Back to Archived Journals » Open Access Bioinformatics » Volume 6
Genome-wide mapping of chromatin state of mouse forelimbs
Authors Eng D, Vogel W, Flann N, Gross M, Kioussi C
Received 11 December 2013
Accepted for publication 16 April 2014
Published 10 September 2014 Volume 2014:6 Pages 1—11
DOI https://doi.org/10.2147/OAB.S59043
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Diana Eng,1 Walter K Vogel,1 Nicholas S Flann,2,3 Michael K Gross,1 Chrissa Kioussi1
1Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, Corvallis, OR, USA; 2Department of Computer Science, Utah State University, Logan, UT, USA; 3Institute for Systems Biology, Seattle, WA, USA
Background: Cell types are defined at the molecular level during embryogenesis by a process called pattern formation and created by the selective utilization of combinations of sequence-specific transcription factors. Developmental programs define the sets of genes that are available to each particular cell type, and real-time biochemical signaling interactions define the extent to which these sets are used at any given time and place. Gene expression is regulated through the integrated action of many cis-regulatory elements, including core promoters, enhancers, silencers, and insulators. The chromatin state in developing body parts provides a code to cellular populations that directs their cell fates. Chromatin profiling has been a method of choice for mapping regulatory sequences in cells that go through developmental transitions.
Results: We used antibodies against histone H3 lysine 4 trimethylations, a modification associated with promoters and open/active chromatin, histone H3 lysine 27 trimethylations associated with Polycomb-repressed regions, and ribonucleic acid polymerase II associated with transcriptional initiation to identify the chromatin state signature of the mouse forelimb during mid-gestation at embryonic day 12. The families of genes marked included those related to transcriptional regulation and embryogenesis. One-third of the marked genes were transcriptionally active, whereas only a small fraction were bivalent marked. Sequence-specific transcription factors that were activated were involved in cell specification, including bone and muscle formation.
Conclusion: Our results demonstrate that embryonic limb cells do not exhibit the plasticity of the embryonic stem cells but rather are programmed for a finer tuning for cell lineage specification.
Keywords: mouse genome, chromatin, forelimb, sequence-specific transcription factors
Introduction
One of the challenges in biology is understanding how one genome can generate an organism composed of hundreds of distinct cell types. The gene expression programs that specify and maintain cell states in mammals are controlled by thousands of transcription factors, cofactors, and chromatin modifiers. Misregulation of these gene expression programs can lead to cellular transformation, organ malfunction, and disease. Identifying the cell state (type) based on the specific combinatorial gene regulatory networks can allow for cell reprogramming when a cell fails to follow the correct route or has lost its molecular memory over time. Embryonic development is the process that normally creates “cell types”. In mammals, the process of specifying cell types occurs during pattern-formation events that require highly complex spatial and temporal gene regulatory networks. A cell can execute the process of staying in or exiting the cell cycle and entering the postmitotic differentiation state by using specific cis-regulatory modules (CRMs). CRMs are several hundred base pairs long, can exert their influence over distances as long as 100 kb, comprise multiple sequence-specific transcription factor (SSTF) binding sites, and can direct the expression of developmental SSTFs and signaling molecules.1 CRMs act as switches to determine “availability” of associated loci for expression.2,3 SSTFs initiate lineage-specific gene expression programs, and epigenetic regulation contributes to stabilization of expression patterns.
