Back to Journals » The Application of Clinical Genetics » Volume 7
Gene mutations that promote adrenal aldosterone production, sodium retention, and hypertension
Authors Moraitis A, Rainey W, Auchus R
Received 15 August 2013
Accepted for publication 16 October 2013
Published 24 December 2013 Volume 2014:7 Pages 1—13
DOI https://doi.org/10.2147/TACG.S35571
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Andreas G Moraitis,1 William E Rainey,1,2 Richard J Auchus1
1Division of Metabolism, Endocrinology, and Diabetes, Department of Internal Medicine, 2Department of Physiology, University of Michigan, Ann Arbor, MI, USA
Abstract: Primary aldosteronism (PA) is the most common form of secondary hypertension, found in about 5% of all hypertension cases, and up to 20% of resistant hypertension cases. The most common forms of PA are an aldosterone-producing adenoma and idiopathic (bilateral) hyperaldosteronism. Rare genetic forms of PA exist and, until recently, the only condition with a known genetic mechanism was familial hyperaldosteronism type 1, also known as glucocorticoid-remediable aldosteronism (FHA1/GRA). FHA type 3 has now been shown to derive from germline mutations in the KCNJ5 gene, which encodes a potassium channel found on the adrenal cells. Remarkably, somatic mutations in KCNJ5 are found in about one-third of aldosterone-producing adenomas, and these mutations are likely to be involved in their pathogenesis. Finally, mutations in the genes encoding an L-type calcium channel (CACNA1D) and in genes encoding a sodium–potassium adenosine triphosphatase (ATP1A1) or a calcium adenosine triphosphatase (ATP2B3) are found in other aldosterone-producing adenomas. These findings provide a working model, in which adenoma formation and/or aldosterone production in many cases derives from increased calcium entry, which drives the pathogenesis of primary aldosteronism.
Keywords: hyperaldosteronism, hereditary, potassium channel, calcium channel
Introduction: hypertension, its causes and consequences
Hypertension, defined as systolic blood pressure (SBP) >140 mmHg, is a physical finding reflecting a variety of genetic and environmental factors, including renal sodium retention and increased vascular resistance. Hypertension affects approximately 85 million people in the US. A National Health Examination Survey (NHANES) spanning from 2005–2006 showed that 29% of US adults 18 years of age and older were hypertensive; 7% of hypertensive adults had never been told that they had hypertension. Of those with hypertension, 78% were aware they were hypertensive, 68% were being treated with antihypertensive agents, and only 64% of treated individuals had controlled hypertension (SBP <125 mmHg). In addition, NHANES from 1999–2006 estimated that 30% of adults 20 years of age and older have prehypertension, which is defined as an untreated SBP of 120–139 mmHg, or an untreated diastolic blood pressure of 80–89 mmHg.1 In the majority of cases, hypertension is idiopathic or “essential,” but a subgroup of approximately 15% has secondary hypertension. Secondary causes of hypertension, such as primary aldosteronism, account for 20% of resistant hypertension, which is defined as a blood pressure >140/90 mmHg, despite the use of three different classes of antihypertensive medications, one of which is a diuretic. The secondary causes of hypertension can be further divided into renal and endocrine causes. Endocrine causes of hypertension can be further subdivided into five categories:
- Apparent mineralocorticoid excess (AME)
- Thyroid dependent
- Parathyroid dependent
- Pituitary dependent
- Adrenal dependent
Primary aldosteronism (PA) accounts for the majority of endocrine causes of hypertension. The diagnosis of PA presents several challenges, especially the selection of patients eligible for surgery. Due to associated increases in morbidity and mortality when hypertension is due to PA, diagnosis and proper treatment of PA is important. The prevalence of cardiovascular complications associated with PA is disproportionately higher than predicted by blood pressure alone. Aldosterone exerts direct effects on the myocardium itself and increases myocardial fibrosis, which reverses significantly after successful management of PA with either an adrenalectomy or with mineralocorticoid receptor antagonist (MRA) treatment.
Besides the cardiovascular complications of PA, patients with aldosterone excess present with an increased incidence of renal, metabolic, and skeletal abnormalities. Several studies have shown an increased incidence of chronic kidney injury in PA patients. Successful treatment with either MRA or adrenalectomy unmasks relative glomerular hyperfiltration associated with PA. Several studies have found an increased incidence of metabolic syndrome in PA patients. Lower adiponectin, as well as lower insulin sensitivity in patients with PA, results from both direct (aldosterone excess) and indirect (hypokalemia) mechanisms. Osteoporosis, hypercalciuria, and nephrolithiasis are more common in patients with PA as compared to patients with essential hypertension. The effects of aldosterone excess on bone metabolism reverse after successful treatment. Consequently, PA should be viewed as a multisystem disease in which – aside from hypertension – the aldosterone excess directly or indirectly affects several other organs.
Review of genetic factors involved in development of hypertension
In addition to the known monogenic forms of hypertension, linkage analysis, candidate gene, and genome-wide association studies have detected suspected loci associated with hypertension cases with familial aggregation. There are currently eight known familial forms of monogenic hypertension. A common feature of these disorders is low plasma renin activity. Hypertension develops as a consequence of increased sodium, chloride, and water reabsorption, and subsequent volume expansion.
Glucocorticoid-remediable aldosteronism (GRA), known also as familial hyperaldosteronism type 1 (FH-1) was the first form of hypertension recognized as a monogenic disorder. GRA accounts for up to 1% of all cases of PA and occurs equally among women and men.2 GRA is inherited as an autosomal dominant trait that presents with significant phenotype variability, even among members of the same family.3 In 1992, Lifton et al4 first described a large family with GRA in which the disease cosegregated with a chimeric gene duplication, resulting from an unequal crossing over between the highly homologous 11β-hydroxylase (CYP11B1) and the aldosterone synthase (CYP11B2) genes. Both genes are located in chromosome 8 in close proximity to one another. In the normal adrenal gland, the synthesis of aldosterone takes place in the zona glomerulosa, because the expression of CYP11B2 is restricted to this layer of the adrenal cortex. The principal stimulators of aldosterone secretion are angiotensin 2 (Ang2) via the renin–angiotensin system and elevated extracellular potassium. On the other hand, cortisol production is restricted to the zona fasciculata of the adrenal cortex. The enzyme CYP11B1 catalyzes the last step of cortisol production, and adrenocorticotropin (ACTH) positively regulates its expression. In patients with GRA, the chimeric gene results from the fusion of the 5′ ACTH-responsive promoter region of the CYP11B1 gene to the 3′ coding sequence of the CYP11B2 gene. As a consequence of this fusion, aldosterone is produced in the zona fasciculata, primarily under ACTH regulation. ACTH-dependent aldosterone regulation results in a circadian pattern of aldosterone production, which parallels that of cortisol.
