Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 19
Genetics of Vitiligo: A Review
Received 6 January 2026
Accepted for publication 23 February 2026
Published 27 February 2026 Volume 2026:19 589298
DOI https://doi.org/10.2147/CCID.S589298
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Honghao He,* Rina Su,* Fang Liu
Department of Dermatology, Venereology and Medicine, Beijing Chao-Yang Hospital, Capital Medical University, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Fang Liu, Department of Dermatology, Venereology and Medicine, Beijing Chao-Yang Hospital, Capital Medical University, No. 8 Gongti South Road, Chaoyang District, Beijing, 100020, People’s Republic of China, Email [email protected]
Abstract: Vitiligo is a common acquired depigmentation skin disease with obvious family aggregation. About 25− 50% of patients have positive family history, which belongs to polygenetic disease. In recent years, through candidate genes and genome-wide association studies, multiple susceptibility gene loci have been found, and studies also show that there is genetic heterogeneity among different populations. Environmental factors can also interact with genetic factors to trigger diseases through various mechanisms. The risk assessment model based on genetic and environmental factors provides a new direction for early screening and personalized prevention and treatment. In the future, we need to combine single cell sequencing and other multi omics technologies to explore the mechanism, develop targeted treatment strategies, and strengthen the application of genetic counseling and preventive measures in high-risk populations.
Keywords: vitiligo, genetics, genome, autoimmune
Introduction
Vitiligo is an autoimmune disorder characterized by the selective destruction of melanocytes in the skin and mucous membranes, presenting as localized or generalized white patches. The global prevalence is approximately 0.5–2%, with no significant gender or geographic differences, though slightly higher rates are observed in regions such as India and the Middle East.1
Vitiligo is primarily classified into two clinical subtypes, they are non-segmental vitiligo (NSV) and segmental vitiligo (SV). NSV is the most common (accounting for 80–90% of cases), exhibiting symmetrical distribution and often associated with autoimmune diseases (eg, thyroid disorders, type 1 diabetes).2 SV is relatively rare, follows a dermatomal distribution, has an earlier onset and is less frequently linked to autoimmune conditions.3,4
Lesions typically present as well-defined milky-white patches, commonly occurring on the face, hands, limbs, and friction-prone areas (eg, elbows, knees). Disease progression is generally categorized as stable or active, with some patients experiencing worsening due to the Koebner phenomenon.5
Although vitiligo is not life-threatening, it profoundly impacts patients’ psychological and social well-being. Due to the visibility of lesions, patients often face social stigma, discrimination, and challenges in employment and relationships.6 Multiple studies indicate that anxiety and depression rates are significantly higher among vitiligo patients, particularly adolescents and females.7 Additionally, individuals with darker skin tones experience greater psychological burden due to the stark contrast of lesions.8 Therefore, vitiligo treatment must address both repigmentation and psychological support.
The etiopathogenesis of vitiligo remains multifactorial, with several competing theories proposed. The autoimmune hypothesis posits that melanocytes are targeted by autoreactive T cells, supported by the frequent association with other autoimmune disorders.9 The oxidative stress theory suggests that excessive reactive oxygen species (ROS) accumulation leads to melanocyte apoptosis.10 Neurohumoral mechanisms involve neuropeptide-mediated melanocyte damage,11 while the melanocyte intrinsic defect theory highlights inherent vulnerabilities in melanocyte survival pathways.12 More recently, viral triggers and genetic predisposition have been integrated into a convergent model, wherein environmental factors interact with susceptibility genes to initiate disease.13 These pathogenic theories provide a framework for understanding the complex genetic and environmental interactions discussed in this review.
Genetic Epidemiology of Vitiligo
Family studies provide direct evidence for the genetic susceptibility of vitiligo. Studies have shown that the risk of vitiligo in first-degree relatives is significantly higher than that in the general population. For example, Alkhateeb et al1 and other scholars’ investigation on 2624 cases of vitiligo probands showed that about 20% of the patients had at least one affected first-degree relative, and the incidence was 10–36 times higher than that of the general population. Zhang et al.14 Reported a 28.3% familial incidence in the Chinese population, suggesting that there may be stronger familial aggregation in the Asian population. There are differences in disease risk among different kinship. The closer the kinship, the higher the risk of disease. The risk of individual with parents’ medical history is about 5–7%; The individual risk of brothers and sisters with medical history is about 6–8%. Nath15 and other studies further proved that if both parents suffer from vitiligo, the risk of offspring will rise to 30–40%, indicating that the disease has a polygenic cumulative effect.
Twin studies provide key insights into the contribution of genetic factors to vitiligo. Alkhateeb’s epidemiological studies1 showed that the comorbidity rate of monozygotic twins (MZ) was 23−26%, and that of dizygotic twins (DZ) was 4−6%. This significant difference strongly supports the role of genetic factors in the pathogenesis of vitiligo. It is worth noting that even identical twins do not show 100% consistency, indicating that environmental factors (such as stress, skin trauma) may trigger the disease in genetically susceptible individuals.16 The heritability estimation based on twin data showed that the heritability of vitiligo was about 75−80%, and the remaining 20−25% was attributed to environmental factors. The heritability of autoimmune subtypes may be higher.17
There are differences in genetic susceptibility to vitiligo among different races and populations. The global prevalence of vitiligo varies by race. The prevalence rate of Caucasian population is 0.5–1%, that of Asian population is 0.5–2%, that of African population is 1–2.5%, and that of certain groups in some parts of India can be as high as 3–4%. Genome-wide association analysis (GWAS) studies highlighted significant ethnic differences in genetic association. European populations were mainly associated with HLA-A*02:01, PTPN22 and TYR alleles. The contribution of HLA-A*30:01 and HLA-DQB1*03:03 alleles was greater in Asian population.8 The African population is associated with a unique slev1 locus. Protective alleles (eg, HLA-A*03:01) are more common in European populations, which may explain its low incidence.18,19
Research Progress
Early Candidate Gene Research
Before the emergence of GWAS technology, there were several different hypotheses about the pathogenesis of vitiligo. The most mainstream theory is the autoimmune theory, with the core idea that melanocytes are mistakenly attacked by the autoimmune system; the oxidative stress theory (accumulation of reactive oxygen species (ROS) in melanocytes leading to apoptosis), neurohumoral theory (neuropeptide mediated melanocyte destruction), and melanocyte intrinsic defect theory (melanocyte intrinsic abnormalities leading to increased vulnerability) are also parallel theories of the same period; the theory of viruses is an emerging hypothesis in recent years, whose core idea is that viral infections trigger cross immune responses. The current academic consensus is the integration theory, which refers to the joint action of multiple factors to cause disease. Based on these hypotheses, the following candidate genes have been selected and studied (Table 1).