The combination of chromatin immunoprecipitation (ChIP) and massive parallel sequencing (ChIP-seq) allows the thorough detection of protein–DNA interactions and enables the identification of regulatory events central to biological processes and disease states.4–7 Histone modifications modulate transcriptional initiation, elongation, and enhancer activity or repression.8 Commonly studied interactions utilizing ChIP-seq include ribonucleic acid (RNA) polymerases, SSTFs, and histone marks.9 The advantage of this system is its high-throughput nature and ability to identify genome-wide de novo molecular interactions. ChIP-seq is one of the primary techniques used in the various Encyclopedia of DNA Elements (ENCODE) projects, with the goal of identifying functional elements in the genomes of humans, with similar consortiums focusing on model organisms.10
Open chromatin is characterized by the presence of histone modifications such as histone H3 lysine 4 trimethylation (H3K4me3), histone H3 lysine 9 acetylation (H3K9ac), and histone H4 acetylation (H4ac), whereas closed chromatin is characterized by the presence of histone H3 lysine 9 trimethylation (H3K9me3) and histone H3 lysine 27 trimethylation (H3K27me3). The presence of H3K4me3 on a promoter is a hallmark of active genes and open chromatin and is highly correlated with the presence of RNA polymerase II (Pol2).11 Conversely, H3K27me3 is a marker of repressed genes, which negatively regulates transcription by promoting heterochromatin formation.12 The methylation of H3K4 and H3K27 is catalyzed by the highly conserved Trithorax (TrxG) and Polycomb (PcG) groups of proteins, respectively, opposing forces to regulate transcription.12,13 However, H3K4me3 and H3K27me3 are not mutually exclusive and can bivalently mark chromatin. Bivalent genes are poised for either activation or repression, are often expressed at a low level, and tend to lose H3K27me3 marks as they differentiate from a stem cell state to a committed state.14 Broad H3K27me3 marks on bivalent genes were associated with a general repression of expression, and more “peak like” marks on the promoter region were often correlated with active gene expression.15 Thus, bivalently marked regions are of particular interest when studying developmental networks.
Methods
Mice
All research was conducted in accordance with Oregon State University Institutional Animal Care and Use Committee (IACUC) approval, under ACUP# 4227. ICR mice from Harlan Laboratories were bred, and females were checked for the presence of a vaginal plug (E0.5). Multiple litters, each containing six to 15 embryos, were isolated at E12.5 and dissected for forelimbs in ice-cold phosphate-buffered saline (PBS). Biopsies were pooled with at least ten embryos per 1.5 mL tube and immediately fixed.
ChIP and ChIP-seq
Biopsies from embryos were crosslinked for 20 minutes in freshly made fixation buffer (1% formaldehyde 100 mM NaCl, 0.5 mM EGTA (ethylene glycol tetraacetic acid), 50 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), pH 8.0) and quenched by the addition of glycine (125 mM). Tissue was washed twice with ice-cold PBS and snap-frozen in liquid nitrogen prior to sonication (14 cycles of 10 seconds at 20% output with 60 seconds on ice between cycles, Branson 450 sonicator in lysis buffer [1% SDS, 10 mM ethylenediaminetetraacetic acid (EDTA) pH 8.0, 50 mM Tris-HCl pH 8.0, protease inhibitors]) to achieve an average sheared fragment size of 300 bp, with the primary smear between 200 bp and 450 bp. A portion of sheared chromatin from each preparation was reserved as the input control. Protein G magnetic beads (NEB S1430S) were prepared by washing three times in PBS with 0.05% bovine serum albumin (BSA), bound overnight to rabbit anti-H3K4me3 (Abcam plc, Cambridge, UK, ab1342), rabbit anti-H3K27me3 (EMD Millipore, Billerica, MA, USA, 07-449), or mouse anti-Pol2 (EMD Millipore, 17-672) at 2 μg/20 μL beads, and washed three times in PBS–BSA. Material for immunoprecipitation (IP) was diluted 1:10 in dilution buffer (1% Triton X-100, 150 mM NaCl, 2 mM EDTA pH 8.0, 20 mM Tris-HCl pH 8.0, protease inhibitors), and 20 μL of protein G magnetic beads were added to each milliliter of sheared chromatin (about 200 μg each) and allowed to rotate overnight at 4°C. The following day, samples were washed once in low-salt wash (1% Triton X-100, 0.1% SDS, 150 mM NaCl, 2 mM EDTA pH 8.0, 10 mM Tris HCl pH 8.0, protease inhibitors), twice in high-salt wash (1% Triton X-100, 0.1% SDS, 500 mM NaCl, 2 mM EDTA pH 8.0, 10 mM Tris HCl pH 8.0, protease inhibitors), twice in lithium wash (100 mM Tris HCl pH 9.0, 500 mM LiCl, 1% NP40, 1% deoxycholic acid), and three times in TE (10 mM Tris HCl pH 8.0, 1 mM EDTA) and eluted in 100 μL of elution buffer containing RNaseA and proteinase K (1% SDS, 100 mM NaHCO3, 10 mg/mL RNaseA, 5 M NaCl, 0.2 mg/mL proteinase K). Elutants and respective input controls were decrosslinked overnight at 65°C, purified on Qiagen Miniprep columns (Qiagen NV, Venlo, The Netherlands), and eluted in ddH2O. One set from each condition was reserved for quantitative polymerase chain reaction validation, and the remaining ChIP DNA was speed vacuum dried. DNA was resuspended in 15 μL of ddH2O, quantified with the Qubit fluorometer (Life Technologies, Carlsbad, CA, USA) or the NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), until at least 10 ng was collected for each sample. Single-end libraries were generated using the Illumina TruSeq ChIP-Seq Library Preparation Kit (Illumina, Inc., San Diego, CA, USA) using 18 cycles for polymerase chain reaction sample enrichment, with the only protocol modification being size selection after fragment amplification. The library size selected was 200–300 bp in length and, after gel purification, size was confirmed with the Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Libraries were then sequenced on a 51 bp single-end run on the Illumina HiSeq 2000.