A second form of Mendelian monogenic hypertension is called AME, which was first described biochemically in 1977.5 Patients with AME present early in life with severe low renin hypertension, failure to thrive, and low birth weight. Polyuria, polydipsia, hypokalemia with metabolic alkalosis (renal concentrating defect due to hypokalemia), hypercalciuria, and nephrocalcinosis are frequently seen. In these patients, cortisol, rather than aldosterone, acts as the mineralocorticoid. The disease was shown to be caused by mutations in the HSD11B2 gene, which encodes the 11β-hydroxysteroid dehydrogenase (11βHSD2) enzyme, which converts cortisol to the inactive metabolite (cortisone) in aldosterone target tissues, thus protecting mineralocorticoid receptor (MR) from inappropriate cortisol binding and activation. The 11βHSD enzyme has two isoforms. The 11βHSD1 isoform is expressed in several human tissues, and mutations in the cognate HSD11B1 gene cause cortisone reductase deficiency rather than AME. The second isoform, 11βHSD2, is primarily expressed in the kidneys and colon, where MR is also found.
Cortisol is secreted in milligram amounts and aldosterone in micrograms amounts daily. MR has equal affinity for both aldosterone and cortisol, but the action of 11βHSD2 protects normal subjects from cortisol intoxication. The 11β-hydroxyl group of aldosterone is bound in a hemiacetal with the 18-oxo group, and thus aldosterone is not metabolized by 11βHSD2. In patients with AME, mutations in the HSD11B2 gene impair enzyme activity, which allows cortisol to saturate MR, leading to clinical and biochemical manifestations of mineralocorticoid excess in the absence of aldosterone.
AME is an autosomal recessive disease that presents phenotypic variability even within the same families. Up to now, more than 30 mutations have been reported in patients with AME, most of them found in exons 3 to 5, with the exception of the Arg74Gly and Pro75, Δ1nt in exon 16 and Leu114, Δ6nt mutant in exon 2.7 A few mutations were found to leave the amino acid sequence unchanged, instead causing aberrant splicing. The diagnosis of AME should be suspected in cases of low renin and low aldosterone hypertension with signs of mineralocorticoid excess, like an increased transtubular potassium gradient. In AME, urinary steroid hormone profiling shows decreased enzyme activity, as shown by an increased ratio of urine-free cortisol to urine cortisone, as well as by the ratio of their metabolites (tetrahydrocortisol/ [tetrahydrocortisone + allo-tetrahydrocortisone]). The disease should be genetically confirmed, because acquired AME and decreased 11βHSD2 activity may result from the ingestion of exogenous (licorice or carbenoxolone) and possibly endogenous (action of glycyrrhetinic acid-like factors) 11βHSD2 inhibitors.8,9
Another two forms of monogenic hypertension include certain subtypes of congenital adrenal hyperplasia (CAH). The CAH subtypes associated with hypertension include CYP11B1 (11β-hydroxylase) and CYP17A1 (17α-hydroxylase/17,20-lyase) deficiencies, which are both transmitted as autosomal recessive traits. The lack of inhibitory feedback by cortisol on the hypothalamus and the pituitary produces an ACTH-driven buildup of cortisol precursors proximal to the enzymatic deficiency. CYP11B1 and CYP17A1 deficiencies cause hypersecretion of the mineralocorticoid 11-deoxycorticosterone (DOC), which results in hypertension and hypokalemia. Up to 5%–8% of all CAH cases are due to CYP11B1 deficiency,10 particularly among Sephardic Jews from Morocco. The CYP11B1 gene is located in the long arm of chromosome 8 and has nine exons. More than 50 mutations have been described in the CYP11B1 gene.11 These mutations are distributed over the entire encoding region, but tend to cluster in exons 2, 6, 7, and 8,12 with the founder mutation (R448H) accounting for most of the mutations in Moroccan Jews.13
Most of the known mutations completely abolish the activity of the enzyme, but the clinical manifestations of the disorder vary substantially. Virilization and hypertension are the main clinical features of CYP11B1 deficiency. Female patients with a CYP11B1 deficiency can present with ambiguous genitalia, and it is not uncommon for a CYP11B1-deficient female to be misassigned as male at birth. Postnatal continuous excess androgen production results in sexual precocity and short adult stature in both sexes. Treatment with glucocorticoids provides cortisol replacement and normalizes ACTH-driven DOC and adrenal androgen excess which, in most cases, leads to the resolution of hypertension. Spironolactone (an MR and androgen receptor antagonist) is a useful adjunct to glucocorticoids, particularly for females with CYP11B1 deficiency, to simultaneously control the consequences of mineralocorticoid and androgen excess.
Mutations in the CYP17A1 gene impair steroid biosynthesis in the adrenals and gonads, resulting in 17α-hydroxylase/17, 20-lyase (P450c17) deficiency, which leads to amenorrhea, sexual infantilism, hypokalemia, and hypertension. To date, nearly 100 different mutations14,15 in the CYP17A1 gene on chromosome 10q24.3 have been identified, leading to varying degrees of inactivation of the 17α-hydroxylase and/or 17,20-lyase activities. Using assays with recombinant enzyme, many of these mutations have been shown to impair 17α-hydroxylase and/or 17, 20-lyase activities; the degree of inactivation depends on the type and location of the mutation in the gene.