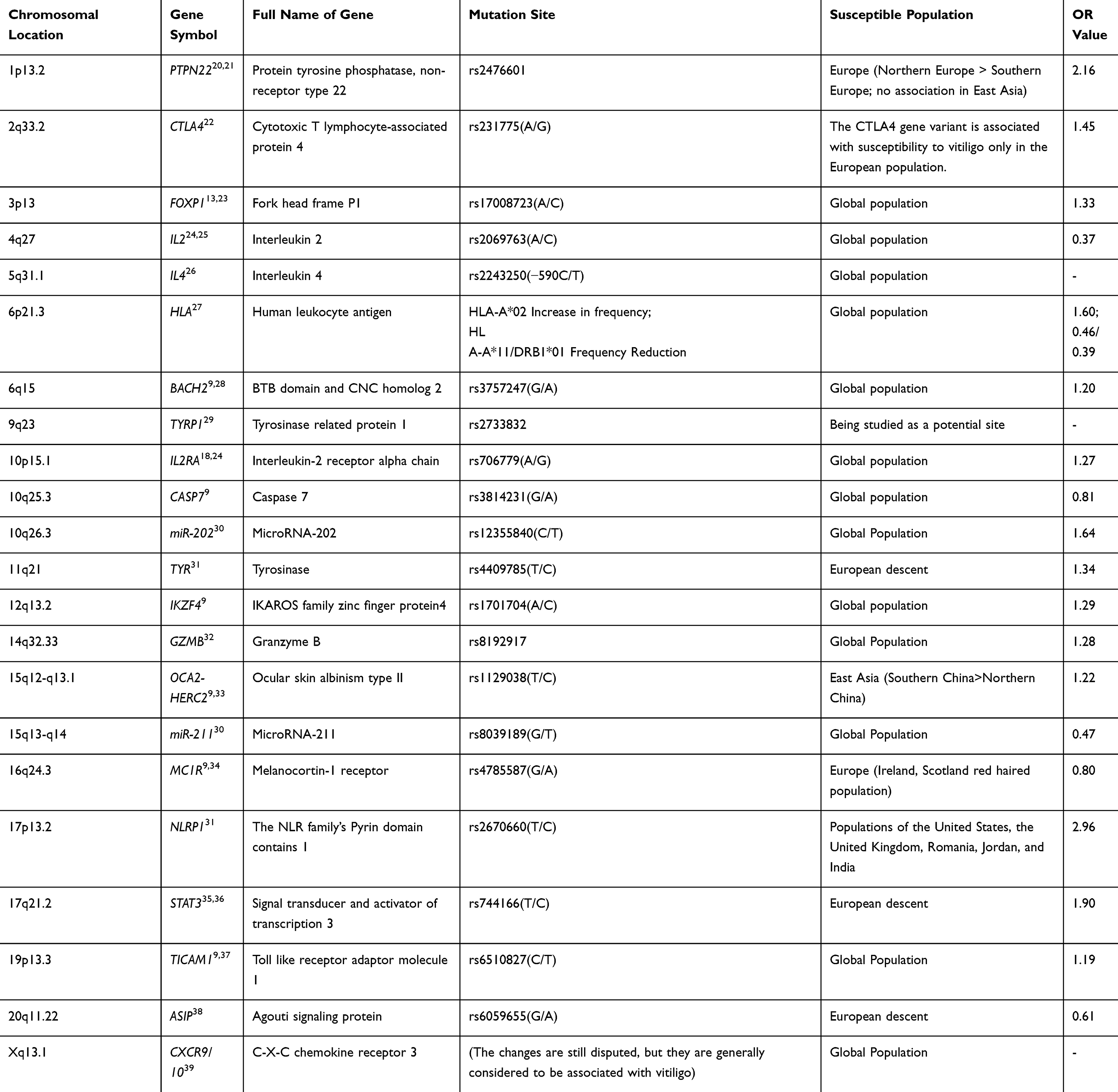 |
Table 1 Abnormal Expression of Vitiligo Candidate Genes |
GWAS Findings
GWAS has identified numerous susceptibility loci (Table 2). Vitiligo susceptibility loci exhibit both consistency and variability across populations.13 Among these, the major histocompatibility complex (MHC) region (6p21.32), particularly HLA-B*33:0140 and HLA-DQB1,8,41 shows significant associations in both European and Asian populations, suggesting that HLA-mediated autoimmune responses are a core pathogenic mechanism. Cross-population shared immune-related genes such as TYK2 (7p15.2), IL12 (12q13.2) and IL23 (9p24.1)42 contribute to disease onset by regulating Th1/Th17 immune responses. Meanwhile, pigment metabolism genes like TYR (11q14.3) and MC1R (16q24.3) are associated with vitiligo risk across all populations, though TYR shows a stronger association in Asians.29
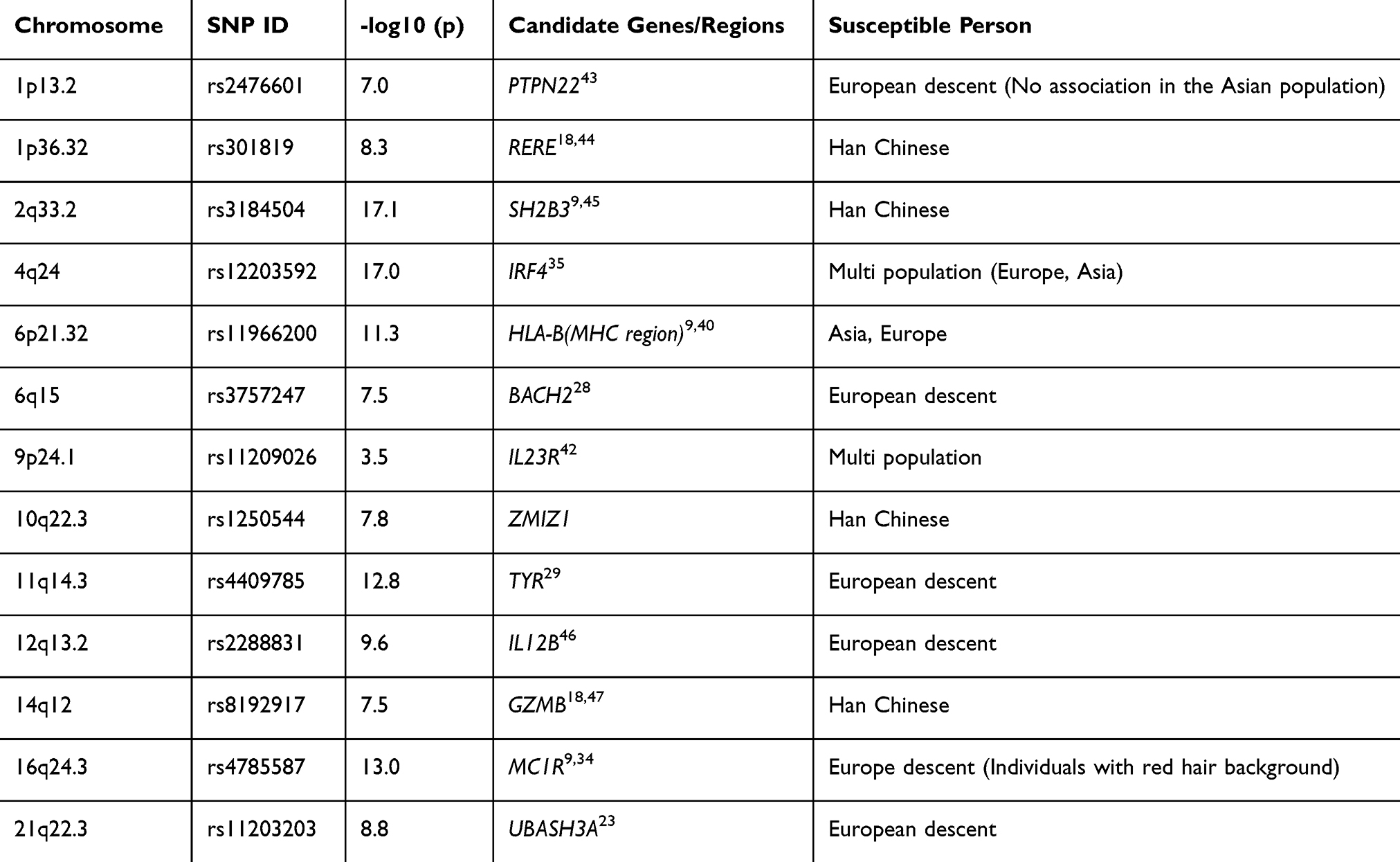 |
Table 2 GWAS Discovers Susceptible Gene Micro Stores |
Population-specific loci include ZMIZ1 (10q22.3)48 and GZMB (14q12),49 which are particularly significant in the Chinese Han population, likely due to their roles in regulating T-cell differentiation and cytotoxicity. In contrast, FOXD3 (1p13.2)42 and PTPN22 (1p13.2)19 are more prominent in European populations, reflecting differences in neural crest development and B-cell regulation.
Functionally, these loci are involved in the pathogenesis through three major pathways. First, HLA mediated antigen presentation activates CD4+T cells, and cytokines such as IL23R/IL12B50 can drive Th17 inflammatory response; Secondly, the mutation of pigment related genes (such as TYR and MC1R) will lead to melanocyte stress and antigen release. In addition, GZMB, which is unique to Asian people, can directly induce melanocyte apoptosis through granzyme B; Finally, the genetic variation of BACH2 (6q15) may destroy immune tolerance. These findings not only reveal the complex genetic background of vitiligo but also provide an important basis for the development of precision medical strategies for different populations.
Genetic Mechanisms
Gene–Environment Interactions
