Back to Journals » The Application of Clinical Genetics » Volume 9
Genetic basis of Cowden syndrome and its implications for clinical practice and risk management
Authors Gammon A, Jasperson K, Champine M
Received 14 February 2014
Accepted for publication 10 May 2014
Published 13 July 2016 Volume 2016:9 Pages 83—92
DOI https://doi.org/10.2147/TACG.S41947
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Amanda Gammon,1 Kory Jasperson,1,2 Marjan Gammon1
1Huntsman Cancer Institute Family Cancer Assessment Clinic Salt Lake City, UT, USA; 2Ambry Genetics Medical Affairs Aliso Viejo, CA USA
Abstract: Cowden syndrome (CS) is an often difficult to recognize hereditary cancer predisposition syndrome caused by mutations in phosphatase and tensin homolog deleted on chromosome 10 (PTEN). In addition to conferring increased cancer risks, CS also predisposes individuals to developing hamartomatous growths in many areas of the body. Due to the rarity of CS, estimates vary on the penetrance of certain phenotypic features, such as macrocephaly and skin findings (trichilemmomas, mucocutaneous papules), as well as the conferred lifetime cancer risks. To address this variability, separate clinical diagnostic criteria and PTEN testing guidelines have been created to assist clinicians in the diagnosis of CS. As knowledge of CS increases, making larger studies of affected patients possible, these criteria continue to be refined. Similarly, the management guidelines for cancer screening and risk reduction in patients with CS continue to be updated. This review will summarize the current literature on CS to assist clinicians in staying abreast of recent advances in CS knowledge, diagnostic approaches, and management.
Keywords: Cowden syndrome, PTEN gene, hereditary cancer, genetic counseling
Introduction
Cowden syndrome (CS), along with Bannayan-Riley-Ruvalcaba syndrome, is part of the phosphatase and tensin homolog deleted on chromosome 10 (PTEN) hamartoma tumor syndrome (PHTS), a disorder primarily predisposing an affected individual to hamartomatous growths and malignancy in multiple organ systems. Increased risks associated with CS include female breast, endometrial, thyroid, colon, and renal cancers. While malignancies associated with CS are generally considered to be adult-onset, some of the other phenotypic features associated with PHTS can be recognized in childhood. CS is a rare condition, affecting approximately one in 200,000 individuals worldwide, although this number may be an underestimate.1 Due to its phenotypic variability, CS can present a dilemma for clinicians, and affected individuals often undergo numerous medical evaluations before a diagnosis is made. Germline mutations in the PTEN gene are known to cause CS. However, studies have shown great variability, with PTEN mutation detection rates ranging from 11% to 80% for patients meeting the clinical diagnostic criteria set forth in 1996.2–4 In this paper, we review the genetics of CS, its associated cancer risks and other clinical manifestations, approaches to diagnosing CS, and current risk management recommendations for patients.
Genetic basis
CS was first recognized as a distinct clinical entity in 1963; however, causative mutations in PTEN were not linked to CS until 1997.5,6 The protein product of PTEN functions as a tumor suppressor through its lipid phosphatase activity regulating the phosphatidylinositol 3-kinase pathway.7,8 Loss of PTEN function results in a downstream effect of increased cell proliferation and survival, leading to tumorigenesis.8 Somatic PTEN mutations are found in a variety of cancers, including breast and endometrial cancers and melanoma.9–11 However, germline PTEN mutations are rare in individuals with these cancers and additional phenotypic features associated with PHTS are almost always identified in these cases.4,12
Within families, PTEN mutations are passed on in an autosomal dominant pattern of inheritance. Thus each child of an individual with a molecular diagnosis of CS has a 50% chance of having CS as well. As many as 45% of cases of CS may be due to de novo PTEN mutations.13 A much smaller number of cases are believed to be due to PTEN mutation mosaicism.14,15 It has been postulated that some cases of CS previously thought to be de novo could have resulted from mosaicism in a parent.14 De novo and mosaic cases of CS may lack a suggestive family history and therefore add to the diagnostic difficulty of this condition.
Given that a significant proportion of individuals meeting clinical diagnostic criteria for Cowden syndrome do not have a detectable PTEN mutation, other genetic causes for CS are also being explored. Germline mutations in SDHB, SDHC, and SDHD have been identified in some individuals meeting clinical diagnostic criteria for CS (or having CS-like features).16,17 Mutations in these three genes are known to cause hereditary pheochromocytoma and paraganglioma syndrome. While paraganglioma and pheochromocytoma are not typical malignancies associated with CS, the two conditions share increased risks for thyroid and renal cancers.16,17 Hypermethylation of the KILLN promoter has also been detected in some individuals meeting clinical CS criteria or having CS-like features.18 This is particularly notable as KILLN and PTEN are expected to share the same promoter. Many of the studies examining the potential role of the SDH genes and KILLN in CS were completed before the recent revisions to the clinical diagnostic criteria for CS. Therefore, it will be important to determine how many individuals with dysfunction in these CS candidate genes meet the revised clinical criteria.