Another form of monogenic hypertension is generalized glucocorticoid resistance, in which impaired cortisol signaling results from target tissue insensitivity to glucocorticoids. This defect results in compensatory activation of the hypothalamic–pituitary–adrenal axis, which leads to increased ACTH secretion. ACTH, in turn, drives increased secretion of cortisol and cortisol precursors such as the mineralocorticoids DOC and corticosterone, as well as the adrenal androgen precursors. The clinical features of generalized glucocorticoid resistance vary substantially, and include hypoglycemia, hypertension, metabolic alkalosis, infertility, hirsutism, menstrual irregularities, and sexual precocity. Some individuals may present with biochemical abnormalities alone, such as low renin and low aldosterone, due to inappropriate MR activation by the excess adrenal production of cortisol and the mineralocorticoids DOC and corticosterone.16
The human glucocorticoid receptor (GR) (NR3C1) gene is located on the long arm of chromosome 5, and consists of nine exons. To date, 15 mutations have been described, and compared with the wild-type (WT) receptor, all mutant receptors demonstrated variable but incomplete reduction in their ability to transactivate glucocorticoid-responsive genes in response to dexamethasone.
The next form of monogenic low renin hypertension, which again involves GR, is called primary generalized glucocorticoid hypersensitivity. It represents the exact opposite of primary generalized glucocorticoid resistance and is characterized by generalized, partial, target-tissue hypersensitivity to glucocorticoids, and compensatory hypoactivation of the hypothalamic–pituitary–adrenal axis. To date, only one case with glucocorticoid hypersensitivity has been described.17 The patient was a 43-year-old female who presented with a long-standing history of visceral obesity, dyslipidemia, type 2 diabetes, and hypertension.
In Liddle’s syndrome, hypertension is induced by excessive renal tubular sodium reabsorption, independent of mineralocorticoid action. Liddle’s syndrome was first described in 1963, as it resembled PA with negligible aldosterone secretion.18 Liddle’s syndrome is an autosomal dominant disorder characterized by early onset hypertension and hypokalemia with suppressed renin and aldosterone secretion. The clinical abnormalities are corrected with a low sodium diet, plus use of antagonists of the epithelial sodium channel (ENaC) in the distal nephron, such as amiloride. Liddle’s syndrome has a heterogeneous genetic background characterized by prolonged residence of ENaC on the cell surface. ENaC is composed of three similar α, β, and γ subunits. Initial genetic and electrophysiological studies had shown that mutations in the β and γ subunits of the amiloride-sensitive ENaC result in constitutive activation of channel activity.19 This elevated ENaC activity is caused by both an increase in the number of channels at the plasma membrane, as well as by a higher mean open probability of each channel (in most cases, from C-terminal truncations), which impair channel internalization. Liddle’s mutations have been also linked to defective sodium-dependent feedback inhibition.20
Another rare form of monogenic hypertension featuring hyperkalemia, metabolic acidosis, and hypertension is pseudohypoaldosteronism type 2 (PHA2), also known as Gordon’s syndrome. Gordon’s syndrome is transmitted as an autosomal dominant disorder. The cause of hypertension has been attributed to increased sodium reabsorption, and the hyperkalemia and acidosis are due to reduced renal potassium and hydrogen ion excretion. Most of the features of PHA2 are chloride dependent, because infusion of sodium bicarbonate corrects all electrolyte abnormalities. Thiazide diuretics, which inhibit sodium reabsorption in the cortical diluting segment in the distal nephron, ameliorate these abnormalities. Initially, mutations in two members of “with no lysine” (WNK) serine–threonine kinases,21 WNK1 and WKN4, were found to be responsible for some forms of PHA2. These WNK mutations alter the function of the outward-rectifying potassium channels (ROMKs), accounting for the somewhat paradoxical hyperkalemia. Most recently, mutations in the kelch-like 3 (KLHL3) and cullin 3 (CUL3)22 genes have been identified as a cause of Gordon’s syndrome in patients lacking mutations in WNK1 and WNK4. KLHL3 mutations are either recessive or dominant, while CUL3 mutations are dominant and predominantly de novo. KLHL3 binds substrate proteins, promoting substrate ubiquitination via interaction of its bric-à-brac 1–tramtrack–broad complex proteins (BTB) domain with CUL3, a component of a cullin/RING E3 ubiquitin ligase complex. Ubiquitination serves diverse functions, as it targets proteins for proteasomal degradation, as well as for nondegradative roles, such as modulation of protein activity, interactions, and localization.
Finally, the last form of monogenic low renin hypertension is familial hyperaldosteronism type 3 (FH-3), a rare form of PA. The genetic cause of FH-3 has been recently described as being associated with germline mutations of the KCNJ5 potassium channels,23 which alter the selectivity of the channels for the potassium ions. This form of hypertension will be described further in this paper.
Involvement of benign adrenal gland tumors in hypertension development
Adrenocortical hypertension represents the majority of cases of endocrine hypertension and it is associated with the hypersecretion of aldosterone, cortisol, or both. Cortisol hypersecretion syndromes are associated with clinical or subclinical features of Cushing’s syndrome. This paper will focus on PA screening, diagnosis, subtype classification, and therapy.
Dr Jerome W Conn presented the first case of PA during his presidential address at the Annual Meeting of the Central Society for Clinical Research in October of 1954. Six months earlier, Dr Stefan Fajans asked Conn to evaluate a 34-year-old woman with a 7-year history of muscle spasms, weakness, episodic paralysis, and tetany, who was hypertensive for at least 4 years. Conn suspected, based on his prior research in sodium balance and acclimatization to tropical weather, that the patient suffered from excess production of the adrenal salt-retaining hormone. He found that her sweat chloride was essentially undetectable, supporting his hypothesis. Conn planned a bilateral adrenalectomy, but the surgeon, William Baum, encountered and removed the right adrenal gland containing a 13 g tumor. Postoperatively, the metabolic and clinical abnormalities reversed almost completely.
Prior studies, in which only patients with spontaneous hypokalemia were screened for PA, found prevalence rates of less than 0.5% of hypertensive patients. After Hiramatsu et al24 introduced the aldosterone-to-renin ratio (ARR) as a screening test, much higher prevalence estimates have emerged, ranging from 5%–13% of all patients with hypertension and approaching 20% in those with resistant hypertension. Most patients with PA lack specific physical findings, and edema is uncommon because of the mineralocorticoid escape phenomenon. When severe, the hypokalemia associated with PA can cause weakness, myalgias, muscle spasms, headaches, and palpitations. Rarely, decreased ionized calcium from hypokalemic metabolic alkalosis causes tetany, as in Conn’s index patient. The serum sodium concentration tends to be high normal or slightly high due to a reset osmostat, which is a useful clinical clue in PA. PA can be associated with secondary hyperparathyroidism in the absence of vitamin D deficiency and with nephrolithiasis.