Environmental factors significantly influence vitiligo onset through gene–environment interactions (G×E), which regulate the expression of susceptibility genes. Ultraviolet (UV) exposure is a classic environmental trigger that activates immune responses via oxidative stress and DNA damage pathways, thereby modulating vitiligo-related genes (eg, TYR, PTPN22, and NLRP1) and contributing to disease manifestation. For instance, UV radiation induces keratinocytes to release pro-inflammatory factors, leading to the exposure of melanocyte-specific antigens and triggering autoimmune attacks.42 Additionally, GWAS studies have revealed that MC1R gene variants (associated with skin pigmentation) synergistically increase vitiligo risk when combined with UV exposure, suggesting a genotype-environment correlation.13
Chemical agents (eg, phenolic compounds, hydroquinone) interfere with melanocyte function through epigenetic modifications or direct toxicity, altering the expression of susceptibility genes. Research indicates that occupational exposure to such chemicals is closely linked to polymorphisms in catalase (CAT) and vitamin D receptor (VDR) genes, which activate oxidative stress pathways and promote melanocyte apoptosis. For example, hydroquinone suppresses the expression of microphthalmia-associated transcription factor (MITF), disrupting melanocyte survival and differentiation—a mechanism particularly pronounced in individuals carrying the PTPN22 risk allele.19
Psychological stress modulates the immune microenvironment via neuroendocrine pathways (eg, Hypothalamic-Pituitary-Adrenal (HPA) axis activation), influencing vitiligo susceptibility genes. Chronic stress elevates cortisol levels, stimulating the release of pro-inflammatory cytokines (eg, IL-6, TNF-α) and exacerbating autoimmune responses. Studies show that individuals with specific variants of the catechol-O-methyltransferase (COMT) gene are more prone to melanocyte-specific T-cell activation under prolonged stress, likely due to epigenetic regulation (eg, DNA methylation).11
Gene Networks and Pathway Abnormalities
Vitiligo is a complex polygenic disorder involving interactions among susceptibility genes across multiple pathways, including cell signaling, immune regulation, and melanocyte development, ultimately leading to melanocyte destruction or dysfunction. Below is a molecular-level summary of key genes and their networks.