Cancer risks and clinical manifestations
Breast
Adenocarcinoma of the breast is the most common malignancy seen among women with CS. The lifetime breast cancer risk estimate has typically been reported to be between 25% and 50% for women with CS (Table 1).1,19 However, some recent studies have postulated a higher lifetime risk for breast cancer, ranging from 77% to 85%.20,21 These recent studies are complicated by ascertainment bias to varying degrees, so as with many of the lifetime cancer risk estimates associated with CS, the exact risks continue to be debated in the literature.22 CS predisposes affected females to develop premenopausal breast cancer and the risk appears to start increasing around age 30 years.20 Bilateral breast cancer risk is also increased in CS, with the risk of a second primary breast cancer in affected women still unknown.1,21 Of the 23 women in Bubien et al’s21 study cohort with pathogenic PTEN mutations who developed breast cancer, nearly half were diagnosed with a contralateral breast cancer.21 To date, only two cases of male breast cancer have been reported in individuals diagnosed with CS.23 In addition, germline PTEN mutations account for relatively few cases of familial breast cancer.4,24,25
 | Table 1 Lifetime cancer risks |
A recent study by Banneau et al examined 15 breast cancers from women with germline PTEN mutations. The average age of breast cancer onset among these 15 women was 42 (range 27–59) years.26 This is in line with previous estimates of the average age of breast cancer onset being between 38 and 46 years in women with CS.1 Of these 15 breast cancers, four were of the invasive apocrine histologic type, eight were invasive ductal carcinoma, one was ductal carcinoma in situ, one was invasive lobular carcinoma, and one was micropapillary carcinoma.26 Eleven of the 15 breast cancers were estrogen receptor-positive.26 Looking at the gene expression signature of these tumors, their findings suggest that CS predisposes to the development of breast cancer with apocrine features.26 Additional molecular and histologic profiling of breast cancers associated with CS may lead to more targeted treatments and chemoprevention measures for affected patients.
While benign breast findings such as fibrocystic breast disease and fibroadenomas have been long considered part of the CS phenotype, recent analysis has called into question whether or not these findings are truly more prevalent in CS compared with the general population.22 Therefore, these findings have been excluded from the more recently published diagnostic criteria for CS.22
Gynecological
Endometrial cancer is the only known gynecologic cancer significantly associated with CS. Reported lifetime risks for endometrial cancer in CS range from 5% to 28%.1,20,22 The risk for endometrial cancer appears to start around age 25 years.20 However, two case reports of endometrial cancer in adolescence have been reported in individuals with CS, with both patients having germline PTEN mutations.27,28 Germline mutations appear to be rare in sporadic endometrial cancers. In one study, a consecutive series of 240 patients with endometrial cancer diagnosed at a single institution were evaluated for germline mutations in PTEN and no deleterious mutations were identified.12
Endometrial cancer occurring prior to age 50 years is a feature commonly seen in families with a different hereditary cancer syndrome known as Lynch syndrome. As Lynch syndrome is far more common than CS, routine screening of all endometrial cancer specimens for Lynch syndrome has been initiated at certain hospitals across the USA.29 The reported instances of strikingly early-onset endometrial cancer in CS should remind clinicians to think beyond Lynch syndrome when evaluating these patients.
Two cases of ovarian tumors (one dysgerminoma and one cystadenoma) have been reported in women with CS.30 Unlike the much more common hereditary breast and ovarian cancer syndrome, the risk of ovarian cancer does not appear to be significantly elevated in CS.
Nonmalignant findings, such as uterine fibroids and genitourinary malformations, have long been thought to be associated with CS. Further review of the recent literature suggests that these features may not be significantly more common in CS than in the general population.22
Thyroid
Benign thyroid findings, including multiple thyroid adenomas and goiter, as well as nonmedullary thyroid cancer, are seen with increased frequency in individuals with CS (Table 2). Prior lifetime risk estimates ranged from 3% to 10% for nonmedullary thyroid cancer in CS.1 Tan et al estimated a 35% lifetime risk for epithelial thyroid cancer in CS patients, although this number may be inflated due to ascertainment bias.20 Milas et al reported thyroid cancer in 32 of 225 (14%) patients with CS and confirmed PTEN mutations.31 Bubien et al reported seeing thyroid cancer in 24 of 140 (17%) CS patients with PTEN mutations.21 The average age of thyroid cancer onset in CS is in the late 30s to early 40s.32,33 However, childhood-onset thyroid cancer has also been reported in individuals with PTEN mutations.32,34 Recent studies support follicular thyroid cancer being the characteristic histopathology seen among CS-related thyroid malignancies, although papillary thyroid cancers are also common.22 CS does not appear to account for a substantial number of thyroid cancer cases in the absence of additional phenotypic features. A 2011 study examined 259 consecutive thyroid cancer cases, of which only two (0.8%) were found to have PTEN mutations. Of note, both cases were of the follicular type.35