ChIP-seq data analysis
Primary Illumina data image analysis, base calling, and filtering were performed by the Casava pipeline (version 1.8.2; Illumina), and low-quality filtered reads were removed from the data set prior to alignment. Filtered reads from the ChIP and input control libraries were aligned to the mouse genome (NCBI37/mm9) reference assembly using Burrows–Wheeler Aligner (version 0.6.2). Model-based Analysis of ChIP-Seq (MACS) (version 2.0.10) was used to identify genomic regions significantly enriched (q≤0.05) in ChIP-seq samples over input controls, and the software “broad peaks” option was used to delineate the mapped extent of histone marks.16 The University of California Santa Cruz (UCSC) Genome Browser was used to visualize the mapping data and peak calls. Homer tools were utilized for gene ontology annotation and comparisons of found peaks.17 DAVID bioinformatics tools were used for gene ontology clustering.18,19 ChIP-seq data from this study are accessible under accession number GSE49010 from NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo).
Results
Genome mapping of mouse embryonic forelimb
Multiple litters of ICR mouse forelimbs at E12.5 were collected and processed for ChIP-seq. High-throughput sequencing produced between 37 million and 47 million reads for each of the three (H3K4me3, H3K27me3, Pol2) ChIP samples. Each input control sample had between 74 million and 90 million reads. After initial quality filtering, about 96% of these reads were mapped to the mouse mm9 reference assembly using Burrows–Wheeler Aligner,20 which resulted in 22 million uniquely mapped reads for ChIP samples and 46 million uniquely mapped reads for input samples (Figure 1A). On average, 64% of the total raw reads were used for analysis. To identify peaks or regions that were enriched in each data set, MACS was utilized with the input as the control to account for local sequencing bias of the genome.16 Histone marks are known to appear over broad regions, compared with transcription factors, and thus the broad peak calling option was turned on for analysis, which resulted in 16,234, 4,442, and 11,425 broad peaks for H3K4me3, H3K27me3, and Pol2, respectively (Figure 1B). The greater number, 16,234 of total peaks representing the 50% in H3K4me3, was expected, as this trend can be seen in the publicly accessible data sets on the UCSC Genome Browser for various cell and tissue types. Genes that exhibited H3K4me3 and Pol2 presence represented 31% of total peaks, whereas genes with both H3K4me3 and H3K27me3 presence (bivalent) represented 7%, and genes with H3K4me3, H3K27me3, and Pol2 presence (trivalent) represented 2% of total peaks. These data suggest that the most developmental regulators are no longer bivalent as in ES cells, which represent 22% of genes,7 and by E12.5 the cells lost their plasticity and cell stages have been determined.