In terms of gene interactions in cell signaling pathways, vitiligo susceptibility genes (such as PTPN22, STAT4 and IFIH1) jointly regulate signal transduction in immune cells and melanocytes. Specifically, the lymphoid specific protein TYRosine phosphatase (LYP) encoded by PTPN22 gene, whose function acquired mutations (such as R620W) can enhance the signal transduction of T cell receptor (TCR), thus promoting the activation of self-reactive T cells.51 At the same time, STAT4 gene mediates the signal transmission of interferon-γ (IFN-γ) through JAK-STAT pathway, up regulates the production of C-X-C Motif Chemokine Ligand 10 (CXCL10) and other chemokines, and then recruits cytotoxic CD8+T cells to attack melanocytes.9 In addition, IFIH1 gene (also known as MDA5) is responsible for detecting viral RNA in cells, stimulating the production of type I interferon by activating Mitochondrial Antiviral Signaling Protein (MAVS)-Interferon Regulatory Factor (IRF)3/7 pathway, and finally amplifying the immune response.52 The abnormal activation of these genes laid the foundation for the immunological pathogenesis of vitiligo.
In the link between immune regulatory genes and the collapse of autoimmune tolerance, the core mechanism of vitiligo is the destruction of melanocytes mediated by autoimmune response, which is mainly driven by key genes such as nlrp1, FOXP3 and CTLA4. First, nlrp1 gene encodes a key component of inflammasome, and its mutation will lead to the excessive release of interleukin-1 β (IL-1 β) and IL-18, thus creating a pro-inflammatory microenvironment.16 More importantly, FOXP3, as the main transcription factor of regulatory T cells (Treg), its reduced expression or dysfunction will weaken the immunosuppressive function, resulting in the escape of self-reactive cells such as MELAN-A-specific CD8+T cells from the tolerance mechanism.53 It is also important that CTLA4 gene maintains peripheral immune tolerance by inhibiting the costimulatory signal of T cells, and its single nucleotide polymorphisms (SNPs) are closely related to the risk of vitiligo.54
As for the role of melanocyte self-development and oxidative stress defense genes, their survival and function depend on key genes such as MITF. MITF is the core regulator of melanocyte development, and its downstream target Tyrosinase (TYR) dominates the biosynthesis of melanin. Therefore, the mutation or low expression of TYR will directly lead to pigmentation dysfunction. Especially, oxidative stress plays a central role in this process.12 The deficiency of catalase (CAT) and superoxide dismutase 2 (SOD2) can significantly increase the sensitivity of melanocytes to hydrogen peroxide (H2O2) and then cause endoplasmic reticulum stress and apoptosis.10 In addition, the genetic variation of vitamin D receptor (VDR) may also interfere with the calcium signal transduction in melanocytes and weaken their antioxidant capacity.55
Finally, these genes are not acting in isolation, but constitute a dynamic gene pathway regulatory network and provide targets for treatment. For example, MITF can up regulate the anti apoptotic protein BCL2 to maintain the survival of melanocytes, and oxidative stress can inhibit the activity of MITF by activating NF-κB pathway, which forms a vicious circle.56 On the other hand, the abnormal expression of immune checkpoint molecules (such as PD-1/PD-L1) will further aggravate T cell-mediated cytotoxicity.57 These in-depth mechanism studies provide directions for the development of targeted therapies, such as the use of JAK inhibitors to block IFN-γ signaling pathway, the use of antioxidants such as N-acetylcysteine, and Treg amplification therapy.58
Epigenetic Factors
The development and progression of vitiligo are not only associated with genetic predisposition but are also significantly influenced by epigenetic modifications (eg, DNA methylation, histone modifications). These modifications regulate gene expression and participate in key pathological processes such as melanocyte dysfunction, autoimmune responses, and oxidative stress.
DNA methylation is one of the most in-depth research fields in the epigenetic mechanism of vitiligo. Studies have shown that melanocytes and surrounding keratinocytes in patients with vitiligo have global hypomethylation, which leads to the up regulation of IFN-γ, CXCL10 and other pro-inflammatory genes, and then recruit self-reactive T cells to attack melanocytes.59 At the same time, hypermethylation in the promoter region of FOXP3 gene (the core gene regulating the function of Treg cells) will inhibit its expression, thus weakening the immune tolerance and promoting the autoimmune response.60 In addition, aberrant methylation of key genes such as MITF and TYR in melanocytes may interfere with melanin synthesis and further aggravate depigmentation.61 These findings jointly reveal the key role of DNA methylation imbalance in vitiligo immune imbalance and melanocyte dysfunction.
In the regulation of histone modification on vitiligo related genes, acetylation, methylation and other modifications regulate the transcriptional activity of susceptible genes by changing the chromatin structure. Specifically, overexpression of histone deacetylases (HDACs) can inhibit MITF and anti apoptotic gene BCL2, thereby promoting melanocyte apoptosis.34 More importantly, in CD8+T cells, the enrichment of inhibitory histone marker H3K27me3 on immune checkpoint genes such as PD-1 may enhance its aggressiveness to melanocytes.62 On the other hand, the decreased activity of histone acetyltransferases (HATS) may lead to the down-regulation of the expression of Nrf2, a key antioxidant factor, and increase the sensitivity of melanocytes to oxidative stress.63 These modifications together shape the epigenetic landscape of vitiligo and continue to affect the progress of the disease.
In view of the reversibility of epigenetic modification, drugs targeting DNA methylation or histone modification (such as HDAC inhibitors and demethylation drugs) are expected to provide new therapeutic strategies. For example, DNA demethylation drug 5-azacytidine can restore the expression of FOXP3, thereby enhancing the function of regulatory T cells.64 HDAC inhibitors (such as vorinostat) may promote the survival of melanocytes by up regulating the expression of MITF. These studies laid a theoretical foundation for the development of epigenetic targeted therapy.65
The current research has formed a more objective system (Figure 1). It still needs to be pointed out that future research should further explore the interaction between environmental factors and epigenomes, which will not only help to deepen the understanding of the pathogenesis of vitiligo, but also provide a new direction for the development of personalized intervention strategies.