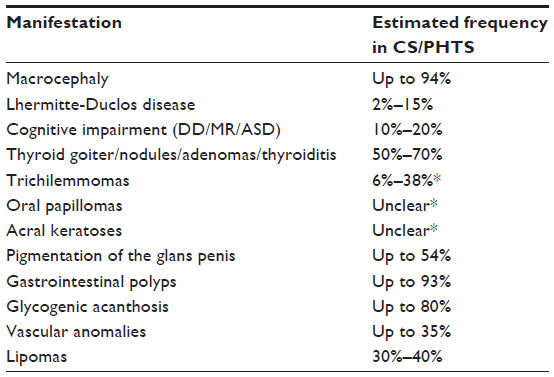 | Table 2 Nonmalignant manifestations |
Benign thyroid disease has been estimated to occur in 50%–70% of individuals with CS.1 In a 2013 study by Bubien et al, multinodular goiter was documented in 62% of patients, while thyroid adenomas were seen in 23% (Table 2).21 From the cohort of PTEN study patients at the Cleveland Clinic, 17 of 64 (27%) PTEN mutation-positive patients with CS had Hashimoto’s thyroiditis and 47 of 64 (73%) had a thyroid goiter.31 Laury et al examined 20 individuals with CS/Bannayan-Riley-Ruvalcaba syndrome with available thyroid evaluation records/biopsies, and found 75% had multiple adenomatous thyroid nodules, 55% had thyroiditis, and 55% had C-cell hyperplasia.32
Gastrointestinal
CS is considered one of the quintessential hamartomatous polyposis syndromes and is characterized by a variety of different polyp types that occur throughout the entire gastrointestinal tract. Colonic polyposis may also be the first sign of CS. The variability in colon polyp types in CS is remarkable and includes hamartomas (these are often juvenile although unspecified hamartomatous polyps also occur), hyperplastic, sessile serrated, ganglioneuromas, adenomas, lipomas, leiomyomas, lymphoid, and inflammatory polyps.22 Many of these polyp types also occur in the stomach.22 Evidence now supports that CS is associated with an increased risk for colorectal cancer. Esophageal polyps in CS are glycogenic acanthosis. Glycogenic acanthosis in addition to gastrointestinal polyps is considered by some to be pathognomonic for CS; however, these would not be considered diagnostic by themselves using the updated criteria.22,36
Colorectal
In one study, 67 patients with PTEN mutations underwent at least one colonoscopy and 62 (93%) were found to have colorectal polyps (Table 2).37 Hyperplastic polyps were the most common, followed by hamartomatous, ganglioneuromatous, adenomatous, and inflammatory polyps.37 Other polyp types include leiomyomas, lipomas, lymphoid, hyperplastic, and sessile serrated polyps.22,37 Colonic polyps numbering in the hundreds may also occur in CS.22 The risk of colorectal cancer is also increased in CS and may occur at young ages.22 The lifetime risk of colorectal cancer in CS is currently estimated to be from 9% to 16% (Table 1).22,37
In a large study of 603 individuals with moderate colonic polyp loads (five or more polyps) and varied histologic types (one or more hamartomatous or hyperplastic/serrated polyps), 13 (2%) were found to have PTEN mutations.38 Individuals with ganglioneuromas or more than two histologic types of polyps were significantly more likely to have a PTEN mutation.38
Upper gastrointestinal
Limited data are available regarding the upper gastrointestinal phenotype in CS. Of 39 patients with CS who underwent at least one esophagogastroduodenoscopy, 26 (67%) had upper gastrointestinal polyps within the esophagus, stomach, and/or duodenum.37 Glycogenic acanthosis was found in eight (21%) of these individuals.37 In another study, all ten individuals with CS who underwent at least one esophagogastroduodenoscopy had gastric (mostly hyperplastic) polyps and nine (90%) had duodenal polyps, three of which had confirmed adenomas.39 Eight (80%) of these individuals also had glycogenic acanthosis and five of these were classified as having severe acanthosis.39
Gastric cancers have been reported in three males with CS, including a 67-year-old with signet ring cell gastric adenocarcinoma arising in a large hyperplastic/hamartomatous polyp, a 73-year-old with synchronous gastric adenocarcinomas arising from an adenomatous polyp, and a 52-year-old with gastric adenocarcinoma.36,37,40 A gastrointestinal stromal tumor of the jejunum was reported in one case, meeting clinical criteria for CS but no PTEN mutation was found.41
Renal
A personal history of renal cell carcinoma is one of the minor criteria included in the National Comprehensive Cancer Network (NCCN Guidelines®) Clinical Practice Guidelines In Oncology for Genetic/Familial High-Risk Assessment: Breast and Ovarian, for genetic testing of CS.42 The risk of renal cell carcinoma in individuals with a PTEN mutation was previously reported as unknown.1 However, a recent study from the Cleveland Clinic reported that the lifetime renal cell carcinoma risk may be as high as 34%.20 Shuch et al recently reported that almost 17% (four of 24 patients) of their National Institutes of Health study cohort from 2008 to 2011 with a germline PTEN mutation had a personal history of renal cell carcinoma while none of them had a reported family history of this cancer.43 Three of these patients were reported to have had solitary renal lesions (two with papillary type 1 and one with clear cell carcinoma) while one presented with bilateral chromophobe renal cell carcinoma.43 Both of these studies have ascertainment biases, so further research is needed to clarify the incidence of renal cancer in CS.