The screening process for the diagnosis of PA, according to the Endocrine Society’s guidelines,25 involves two simultaneous blood tests: the plasma (or serum) aldosterone concentration (PAC) and the plasma renin activity (PRA), which are used to calculate the ARR:
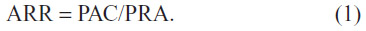
Screening may be performed while the patient is taking any antihypertensive drugs except for MRAs, and plasma samples are best collected in the morning after at least 2 hours of ambulation followed by 5 minutes of sitting. Although many medications can influence the ARR (Table 1), most drugs tend to raise PRA, and thus lower the ARR. Consequently, if the PRA is suppressed (<1 ng/mL/hour), a positive screening test is generally valid, despite the use of medications, as will be explained. Nevertheless, the ARR is a case detection test only, which should be repeated if the initial results are inconclusive or if the results are difficult to interpret because of suboptimal sampling conditions. Preferred medications to use during screening and confirmatory testing for PA, which minimally influence PAC and PRA, include verapamil, doxazosin, and terazosin, and moderate doses of hydralazine. A positive ARR is generally considered when values are >20–40, and the specificity is improved if the PAC is also >10–15 ng/dL, particularly when the PRA is <1. Due to conflicting results among different studies, the Endocrine Society guidelines do not provide firm cut-off values for ARR and PAC, and the clinician should use ARR screening to solely determine for which patients they will pursue confirmatory testing.
 | Table 1 The influence of medications and factors on PAC, PRA, and ARR |
Screening for PA is indicated in patients who demonstrate:
- Hypertension with spontaneous or easily-induced hypokalemia;
- Hypertension with adrenal adenoma;
- Resistant hypertension (uncontrolled on three drugs, including a diuretic);
- Suspicion of secondary hypertension;
- Onset of hypertension at age <30 years;
- Stage 3 hypertension (>160/100 mmHg); and/or
- Hypertension and a family history of PA.
Table 2 summarizes the four commonly used confirmatory tests (oral sodium loading, normal saline infusion, fludrocortisone suppression, and captopril challenge). Currently, there is insufficient direct evidence to recommend one test over the others, so the choice is generally determined by considering cost, patient compliance, laboratory capabilities, and local expertise. The captopril suppression test is best used for confirmatory testing in patients with high daily sodium intake.
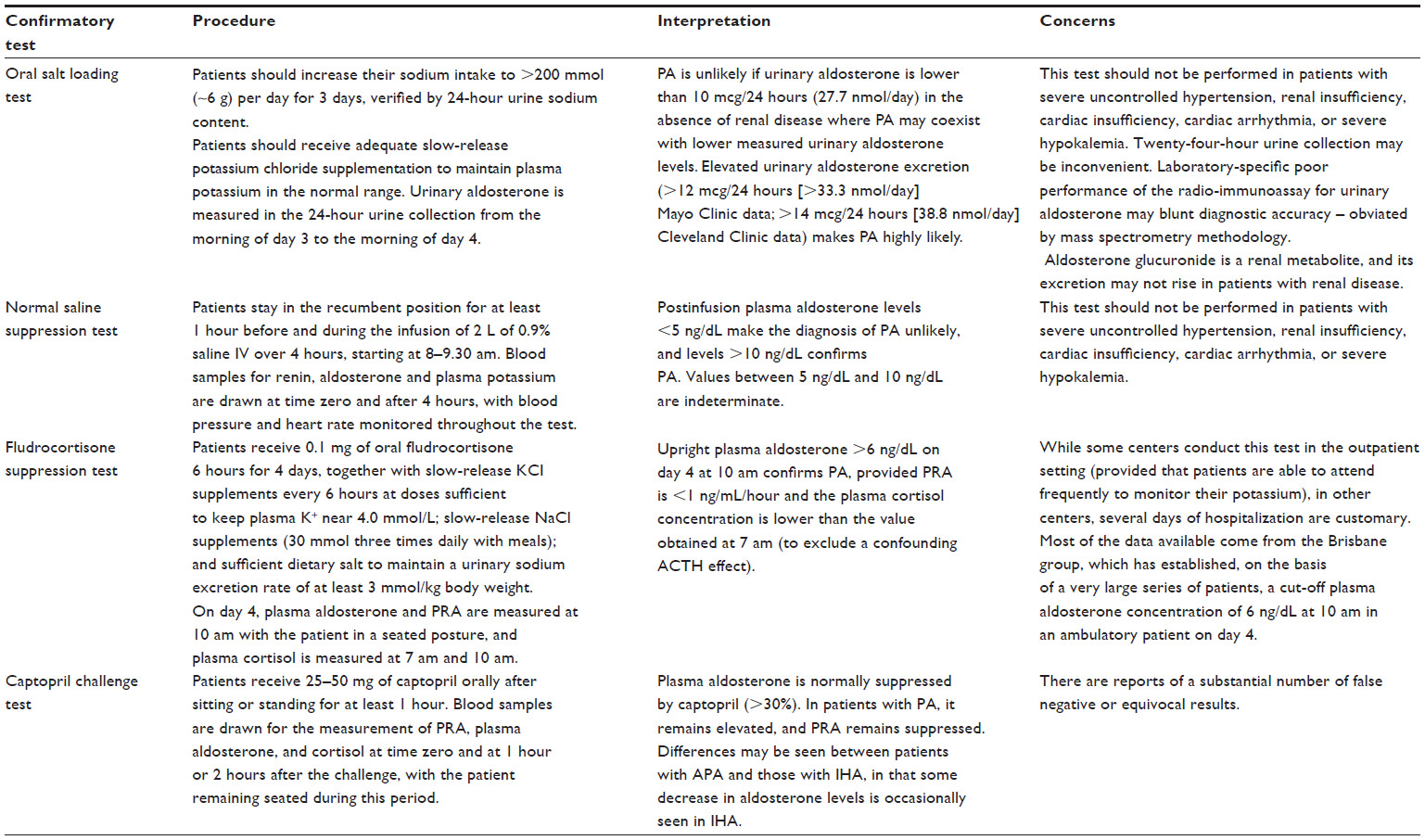 | Table 2 The four commonly used confirmatory tests for diagnosing PA |
Following confirmation of PA after dynamic testing, patients with PA should undergo a computed tomography of the adrenals to rule out large masses, which might represent adrenocortical carcinomas, and also to image the adrenal veins in preparation for the next step of the investigation. Localization of the source of the excess aldosterone production guides the management of patients who are candidates for surgical adrenalectomy. The gold standard for the differentiation of unilateral versus bilateral disease is adrenal venous sampling (AVS). Some AVS controversies remain, including the use of cosyntropin and the best diagnostic criteria.26,27 Certain antihypertensive medications, which raise PRA and thus induce bilateral aldosterone production, should be avoided to prevent spurious results. MRAs (spironolactone and eplerenone) in particular should be discontinued for up to 6 weeks before AVS, and documentation of suppressed PRA before sampling is advised for shorter durations of MRA withdrawal. Preferred medications include extended-release verapamil, α-adrenergic blockers, and hydralazine. Hypokalemia should be corrected before the procedure, and the patient should be fasting and supine for at least 1 hour before AVS.28
The selectivity index (SI) is used to determine if cannulation of the adrenal veins was adequate. The SI is the ratio of cortisol concentrations from the adrenal vein sample compared to the peripheral (usually inferior vena cava [IVC]) sample for both right- and left-sided samples. With cosyntropin stimulation, a ratio of 5:1 indicates successful cannulation of the adrenal vein, although ratios as low as 3:1 have been applied. Without cosyntropin stimulation, a ratio of 3:1 suggests successful cannulation, although lower cut-offs have been suggested.28 If bilateral cannulation is successful, the source(s) of aldosterone are deduced from calculations of the aldosterone/cortisol (A:C) ratios, which corrects the aldosterone concentration for inevitable dilution with peripheral blood (Table 3). The