and mutations in RNA splicing genes, RBM10 (8%) and U2AF1 (3%), were also identified (all gene names with symbols discussed can be found in Table S1). Figure 1A shows the frequency of molecular alterations that will be discussed in this review as established or emerging therapeutic targets. Common alterations in key pathways were also identified: mitogen-activated protein kinases (MAPK) activation (76%); phosphatidylinositol 3-kinases (PI3K)–AKT–mammalian target of rapamycin (MTOR) activation (25%); TP53 alteration (63%); cell-cycle regulation dysfunction (64%); oxidative stress pathway modification (22%); and mutations in chromatin or RNA splice factors (49%).10 As a growing number of these genes and pathways are therapeutically targetable, identification of genomic alterations in an individual tumor should predict which therapy is more likely to elicit a response.
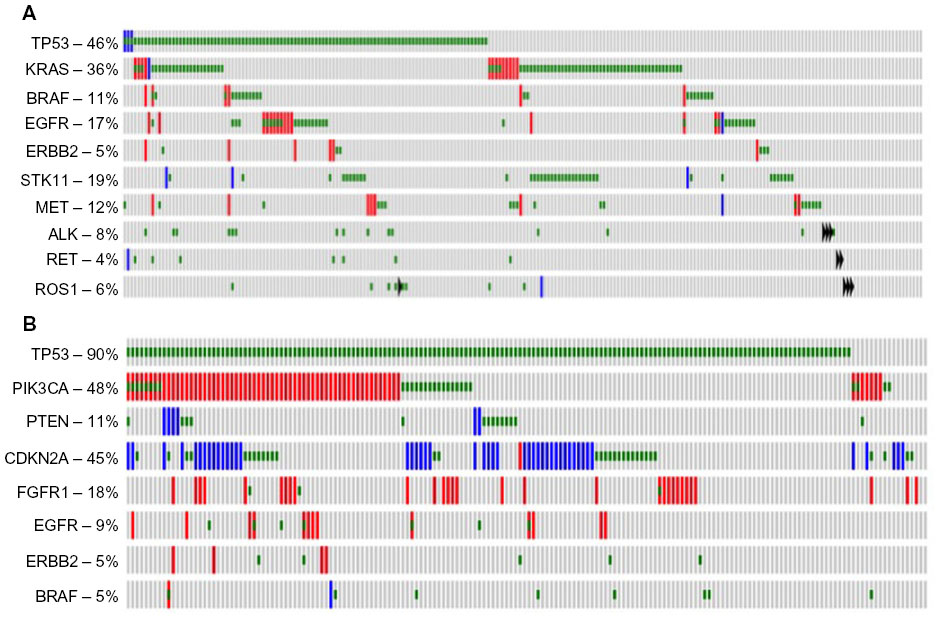 | Figure 1 Frequency of selected molecular alterations in lung adenocarcinomas and squamous cell carcinomas. |
Squamous cell lung carcinoma
SCC is a distinct subtype of NSCLC occurring in approximately 30% of cases and is the second most common type of NSCLC behind adenocarcinoma. Common driver mutations dictating therapeutic selection – such as EGFR and ALK – while prominent in adenocarcinoma, are rarely found in SCC and targeting agents for these mutations are mostly ineffective in SCC.5,6 Until recently, the molecular drivers of SCC remained unknown and few targeting agents have been in development. In 2012, TCGA published a study profiling the genetics of 176 SCC samples and found that SCC had a high mutation rate of 8.1 mutations per megabase with the most frequent mutation found in TP53 (83%).12 Additionally, they found nine other significant mutated genes: CDCKN2A (15%); PTEN (8%); PIK3CA (16%); KEAP (12%); MLL2 (20%); HLA-A (3%); NFE2L2 (15%); NOTCH1 (8%); and RB1 (7%). Figure 1B shows the frequency of molecular alterations that will be discussed in this review as established or emerging therapeutic targets. Notably, these mutations represent a set of frequently mutated pathways including cell-cycle control, oxidative stress, cell survival, apoptotic control, and squamous cell differentiation. This study suggested that ~64% of the cases contained a targetable genomic alteration (defined by a US Food and Drug Administration-approved agent and the mutation present in the RNA).12 These data provide important information regarding the molecular drivers of lung SCC and allow for the development of targeted therapeutic opportunities.
Drug targets and precision treatment strategies
Standard of care
While targeted therapies are approved for treatment of the small population of adenocarcinoma patients carrying specific mutations, the bulk of treatment options for patients with SCC, large-cell carcinoma, and LAC are focused upon standard treatment with cytotoxic drugs and surgical resection. Pathological diagnosis and staging are critical in determining the course of treatment. Current treatment recommendations indicate surgical resection without chemotherapy for patients with early-stage, nonmetastatic disease (stage IA–IB), surgical resection with postoperative chemotherapy for patients with local metastatic disease (stage IIA–IIIB), and chemotherapy for patients with nonresectable, metastatic NSCLC (stage IV).13,14 Platinum-based chemotherapy (cisplatin, carboplatin), combined with antimitotics (vinorelbine, vinblastine, docetaxel, and paclitaxel) or antimetabolites (gemcitabine, pemetrexed), is the treatment of choice for patients with stage IIA–IV, shown to provide significant overall survival benefits in several clinical trials.13,14 Concurrent thoracic radiation therapy for patients has been found to have benefit for patients with metastatic disease,15,16 and is particularly beneficial for patients harboring brain metastasis.17 More recent data found that a combination of cisplatin with pemetrexed provided improved survival for patients with nonsquamous NSCLC histology (LAC, large-cell carcinoma), compared to cisplatin with gemcitabine,18,19 leading to the current recommendation of this combination for nonsquamous tumors. As such, defining the NSCLC histological subtype through pathological review and the routine application of immunohistochemical staining for specific markers of SCC (p63, cytokeratins 5 and 6) and LAC (thyroid transcription factor-1, napsin A, cytokeratin 7) has become the standard of care.20,21
Defined molecular targets in LAC
EGFR
EGFR, a member of the ERBB family of receptors, is a transmembrane receptor with an intracellular tyrosine kinase domain. Induction of EGFR phosphorylation by ligand (EGF) binding activates downstream pathways such as RAS–RAF–MEK–ERK–MAPK and PI3K–AKT–MTOR,11 which function in modulating normal cell growth and survival. Mutations that alter the kinase activity of EGFR lead to abnormal activation of the receptors, even in the absence of a ligand, leading to increased cell proliferation, evasion of apoptosis, angiogenesis, and metastasis.22 As such, considerable efforts have been directed at developing therapeutic compounds that inhibit EGFR signaling. The intracellular tyrosine kinase domain (activating domain) has been a key target for inhibitory drugs. First-generation reversible tyrosine kinase inhibitors (TKI), erlotinib and gefitinib, are small-molecule drugs that competitively bind the adenosine triphosphate (ATP) pocket within the EGFR tyrosine kinase domain and inhibit its kinase activity. For patients with EGFR-activating mutations, both erlotinib and gefitinib are found to increase progression-free survival time and response rates,23,24 and they are currently frontline therapeutics in LAC with EGFR mutations.25 Monoclonal antibodies (mABs) have also been used to bind to the extracellular component of the EGFRs and prevent ligand binding. mABs (cetuximab and panitumumab) are not only used to block ligand binding, but they are also used to promote endocytosis of the receptor and mediate complement cascade cytotoxicity of the cancer cell.26 mABs, however, have only improved overall survival by approximately 1 month and have not demonstrated a statistically significant difference in progression-free survival in comparison to standard chemotherapy; they are not currently the standard of care for LAC patients.27–29
Despite the demonstrated progress using EGFR TKIs, drug resistance has become a challenging hurdle for precision medicine in EGFR-mutant patients. LAC patients whose tumors carry EGFR mutations will ultimately develop resistance to TKIs25 and, moreover, some EGFR-positive patients fail to display an initial response to TKIs, attributed to the presence of resistance mutations to small-molecule inhibitors at the time of diagnosis. Acquired resistance mechanisms have been grouped into four categories: 1) mutation of EGFR to a drug-resistant state; 2) oncogenic shift, or activation of an alternative signaling pathway; 3) impairment of TKI-mediated apoptosis; and 4) histological transformation to small-cell lung cancer or epithelial–mesenchymal transition.30,31 The mutation of threonine 790 to a methionine (T790M) is the most common mechanism for acquired resistance to EGFR TKIs, resulting in increased kinase affinity for ATP, thus decreasing the sensitivity to ATP-competitive inhibitors.32 Several second-generation EGFR TKIs have been developed to combat T790M resistance. These second-generation EGFR TKIs differ from erlotinib and gefitinib by being both mutant-selective and irreversibly binding into the ATP pocket of EGFR. Several of these second-generation EGFR TKIs have entered the clinic (Table 1), demonstrating significant responses rates and progression-free survival as a first-line treatment,33 as well as showing promise in patients with acquired resistance.34 Alternative pathways have been clinically validated to cause resistance, such as activation of BRAF, allowing for continual activation of downstream effectors despite EGFR inhibition.35 MET expression after TKI resistance has been proposed to downregulate the expression of BIM (BCL2L11), which is involved in apoptosis.36 The epithelial–mesenchymal transition, characterized by a loss of E-cadherin expression and the increased expression of fibronectin and vimentin, is associated with EGFR TKI resistance.37 The transition to small-cell lung cancer from TKI-resistant EGFR adenocarcinoma has been demonstrated.38 Although less commonplace than the EGFR T790M mutation, therapeutic approaches and clinical trials are ongoing (Table 1) to overcome these resistance mechanisms.