Skin
While particular mucocutaneous lesions are rare in the general population, certain dermatologic findings, including multiple trichilemmomas, oral papilloma, and acral keratosis, are often the initial indicator for dermatologists to seek out a high-risk genetics evaluation of CS for their patients.44 These lesions are identified in many patients with a germline PTEN mutation, although early prevalence estimates may be inflated due to the original focus on dermatologic manifestations in the diagnostic work-up for CS.45
Mucocutaneous lesions have been reported as presenting predominantly in the second decade of life.19 However, clinical judgment should be used in determining the clinical criterion for CS with regard to skin findings, as a sufficient body of research is not currently available to accurately describe the number of lesions necessary to raise suspicion. Trichilemmomas, when seen in multiples (three or more lesions) in particular, are one of the hallmark features of CS, making it a highly suspicious clinical indicator of CS. Trichilemmomas are a benign hamartoma of the outer sheath of hair follicles that are most often located on the face in individuals with a PTEN mutation. Prior studies suggest that these trichilemmomas may also be present in other areas, such as the hairline, neck, axillae, and hands.46,47 Trichilemmomas may mimic other more common dermatologic findings, such as viral warts or cornu cutaneum. Therefore, it is recommended that at least one lesion suspicious for trichilemmoma be confirmed via biopsy. Studies have reported varying degrees of prevalence of trichilemmoma in patients with CS, ranging from 6% to 38%.21,22 The true penetrance of these lesions is unclear, because not all studies have involved confirmation of trichilemmoma pathology by a formal dermatology evaluation and/or biopsy (Table 2).22
Other clinically significant dermatologic features of CS include oral papilloma and benign acral keratosis. Acral keratotic lesions can occur on the dorsal feet or hands. They are also generally identified on the palmoplantar surfaces, mimicking the appearance of warts.46 The age of onset and penetrance of these lesions is still unknown. Finally, macular pigmentation of the glans penis is a major diagnostic criterion for CS in male patients; the prevalence is unclear, but recent studies have reported finding genital pigmentation in 46%–54% of their male patients with PTEN mutations.21,48 Current major criteria related to skin lesions warranting genetic testing for CS according to the NCCN guidelines include: one biopsy-proven trichilemmoma, multiple palmoplantar keratosis, multifocal or extensive oral mucosal papillomatosis, and multiple cutaneous facial papules (often verrucous). Minor dermatologic criteria for genetic testing include the presence of lipomas and testicular lipomatosis.42 It is important to note that lipomas are also seen in approximately 30%–40% of patients with CS.19,49
Finally, the lifetime risk for cutaneous melanoma has not been reported in individuals with a known PTEN mutation, although various case reports have suggested its association with this syndrome. Recent data have supported a projected lifetime risk of 6% for melanoma in individuals with a germline PTEN mutation (Table 1).20 It has been suggested that additional research is still needed before adding melanoma to the diagnostic criteria for this syndrome.22
Brain/cognitive
Macrocephaly
Macrocephaly is one of the most consistent features of CS (Table 2). It is typically defined as having a head circumference two or more standard deviations above the mean.50 A recent study from France looking at patients with documented PTEN mutations identified macrocephaly in 113 of 122 (93%) patients.21 Likewise, a 2011 study from the Cleveland Clinic examining 161 patients with PTEN mutations found that 152 (94%) patients had macrocephaly.50 In their cohort, the average adult female head circumference was measured at 60 cm, while the average male head circumference was 62.8 cm.50 The high incidence of macrocephaly in CS patients further emphasizes the importance of routine measurement of occipital-frontal circumference when evaluating a patient for CS.
Lhermitte-Duclos disease
Lhermitte-Duclos disease (LDD) is a dysplastic gangliocytoma of the cerebellum, ie, a hamartomatous growth in the brain. Symptoms of LDD include ataxia, seizures, headaches, and vision disturbance.51,52 Frequency estimates have ranged from 2% to 15% among CS study cohorts and cases reported in the literature.2,22,53 The frequency of LDD in CS is unclear because routine screening for LDD is not generally recommended for asymptomatic individuals.22,51 A 2005 study looked at brain magnetic resonance imaging findings from 20 individuals with CS, none of whom reported clinical symptoms of LDD. Three of the 20 (15%) patients were found to have LDD.51 All 20 patients met clinical diagnostic criteria for CS, 16 of whom also had an identifiable PTEN mutation via sequencing.51 Onset of LDD in individuals with CS is typically in adulthood, although a few pediatric onset cases have been reported.22,51 Cases of meningioma occurring in individuals with CS have also been reported, but it is unclear if the incidence in CS is truly higher than what would be expected in the general population.22,51
Cognitive impairment
Varying degrees of cognitive impairment have been reported in individuals with CS, ranging from no detectable impairment to the presence of autism spectrum disorder, developmental delay, or mental retardation (Table 2).54 Developmental delay or mental retardation has been reported in up to 20% of CS patients.22 Among 144 PTEN mutation-positive patients with appropriate clinical records, Bubien et al identified 15 patients (10%) with mental retardation with or without a diagnosis of autism.21 Some children whose only features of PHTS appear to be macrocephaly and autism have also been found to have pathogenic PTEN mutation.22 It is unclear whether or not more features consistent with CS will emerge in these children as they age.