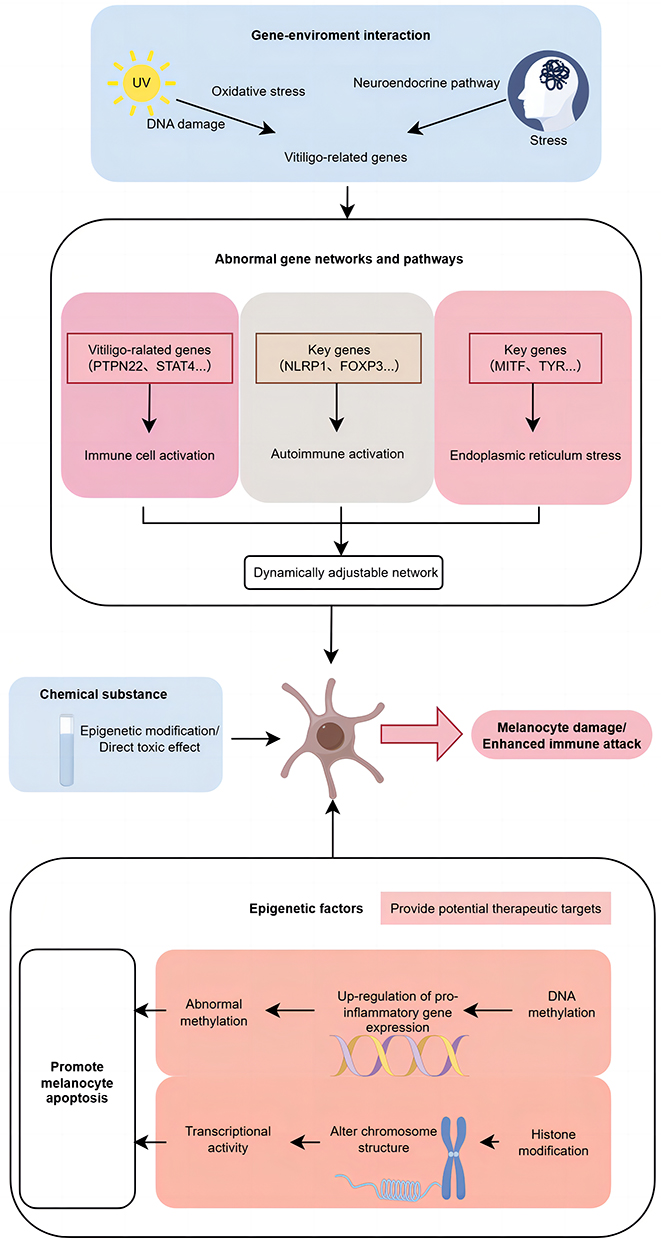 |
Figure 1 A proposed mechanistic framework for vitiligo pathogenesis integrating genetic susceptibility, environmental triggers, and epigenetic dysregulation. |
Establishment and Application of Genetic Risk Assessment Models for Vitiligo
Vitiligo risk assessment models integrate GWAS-identified susceptibility genes (eg, NLRP1, PTPN22, TYR) and environmental exposure data (UV radiation, chemical contact, psychological stress, etc).66 These models use multivariate regression or machine learning algorithms to calculate individual disease risk. Clinical applications include screening high-risk populations (eg, those with a family history) and formulating personalized prevention strategies (eg, enhanced photoprotection or antioxidant therapy).8 Early intervention based on these models may reduce disease risk by 30–50%.67
However, the current model shows obvious limitations. Genetic factors accounted for only 25% of the disease risk.68 Due to the need for real-time tracking, environmental assessment faces challenges in data integration.69 The molecular mechanism of gene environment interaction has not been fully elucidated.
Future Research Directions and Perspectives
Application of Novel Technologies in Vitiligo Genetic Research
Emerging technologies are reshaping vitiligo research. Single-cell RNA sequencing (scRNA-seq) has uncovered aberrant interactions between CD8+ T cells and melanocytes in lesions, identifying novel immune-related gene clusters (eg, CXCL10+, CD8+, T-cell, subsets).65 Multi-omics integration (epigenomics, proteomics) with GWAS revealed that noncoding variants (eg, rs11966200) may regulate MITF expression, affecting melanocyte function.13 Besides, CRISPR-Cas9 generated NLRP1-mutant mouse models, confirming its role in inflammasome-mediated autoimmunity.70 These approaches promise to uncover new therapeutic targets and advance precision medicine.
Implications of Genetic Research for Vitiligo Treatment
Targeted therapies based on genetic insights are gaining traction. JAK inhibitors (eg, tofacitinib), which block the JAK-STAT pathway (linked to STAT4/IFNGR2), significantly improve repigmentation.71 Besides, MITF agonists (eg, α-MSH analogs) aim to restore melanocyte function in patients with low MITF expression.72
In terms of individual treatment, HLA-DRB1*04 carriers respond better to immunotherapy (eg, PD-1 inhibitors), while TYR mutation patients may benefit from antioxidant combos.73
Genetic Insights for Vitiligo Prevention
Polygenic risk scores (eg, based on PTPN22/IL2RA SNPs) identify high-risk individuals (70% accuracy for family history-positive cases).8
Epigenetic clock analysis suggests UV protection may delay DNA methylation aging in high-risk children, reducing disease risk.61 Prospective cohort studies are needed to validate prevention strategies.
The Evolving Role of Genetic Counseling
Genetic counseling for vitiligo is shifting from empirical judgment to a precise model based on molecular genetics. First, through the gene screening of high-risk groups (such as those with family history) and the evaluation combined with polygene risk model.74 Secondly, genotypes are used to predict disease progression and drug response, for example, JAK-STAT pathway variation is used to predict the sensitivity to JAK inhibitors.75 Finally, provide personalized lifestyle suggestions and early intervention strategies. With the improvement of the accessibility of gene testing, genetic counseling based on genome-wide association analysis (GWAS) will gradually become the core component of clinical management74 and promote the transformation of diagnosis and treatment strategies from passive treatment to active prevention.
Conclusion
This review synthesizes current knowledge on the genetic architecture of vitiligo, underscoring its high heritability (~80%) and polygenic nature. Family and twin studies robustly support a strong genetic component, while GWAS has identified numerous susceptibility loci within immune regulation (eg, HLA, PTPN22), pigmentation (eg, TYR, MC1R), and apoptosis pathways, with notable ethnic heterogeneity. Gene–environment interactions, particularly involving UV exposure, chemical agents, and psychological stress, modulate disease expression through epigenetic and transcriptional mechanisms.
Emerging technologies—such as single-cell sequencing, multi-omics integration, and CRISPR-based models—are deepening our understanding of vitiligo pathogenesis and revealing novel therapeutic targets. The development of polygenic risk scores and epigenetic-based interventions holds promise for personalized prevention and treatment strategies. Moving forward, integrating genetic counseling with molecular profiling will facilitate early identification of high-risk individuals and enable precision medicine approaches, ultimately improving clinical outcomes and quality of life for patients with vitiligo.