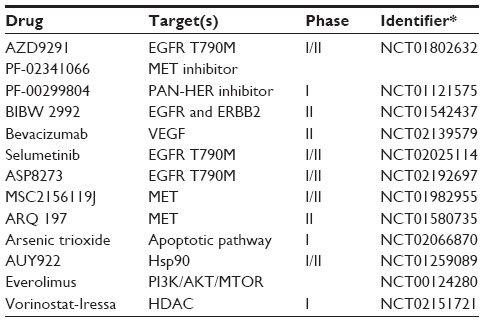 | Table 1 Selected clinical trials for EGFR-resistant NSCLC |
Anaplastic lymphoma receptor tyrosine kinase (ALK)
ALK is a receptor tyrosine kinase in the insulin receptor superfamily. Although its role in normal tissues is not well characterized, activating mutations, transforming rearrangements leading to gene fusions (most common), and aberrant expression of the ALK gene have been described in several different tumor types: anaplastic large-cell lymphoma; neuroblastoma; glioblastoma; colorectal cancer; ovarian cancer; inflammatory myofibroblastic tumor; and NSCLC.6,39–41 In NSCLC, ALK rearrangements produce an inversion in which one of several possible 5′ fusion partners and its promoter region are moved upstream of the ALK kinase domain, resulting in a fusion gene.42,43 In 2%–7% of NSCLC patients, the inversion event fuses ALK with echinoderm microtubule-associated protein-like 4 (EML4), though fusions with KIF5B and TFG have also been described.6 The resulting fusion gene encodes a protein with a ligand-independent, constitutively active kinase domain capable of driving tumor progression, proliferation, survival, and migration through the downstream activation of the MAPK, JAK–STAT, and PI3K/AKT pathways.6,42–44 The oncogenic capabilities of EML4–ALK have been described in vitro and in vivo, and they have been successfully suppressed by certain small-molecule inhibitors to ALK.43,45,46
These characteristics have led to the clinical application of small molecules to inhibit EML4–ALK fusions present in some LAC patients. Crizotinib (PF-02341066), a clinically approved, well-tolerated small-molecule inhibitor to the tyrosine kinase activity of ALK and hepatocyte growth factor receptor (HGFR/MET) is the first ALK inhibitor developed, and it remains a standard TKI for ALK-positive patients.42,47 A clinical study by Kwak et al6 involving 82 patients with ALK-positive NSCLC evaluated the effectiveness of ALK inhibition with crizotinib, where 57% of patients showed a complete or partial response (1/46 complete; 45/46 partial); a partial response by Response Evaluation Criteria in Solid Tumors (RECIST) criteria was defined by at least a 30% change in tumor burden, while 33% (27 patients) had stable disease. A Phase I trial involving 149 ALK translocation-positive NSCLC patients showed a reduction in tumor size by >90%, with 61% displaying an objective response.42 ALK-positive NSCLC patients have shown both increased progression-free survival and response rates with crizotinib in Phase I and II clinical trials.47 As crizotinib inhibits multiple tyrosine kinases, it is also being used clinically to treat adenocarcinomas that harbor a ROS1 rearrangement, a genomic aberration observed in 2% of NSCLC patients.48
To date, numerous second-generation small-molecule inhibitors of ALK are currently being developed or are undergoing clinical trials to improve efficacy and combat crizotinib resistance (Table 2). Ceritinib (LDK378), clinically approved in 2014, is a second-generation ALK inhibitor shown to overcome crizotinib resistance in preclinical and Phase I clinical trials of NSCLC patients harboring ALK rearrangements.42,49 In a recent Phase I/II clinical trial,50 crizotinib-naïve NSCLC patients with ALK rearrangements were treated with alectinib (CH5424802), a well-tolerated, selective ALK inhibitor with 93.5% having an objective response. A dual ALK and EGFR inhibitor, AP26113 showed anti-ALK kinase activity in a Phase I/II study.42 Inhibitors of heat shock protein 90 (Hsp90), a chaperone protein involved in ALK synthesis, have shown promise in reducing ALK protein levels in preliminary studies of ALK rearranged NSCLC.42 Specifically, ganetespib (STA-9090), a Hsp90 inhibitor, has shown efficacy in a Phase IIb/III study in combination with docetaxel.42
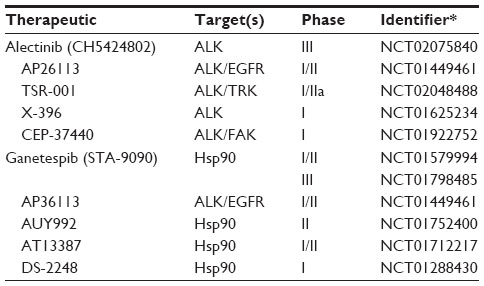 | Table 2 Selected clinical trials using ALK inhibitors |
The ret proto-oncogene (RET)
RET, the ret proto-oncogene of the cadherin superfamily, encodes a receptor tyrosine kinase involved in neural crest development, growth, and differentiation.51 RET mutations are implicated in several different diseases including multiple endocrine neoplasia (types IIA and IIB), Hirschsprung disease, medullary thyroid carcinoma, and NSCLC.51
In a study involving 1,876 patients with lung carcinomas, fluorescent in situ hybridization and reverse transcriptase polymerase chain reaction were used to detect RET gene rearrangements. A total of 1.2% (number =22) of cases were found to be positive for RET rearrangement, and all cases were LAC.51 RET rearrangement is correlated with younger patients (<60 years of age), adenocarcinomas with no other known oncogenic drivers, small primary tumors, and a history of nonsmoking.51–53 In NSCLC, chromosomal rearrangements result in the fusion of RET’s C-terminal region to the N-terminal of several proteins (KIF5B, CCD6, NCOA4, TRIM33), resulting in constitutive activation of the RET kinase domain51 and oncogenic activity.53 In 19 of the 22 cases, RET was fused with KIF5B, and in 3/22 cases, the fusion partner was CCD6.51 KIF5B is the most prevalent fusion partner, though RET fusions with NCOA4 and TRIM33 have been identified.52,54
To date, limited treatment options are available for patients harboring RET rearrangements. Carbozantinib (XL-184), a multi-TKI and RET inhibitor, is undergoing Phase II clinical trials to determine its efficacy in NSCLC patients with RET fusion-positive advanced NSCLC (NCT01639508). Clinical data available on the first three patients treated with carbozantinib indicated a partial response in two of the three patients, and one had prolonged stable disease for 31 weeks. All three were progression free during treatment.31
Vandetanib is a RET/VEGF/EGFR inhibitor approved for the treatment of medullary thyroid cancer. It was shown to decrease metastasis size and led to remission in a 58-year-old patient with metastasized LAC.53 A Phase II clinical trial is currently recruiting patients to study the safety and efficacy of vandetanib in advanced NSCLC patients with RET gene rearrangements (NCT01823068). Other small-molecule inhibitors currently being investigated for efficacy in RET-positive LAC patients include ponatinib (Phase II), levatinib/E7080 (Phase II), MGCD516 (Phase I/Ib), and sunitinib (Phase II).