Vascular
Certain types of vascular malformations have also been seen in individuals with CS, including arteriovenous malformations and hemangiomas (Table 2). Bubien et al reported finding vascular malformations in 48 (35%) of their 136 patients with PTEN mutations and sufficient documentation.21 In a 2005 brain magnetic resonance imaging study by Lok et al, vascular malformations in the brain were identified in six of 20 patients with CS, including venous angiomas and two cavernous angiomas.51
Clinical evaluation and genetic counseling
Having access to a multidisciplinary team of specialists (ie, genetics, gastroenterology, dermatology, neurology) can be of great value when evaluating a patient for CS. The clinical evaluation should include genetic counseling, review of medical and family history, and a physical examination. Two sets of guidelines are available to assist in this assessment, ie, clinical diagnostic criteria for CS and PTEN genetic testing criteria.
Clinical diagnostic criteria for CS were first proposed in 1996 by the International Cowden Syndrome Consortium.55 These criteria were revised in 2000, and features such as LDD and autism were later included.52,56 The most recent revisions proposed by Pilarski et al were prompted by the significant number of individuals who met the prior diagnostic criteria primarily based on features common in the general population; these individuals were also frequently found not to have PTEN mutations (ie, women with breast and thyroid cancer but no other features).22 By making the diagnostic criteria more stringent, the number of PTEN mutations identified in individuals meeting these criteria is expected to increase. The newly proposed criteria are summarized in Table 3, and have also been recently incorporated into the 2014 NCCN guidelines on CS/PHTS.42 Performing PTEN genetic testing in an individual who already meets the clinical diagnostic criteria is still incredibly valuable. This is because identifying a pathogenic PTEN mutation allows for targeted genetic testing in family members and initiation of cancer screening and risk reduction in relatives who test positive and are therefore at increased risk. Knowing the PTEN mutation also allows assisted reproductive technologies such as preimplantation genetic diagnosis to be utilized to avoid CS in the future offspring of patients with CS.
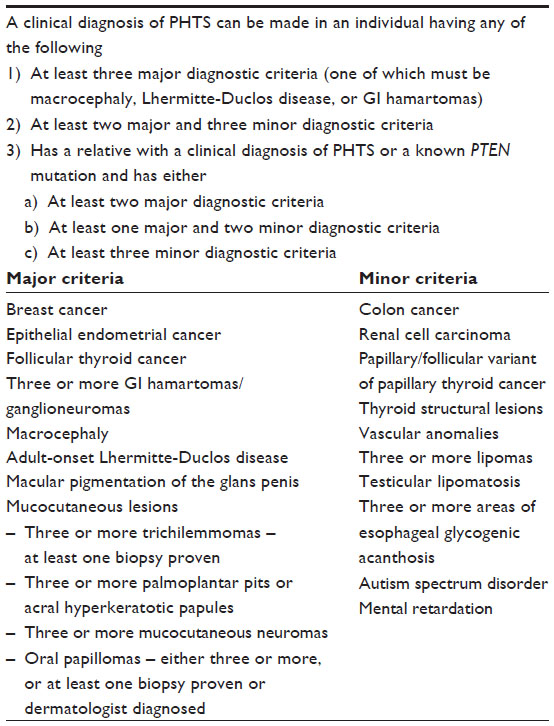 | Table 3 Clinical diagnostic criteria |
The NCCN has proposed separate guidelines for PTEN testing, as summarized in Table 4.42 It is important to note that the guidelines for PTEN testing are far less stringent than the clinical diagnostic criteria for CS. Given that the CS phenotype is quite variable between affected individuals, the more relaxed testing guidelines help to account for the limitations inherent in assessment of CS. Researchers at the Cleveland Clinic and Ohio State University have also created online PTEN mutation prediction models.2,57 By entering a patient’s phenotypic information, these models can provide an estimation of the likelihood of a patient testing positive for a pathogenic PTEN mutation. It is recommended that the same management recommendations be followed for individuals who do not currently meet diagnostic criteria for CS, but in whom a PTEN mutation is identified, as for individuals with a clinical diagnosis of CS.
 | Table 4 Genetic testing criteria (NCCN criteria) |
When evaluating a patient for CS, the patient’s medical and physical phenotype should be compared with the most current clinical diagnostic criteria for CS, as well as the guidelines for PTEN testing. Genetic testing should be offered when appropriate. The physical examination should be tailored to assess for features of CS, including a skin examination and measurement of head circumference. Having access to a dermatologist familiar with CS, as well as a dedicated gastrointestinal pathologist to review any gastrointestinal biopsies/polyps, can be of significant help when evaluating patients who nearly meet the clinical diagnostic criteria and/or PTEN testing guidelines for CS.57
Genetic testing techniques related to evaluation of hereditary cancer have changed significantly over the past few years. Next-generation sequencing techniques have been shown to be more sensitive in identifying low-level mosaicism in blood lymphocytes than traditional Sanger sequencing.15 This new testing technology has the potential to improve mutation detection rates in CS and many other genetic diseases. Next-generation sequencing has also allowed for the development of multigene panels. These panels can focus on hereditary risk for a specific type of cancer (eg, breast, colon), or can include genes that predispose to a wide variety of cancers. The number of genes included in each panel and the magnitude of cancer risk that mutations in these genes confer can also vary significantly depending on the laboratory offering the test.