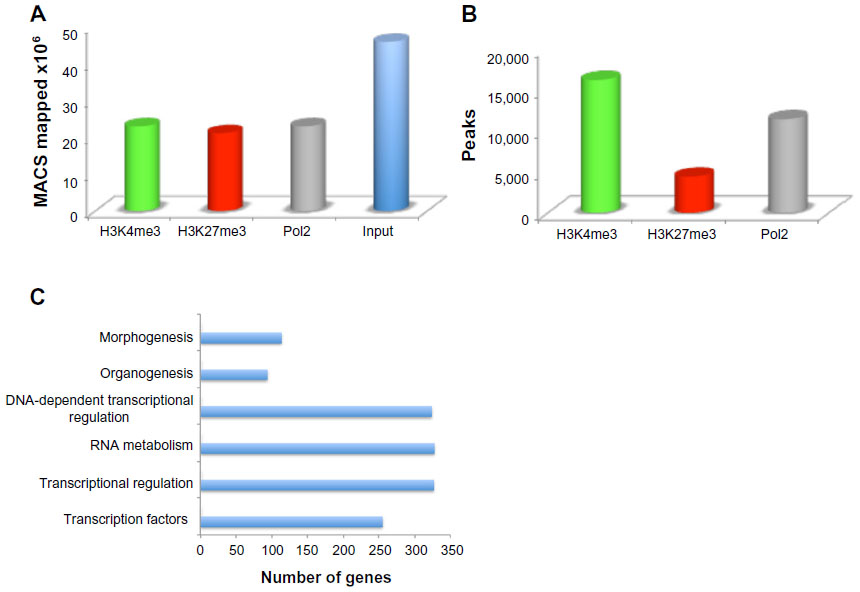 | Figure 1 Mapping the mouse embryonic forelimb. |
Chromatin state of mouse embryonic forelimb
Homer tools were used to characterize the found peaks, the distance to the nearest transcriptional start site (TSS), defined as the start of a RefSeq gene, and the region in which the peak was present (Figure 2A).17 The centers of peaks for all three marks were detected at or just downstream of (+200 for Pol2 and +500 for H3K4me3 and H3K27me3) the TSS. The Pol2 marks were more tightly distributed near the TSS, whereas H3K27me3 marks often covered a much broader region over a gene. About 60% of the H3K4me3 and H3K27me3 peaks occurred over the body of a gene (intron, exon, 3′ UTR, 5′ UTR), and 60% of the Pol2 peaks occurred on the promoter regions. Homer tools were also used to characterize the occurrence of cobound peaks, where one or more mark(s) overlapped (Figure 2B), with approximately 2,400 bivalent peaks. Bivalent domains are defined by the coexistence of the permissive (H3K4me3) and the repressive (H3K27me3) histone mark and are important in pluripotency and embryogenesis by keeping transcriptional regulators and developmental genes in a silenced state poised for activation upon differentiation.21 DAVID bioinformatics clustering tools were applied to categorize and identify the gene families with bivalent chromatin marks.19 To filter out potential false positives, we identified the bivalent marks within 20 kb of a transcriptional start of a gene. The enriched gene families included genes involved primarily in transcription and transcriptional regulation (80%) and in embryogenesis (15%) (Figure 1C). The presence of H3K4me3 was primarily noted in the promoter regions within the genes that are marked with it (Figure 2B), whereas H3K27me3 was highly present in the intron of the genes that was detected (Figure 2B). Half of the genes exhibited H3K4me3 marks, 36% of them Pol2 marks, and only 14% of them H3K27me3 marks, at this developmental stage. The presence of Pol2 on specific genes is often correlated with active transcription, which requires open and accessible chromatin. Over 95% of all Pol2 marks occurred in association with the H3K4me3 (Figure 1B). There were only four occurrences where a Pol2 mark occurred in association with only one H3K27me3 mark. However, the presence of Pol2 does not conclude that the gene is transcriptionally engaged, as additional assays can be included, such as RNA-seq, to verify the actively transcribed genes.
 | Figure 2 Characterization of H3K4me3, H3K27me3, and Pol2 marks in respect of location on the genome and nearest gene. |
Signaling molecules in mouse embryonic forelimb
The mouse forelimb has a self-regulatory signaling system that involves several networks to regulate cell patterning, commitment, and differentiation. Fibroblast growth factors (FGFs) that are expressed in the apical ectodermal ridge (AER) induce limb bud formation22,23 and pattern the distal forelimb.24 Sonic hedgehog (Shh) from the zone of polarizing area specifies the pattern formation via a concentration gradient.25 BMP (bone morphogenetic protein) signals from the distal part of the forelimb antagonize the effect of FGF signaling from the AER.26 By E12.5 the patterning events have been finalized and the signaling molecules should be poised for deactivation. Different network kernels that will guide the cell types toward their next developmental state are activated. Data from our analysis indicated that Shh and FGF8 were not significantly marked by any of the histone trimethylases or by Pol2, but rather were maintained in a bivalent state (Figure 3). However, Gli3, a key component of the Shh signaling, which results in formation of additional digits during limb bud development and acts as a gatekeeper for the exit to chondrogenic differentiation,27 was marked for H3K4me3 and Pol2 (Figure 3). FGF10 exhibited H3K4me3 marks, as its expression remains in the expanding mesenchyme at this stage (Figure 3). BMP4 is expressed in limb mesoderm and regulates digit number and identity,28 and thus was marked for both H3K4me3 and Pol2 (Figure 3). Grem1 is a BMP antagonist and contributes to maintain FGF expression in AER29 and was poised with low marks for H3K4me3 and H3K27me3 (data not shown).