Defined molecular targets in SCC
While molecular targets for SCC have been limited in the past, current research has identified several notable targets including the fibroblast growth factor receptor (FGFR) family kinases and the PI3K/AKT pathway.
Fibroblast growth factor 1 (FGFR1)
FGFR are a family of tyrosine kinases that, under normal cellular function, play an important role in development, angiogenesis, and proliferation.55 Of the four FGFRs (FGFR1–4), FGFR1 has been found to be frequently deregulated in SCC by amplification or receptor activation.55,56 One study found amplifications in chromosome 8p12 in 22% of SCC patients.56 Additionally, FGFR1 amplification and high serum basic fibroblast growth factor (bFGF) levels have been associated with poor prognosis57,58 and increased proliferative rate.59 The oncogenic role of FGFR1 was demonstrated in preclinical studies of FGFR1-amplified cell lines. Inhibition of FGFR1 signaling through decreased FGFR1 expression via FGFR1-specific small hairpin (sh)RNA,56 or the inhibition of bFGF through neutralizing mABs,59 results in growth inhibition. Similarly, multitargeted small-molecule inhibitors like nintedanib,60 ponatinib,61 and the FGFR-specific inhibitor AZD454762 have demonstrated antitumor and antiangiogenic effects in FGFR-amplified preclinical studies, which has led to clinical studies of FGFR1 inhibitors (Table 3).
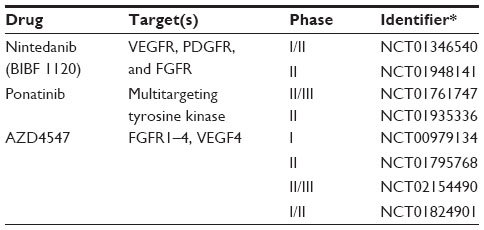 | Table 3 Selected clinical trials for FGFR1 in SCC |
Nintedanib (BIBF 1120) is a multitargeting TKI that blocks vascular endothelial growth factor receptor, platelet-derived growth factor receptor, and FGFR signaling.60 It is currently in a Phase I/II clinical trial (NCT01346540) in combination with platinum-based chemotherapy for recurrent SCC NSCLC patients. A completed Phase I trial employing nintedanib with carboplatin/paclitaxil in advanced NSCLC found partial responses in 27% of patients; however, two of three patients with squamous histology responded.63 Ponatinib is another multi-targeting TKI, initially developed to target aberrant BCR-ABL, but has since been found to also inhibit FGFR and preferentially inhibit growth of FGFR1-amplified primary lung cancer cells.64 Ponatinib is currently in Phase II/III clinical trials (NCT01761747) for SCC NSCLC or for SCC of the head and neck with confirmed FGFR1 amplifications; however, this trial has currently suspended enrollment due to an increased risk of blood clots.
AZD4547 is a TKI specific to FGFR1–4, but it also results in mild inhibition of VEGF4.62 In preclinical models, AZD4547 showed cytotoxic and cytostatic effects in cell lines with FGFR1 amplifications.62 A current Phase II clinical trial (NCT01795768) is examining AZD4547 in breast, squamous lung, and stomach cancers with FGFR1 or 2 amplifications. This trial is specifically looking at tumor growth and tumor ERK1/2 phosphorylation as a proof-of-concept study. Notably, a closed Phase I clinical trial (NCT00979134) found partial response in lung SCC patients with high FGFR amplification and mild, reversible adverse effects to AZD4547 treatment.
Phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PIK3CA)
The PI3K are a family of lipid kinases that are generally activated by receptor tyrosine kinases and contribute to cell proliferation, growth, and differentiation. The kinases are heterodimers consisting of a 58 KDa regulatory subunit (PIK3R1, 2, or 3) and a 110 KDa catalyic subunit (PIK3CA, B, or D) which, when mutated, are implicated in a number of cancers including lung SCC.65 Upon activation, PI3K functions through activating the AKT/MTOR pathway to drive cell growth and survival. The tumor suppressor, PTEN, is one of the main antagonistic regulators of the PI3K, and mutational inactivation of PTEN leads to hyperactivation of PI3K signaling and increased cell growth.66 Deletion of PTEN and LKB1 in GEMMs induces NSCLC tumors of SCC histology.67 Current TCGA data revealed mutations or amplifications in the PI3K/AKT pathway in 43% of lung SCC samples including 16% with altered PIK3CA, 16% AKT3 mutations, and 15% PTEN alterations.12 Additionally PIK3CA is amplified in 33%–43% of SCC cases.68 However, because of the high mutation rate of SCC, it is possible that many of these mutations are passenger mutations rather than drivers. One study found that samples with alterations in PIK3CA had no other common driver mutations (eg, KRAS and EGFR), thus indicating a potential therapeutic response from targeting the PI3K pathway in SCC.68
PI3K inhibitors have been developed for clinical trials in other cancers, but yet they are still limited for lung SCC. Current PI3K inhibitors including pan-PI3K inhibitors, isoform-specific PI3K inhibitors, AKT inhibitors, MTOR inhibitors, and dual PI3K/MTOR inhibitors are currently in clinical trials (Table 4), and they are being evaluated alone and in combination with standard platinum-based chemotherapy treatments.69
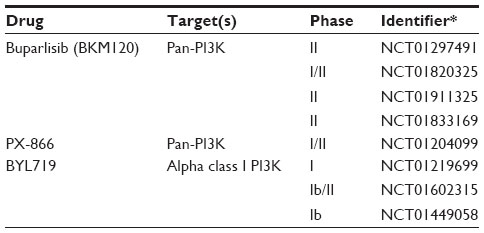 | Table 4 Selected clinical trials for PIK3CA in SCC |
Two pan-PI3K inhibitors, buparlisib (BKM120) and PX-866, are being evaluated in SCC lung cancer. The pan-PI3K inhibitor buparlisib binds to the ATP-binding site in the lipid kinase subunit of all PI3K isoforms; in preclinical trials, it has shown antiangiogenic and antiproliferative effects preferentially in PIK3CA mutated cell lines.70 Phase I trials in Japanese patients with advanced solid tumors demonstrated stable disease and partial responses to buparlisib treatment alone.71 Phase II trials have been initiated in patients with pretreated metastatic NSCLC (including SCC) with an activated PI3K pathway; however, no results have been reported (NCT01297491, NCT01820325). PX-866 is an irreversible pan-class I PI3K inhibitor that has shown lasting PI3K inhibition and antitumor effects in vivo in PIK3CA-mutated SCC of the head and neck.72,73 In Phase I and II clinical trials for PX-866, alone or in combination, for patients with advanced solid tumors, it was found that 79%–85% of patients had stable disease for the period of the trial.74 However, these trials saw no correlation between PIK3CA status and response rate. An ongoing Phase I and II clinical trial (NCT01204099) in NSCLC and SCC of the head and neck is evaluating the response of PX-866 and docetaxel combination therapy. Preliminary data from this study found that the treatment was well tolerated, and patients with PIK3CA mutations maintained progression-free disease longer than those with KRAS or both KRAS and PIK3CA mutations.