greater A:C ratio is divided by the lower A:C ratio to calculate the lateralization index (LI). Under cosyntropin stimulation, a LI of >4:1 demonstrates lateralization to the higher side; a LI <2:1 indicates bilateral disease; and LI values 2–4:1 are inconclusive.26,28 The example given in Table 3 yields SI values of 30–40, which indicates a successful study. The A:C ratios of 0.25 on the right, 10 on the left, and 1.1 from the IVC yields a LI of 10/0.25=40, confidently lateralizing aldosterone production to the left adrenal. If the LI is between 2 and 4, we look for “contralateral suppression,” meaning that the A:C ratio in the lower adrenal vein specimen is lower than in the IVC, which serves as evidence that the lower side is not producing significant aldosterone. In the example outlined in Table 3, the A:C ratio in the IVC is significantly higher than in the right adrenal, reinforcing the idea that the left adrenal is the source of aldosterone. Alternatively, measuring 18-hydroxycorticosterone29 or 18-oxo-cortisol, could be considered if assays are available.30 The true sensitivity and specificity of the test is difficult to determine, as calculating these parameters would require performing an adrenalectomy on all patients to verify the diagnosis.
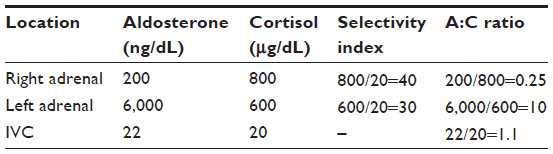 | Table 3 Example of AVS data and A:C ratios |
At centers with experienced AVS interventional radiologists, the complication rate is 2.5% or lower. The success rates for cannulating the right adrenal vein in centers without experienced radiologists in AVS is not higher than 70%–80%. With experience, the success rate increases to 90%–96%. The addition of rapid intraprocedural measurement of adrenal vein cortisol concentrations has facilitated improved success of AVS catheter placement.31,32
AVS differentiates patients with bilateral aldosterone production from those with unilateral disease who are thus candidates for a unilateral adrenalectomy. During the investigation for PA, however, all different subtypes of PA should be considered prior to selecting patients for AVS. There are eight different subtypes of PA, which are listed in order from the most common to the least common:
- Idiopathic nonfamilial bilateral adrenal hyperplasia;
- Sporadic unilateral aldosterone-producing adenomas (APA);
- Renin/Ang2-unresponsive APA;
- Renin/Ang2-responsive APA (renin-responsive APA);
- Unilateral (primary) adrenal hyperplasia, ACTH-responsive;33
- Familial hyperaldosteronism;
- Type 1 (FH-1);
- Type 2 (FH-2);
- Type 3 (FH-3);
- Aldosterone-producing adrenocortical carcinomas;
- Ectopic aldosterone production from malignant ovarian tumors;34,35 and
- ACTH-independent bilateral macronodular adrenal hyperplasia associated with Cushing’s syndrome and hyperaldosteronism.
The family history of the patient undergoing evaluation for PA and the associated radiographic findings should be carefully reviewed before pursuing lateralization studies. In patients with a high suspicion for familial forms of PA, especially FH-1 and FH-3, the diagnosis can be confirmed by genetic testing, and AVS should be avoided.
Recent scientific studies in hypertension genetics
Over the last 2 years, great advances have been made in the genetics of PA. In early 2011, Choi et al23 performed whole-exome capture and sequencing on four APA–blood pairs from unrelated subjects without loss of heterozygosity segments. In two out of the four tumors, somatic mutations were found in the KCNJ5 (Kir3.4) gene, which encodes an inward-rectifying potassium channel. Two mutations were found, the G151R and L168R mutations. Mutant allele frequencies in tumor deoxyribonucleic acid (DNA) and complementary DNA were consistent with heterozygous mutations in the tumor cells. The WT amino acids, G151 and L168, lie at highly conserved positions in and near the ion selectivity filter, and these mutations reduce channel selectivity. KCNJ5 exists both as homotetramers and heterotetramers with KCNJ3 (KCNJ3 is inactive as a homotetramer), and the heterotetramers are more active than homotetramers. Electrophysiological studies conducted in HEK-293T cell lines expressing WT or mutant KCNJ5 with KCNJ3 demonstrated loss of selectivity of the KCNJ5/KCNJ3 channel and increased sodium permeability, resulting in continuous membrane depolarization. More recent studies have demonstrated that expression of mutant KCNJ5 in the adrenal cells causes depolarization, increased intracellular calcium, aldosterone production, and CYP11B2 expression.36,37 (Figure 1). Chronic calcium stimulation promotes increased proliferation in glomerulosa and other cell types, which might promote clonal expansion of cells harboring these somatic mutations and adenoma formation.