 | Figure 3 Chromatin state of signaling molecules. |
SSTFs in mouse embryonic forelimb
The limb bud emerges from the mesenchymal cells covered with a layer of ectodermal cells. Within days these cell populations give rise to skeletal elements, muscles, and connective tissues. The T-box transcription factors Tbx5 and Tbx4 are expressed and regulate the formation in forelimbs and hind limbs, respectively.30 Our ChIP-seq data have shown that Tbx5 exhibited H3K4me3 with small traces of Pol2, whereas Tbx4 exhibited H3K27me3 (Figure 3), further supporting the limb specification properties of these markers. The aristaless-like homeobox 4 (Alx4) is involved in pattering the structures of the forelimb at earlier stages and exhibited marks for both H3K4me3 and H3K27me3 (Figure 4). Two families of Hox genes, Hoxa and Hoxd, are involved in forelimb development, and their mutations display a range of phenotypes, including prenatal and postnatal lethality and developmental defects in skeletal, muscular, and other systems. Our ChIP-seq data indicated that members of the Hoxa cluster, a9, a10, a11, and a13, and the Hoxd clusters d3, d8, d9, d10, d11, d12, and d13 exhibited both H3K4me3 and Pol2 (Figure 4), suggesting that at E12.5 the initial patterning of forelimb is still an ongoing process to further define the anterior–posterior and dorsal–ventral borders during cell growth and specification.
 | Figure 4 Chromatin state of forelimb-specific sequence-specific transcription factors. |
Osteogenesis is a highly regulated developmental process with Runx2 as a master regulator of the transcriptional program essential for bone formation.31 Runx2 was marked for H3K4me3 but not Pol2 (Figure 5A), suggesting that the osteogenic program has been initiated before E12.5. The Sry-related transcription factor Sox9 is essential for chondrogenesis of the mesenchymal stem cells in the limb.32 Sox9 was marked with both H3K4me3 and Pol2 (Figure 5A), suggesting that the chondrogenic program has been activated. Formation of adipocytes will follow gene regulators including peroxisome proliferator-activated receptor gamma (PPARγ) and the CCAAT/enhancer binding protein (CEBPδ) only H3K4me3 marks (Figure 5B). Both genes were poised for activation in later developmental stages.
 | Figure 5 Chromatin state of sequence-specific transcription factors involved in osteogenesis (A) and adipogenesis (B). |
Muscle SSTFs in mouse embryonic forelimb