The alpha class I PI3K isoform-specific PI3K inhibitor, BYL719, may be a promising treatment for patients with PI3KCA mutations and copy number gain.75,76 In initial clinical trials (NCT01219699), BYL719 was well tolerated and showed preliminary efficacy in patients with PI3KCA-mutated solid tumors. Although no clinical trials are underway for lung SCC patients, there are studies in progress for previously treated head and neck SCC (NCT01602315) and esophageal SCC (NCT01449058). However, preclinical data also found that although PIK3CA-mutated cell lines are sensitive to the treatment, PTEN inactivation is associated with insensitivity to BYL719,75 indicating that patients with lung SCC may benefit from a pan-PI3K inhibitor due to the frequency of PTEN mutations.
Emerging targets in LAC
Kirsten rat sarcoma viral oncogene homolog (KRAS)
Across 230 primary LAC, 33% were observed to harbor a KRAS mutation by NGS in TCGA.11 As such, KRAS mutation represents one of the most frequent alterations in this tumor type. KRAS mutations are prevalent in other malignancies such as pancreatic cancer; where KRAS mutations are present in up to 95% of cases.77 As a guanosine triphosphatase (GTPase), KRAS functions as a molecular switch that, once activated, functions to propagate signal transduction pathways. KRAS signaling associates with numerous tumor-related signaling pathways including MAPK signaling, PI3K/AKT signaling, and RAC and RAL signaling.78 The prevalence of KRAS mutations in many tumor types, including LAC, has made it an attractive therapeutic target. However, despite early hopes based upon in vitro and in vivo experiments using mutants of the KRAS homolog, HRAS, direct targeting of KRAS has been unsuccessful to date. More recently, a better understanding of mutant KRAS signaling and KRAS function has led to novel therapeutic strategies for this molecular subgroup.
To date, KRAS mutation status has not been proven to be prognostic to treatment with adjuvant chemotherapy.79 There are, however, multiple clinical trials that are recruiting and running, and which are targeting lung tumors with activating KRAS mutations through the immunological targeting of mutant KRAS, RAS-related downstream signaling, and G2 checkpoint inhibitors (Table 5). In 2004, Lu et al80 described a yeast-based immunotherapy in which yeast expressing mutant RAS proteins could illicit tumor killing in lung cancers harboring the KRAS mutation. The use of this immunogenic therapy (GI-4000) is currently in clinical trial for KRAS-mutated lung cancer (NCT00655161). A Phase II study of the GI-4000 KRAS vaccine in patients with LAC harboring common KRAS mutations demonstrated that GI-4000 could be tolerated and elicit an immunogenic response in patients.81
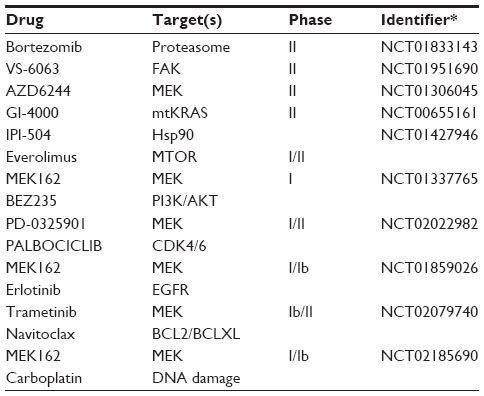 | Table 5 Selected clinical trials for mtKRAS tumors |
Another therapeutic avenue to combat KRAS-driven lung cancer is the inhibition of downstream signaling pathways – more specifically, MAPK and PI3K. In vivo studies with KRAS-driven lung tumorigenesis have demonstrated therapeutic responses with combinations of MEK inhibitors and PI3K inhibitors. In 2008, Engelman et al82 demonstrated synergistic tumor shrinkage in mutant KRAS cancers employing NVP-BEZ235, a PI3K and MTOR inhibitor, and ARRY-142886, a MEK inhibitor. There are currently several clinical trials exploring both the safety and efficacy of combinations of MEK inhibitors and PI3K inhibitors.83 In vivo mouse studies have highlighted other therapeutic combinations with MEK inhibition that are capable of suppressing KRAS-driven tumorigenesis, including MEK with BCLXL inhibitors,84 MEK and insulin-like growth factor 1 receptor inhibitors,85 and MEK with JAK/TBK1 inhibitors.86 However, it is important to consider the contributions of other tumor-associated mutations, as mutations to the STK11/LKB1 tumor suppressor reduced the efficacy of MEK inhibition, while TP53 did not within murine genetically engineered mouse models (GEMM) expressing mutant KRAS.87 Regardless, as the signaling pathways governed by KRAS are better understood, novel therapeutic strategies will continue to improve responses against KRAS-driven lung tumorigenesis.
A third avenue to inhibit KRAS-driven tumorigenesis employs G2 checkpoint inhibitors. The G2/M checkpoint serves to ensure DNA integrity prior to the cell entering mitosis. The inhibition of proteins that govern the G2 checkpoint such as ATM, CHK1/2, PLK1, and WEE1 has attracted significant interest toward cytotoxic treatment. PLK1 is overexpressed in KRAS-mutant tumors, and these tumors were sensitive to PLK inhibitors.88 Recently, Weisberg et al89 demonstrated that WEE1 inhibition combined with MTOR inhibitors selectively inhibited tumors that were positive for mutant RAS. The WEE1 inhibitor AZD1775 is currently in clinical trials in combination with DNA-damaging agents in pancreatic cancer, which is a KRAS-driven tumor type (NCT02037230). Lastly, the use of Hsp90 inhibitors in KRAS-driven tumorigenesis is under investigation. In vivo studies have suggested that mutant KRAS tumors are sensitive to Hsp90 inhibitors.83 Of interest, the efficacy of an Hsp90 inhibitor in NRAS-driven melanoma was dependent on inhibition of WEE1.90 Thus, a better understanding of the role of mutant RAS in the regulation of G2 and DNA damage checkpoints may facilitate new therapeutic strategies in RAS-driven tumors.
Tumor protein p53 (TP53)
The tumor suppressor, TP53, is one of the most frequent genes mutated in cancer. TP53 is altered (mutated or deleted) in 46% of LAC and 90% of SCC according to TCGA.11,12 While the frequency of TP53 alteration is well recognized, therapeutic options based on this alteration have been scarce in lung cancer. One reason for this is an incomplete understanding of TP53 biology in the context of TP53 mutation. TP53 is mutated across the entire coding sequence of the gene leading to everything from TP53 deletions to oncogenic, gain-of-function mutations.91 In situations where the TP53 protein is deleted, reintroduction of the wild type gene has been considered as a therapeutic strategy. Gene transfer of wild type TP53 by retroviral vector was used in lung cancer clinical trials as early as 1996.92 This Phase I study showed no toxic effects of the vector, and tumor regression was noted in three of nine patients. Since then, a number of clinical studies have attempted to reestablish wild type TP53 function by gene transfer.93
Small molecules that inhibit the growth of cancer cells harboring TP53 mutations are also being explored. PRIMA-1 selectively inhibits the growth of mutant TP53 cells by restoring TP53 to a wild type conformation.94 This restoration improves the therapeutic efficacy of DNA-damaging agents such as cisplatin,95 radiation,96 adriamycin,97 and other chemotherapeutic drugs.98 The safety and effectiveness of APR-246, an analog of PRIMA-1, is currently under investigation in clinical trials (NCT02098343) (Table 6).