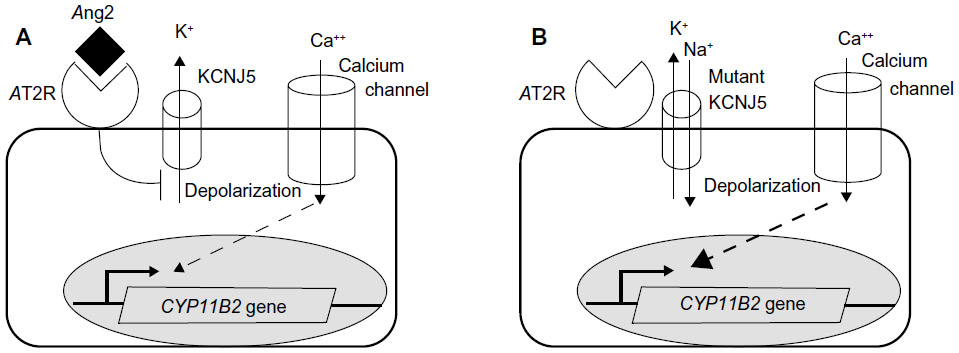 | Figure 1 A normal adrenal zona glomerulosa cell and a cell with a KCNJ5 mutation. |
These investigators then screened for germline KCNJ5 mutations in familial forms of PA, which revealed a third heterozygous T158A mutation. This mutation cosegregated with a form of FH-3: PA associated with bilateral adrenal hyperplasia. This threonine lies in the loop between the selectivity filter and the second transmembrane domain; its hydroxyl group forms hydrogen bonds with conserved residues in the loop between the first transmembrane domain and the pore helix, which constrains the structure. The T158A mutation eliminates these hydrogen bonds, and electrophysiological studies of the mutant channel again showed loss of channel selectivity associated with increased sodium permeability and continuous membrane depolarization.
Following this discovery, Scholl et al38 conducted genetic screening for germline KCNJ5 mutations in four families with familial, non-FH-1 PA. Direct sequencing of the KCNJ5 gene revealed that all affected members in the four relatives not only had heterozygous mutations in KCNJ5, but all had mutations that altered the same amino acid, G151, which is one of the two positions showing recurrent somatic mutation in aldosterone-producing adenomas. Two affected relatives had inherited identical mutations as one of the recurrent somatic mutations found in APAs, substituting arginine for glycine at position 151 (G151R). The other two kindred had a different mutation, resulting in the substitution of glutamate for glycine (G151E). A review of the clinical features of the affected members of certain families illustrates strikingly different presentations for individuals with either G151R or G151E mutations. Patients with the G151E mutation showed aldosteronism early in life, but did not have progressive disease. These patients had remarkable responsiveness to single-agent treatment with spironolactone, leading to normalization of blood pressure and potassium in all subjects, with effectiveness persisting well into adulthood. Moreover, none of these affected subjects showed adrenal enlargement on computed tomography at ages up to 37 years. In contrast, patients with the G151R mutation presented with clinical features similar to those noted among the family described initially with the germline T158A mutation in KCNJ5. These patients had severe aldosteronism, which was poorly responsive to spironolactone and that worsened with age. Among the patients with G151R mutations with follow-up, all required the radical intervention of bilateral adrenalectomy to achieve control of hypertension and hypokalemia at very young ages (range: 1–4 years).
To further determine the pathophysiology of the distinctive clinical presentation of patients with germline KCNJ5 mutations, enhanced green fluorescent protein (eGFP)-tagged KCNJ5-bearing WT sequence, the G151R mutation, or the G151E mutation were expressed in the mammalian HEK-293T cell line to determine cell survival rates over time. By measuring the percentage of eGFP-positive cells by flow cytometry at 12 hours, 24 hours, and 36 hours after the transfection, cells transfected with the KCNJ5 WT showed increasing percentages of eGFP-positive cells over this time period, whereas cells transfected with KCNJ5 G151R (associated with adrenocortical hyperplasia) exhibited a smaller increase in the percentage of eGFP-positive cells at 24 hours and a subsequent decrease by 36 hours. In contrast, cells transfected with KCNJ5 G151E (not associated with adrenocortical hyperplasia) produced a much more extreme outcome, with very few eGFP-positive cells detected at any time after transfection. The experiment was repeated by substituting choline for sodium in the culture medium. The sodium-free culture medium did not alter the percentage of cells expressing KCNJ5 WT at any time point. In contrast, the percentage of eGFP-positive cells after transfection with both mutant constructs showed a significant increase at 24 hours and 36 hours. Most strikingly, the fraction of cells expressing KCNJ5 G151E increased nearly sevenfold at 24 hours and more than eightfold at 36 hours.
These in vitro effects suggest that the absence of hyperplasia in subjects with G151E mutations is attributable to increased cell death in cells expressing this mutation in vivo, limiting the glomerulosa cell mass, and likely accounting for the absence of progressive disease. These cells are, nonetheless, collectively producing large amounts of aldosterone, sufficient enough to cause hypertension. It is interesting that patients with these two different G151 mutations present at similar ages but diverge in severity thereafter, likely due to the increased mass of aldosterone-producing cells among those bearing the KCNJ5 G151R mutation compared to those with the G151E mutation.