Specific transcription factors are required for molecular specification, movement, and myogenic progression during muscle development. The expression of the homeo-paired domain Pax3 transcription factor in the dermomyotome precedes the expression of the muscle regulatory factors (MRFs), a family of four transcription factors, including Myod, Myf5, Myog, and MRF4. Myog expression is required for classic muscle differentiation and precedes the expression of proteins of the contractile apparatus. Pax3 and Pax733 or Myod and Myf534 can contribute to different muscle lineages. The homeodomain transcription factor Pitx2 marks all muscles,35 whereas Pitx1 is expressed only in the hind limb muscles36 and Pitx3 marks muscle cells at later developmental stages.37 Cells expressing Pax3 and Pax7 mark the muscle progenitor pool38,39 and were bivalently marked (Figure 6). Pax3 is required for the delamination of muscle precursors from the dermomyotome, and at this stage of development it is likely that Pax3 is no longer actively transcribed but rather repressed for the next stage of myogenesis to proceed. Similarly, Pax7 begins to be expressed at E12.5 and is essential for postnatal myogenesis.40 The bivalent marks indicate that Pax7 was poised for repression during embryonic myogenesis (E9–E12) and activation during fetal myogenesis (E13–E16). The bivalently marked regions upstream of Pax3 and Pax7 likely lie on enhancer or regulatory regions. The marked region 7 kb upstream of Pax3 is a known enhancer/regulator region involved in Pax3 expression in the ventral–lateral lip of interlimb somites.41 The H3K27me3 mark in the fourth intron of Pax3 also lies on a known regulatory region.42
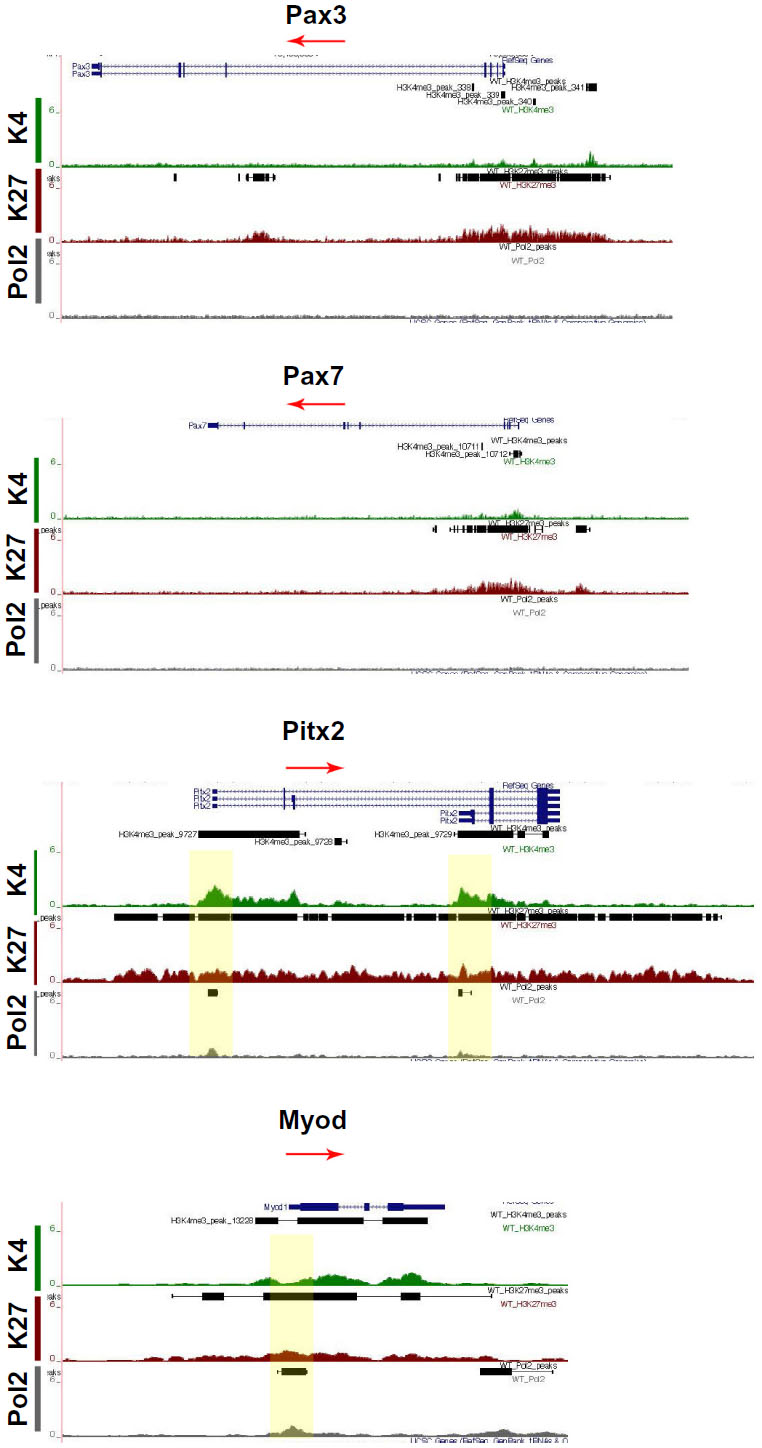 | Figure 6 Chromatin state of sequence-specific transcription factors involved in myogenesis. |
Mapped reads on the UCSC Genome Browser indicated that all four isoforms of Pitx2 were bivalently marked and occupied by Pol2 (Figure 6). Pitx1 was highly marked with H3K27me3, concurrent with its specific expression in the hind limb, and thus would not be available in the forelimbs (data not shown). Pitx3 was bivalently marked for H3K4me3 and H3K27me3 but not Pol2 (data not shown).37 Of the four MRFs, only Myod was bivalently marked with Pol2 mark, indicating that the cells have entered and been specified to the myogenic program (Figure 6). The other three factors were marked with H3K4me3, indicating that the genes could be available for transcription at later developmental stages, which is in accord with their expression profiles.43
Discussion