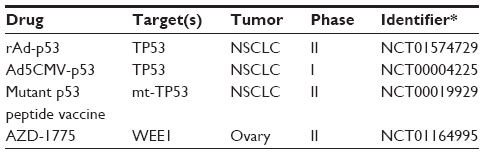 | Table 6 Selected clinical trials targeting TP53 |
Another way in which mutant TP53 is being targeted therapeutically is the development of tumor vaccines. The ability to therapeutically harness the host immune response is under intense investigation toward controlling tumor growth. Tumor vaccines that attempt to use tumor-specific antigens to activate the immune system to target tumor cells are a therapeutic approach that have long been under development. For TP53, it was demonstrated that vaccines could be produced against mutant TP53 and effectively inhibit tumor growth in vivo more than 15 years ago.99 The targeting of mutant TP53 epitopes or TP53 overexpression in tumors by vaccine has shown promise in a number of in vivo settings.93 Currently, TP53 vaccines such as p53-SLP® are in clinical trials.100,101 The use of immunotherapy targeting TP53 in combination with cytotoxic chemotherapy is now under investigation (Table 6). More recently has been the development of therapeutics (anti-CTLA4 and anti-PD-1/PD-L1 antibodies), which are designed to inhibit immune blockade observed in tumors, thus enhancing the immune response.102 Several clinical trials utilizing this therapeutic approach are currently underway, and early results are promising. Clinical trials specific to lung cancers currently employ anti-CTLA4 antibodies (Ipilimumab) in combination with radiation (NCT02221739) or platinum-based therapies (NCT01331525). The anti-PD-1 antibodies, nivolumab (BMS-936558) or pembrolizumab (MK-3475), are being used in clinical trials alone (NCT02259621) or in combination with chemotherapy (NCT02039674) in lung cancer patients. However, as with other targeted therapies, biomarkers for these targets would be invaluable, allowing the tailoring of anti-CTLA4 or anti-PD-1/PD-L1 therapies toward patients who would benefit most.
As is the case with mutant KRAS tumors, a mechanistic understanding of mutant TP53 signaling is also being exploited in lung cancer therapeutics. TP53 is a significant player in the DNA damage response pathway in cells, suggesting that inhibition of the DNA damage response checkpoint proteins may have therapeutic value in mutant TP53 settings. The WEE1 inhibitor, AZD1775 (formerly MK1775), displays preferential effectiveness in mutant TP53 cell lines.103,104 A current clinical trial is recruiting ovarian cancer patients with mutated TP53 to be treated with AZD1775 and carboplatin (NCT01164995).
Serine/threonine kinase 11 (STK11)
STK11/LKB1 (serine/threonine kinase 11/Liver kinase B1) was originally identified as the causative gene mutated in the familial cancer disease, Peutz–Jeghers’ syndrome.105 Subsequent investigations for sporadic LKB1 mutations found a high prevalence for LKB1 inactivation in NSCLC relative to other solid tumors.106–108 More recent TCGA analyses of SCC and LAC tumors show that DNA mutations to LKB1 are primarily a characteristic of LAC.10,11,12 The detection of LKB1 inactivation has been difficult, with studies placing the frequency of LKB1 mutations between 15%–30% of LAC tumors106–108 and homozygous deletion, and the loss of heterozygosity of the LKB1 locus of chromosome 19p at 89%,109 suggesting that the true frequency of LKB1 inactivation in LAC is high. LKB1 inactivation commonly occurs in concert with activating KRAS mutations (mtKRAS) (10% of patients), and mtKRAS synergizes with the biallelic deletion of LKB1 in GEMMs to produce highly metastatic, aggressive tumors in the lung107 that are genetically distinct from other NSCLC tumors harboring mtKRAS alone.110 Although the broad regulatory functions of LKB1 are still being elucidated,105 efforts have been made to define possible treatment options using a well-characterized GEMM of mtKRAS/LKB1-deficiency (mtKRAS/LKB1null).87,107,110–112
Using gene expression analysis of GEMM mtKRAS/LKB1null NSCLC tumors, Carretero et al110 identified and demonstrated that the SRC kinase is a putative target in mtKRAS/LKB1null NSCLC and when combined with inhibitors to the RAS–MAPK and PI3K–AKT pathways, SRC inhibition induced significant tumor regression. Perhaps more interestingly, GEMM mtKRAS/LKB1null NSCLC tumors display increased activation of the PI3K pathway with reduced activation of the RAS–MAPK pathway, which is thought to manifest into resistance to MEK inhibition.87 Similarly, synthetic lethal RNA interference (RNAi) screens using tumor cell lines generated from GEMM mtKRAS/LKB1null NSCLC tumors identified Dtymk, an enzyme responsible for dTTP biosynthesis, as a potential target in LKB1-deficient NSCLC.113
Concurrent to these efforts are studies aimed toward taking advantage of hypersensitivity to stress present within LKB1-deficient cells. Substantial data indicate that inactivation of LKB1 renders cells unable to respond to stress resulting from a variety of sources.111,114,115 In particular, there is an appreciation that LKB1 is critical in mediating the effects of metformin and its analogs.105,116 Recruitment is underway to assess whether LKB1 gene status will determine the response to metformin plus standard therapy in LAC (clinical trial NCT01578551). Critically, treatment with phenformin, a more potent analog of metformin, significantly perturbs the growth of tumors and improves survival in the mtKRAS/LKB1null NSCLC GEMM, compared to GEMMs harboring mtKRAS alone or mtKRAS with TP53 deletion.112 In our laboratories, we found that mtKRAS/LKB1null NSCLC cells are hypersensitive to the disruption of protein synthesis within the endoplasmic reticulum (ER), and subsequent activation of ER stress responses by an aggravator of ER stress can reduce NSCLC tumor growth in the mtKRAS/LKB1null NSCLC GEMM.111 Given the potentially high incidence of LKB1 inactivation in LAC, the next step will be to develop and validate these approaches within the mtKRAS/LKB1null NSCLC GEMM toward patient clinical trials.
Other molecular targets in LAC
While a significant amount of attention has been paid with respect to EGFR and ALK toward precision medicine strategies in LAC, other molecular targets are being explored. Figure 2 depicts selected molecular alterations in LAC with targeted therapeutic opportunities. Mutations in BRAF occur in ~10% of adenocarcinoma patients according to TGCA.10,11 The prevalence of mutant BRAF in melanoma led to BRAF inhibitor development with clinical successes in BRAF mutant cases.117 LAC cases with mutant BRAF (V600E) have responded to the BRAF inhibitor, vemurafenib.118 Genomic alterations (mutations and amplifications) in hepatocyte growth factor receptor (HGFR/MET) occur in 12% of LAC cases.11 The amplification of MET is a known mechanism of EGFR TKI resistance, as well as a risk factor for metastasis.119,120 Inhibitors against MET are under clinical development toward the treatment of patients with aberrant MET expression.119,120 In particular are ongoing clinical trials to determine the safety of the MET inhibitor, RO5490258, also known as MET-Mab, in NSCLC (NCT01496742).