Subsequently, new somatic mutations were identified in roughly 7% of aldosterone-producing adenomas.39 Somatic mutations in the P-type adenosine triphosphatase (ATPase) gene family, ATP1A1 (encoding a sodium/potassium ATPase α-subunit) and ATP2B3 (encoding a sarcoplasmic reticulum calcium ATPase) were identified by exome sequencing in these tumors. Subsequently, screening for mutations in 308 aldosterone-producing adenomas found ATP1A1 or ATP2B3 mutations in 21 (7%) and KCNJ5 mutations in 118 (38%) of cases. Concomitant KCNJ5 and ATP1A1 or ATP2B3 mutations within the same tumor were not observed. KCNJ5 mutations have been found in 30%–40% of aldosterone-producing adenomas, with higher prevalence noted in women, and the highest prevalence found in Japan.36 The sodium/potassium ATPase, of which ATP1A1 encodes the α-subunit, exchanges three cytoplasmic sodium ions for two extracellular potassium ions using the energy from ATP hydrolysis. The sodium and potassium gradients and their conductances generate the membrane potential, with the dominant potassium conductance maintaining a negative resting potential. The specific antagonist, ouabain, inhibits the sodium/potassium ATPases and causes dose-dependent stimulation of aldosterone release from rat glomerulosa cells and rat glomerulosa cell growth in vivo.40 Ang2 lowers sodium/potassium ATPase activity, indicating the potential contribution of this enzyme to angiotensin-dependent aldosterone release.41 ATP2B3 belongs to the ATPase gene family that encodes plasma membrane calcium ATPase, which is essential for pumping calcium ions from the cytoplasm into intracellular stores. The three-dimensional structure of the ATP2B3 is not known; however, from the structure of the homologous rabbit sarcoplasmic reticulum type calcium ATPase, it became apparent that the deletions in ATP2B3 associated with aldosterone-producing adenomas alter the M4 transmembrane helix, causing a major distortion in the binding site for calcium ions. Electrophysiological examination of primary cultured adenoma cells with different underlying mutations showed substantially higher depolarization in ATPase (ATP1A1 or ATP2B3)-mutant cells compared to cells from normal adjacent tissue. ATP1A1 or ATP2B3 germline mutations were not found in familial cases of PA, while KCNJ5 germline mutations have been found in cases of familial hyperaldosteronism type 3 alone.
Mutations in ATP1A1, ATP2B3, and KCNJ5 are found in <50% of aldosterone-producing adenomas, suggesting that mutations in other ion transport proteins are responsible for additional cases. Consistent with this prediction, two groups have now reported somatic mutations in CACNA1D, encoding an L-type calcium channel, in APAs.42,43 These findings suggest that the pathogenesis of many APAs derives from somatic gene mutations, which alter intracellular calcium homeostasis (Figure 2).
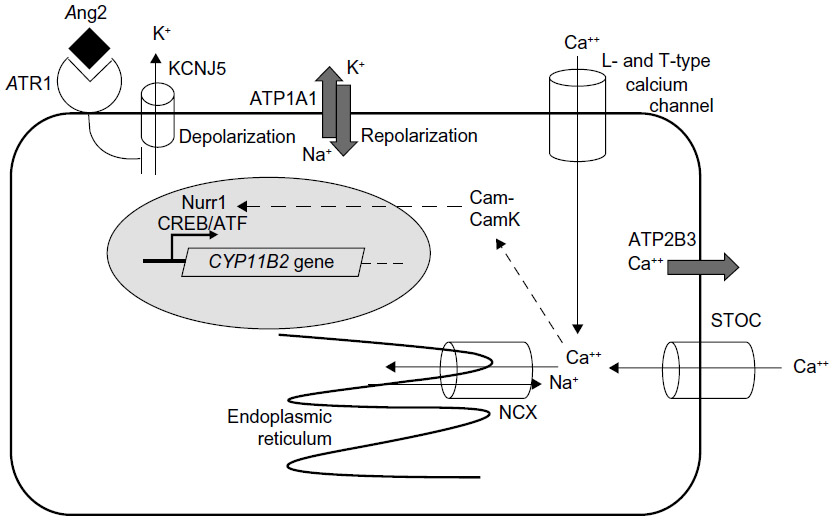 | Figure 2 Ion fluxes in zona glomerulosa cells controlling aldosterone production and points where intracellular calcium might be regulated. |
Effective management of hypertension through lifestyle modification and therapy – potential role of screening for genetic susceptibility
The management of patients with PA with bilateral disease, or of patients with aldosterone-producing adenomas, who prefer medical over surgical therapy include the use of MRA drugs, spironolactone and eplerenone, as well as dietary changes. Spironolactone antagonizes MR in aldosterone-sensitive distal tubular sites of the nephron, which reduces ENaC action and thus increases sodium excretion and potassium retention. Because spironolactone also antagonizes the androgen and progesterone receptors, its use can cause gynecomastia and sexual dysfunction in men, as well as breast tenderness and vaginal spotting or irregular menses in premenopausal women. Spironolactone has a complex pattern of metabolism and a long half-life (greater than 12 hours in healthy individuals, 24 hours in heart failure patients, and up to 58 hours in patients with cirrhotic ascites). In the liver, spironolactone is converted to two active metabolites, 7α-thiomethylspironolactone and canrenone, which are responsible for its prolonged pharmacological effect.
Eplerenone is a selective MRA, which lacks many of the side effects of spironolactone; however, eplerenone appears to be less potent than spironolactone in reducing blood pressure, at least when used at recommended doses of 50 mg once or twice daily. Eplerenone does not have active metabolites, and because its half-life at steady state is 3–4 hours, eplerenone is most effective when administered twice a day. Eplerenone is metabolized in the liver largely by CYP3A4, explaining why compounds that affect CYP3A4 function can change drug exposure.
Amiloride is also used in the management of patients with PA. Amiloride directly blocks ENaC, thereby inhibiting sodium reabsorption in the late distal convoluted tubules, connecting tubules, and collecting ducts in the kidneys. Although amiloride is useful for controlling the hypokalemia of PA, particularly during diagnostic evaluation, it is inferior to MRA in terms of blood pressure control. Furthermore, amiloride does not block MR in the extra-renal organs, which is likely to allow for greater end-organ damage than MRA therapy.