Chromatin profiling is a powerful indicator for detection of the genome’s regulatory activity. Genome-wide analysis of all cell types of an organ, the forelimb in our studies, at different developmental stages will provide a detailed map of transcriptional activity and generate predictions of cell fate. Developmental regulatory genes have both activating (H3K4me3) and repressive (H3K27me3) histone modifications in embryonic forelimb. This bivalent configuration can maintain cell state commitment in a poised state. We have applied a ChIP-seq approach to investigate chromatin in mouse forelimbs at E12.5, a critical stage for growth and cell lineage specification. We report that almost one-third (31%) of the mapped genes were transcriptionally active by exhibiting H3K4me3 and Pol2 marks, 4% were repressed by exhibiting H3K27me3 marks, 9% were poised by exhibiting H3K4me3 marks, 7% were in a bivalent state exhibiting H3K4me3 and H3K27me3 marks, and only 2% were in a trivalent state exhibiting all three marks H3K4me3, H3K27me3, and Pol2. The active presence (50%) of H3K4me3 in the promoters can be explained by the active remodeling of the limb at this stage. The repressed genes are not expressed in the forelimb. The patterning genes have already established all axes, anterior–posterior, dorsal–ventral, distal–proximal, and signaling molecules, and SSTFs involved in lineage determination, including osteogenesis/chondrogenesis, myogenesis, and adipogenesis, have been activated or remained poised for the next regulatory kernel to be established. The small percentage of bivalent and the even smaller trivalent domains suggest that the cells have lost the plasticity and pluripotency that characterizes the ES cells. Limb cells at E12.5 are prepared for a fine-tune gene expression to produce more specific lineages with unique characteristics and functions rather than a standby bivalent state that allows them to be self-renewed and differentiated. Our observations suggest that mesoderm-, mesenchyme-, and ectoderm-derived cells could be altered though modulation of distinct chromatin states.
Acknowledgments
We thank Mark Dasenko for CGRB Core Laboratories support, Roy Brown for computational support, and Denny Weber for help with editing the manuscript. This research was supported by the College of Pharmacy at Oregon State University, the NIH-NIAMS AR054406, and MRF grants to CK.
Author contribution
Substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; DE, WKV, MGK, NSF, CK Drafting the article or revising it critically for important intellectual content; DE, WKV, MGK, NSF, CK. Final approval of the version to be published; DE, WKV, MGK, NSF, CK. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. DE, WKV, MGK, NSF, CK.
Disclosure
The authors report no conflicts of interest in this work.