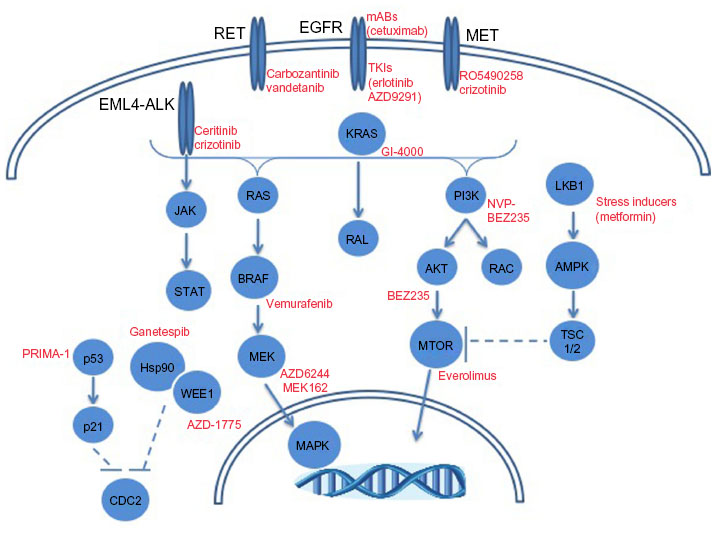 | Figure 2 Molecular alterations in lung adenocarcinomas with targeted therapeutic opportunities. |
Emerging molecular targets in SCC
While FGFR and PI3K inhibitors are being explored clinically in squamous cell lung cancer, other molecular alterations may provide therapeutic opportunities. Figure 3 depicts frequent alterations in SCC with targeted therapeutic opportunities. The high prevalence of TP53 mutations in this histologic subtype may dictate the use of TP53 therapeutics, as has been discussed. The frequency of genomic alterations (mutation or amplification) in EGFR, ERRB2, KRAS, BRAF, and MET ranges between 3%–9% in squamous cell lung cancer.12 Though modest, these patient tumors could be treated with targeted therapeutic strategies discussed previously in this review. A more thorough understanding of the molecular drivers of squamous cell lung cancer is going to be necessary to improve precision medicine-based therapeutic strategies.
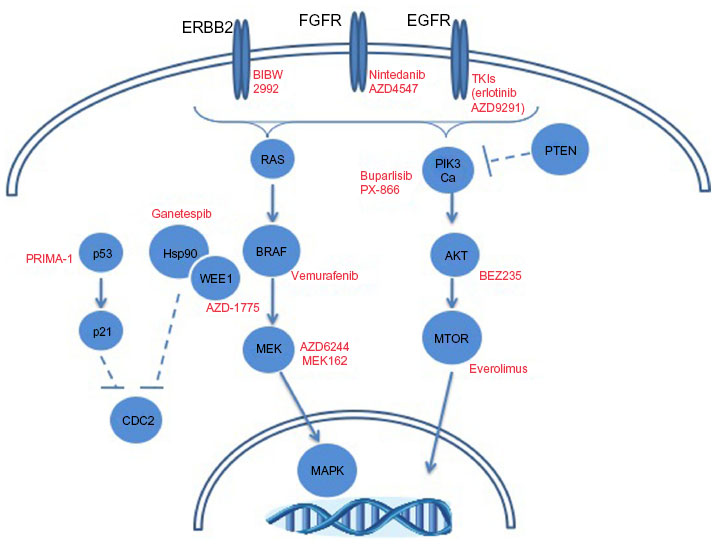 | Figure 3 Molecular alterations in squamous cell lung carcinomas with targeted therapeutic opportunities. |
Conclusion
NSCLC remains a leading cause of cancer-related mortality in the US and throughout the world. New genomic technologies have begun to shed light on the genomic alterations and pathways that drive NSCLC and provide rationale for therapeutic intervention based on genomic aberrations. Despite demonstrated successes using genomic alterations to dictate therapy (such as EGFR mutations in NSCLC), there are significant challenges associated with the clinical use of precision medicine. First, while cost and time-to-results have improved, they are still hurdles to clinical adoption. Second, tumor heterogeneity presents a challenge to precision medicine, as the tumor piece providing the genomic alterations may not fully represent the bulk tumor. This has been cited as a potential concern for therapeutic resistance.121 Strategies for validation have been noted as a potential complication for the clinical adoption of NGS. Another challenge posed by the use of whole-genome sequencing in the clinical setting is providing genomic alterations (both somatic and germline) that are not currently actionable targets, or deciding which of several alterations to target.
While success has been demonstrated in LAC patients with mutated EGFR or gene rearrangements in ALK, therapeutic resistance is pervasive in these subgroups. More importantly, the majority of NSCLC patients do not harbor either of these alterations, indicating a demonstrable need to develop novel therapies that address the bulk of NSCLC tumors. Novel therapeutic strategies are currently in preclinical and clinical development toward targeting recurrent alterations in NSCLC, such as TP53 and KRAS, as well as targeting specific resistance phenotypes. With improvements in our understanding of the molecular tumorigenesis in NSCLC, heavily influenced by next-generation sequence technologies and improved targeted therapeutics, the future of precision medicine in NSCLC should significantly impact NSCLC mortality.
Acknowledgments
The authors would like to thank the Virginia G Piper Charitable Trust (TGW) and the American Lung Association and St Joseph’s Foundation of St Joseph’s Hospital and Medical Center (LJI) for providing funding support.
Disclosure
The authors report no conflicts of interest in this work.