What is Already Known?
Vitiligo is a polygenic autoimmune disease with high heritability (~80%). GWAS has identified numerous susceptibility loci in HLA, immune regulation (eg, PTPN22), and pigmentation (eg, TYR) pathways, showing significant ethnic heterogeneity. Environmental triggers like UV exposure interact with genetic factors.
What Does This Study Add?
This review synthesizes recent advances, highlighting population-specific genetic risks, gene–environment interaction mechanisms, and epigenetic dysregulation. It outlines future directions, including polygenic risk scores, novel therapeutics (eg, JAK inhibitors), and the application of single-cell technologies for precision.
Generative AI Statement
The authors declare that no Generative AI was used in the creation of this manuscript. Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Funding
This work was supported by National Natural Science Foundation of China (NSFC) (82273551) and Undergraduate innovation program of Capital Medical University (No. XSKY2025295).
Disclosure
The authors declare that there are no conflicts of interest regarding the publication of this article.
References
1. Alkhateeb A, Fain PR, Thody A, et al. Epidemiology of vitiligo and associated autoimmune diseases in Caucasian probands and their families. Pigment Cell Res. 2003;16(3):208–12. doi:10.1034/j.1600-0749.2003.00032.x
2. Ezzedine K, Lim HW, Suzuki T, et al. Revised classification/nomenclature of vitiligo and related issues: the vitiligo global issues consensus conference. Pigm Cell Melanoma Res. 2012;25(3):E1–13. doi:10.1111/j.1755-148X.2012.00997.x
3. van Geel N, Speeckaert R. Segmental Vitiligo. Dermatol Clin. 2017;35(2):145–150. doi:10.1016/j.det.2016.11.005
4. de Barros JC, Machado Filho CD, Abreu LC, et al. A study of clinical profiles of vitiligo in different ages: an analysis of 669 outpatients. Int J Dermatol. 2014;53(7):842–848. doi:10.1111/ijd.12055
5. Abdel-Malek ZA, Jordan C, Ho T, et al. The enigma and challenges of vitiligo pathophysiology and treatment. Pigm Cell Melanoma Res. 2020;33(6):778–787. doi:10.1111/pcmr.12878
6. Sanchez DP, Sonthalia S. Koebner Phenomenon. In: StatPearls. Treasure Island (FL): StatPearls Publishing Copyright © 2025, StatPearls Publishing LLC; 2025.
7. Ezzedine K, Eleftheriadou V, Whitton M, et al. Vitiligo. Lancet. 2015;386(9988):74–84. doi:10.1016/S0140-6736(14)60763-7
8. Roberts GHL, Santorico SA, Spritz RA. The genetic architecture of vitiligo. Pigm Cell Melanoma Res. 2020;33(1):8–15. doi:10.1111/pcmr.12848
9. Jin Y, Birlea SA, Fain PR, et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat Genet. 2012;44(6):676–680. doi:10.1038/ng.2272
10. Schallreuter KU, Bahadoran P, Picardo M, et al. Vitiligo pathogenesis: autoimmune disease, genetic defect, excessive reactive oxygen species, calcium imbalance, or what else? Exp Dermatol. 2008;17(2):139–140; discussion141–160. doi:10.1111/j.1600-0625.2007.00666_1.x
11. Li K, Li C, Gao L, et al. A functional single-nucleotide polymorphism in the catechol-O-methyltransferase gene alter vitiligo risk in a Chinese population. Arch Dermatol Res. 2009;301(9):681–687. doi:10.1007/s00403-008-0920-8
12. Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med. 2006;12(9):406–414. doi:10.1016/j.molmed.2006.07.008
13. Jin Y, Andersen G, Yorgov D, et al. Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nat Genet. 2016;48(11):1418–1424. doi:10.1038/ng.3680
14. Zhang XJ, Liu HS, Liang YH, et al. Association of HLA class I alleles with vitiligo in Chinese Hans. J Dermatol Sci. 2004;35(2):165–168. doi:10.1016/j.jdermsci.2004.05.003
15. Nath SK, Kelly JA, Namjou B, et al. Evidence for a susceptibility gene, SLEV1, on chromosome 17p13 in families with vitiligo-related systemic lupus erythematosus. Am J Hum Genet. 2001;69(6):1401–1406. doi:10.1086/324470
16. Sandru F, Carsote M, Albu SE, et al. Vitiligo and chronic autoimmune thyroiditis. J Med Life. 2021;14(2):127–130. doi:10.25122/jml-2019-0134
17. Krüger C, Schallreuter KU. A review of the worldwide prevalence of vitiligo in children/adolescents and adults. Int J Dermatol. 2012;51(10):1206–1212. doi:10.1111/j.1365-4632.2011.05377.x
18. Jin Y, Birlea SA, Fain PR, et al. Variant of TYR and autoimmunity susceptibility loci in generalized vitiligo. N Engl J Med. 2010;362(18):1686–1697. doi:10.1056/NEJMoa0908547
19. Tizaoui K, Shin JI, Jeong GH, et al. Genetic polymorphism of PTPN22 in autoimmune diseases: a comprehensive review. Medicina. 2022;58(8):1034. doi:10.3390/medicina58081034
20. Cantón I, Akhtar S, Gavalas NG, et al. A single-nucleotide polymorphism in the gene encoding lymphoid protein tyrosine phosphatase (PTPN22) confers susceptibility to generalised vitiligo. Genes Immun. 2005;6(7):584–587. doi:10.1038/sj.gene.6364243
21. LaBerge GS, Bennett DC, Fain PR, et al. PTPN22 is genetically associated with risk of generalized vitiligo, but CTLA4 is not. J Invest Dermatol. 2008;128(7):1757–1762. doi:10.1038/sj.jid.5701233
22. Salinas-Santander MA, Suárez-Valencia VJ, Ángel-Martínez MD, et al. Association between the CTLA4 +49A/G (rs231775) and CT60 (rs3087243) gene variants with vitiligo: study on a Mexican population. An Bras Dermatol. 2022;97(6):710–715. doi:10.1016/j.abd.2021.10.012