Since PTEN mutations confer increased risks for multiple common cancers (ie, breast, colon, endometrial), PTEN is commonly included in many hereditary cancer gene panels.15 The widespread presence of PTEN in these multigene panels will mean that more individuals who do not meet the NCCN guidelines for PTEN testing will be tested (eg, women with premenopausal breast cancer who have no other features of CS). One outcome may be that the phenotypic spectrum of CS/PHTS will increase as pathogenic PTEN mutations are identified in individuals who do not have the classical features of CS. On the other hand, while it has been suspected for many years that CS is underdiagnosed, wider uptake of PTEN testing via gene panels may provide more evidence that CS is truly rare. In either scenario, lifetime cancer risk estimates, as well as screening/risk reduction recommendations for individuals with CS, will continue to be refined with this new influx of information.
The genetic counseling process becomes increasingly important with the availability of next-generation sequencing panels. Testing multiple genes simultaneously increases the probability of identifying unexpected clinically relevant results, as well as variants of uncertain significance. Having a trained genetic counselor involved in the CS assessment and genetic testing process provides patients and their physicians with assistance in interpreting difficult results as well as access to appropriate research studies and continuous follow-up support resources.57
Management
The increased risk for malignancy in individuals with CS warrants tailored surveillance and recommendations for risk reduction. These are the primary longitudinal care recommendations for individuals with CS. The NCCN has determined a set of management guidelines for individuals with CS (summarized in Table 5), which are reviewed annually.42 These screening and risk reduction interventions are recommended for individuals with a known pathogenic mutation in PTEN or who meet the current diagnostic criteria for CS in the absence of an identifiable mutation. All of these recommendations can be further tailored to the individual patient’s personal and family history of cancer, which may warrant earlier or more frequent screening, or screening for additional cancers than are typically associated with CS.42 Some researchers have proposed more stringent and intensive screening and recommendations for risk reduction based on the higher lifetime cancer risks reported from their studies, as well as the reports of childhood-onset thyroid cancers in some affected individuals.20,21
 | Table 5 NCCN management recommendations with ages of initiation |
Beyond screening, risk reduction measures are available for some of the increased cancer risks associated with CS. Prophylactic mastectomy and/or hysterectomy can be considered for reduction of a woman’s risk of breast and endometrial cancers, respectively. The benefits, limitations, and risks of these surgeries should be discussed thoroughly with women diagnosed to have CS. Colonoscopy provides both screening and risk reduction with regard to colorectal cancer. While individuals with CS may develop numerous gastrointestinal polyps even at young ages, the lifetime risk of developing gastrointestinal cancer appears to be much lower than is seen in other hereditary polyposis conditions, such as familial adenomatous polyposis and Peutz-Jeghers syndrome. Individuals with CS should be encouraged to participate in clinical trials and other research studies, as more data are needed regarding the optical management of CS.
Conclusion
The diagnostic complexity surrounding CS highlights the need for continued international collaboration to assess cohorts of individuals with CS and further define the penetrance of both malignant and nonmalignant features. It is very difficult to follow a large enough cohort of patients with CS to make highly reliable estimates of phenotype penetrance. Having a cohort that includes not only probands presenting with features of CS and who have an identifiable PTEN mutation, but also a large number of their PTEN-positive at-risk relatives is ideal. These relatives may include more individuals at the mild end of the phenotypic spectrum, who were not identified for CS evaluation until their relative was already diagnosed. Also, as broad genetic testing technologies like whole exome and whole genome sequencing become more affordable and more clinically available to the general population, our knowledge about both rare hereditary diseases like CS and common diseases will increase. In the interim, continued collaboration between health care providers in multiple specialties will help to provide a more complete diagnostic evaluation and more comprehensive follow-up care for patients with CS.
Disclosure
K Jasperson is an employee of Ambry Genetics. The authors report no other conflicts of interest in this work.