References
Allemani C, Weir HK, Carreira H, et al. Global surveillance of cancer survival 1995-2009: analysis of individual data for 25 676 887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet. Epub Nov 26, 2014. | |
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | |
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | |
Little AG, Gay EG, Gaspar LE, Stewart AK. National survey of non-small cell lung cancer in the United States: epidemiology, pathology and patterns of care. Lung Cancer. 2007;57(3):253–260. | |
Giaccone G. Epidermal growth factor receptor inhibitors in the treatment of non-small-cell lung cancer. J Clin Oncol. 2005;23(14):3235–3242. | |
Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. | |
Thomas RK, Baker AC, Debiasi RM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39(3):347–351. | |
Su Z, Dias-Santagata D, Duke M, et al. A platform for rapid detection of multiple oncogenic mutations with relevance to targeted therapy in non-small-cell lung cancer. J Mol Diagn. 2011;13(1):74–84. | |
Li T, Kung HJ, Mack PC, Gandara DR. Genotyping and genomic profiling of non-small-cell lung cancer: implications for current and future therapies. J Clin Oncol. 2013;31(8):1039–1049. | |
Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–1075. | |
Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. | |
Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–525. | |
Azzoli CG, Temin S, Aliff T, et al; American Society of Clinical Oncology. 2011 Focused Update of 2009 American Society of Clinical Oncology Clinical Practice Guideline Update on Chemotherapy for Stage IV Non-Small-Cell Lung Cancer. J Clin Oncol. 2011;29(28):3825–3831. | |
Pisters KM, Evans WK, Azzoli CG, et al; Cancer Care Ontario; American Society of Clinical Oncology. Cancer Care Ontario and American Society of Clinical Oncology adjuvant chemotherapy and adjuvant radiation therapy for stages I-IIIA resectable non small-cell lung cancer guideline. J Clin Oncol. 2007;25(34):5506–5518. | |
Curran WJ Jr, Paulus R, Langer CJ, et al. Sequential vs concurrent chemoradiation for stage III non-small cell lung cancer: randomized phase III trial RTOG 9410. J Natl Cancer Inst. 2011;103(19):1452–1460. | |
Su SF, Hu YX, Ouyang WW, et al. Overall survival and toxicities regarding thoracic three-dimensional radiotherapy with concurrent chemotherapy for stage IV non-small cell lung cancer: results of a prospective single-center study. BMC Cancer. 2013;13:474. | |
Owonikoko TK, Arbiser J, Zelnak A, et al. Current approaches to the treatment of metastatic brain tumours. Nat Rev Clin Oncol. 2014;11(4):203–222. | |
Rossi A, Ricciardi S, Maione P, de Marinis F, Gridelli C. Pemetrexed in the treatment of advanced non-squamous lung cancer. Lung Cancer. 2009;66(2):141–149. | |
Scagliotti GV, Parikh P, von Pawel J, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26(21):3543–3551. | |
Langer CJ, Besse B, Gualberto A, Brambilla E, Soria JC. The evolving role of histology in the management of advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(36):5311–5320. | |
Noh S, Shim H. Optimal combination of immunohistochemical markers for subclassification of non-small cell lung carcinomas: A tissue microarray study of poorly differentiated areas. Lung Cancer. 2012;76(1):51–55. | |
Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358(11):1160–1174. | |
Rosell R, Carcereny E, Gervais R, et al; Spanish Lung Cancer Group in collaboration with Groupe Français de Pneumo-Cancérologie and Associazione Italiana Oncologia Toracica. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC):a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. | |
Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802):a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735–742. | |
Chong CR, Jänne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19(11):1389–1400. | |
Campoli M, Ferris R, Ferrone S, Wang X. Immunotherapy of malignant disease with tumor antigen-specific monoclonal antibodies. Clin Cancer Res. 2010;16(1):11–20. | |
Lynch TJ, Patel T, Dreisbach L, et al. Cetuximab and first-line taxane/carboplatin chemotherapy in advanced non-small-cell lung cancer: results of the randomized multicenter phase III trial BMS099. J Clin Oncol. 2010;28(6):911–917. | |
Mitsudomi T, Morita S, Yatabe Y, et al; West Japan Oncology Group. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405):an open label, randomised phase 3 trial. Lancet Oncol. 2010;11(2):121–128. | |
Pirker R, Pereira JR, Szczesna A, et al; FLEX Study Team. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX):an open-label randomised phase III trial. Lancet. 2009;373(9674):1525–1531. | |
Lee JK, Shin JY, Kim S, et al. Primary resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in patients with non-small-cell lung cancer harboring TKI-sensitive EGFR mutations: an exploratory study. Ann Oncol. 2013;24(8):2080–2087. | |
Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. | |
Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105(6):2070–2075. | |
Dienstmann R, De Dosso S, Felip E, Tabernero J. Drug development to overcome resistance to EGFR inhibitors in lung and colorectal cancer. Mol Oncol. 2012;6(1):15–26. | |
Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4(9):1046–1061. | |
Ohashi K, Sequist LV, Arcila ME, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A. 2012;109(31):E2127–E2133. | |
Garofalo M, Romano G, Di Leva G, et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat Med. 2012;18(1):74–82. | |
Uramoto H, Iwata T, Onitsuka T, Shimokawa H, Hanagiri T, Oyama T. Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res. 2010;30(7):2513–2517. | |
Alam N, Gustafson KS, Ladanyi M, et al. Small-cell carcinoma with an epidermal growth factor receptor mutation in a never-smoker with gefitinib-responsive adenocarcinoma of the lung. Clin Lung Cancer. 2010;11(5):E1–E4. | |
Lipson D, Capelletti M, Yelensky R, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18(3):382–384. | |
Pulford K, Lamant L, Espinos E, et al. The emerging normal and disease-related roles of anaplastic lymphoma kinase. Cell Mol Life Sci. 2004;61(23):2939–2953. | |
Ren H, Tan ZP, Zhu X, et al. Identification of anaplastic lymphoma kinase as a potential therapeutic target in ovarian cancer. Cancer Res. 2012;72(13):3312–3323. | |
Gridelli C, Peters S, Sgambato A, Casaluce F, Adjei AA, Ciardiello F. ALK inhibitors in the treatment of advanced NSCLC. Cancer Treat Rev. 2014;40(2):300–306. | |
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. | |
Casaluce F, Sgambato A, Maione P, et al. ALK inhibitors: a new targeted therapy in the treatment of advanced NSCLC. Target Oncol. 2013;8(1):55–67. | |
Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27(26):4247–4253. | |
Soda M, Takada S, Takeuchi K, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci U S A. 2008;105(50):19893–19897. | |
Bowles DW, Weickhardt AJ, Doebele RC, Camidge DR, Jimeno A. Crizotinib for the treatment of patients with advanced non-small cell lung cancer. Drugs Today (Barc). 2012;48(4):271–282. | |
Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30(8):863–870. | |
Friboulet L, Li N, Katayama R, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4(6):662–673. | |
Kodama T, Tsukaguchi T, Yoshida M, Kondoh O, Sakamoto H. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett. 2014;351(2):215–221. | |
Tsuta K, Kohno T, Yoshida A, et al. RET-rearranged non-small-cell lung carcinoma: a clinicopathological and molecular analysis. Br J Cancer. 2014;110(6):1571–1578. | |
Drilon A, Wang L, Hasanovic A, et al. Response to Cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov. 2013;3(6):630–635. | |
Gautschi O, Zander T, Keller FA, et al. A patient with lung adenocarcinoma and RET fusion treated with vandetanib. J Thorac Oncol. 2013;8(5):e43–e44. | |
Wang R, Hu H, Pan Y, et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol. 2012;30(35):4352–4359. | |
Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–129. | |
Weiss J, Sos ML, Seidel D, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med. 2010;2(62):62ra93. | |
Bremnes RM, Camps C, Sirera R. Angiogenesis in non-small cell lung cancer: the prognostic impact of neoangiogenesis and the cytokines VEGF and bFGF in tumours and blood. Lung Cancer. 2006;51(2):143–158. | |
Iwasaki A, Kuwahara M, Yoshinaga Y, Shirakusa T. Basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF) levels, as prognostic indicators in NSCLC. Eur J Cardiothorac Surg. 2004;25(3):443–448. | |
Kuhn H, Köpff C, Konrad J, Riedel A, Gessner C, Wirtz H. Influence of basic fibroblast growth factor on the proliferation of non-small cell lung cancer cell lines. Lung Cancer. 2004;44(2):167–174. | |
Hilberg F, Roth GJ, Krssak M, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68(12):4774–4782. | |