23. Jin Y, Birlea SA, Fain PR, et al. Common variants in FOXP1 are associated with generalized vitiligo. Nat Genet. 2010;42(7):576–578. doi:10.1038/ng.602
24. Guttman-Yassky E, Del Duca E, Da Rosa JC, et al. Improvements in immune/melanocyte biomarkers with JAK3/TEC family kinase inhibitor ritlecitinib in vitiligo. J Allergy Clin Immunol. 2024;153(1):161–172.e168. doi:10.1016/j.jaci.2023.09.021
25. Martins LT, Frigeri HR, de Castro CCS, et al. Association study between vitiligo and autoimmune-related genes CYP27B1, REL, TNFAIP3, IL2 and IL21. Exp Dermatol. 2020;29(6):535–538. doi:10.1111/exd.14100
26. Pehlivan S, Ozkinay F, Alper S, et al. Association between IL4 (−590), ACE (I)/(D), CCR5 (Delta32), CTLA4 (+49) and IL1-RN (VNTR in intron 2) gene polymorphisms and vitiligo. Eur J Dermatol. 2009;19(2):126–128. doi:10.1684/ejd.2008.0578
27. Hayran Y, Özge Ergen G, Özmen F. The relationship between non-segmental vitiligo, HLA genotype and oxidative stress. Int J Clin Pract. 2021;75(3):e14024. doi:10.1111/ijcp.14024
28. Fichna M, Żurawek M, Słomiński B, et al. Polymorphism in BACH2 gene is a marker of polyglandular autoimmunity. Endocrine. 2021;74(1):72–79. doi:10.1007/s12020-021-02743-9
29. Tan IJ, Podwojniak A, Parikh A, et al. Precision dermatology: a review of molecular biomarkers and personalized therapies. Curr Issues Mol Biol. 2024;46(4):2975–2990. doi:10.3390/cimb46040186
30. Shahroudi MJ, Rezaei M, Mirzaeipour M, et al. Association between miR-202, miR-211, and miR-1238 gene polymorphisms and risk of vitiligo. Arch Dermatol Res. 2024;316(5):118. doi:10.1007/s00403-024-02847-y
31. Męcińska-Jundziłł K, Tadrowski T, Jundziłł A, et al. Evaluation of polymorphisms and expression of PTPN22, NLRP1 and TYR genes in vitiligo patients. Postepy Dermatol Alergol. 2023;40(2):225–233. doi:10.5114/ada.2023.126314
32. Giri PS, Bharti AH, Dwivedi M. Decreased GZMB, NRP1, ITPR1, and SERPINB9 transcripts lead to reduced regulatory T cells suppressive capacity in generalized vitiligo patients. J Immunol Res. 2022;2022:3426717. doi:10.1155/2022/3426717
33. Ma B, Wang X. Unravelling the molecular mechanisms and immune landscape of vitiligo: a comprehensive bioinformatics study on melanogenesis-related genes. Ann Hum Biol. 2025;52(1):2468692. doi:10.1080/03014460.2025.2468692
34. Yardman-Frank JM, Fisher DE. Skin pigmentation and its control: from ultraviolet radiation to stem cells. Exp Dermatol. 2021;30(4):560–571. doi:10.1111/exd.14260
35. Gay-Mimbrera J, Lozano-Ojalvo D, Gómez-Arias PJ, et al. Comprehensive single-cell chromatin and transcriptomic profiling of peripheral immune cells in nonsegmental vitiligo. Br J Dermatol. 2025;193(1):115–124. doi:10.1093/bjd/ljaf041
36. Venkatesh V, Mendez DC, Rajashekar TS, et al. Novel association between STAT3 gene variant and vitiligo: a case-control study. Indian J Dermatol. 2022;67(2):133–137. doi:10.4103/ijd.ijd_515_21
37. Cheng L, Liang B, Tang XF, et al. Validation of susceptibility loci for vitiligo identified by GWAS in the Chinese Han population. Front Genet. 2020;11:542275. doi:10.3389/fgene.2020.542275
38. Rashid S, Molotkov I, Klebanov N, et al. Mendelian randomization analysis reveals inverse genetic risks between skin cancers and vitiligo. JID Innov. 2023;3(6):100217. doi:10.1016/j.xjidi.2023.100217
39. Liu H, Wang Y, Le Q, et al. The IFN-γ-CXCL9/CXCL10-CXCR3 axis in vitiligo: pathological mechanism and treatment. Eur J Immunol. 2024;54(4):e2250281. doi:10.1002/eji.202250281
40. Bowcock AM, Fernandez-Vina M. Targeting skin: vitiligo and autoimmunity. J Invest Dermatol. 2012;132(1):13–15. doi:10.1038/jid.2011.353
41. Bouayad A, Benzekri L. Thyroid autoimmunity in relation to HLA-DRB1 and HLA-DQB1 polymorphism in nonsegmental vitiligo: a cross-sectional-study. Am J Transl Res. 2024;16(2):524–530. doi:10.62347/LDYE8203
42. Gholijani N, Yazdani MR, Dastgheib L. Predominant role of innate pro-inflammatory cytokines in vitiligo disease. Arch Dermatol Res. 2020;312(2):123–131. doi:10.1007/s00403-019-01996-9
43. Thomas SM, Veerabathiran R. Deciphering autoimmune susceptibility: a meta-analysis of PTPN22 gene variants. Immunol Res. 2025;73(1):59. doi:10.1007/s12026-025-09614-9
44. Dutta T, Mitra S, Saha A, et al. A comprehensive meta-analysis and prioritization study to identify vitiligo associated coding and non-coding SNV candidates using web-based bioinformatics tools. Sci Rep. 2022;12(1):14543. doi:10.1038/s41598-022-18766-9
45. Bharti N, Banerjee R, Achalare A, et al. Estimation of genetic variation in vitiligo associated genes: population genomics perspective. BMC Genom Data. 2024;25(1):72. doi:10.1186/s12863-024-01254-6
46. Chen M, Zhu H, Mao YJ, et al. Regulation of IL12B expression in human macrophages by TALEN-mediated epigenome editing. Curr Med Sci. 2020;40(5):900–909. doi:10.1007/s11596-020-2249-2
47. Gleave A, Granville DJ. Granzyme B in autoimmune skin disease. Biomolecules. 2023;13(2):388. doi:10.3390/biom13020388
48. Lu W, Chen Z, Xu H, et al. Decreased ZMIZ1 suppresses melanogenesis in vitiligo by regulating mTOR/AKT/GSK-3β-mediated glucose uptake. Vitro Cell Dev Biol Anim. 2024;60(1):67–79. doi:10.1007/s11626-023-00837-4
49. El-Taweel AE, Abdelrahman A, Hegazy H, et al. Granzyme B gene polymorphisms are associated with severe non-segmental vitiligo. J Clin Aesthet Dermatol. 2023;16(2):55–59.