References
Pilarski R. Cowden syndrome: a critical review of the clinical literature. J Genet Couns. 2009;18(1):13–27. | |
Pilarski R, Stephens JA, Noss R, Fisher JL, Prior TW. Predicting PTEN mutations: an evaluation of Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome clinical features. J Med Genet. 2011;48(8):505–512. | |
Marsh DJ, Coulon V, Lunetta KL, et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet. 1998;7(3):507–515. | |
Tsou HC, Teng DH, Ping XL, et al. The role of MMAC1 mutations in early-onset breast cancer: causative in association with Cowden syndrome and excluded in BRCA1-negative cases. Am J Hum Genet. 1997;61(5):1036–1043. | |
Lloyd KM 2nd, Dennis M. Cowden’s disease. A possible new symptom complex with multiple system involvement. Ann Intern Med. 1963;58:136–142. | |
Liaw D, Marsh DJ, Li J, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16(1):64–67. | |
Myers MP, Stolarov JP, Eng C, et al. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci U S A. 1997;94(17):9052–9057. | |
Squarize CH, Castilho RM, Gutkind JS. Chemoprevention and treatment of experimental Cowden’s disease by mTOR inhibition with rapamycin. Cancer Res. 2008;68(17):7066–7072. | |
Tashiro H, Blazes MS, Wu R, et al. Mutations in PTEN are frequent in endometrial carcinoma but rare in other common gynecological malignancies. Cancer Res. 1997;57(18):3935–3940. | |
Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet. 2002;32(3):355–357. | |
Gast A, Scherer D, Chen B, et al. Somatic alterations in the melanoma genome: a high-resolution array-based comparative genomic hybridization study. Genes Chromosomes Cancer. 2010;49(8):733–745. | |
Black D, Bogomolniy F, Robson ME, Offit K, Barakat RR, Boyd J. Evaluation of germline PTEN mutations in endometrial cancer patients. Gynecol Oncol. 2005;96(1):21–24. | |
Mester J, Eng C. Estimate of de novo mutation frequency in probands with PTEN hamartoma tumor syndrome. Genet Med. 2012;14(9):819–822. | |
Gammon A, Jasperson K, Pilarski R, Prior T, Kuwada S. PTEN mosaicism with features of Cowden syndrome. Clin Genet. 2013;84(6):593–595. | |
Pritchard CC, Smith C, Marushchak T, et al. A mosaic PTEN mutation causing Cowden syndrome identified by deep sequencing. Genet Med. 2013;15(12):1004–1007. | |
Ni Y, He X, Chen J, et al. Germline SDHx variants modify breast and thyroid cancer risks in Cowden and Cowden-like syndrome via FAD/NAD-dependant destabilization of p53. Hum Mol Genet. 2012;21(2):300–310. | |
Ni Y, Zbuk KM, Sadler T, et al. Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am J Hum Genet. 2008;83(2):261–268. | |
Bennett KL, Mester J, Eng C. Germline epigenetic regulation of KILLIN in Cowden and Cowden-like syndrome. JAMA. 2010;304(24):2724–2731. | |
Starink TM, van der Veen JP, Arwert F, et al. The Cowden syndrome: a clinical and genetic study in 21 patients. Clin Genet. 1986;29(3):222–233. | |
Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18(2):400–407. | |
Bubien V, Bonnet F, Brouste V, et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet. 2013;50(4):255–263. | |
Pilarski R, Burt R, Kohlman W, Pho L, Shannon KM, Swisher E. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst. 2013;105(21):1607–1616. | |
Fackenthal JD, Marsh DJ, Richardson AL, et al. Male breast cancer in Cowden syndrome patients with germline PTEN mutations. J Med Genet. 2001;38(3):159–164. | |
Guenard F, Labrie Y, Ouellette G, et al. Germline mutations in the breast cancer susceptibility gene PTEN are rare in high-risk non-BRCA1/2 French Canadian breast cancer families. Fam Cancer. 2007;6(4):483–490. | |
Blanco A, Grana B, Fachal L, et al. Beyond BRCA1 and BRCA2 wild-type breast and/or ovarian cancer families: germline mutations in TP53 and PTEN. Clin Genet. 2010;77(2):193–196. | |
Banneau G, Guedj M, MacGrogan G, et al. Molecular apocrine differentiation is a common feature of breast cancer in patients with germline PTEN mutations. Breast Cancer Res. 2010; 12(4):R63. | |
Schmeler KM, Daniels MS, Brandt AC, Lu KH. Endometrial cancer in an adolescent: a possible manifestation of Cowden syndrome. Obstet Gynecol. 2009;114(2 Pt 2):477–479. | |
Baker WD, Soisson AP, Dodson MK. Endometrial cancer in a 14-year-old girl with Cowden syndrome: a case report. J Obstet Gynaecol Res. 2013;39(4):876–878. | |
Bellcross C, Duquette D, Hampel H, Jasperson K, Mange S. Routine screening for Lynch syndrome: data from the Lynch Syndrome Screening Network 2013. J Genet Counsel. 2013;22(6):1. | |
Cho MY, Kim HS, Eng C, et al. First report of ovarian dysgerminoma in Cowden syndrome with germline PTEN mutation and PTEN-related 10q loss of tumor heterozygosity. Am J Surg Pathol. 2008;32(8):1258–1264. | |