Gozgit JM, Wong MJ, Moran L, et al. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol Cancer Ther. 2012;11(3):690–699. | |
Gavine PR, Mooney L, Kilgour E, et al. AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 2012;72(8):2045–2056. | |
Doebele RC, Conkling P, Traynor AM, et al. A phase I, open-label dose-escalation study of continuous treatment with BIBF 1120 in combination with paclitaxel and carboplatin as first-line treatment in patients with advanced non-small-cell lung cancer. Ann Oncol. 2012;23(8):2094–2102. | |
Ren M, Hong M, Liu G, et al. Novel FGFR inhibitor ponatinib suppresses the growth of non-small cell lung cancer cells overexpressing FGFR1. Oncol Rep. 2013;29(6):2181–2190. | |
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–644. | |
Zito CR, Jilaveanu LB, Anagnostou V, et al. Multi-level targeting of the phosphatidylinositol-3-kinase pathway in non-small cell lung cancer cells. PLoS One. 2012;7(2):e31331. | |
Xu C, Fillmore CM, Koyama S, et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell. 2014;25(5):590–604. | |
Yamamoto H, Shigematsu H, Nomura M, et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 2008;68(17):6913–6921. | |
Beck JT, Ismail A, Tolomeo C. Targeting the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway: an emerging treatment strategy for squamous cell lung carcinoma. Cancer Treat Rev. 2014;40(8):980–989. | |
Maira SM, Pecchi S, Huang A, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11(2):317–328. | |
Bendell JC, Rodon J, Burris HA, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30(3):282–290. | |
Ihle NT, Williams R, Chow S, et al. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther. 2004;3(7):763–772. | |
Keysar SB, Astling DP, Anderson RT, et al. A patient tumor transplant model of squamous cell cancer identifies PI3K inhibitors as candidate therapeutics in defined molecular bins. Mol Oncol. 2013;7(4):776–790. | |
Hong DS, Bowles DW, Falchook GS, et al. A multicenter phase I trial of PX-866, an oral irreversible phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2012;18(15):4173–4182. | |
Fritsch C, Huang A, Chatenay-Rivauday C, et al. Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;13(5):1117–1129. | |
Furet P, Guagnano V, Fairhurst RA, et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg Med Chem Lett. 2013;23(13):3741–3748. | |
Biankin AV, Waddell N, Kassahn KS, et al; Australian Pancreatic Cancer Genome Initiative. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405. | |
Khosravi-Far R, Der CJ. The Ras signal transduction pathway. Cancer Metastasis Rev. 1994;13(1):67–89. | |
Shepherd FA, Domerg C, Hainaut P, et al. Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol. 2013;31(17):2173–2181. | |
Lu Y, Bellgrau D, Dwyer-Nield LD, et al. Mutation-selective tumor remission with Ras-targeted, whole yeast-based immunotherapy. Cancer Res. 2004;64(15):5084–5088. | |
Chaft JE, Litvak A, Arcila ME, et al. Phase II study of the GI-4000 KRAS vaccine after curative therapy in patients with stage I–III lung adenocarcinoma harboring a KRAS G12C, G12D, or G12V mutation. Clin Lung Cancer. 2014;15(6):405–410. | |
Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–1356. | |
Ebi H, Faber AC, Engelman JA, Yano S. Not just gRASping at flaws: finding vulnerabilities to develop novel therapies for treating KRAS mutant cancers. Cancer Sci. 2014;105(5):499–505. | |
Corcoran RB, Cheng KA, Hata AN, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23(1):121–128. | |
Chen R, Sweet-Cordero EA. Two is better than one: combining IGF1R and MEK blockade as a promising novel treatment strategy against KRAS-mutant lung cancer. Cancer Discov. 2013;3(5):491–493. | |
Zhu Z, Aref AR, Cohoon TJ, et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014;4(4):452–465. | |
Chen Z, Cheng K, Walton Z, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature. 2012;483(7391):613–617. | |
Luo J, Emanuele MJ, Li D, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–848. | |
Weisberg E, Nonami A, Chen Z, et al. Identification of Wee1 as a novel therapeutic target for mutant RAS-driven acute leukemia and other malignancies. Leukemia. Epub May 5, 2014. | |
Haarberg HE, Paraiso KH, Wood E, et al. Inhibition of Wee1, AKT, and CDK4 underlies the efficacy of the HSP90 inhibitor XL888 in an in vivo model of NRAS-mutant melanoma. Mol Cancer Ther. 2013;12(6):901–912. | |
Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2(1):a001008. | |
Roth JA, Nguyen D, Lawrence DD, et al. Retrovirus-mediated wild-type p53 gene transfer to tumors of patients with lung cancer. Nat Med. 1996;2(9):985–991. | |
Lane DP, Cheok CF, Lain S. p53-based cancer therapy. Cold Spring Harb Perspect Biol. 2010;2(9):a001222. | |
Bykov VJ, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8(3):282–288. | |
Bykov VJ, Zache N, Stridh H, et al. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene. 2005;24(21):3484–3491. | |
Supiot S, Zhao H, Wiman K, Hill RP, Bristow RG. PRIMA-1(met) radiosensitizes prostate cancer cells independent of their MTp53-status. Radiother Oncol. 2008;86(3):407–411. | |
Magrini R, Russo D, Ottaggio L, Fronza G, Inga A, Menichini P. PRIMA-1 synergizes with adriamycin to induce cell death in non-small cell lung cancer cells. J Cell Biochem. 2008;104(6):2363–2373. | |
Izetti P, Hautefeuille A, Abujamra AL, et al. PRIMA-1, a mutant p53 reactivator, induces apoptosis and enhances chemotherapeutic cytotoxicity in pancreatic cancer cell lines. Invest New Drugs. 2014;32(5):783–794. | |
Mayordomo JI, Loftus DJ, Sakamoto H, et al. Therapy of murine tumors with p53 wild-type and mutant sequence peptide-based vaccines. J Exp Med. 1996;183(4):1357–1365. | |
Leffers N, Lambeck AJ, Gooden MJ, et al. Immunization with a P53 synthetic long peptide vaccine induces P53-specific immune responses in ovarian cancer patients, a phase II trial. Int J Cancer. 2009;125(9):2104–2113. | |
Speetjens FM, Kuppen PJ, Welters MJ, et al. Induction of p53-specific immunity by a p53 synthetic long peptide vaccine in patients treated for metastatic colorectal cancer. Clin Cancer Res. 2009;15(3):1086–1095. | |
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. | |
Hirai H, Iwasawa Y, Okada M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8(11):2992–3000. | |
Rajeshkumar NV, De Oliveira E, Ottenhof N, et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17(9):2799–2806. | |
Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9(8):563–575. | |
Avizienyte E, Loukola A, Roth S, et al. LKB1 somatic mutations in sporadic tumors. Am J Pathol. 1999;154(3):677–681. | |
Ji H, Ramsey MR, Hayes DN, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448(7155):807–810. | |
Sanchez-Cespedes M, Parrella P, Esteller M, et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002;62(13):3659–3662. | |
Gill RK, Yang SH, Meerzaman D, et al. Frequent homozygous deletion of the LKB1/STK11 gene in non-small cell lung cancer. Oncogene. 2011;30(35):3784–3791. | |
Carretero J, Shimamura T, Rikova K, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell. 2010;17(6):547–559. | |
Inge LJ, Friel JM, Richer AL, et al. LKB1 inactivation sensitizes non-small cell lung cancer to pharmacological aggravation of ER stress. Cancer Lett. 2014;352(2):187–195. | |
Shackelford DB, Abt E, Gerken L, et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013;23(2):143–158. | |
Liu Y, Marks K, Cowley GS, et al. Metabolic and functional genomic studies identify deoxythymidylate kinase as a target in LKB1-mutant lung cancer. Cancer Discov. 2013;3(8):870–879. | |
Bungard D, Fuerth BJ, Zeng PY, et al. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329(5996):1201–1205. | |
Shaw RJ, Kosmatka M, Bardeesy N, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101(10):3329–3335. | |
Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310(5754):1642–1646. | |
Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14(7):455–467. | |
Robinson SD, O’Shaughnessy JA, Cowey CL, Konduri K. BRAF V600E-mutated lung adenocarcinoma with metastases to the brain responding to treatment with vemurafenib. Lung Cancer. 2014;85(2):326–330. | |
Feng Y, Thiagarajan PS, Ma PC. MET signaling: novel targeted inhibition and its clinical development in lung cancer. J Thorac Oncol. 2012;7(2):459–467. | |
Sadiq AA, Salgia R. MET as a possible target for non-small-cell lung cancer. J Clin Oncol. 2013;31(8):1089–1096. | |
Burrell RA, Swanton C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol Oncol. 2014;8(6):1095–1111. | |
cbioportal.org [homepage on the Internet]. cBioPortal for Cancer Genomics. Available from: http://www.cbioportal.org/. Accessed January 27, 2015. |
Supplementary materials
 | Table S1 HUGO approved name and symbol |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.