50. Hirabayashi M, Tamura C, Sanbo M, et al. Ability of tetraploid rat blastocysts to support fetal development after complementation with embryonic stem cells. Mol Reprod Dev. 2012;79(6):402–412. doi:10.1002/mrd.22043
51. Menard L, Saadoun D, Isnardi I, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest. 2011;121(9):3635–3644. doi:10.1172/JCI45790
52. Tsai TF, Ng CY. COVID-19 vaccine-associated vitiligo: a cross-sectional study in a tertiary referral center and systematic review. J Dermatol. 2023;50(8):982–989. doi:10.1111/1346-8138.16799
53. Klarquist J, Eby JM, Henning SW, et al. Functional cloning of a gp100-reactive T-cell receptor from vitiligo patient skin. Pigm Cell Melanoma Res. 2016;29(3):379–384. doi:10.1111/pcmr.12458
54. Birlea SA, Jin Y, Bennett DC, et al. Comprehensive association analysis of candidate genes for generalized vitiligo supports XBP1, FOXP3, and TSLP. J Invest Dermatol. 2011;131(2):371–381. doi:10.1038/jid.2010.337
55. Li K, Shi Q, Yang L, et al. The association of vitamin D receptor gene polymorphisms and serum 25-hydroxyvitamin D levels with generalized vitiligo. Br J Dermatol. 2012;167(4):815–821. doi:10.1111/j.1365-2133.2012.11132.x
56. Strub T, Giuliano S, Ye T, et al. Essential role of microphthalmia transcription factor for DNA replication, mitosis and genomic stability in melanoma. Oncogene. 2011;30(20):2319–2332. doi:10.1038/onc.2010.612
57. Richmond JM, Strassner JP, Zapata L Jr, et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci Transl Med. 2018;10(450). doi:10.1126/scitranslmed.aam7710
58. Craiglow BG, King BA. Tofacitinib citrate for the treatment of vitiligo: a pathogenesis-directed therapy. JAMA Dermatol. 2015;151(10):1110–1112. doi:10.1001/jamadermatol.2015.1520
59. Samuel N, Wilson G, Lemire M, et al. Genome-wide DNA methylation analysis reveals epigenetic dysregulation of MicroRNA-34A in TP53-associated cancer susceptibility. J Clin Oncol. 2016;34(30):3697–3704. doi:10.1200/JCO.2016.67.6940
60. Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol. 2018;19(7):665–673. doi:10.1038/s41590-018-0120-4
61. Li S, Kang P, Zhang W, et al. Activated NLR family pyrin domain containing 3 (NLRP3) inflammasome in keratinocytes promotes cutaneous T-cell response in patients with vitiligo. J Allergy Clin Immunol. 2020;145(2):632–645. doi:10.1016/j.jaci.2019.10.036
62. Richmond JM, Bangari DS, Essien KI, et al. Keratinocyte-derived chemokines orchestrate T-cell positioning in the epidermis during vitiligo and may serve as biomarkers of disease. J Invest Dermatol. 2017;137(2):350–358. doi:10.1016/j.jid.2016.09.016
63. He Y, Li S, Zhang W, et al. Dysregulated autophagy increased melanocyte sensitivity to H(2)O(2)-induced oxidative stress in vitiligo. Sci Rep. 2017;7:42394. doi:10.1038/srep42394
64. Yang L, Yang S, Lei J, et al. Role of chemokines and the corresponding receptors in vitiligo: a pilot study. J Dermatol. 2018;45(1):31–38. doi:10.1111/1346-8138.14004
65. Wang X, Wu Y, Du P, et al. Study on the mechanism of miR-125b-5p affecting melanocyte biological behavior and melanogenesis in vitiligo through regulation of MITF. Dis Markers. 2022;2022:6832680. doi:10.1155/2022/6832680
66. Harris JE. Chemical-induced vitiligo. Dermatol Clin. 2017;35(2):151–161. doi:10.1016/j.det.2016.11.006
67. Ezzedine K, Peeva E, Yamaguchi Y, et al. Efficacy and safety of oral ritlecitinib for the treatment of active nonsegmental vitiligo: a randomized phase 2b clinical trial. J Am Acad Dermatol. 2023;88(2):395–403. doi:10.1016/j.jaad.2022.11.005
68. Spritz RA. The genetics of generalized vitiligo. Curr Dir Autoimmun. 2008;10:244–257.
69. Boniface K, Seneschal J, Picardo M, et al. Vitiligo: focus on clinical aspects, immunopathogenesis, and therapy. Clin Rev Allergy Immunol. 2018;54(1):52–67. doi:10.1007/s12016-017-8622-7
70. Zhang YX, Liu LP, Jin M, et al. Direct reprogramming of mouse fibroblasts into melanocytes. J Vis Exp. 2021; 174. doi:10.3791/62911
71. Liu LY, He SJ, Chen Z, et al. The role of regulatory cell death in vitiligo. DNA Cell Biol. 2024;43(2):61–73. doi:10.1089/dna.2023.0188
72. Riding RL, Richmond JM, Fukuda K, et al. Type I interferon signaling limits viral vector priming of CD8 + T cells during initiation of vitiligo and melanoma immunotherapy. Pigm Cell Melanoma Res. 2021;34(4):683–695. doi:10.1111/pcmr.12935
73. Teulings HE, Limpens J, Jansen SN, et al. Vitiligo-like depigmentation in patients with stage III-IV melanoma receiving immunotherapy and its association with survival: a systematic review and meta-analysis. J Clin Oncol. 2015;33(7):773–781. doi:10.1200/JCO.2014.57.4756
74. Spritz RA, Santorico SA. The Genetic Basis of Vitiligo. J Invest Dermatol. 2021;141(2):265–273. doi:10.1016/j.jid.2020.06.004
75. Miot HA, Criado PR, de Castro CCS, et al. JAK-STAT pathway inhibitors in dermatology. An Bras Dermatol. 2023;98(5):656–677. doi:10.1016/j.abd.2023.03.001
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.