References
Davidson EH, Rast JP, Oliveri P, et al. A genomic regulatory network for development. Science. 2002;295(5560):1669–1678. | |
Longabaugh WJ, Davidson EH, Bolouri H. Computational representation of developmental genetic regulatory networks. Dev Biol. 2005; 283(1):1–16. | |
Bolouri H, Davidson EH. The gene regulatory network basis of the “community effect,” and analysis of a sea urchin embryo example. Dev Biol. 2010;340(2):170–178. | |
Robertson G, Hirst M, Bainbridge M, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4(8):651–657. | |
Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316(5830):1497–1502. | |
Barski A, Cuddapah S, Cui K, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. | |
Mikkelsen TS, Ku M, Jaffe DB, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007; 448(7153):553–560. | |
Birney E, Stamatoyannopoulos JA, Dutta A, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. | |
Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10(10):669–680. | |
Dunham I, Kundaje A, Aldred SF, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. | |
Heintzman ND, Stuart RK, Hon G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39(3):311–318. | |
Ringrose L, Paro R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet. 2004;38:413–443. | |
Boyer LA, Plath K, Zeitlinger J, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441(7091):349–353. | |
Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–326. | |
Young MD, Willson TA, Wakefield MJ, et al. ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res. 2011;39(17):7415–7427. | |
Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. | |
Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–589. | |
Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. | |
Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. | |
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. | |
Voigt P, Tee WW, Reinberg D. A double take on bivalent promoters. Genes Dev. 2013;27(12):1318–1338. | |
Xu X, Weinstein M, Li C, et al. Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development. 1998;125(4):753–765. | |
Sekine K, Ohuchi H, Fujiwara M, et al. Fgf10 is essential for limb and lung formation. Nat Genet. 1999;21(1):138–141. | |
Maden M. Intercalary regeneration in the amphibian limb and the rule of distal transformation. J Embryol Exp Morphol. 1980;56:201–209. | |
Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, Tabin CJ. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell. 2004;118(4):517–528. | |
Pizette S, Niswander L. BMPs negatively regulate structure and function of the limb apical ectodermal ridge. Development. 1999;126(5):883–894. | |
Lopez-Rios J, Speziale D, Robay D, et al. GLI3 constrains digit number by controlling both progenitor proliferation and BMP-dependent exit to chondrogenesis. Dev Cell. 2012;22(4):837–848. | |
Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, Tabin CJ. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006;2(12):e216. | |
Zuniga A, Haramis AP, McMahon AP, Zeller R. Signal relay by BMP antagonism controls the SHH/FGF4 feedback loop in vertebrate limb buds. Nature. 1999;401(6753):598–602. | |
Hasson P, DeLaurier A, Bennett M, et al. Tbx4 and tbx5 acting in connective tissue are required for limb muscle and tendon patterning. Dev Cell. 2010;18(1):148–156. | |
Long F. Building strong bones: molecular regulation of the osteoblast lineage. Nat Rev Mol Cell Biol. 2012;13(1):27–38. | |
Cheng A, Genever PG. SOX9 determines RUNX2 transactivity by directing intracellular degradation. J Bone Miner Res. 2010;25(12):2680–2689. | |
Relaix F, Rocancourt D, Mansouri A, Buckingham M. Divergent functions of murine Pax3 and Pax7 in limb muscle development. Genes Dev. 2004;18(9):1088–1105. | |
Kablar B, Krastel K, Ying C, Asakura A, Tapscott SJ, Rudnicki MA. MyoD and Myf-5 differentially regulate the development of limb versus trunk skeletal muscle. Development. 1997;124(23):4729–4738. | |
Shih HP, Gross MK, Kioussi C. Expression pattern of the homeodomain transcription factor Pitx2 during muscle development. Gene Expr Patterns. 2007;7(4):441–451. | |
Szeto DP, Rodriguez-Esteban C, Ryan AK, et al. Role of the Bicoid-related homeodomain factor Pitx1 in specifying hindlimb morphogenesis and pituitary development. Genes Dev. 1999;13(4):484–494. | |
L’Honore A, Coulon V, Marcil A, et al. Sequential expression and redundancy of Pitx2 and Pitx3 genes during muscle development. Dev Biol. 2007;307(2):421–433. | |
Kassar-Duchossoy L, Giacone E, Gayraud-Morel B, Jory A, Gomes D, Tajbakhsh S. Pax3/Pax7 mark a novel population of primitive myogenic cells during development. Genes Dev. 2005;19(12):1426–1431. | |
Relaix F, Rocancourt D, Mansouri A, Buckingham M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature. 2005;435(7044):948–953. | |
Murphy M, Kardon G. Origin of vertebrate limb muscle: the role of progenitor and myoblast populations. Curr Top Dev Biol. 2011;96:1–32. | |
Brown CB, Engleka KA, Wenning J, Min Lu M, Epstein JA. Identification of a hypaxial somite enhancer element regulating Pax3 expression in migrating myoblasts and characterization of hypaxial muscle Cre transgenic mice. Genesis. 2005;41(4):202–209. | |
Degenhardt KR, Milewski RC, Padmanabhan A, et al. Distinct enhancers at the Pax3 locus can function redundantly to regulate neural tube and neural crest expressions. Dev Biol. 2010;339(2):519–527. | |
Hasty P, Bradley A, Morris JH, et al. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature. 1993;364(6437):501–506. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.