Milas M, Mester J, Metzger R, et al. Should patients with Cowden syndrome undergo prophylactic thyroidectomy? Surgery. 2012;152(6):1201–1210. | |
Laury AR, Bongiovanni M, Tille JC, Kozakewich H, Nose V. Thyroid pathology in PTEN-hamartoma tumor syndrome: characteristic findings of a distinct entity. Thyroid. 2011;21(2):135–144. | |
Ngeow J, Mester J, Rybicki LA, Ni Y, Milas M, Eng C. Incidence and clinical characteristics of thyroid cancer in prospective series of individuals with Cowden and Cowden-like syndrome characterized by germline PTEN, SDH, or KLLN alterations. J Clin Endocrinol Metab. 2011;96(12):E2063–E2071. | |
Smith JR, Marqusee E, Webb S, et al. Thyroid nodules and cancer in children with PTEN hamartoma tumor syndrome. J Clin Endocrinol Metab. 2011;96(1):34–37. | |
Nagy R, Ganapathi S, Comeras I, et al. Frequency of germline PTEN mutations in differentiated thyroid cancer. Thyroid. 2011;21(5):505–510. | |
Al-Thihli K, Palma L, Marcus V, et al. A case of Cowden’s syndrome presenting with gastric carcinomas and gastrointestinal polyposis. Nat Clin Pract Gastroenterol Hepatol. 2009;6(3):184–189. | |
Heald B, Mester J, Rybicki L, Orloff MS, Burke CA, Eng C. Frequent gastrointestinal polyps and colorectal adenocarcinomas in a prospective series of PTEN mutation carriers. Gastroenterology. 2010;139(6):1927–1933. | |
Ngeow J, Heald B, Rybicki LA, et al. Prevalence of germline PTEN, BMPR1A, SMAD4, STK11, and ENG mutations in patients with moderate-load colorectal polyps. Gastroenterology. 2013;144(7):1402–1409.e1–e5. | |
Levi Z, Baris HN, Kedar I, et al. Upper and lower gastrointestinal findings in PTEN mutation-positive Cowden syndrome patients participating in an active surveillance program. Clin Trans Gastroenterol. 2011;2:e5. | |
Marques M, Ramalho R, Baldaque-Silva F, Macedo G. Novel mutation identified in Cowden syndrome presenting as a gastric adenocarcinoma. Clin Res Hepatol Gastroenterol. 2013;37(6):e131–e132. | |
Nakamura M, Hirooka Y, Yamamura T, et al. Cowden syndrome complicated by a gastrointestinal stromal tumor. Dig Endosc. Epub 2013 September 30. | |
Daly M, Pilarski R, Axilbund J, et al. Genetic/Familial High-Risk Assessment: Breast and Ovarian. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines). 2015; v2.2015:77. Available from: http://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. | |
Shuch B, Ricketts CJ, Vocke CD, et al. Germline PTEN mutation Cowden syndrome: an underappreciated form of hereditary kidney cancer. J Urol. 2013;190(6):1990–1998. | |
Meotti CD, Pulga RF, Fernandes Kde A, Gusmao PR, Fernandes Kde A, Rocha AR. Do you know this syndrome? An Bras Dermatol. 2013;88(5):832–834. | |
Shah KR, Boland CR, Patel M, Thrash B, Menter A. Cutaneous manifestations of gastrointestinal disease: part I. J Am Acad Dermatol. 2013;68(2):189.e181–121. | |
Masmoudi A, Chermi ZM, Marrekchi S, et al. Cowden syndrome. J Dermatol Case Rep. 2011;5(1):8–13. | |
Porter S, Cawson R, Scully C, Eveson J. Multiple hamartoma syndrome presenting with oral lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;82(3):295–301. | |
Tan MH, Mester J, Peterson C, et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet. 2011;88(1):42–56. | |
Salem OS, Steck WD. Cowden’s disease (multiple hamartoma and neoplasia syndrome). A case report and review of the English literature. J Am Acad Dermatol. 1983;8(5):686–696. | |
Mester JL, Tilot AK, Rybicki LA, Frazier TW 2nd, Eng C. Analysis of prevalence and degree of macrocephaly in patients with germline PTEN mutations and of brain weight in PTEN knock-in murine model. Eur J Hum Genet. 2011;19(7):763–768. | |
Lok C, Viseux V, Avril MF, et al. Brain magnetic resonance imaging in patients with Cowden syndrome. Medicine (Baltimore). 2005;84(2):129–136. | |
Zhou XP, Marsh DJ, Morrison CD, et al. Germline inactivation of PTEN and dysregulation of the phosphoinositol-3-kinase/Akt pathway cause human Lhermitte-Duclos disease in adults. Am J Hum Genet. 2003;73(5):1191–1198. | |
Riegert-Johnson DL, Gleeson FC, Roberts M, et al. Cancer and Lhermitte-Duclos disease are common in Cowden syndrome patients. Hered Cancer Clin Pract. 2010;8(1):6. | |
Busch RM, Chapin JS, Mester J, et al. Cognitive characteristics of PTEN hamartoma tumor syndromes. Genet Med. 2013;15(7):548–553. | |
Nelen MR, Padberg GW, Peeters EA, et al. Localization of the gene for Cowden disease to chromosome 10q22–23. Nat Genet. 1996;13(1):114–116. | |
Eng C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet. 2000;37(11):828–830. | |
Mester JL, Moore RA, Eng C. PTEN germline mutations in patients initially tested for other hereditary cancer syndromes: would use of risk assessment tools reduce genetic testing? Oncologist. 2013;18(10):1083–1090. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.