Normalization of blood pressure is not the only goal of treatment for PA; rather, as for all patients with hypertension, the ultimate treatment goal is to prevent end-organ complications. The dose of MRA should be titrated not only to normalize blood pressure and potassium, but also to maintain nonsuppressed plasma renin activity. Other biochemical markers that indicate adequate MR blockade include reversal of secondary hyperparathyroidism, metabolic alkalosis, and hypocalcaemia. Improvement of albuminuria is another marker of successful MR blockade; however, surgical or medical treatment of PA can cause a slight rise in creatinine and worsening of the estimated glomerular filtration rate, due to the accompanying reduction in hyperfiltration. In addition to the effect of aldosterone on the kidneys, aldosterone also has central effects that increase blood pressure. Patients with PA have increased sympathetic nerve activity, which normalizes after adrenalectomy.44 A similar lowering of sympathetic nerve activity occurs in essential hypertensives treated with spironolactone.45,46
Salt intake significantly contributes to the blood pressure in PA, as well as in essential hypertension. Aldosterone excess in combination with high salt intake promotes target-organ damage through proinflammatory and profibrotic effects, independent of blood pressure elevation.47,48 Aldosterone excess induces diffuse myocardial fibrosis, left ventricular dilatation, and hypertrophy,49 and most experimental studies have indicated that high dietary salt is necessary for aldosterone to exert its effects on target organs.50 These studies have demonstrated a positive correlation between proteinuria and dietary salt in hypertensive patients with aldosterone excess, but not in patients with normal aldosterone, despite similar blood pressure. A more recent study demonstrated a positive correlation between increased levels of urinary sodium with left ventricular thickness and mass in patients with PA, but not in patients with essential hypertension.51 All these data suggest that the aldosterone excess in patients with PA has the most pronounced target-organ damage when dietary salt intake is high. Conversely, many of the pathological effects of hyperaldosteronism are not readily apparent in low salt societies. Consequently, moderation of sodium intake is recommended when PA is managed medically, even with MRA drugs.
Conclusion
Endocrine hypertension accounts for almost 20% of all cases of hypertension. Screening for secondary causes of hypertension should be considered in all patients with hypertension, especially before the institution of an antihypertensive agent. The presence of suppressed plasma renin activity identifies patients for endocrine hypertension screening, as well for the choice of appropriate antihypertension agents. For patients with low-renin hypertension, evaluation by an endocrine hypertension specialist should be considered, irrespective of the severity of hypertension, particularly among those patients with resistant hypertension. The significantly higher end-organ damage associated with PA compared to those with essential hypertension demonstrates the opportunity to limit the burden of hypertension-associated morbidity, which might be realized by taking a more thoughtful approach to all hypertensive patients. The perceived high cost associated with subtype classification, the lack of skilled interventional radiologists to perform AVS, and the decreased awareness of the high prevalence of PA among physicians who manage patients with hypertension are the main reasons for the disappointingly low rates of screening and appropriate therapy. These concerns must be weighed against the substantial benefits associated with case detection and targeted treatment of patients who have been properly diagnosed and classified.
The discovery of new genetic mutations associated with PA has provided fundamental insights into the field of endocrine hypertension. The identification of germline KCNJ5 mutations in Mendelian forms of endocrine hypertension has advanced our understanding of the pathogenesis of aldosterone-producing adenomas and genetic forms of adrenal hyperplasia, which has important implications for diagnosis and treatment. With the broader availability of whole-exome sequencing, the genetic basis of more subtypes of PA will likely be identified, and biomarkers for each subtype might become available. We predict that, in the near future, subtype classification of PA will be based on broad genetic and biomarker testing, which will reduce the reliance on AVS for the initiation of targeted therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Roger VL, Go AS, Lloyd-Jones DM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics – 2012 update: a report from the American Heart Association. Circulation. 2012;125(1):e2–e220. | |
Jackson RV, Lafferty A, Torpy DJ, Stratakis C. New genetic insights in familial hyperaldosteronism. Ann N Y Acad Sci. 2002;970:77–88. | |
Fallo F, Pilon C, Williams TA, et al. Coexistence of different phenotypes in a family with glucocorticoid-remediable aldosteronism. J Hum Hypertens. 2004;18(1):47–51. | |
Lifton RP, Dluhy RG, Powers M, et al. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature. 1992;355(6357):262–265. | |
New MI, Levine LS, Biglieri EG, Pareira J, Ulick S. Evidence for an unidentified steroid in a child with apparent mineralocorticoid hypertension. J Clin Endocrinol Metab. 1977;44(5):924–933. | |
Quinkler M, Bappal B, Draper N, et al. Molecular basis for the apparent mineralocorticoid excess syndrome in the Oman population. Mol Cell Endocrinol. 2004;217(1–2):143–149. | |
Odermatt A, Dick B, Arnold P, et al. A mutation in the cofactor-binding domain of 11beta-hydroxysteroid dehydrogenase type 2 associated with mineralocorticoid hypertension. J Clin Endocrinol Metab. 2001;86(3):1247–1252. | |
Hammer F, Stewart PM. Cortisol metabolism in hypertension. Best Pract Res Clin Endocrinol Metab. 2006;20(3):337–353. | |
Quinkler M, Stewart PM. Hypertension and the cortisol-cortisone shuttle. J Clin Endocrinol Metab. 2003;88(6):2384–2392. | |
Zachmann M, Tassinari D, Prader A. Clinical and biochemical variability of congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency. A study of 25 patients. J Clin Endocrinol Metab. 1983;56(2):222–229. | |
White PC, Curnow KM, Pascoe L. Disorders of steroid 11 beta-hydroxylase isozymes. Endocr Rev. 1994;15(4):421–438. | |
Curnow KM, Slutsker L, Vitek J, et al. Mutations in the CYP11B1 gene causing congenital adrenal hyperplasia and hypertension cluster in exons 6, 7, and 8. Proc Natl Acad Sci U S A. 1993;90(10):4552–4556. | |
White PC, Dupont J, New MI, Leiberman E, Hochberg Z, Rösler A. A mutation in CYP11B1 (Arg-448----His) associated with steroid 11 beta-hydroxylase deficiency in Jews of Moroccan origin. J Clin Invest. 1991;87(5):1664–1667. | |
Auchus RJ. Genetic deficiencies of cytochrome P450c17 (CYP17A1): combined 17-hydroxylase/17,20-lyase deficiency and isolated 17,20-lyase deficiency. In: New MI, Parsa A, Hammer GD, editors. Genetic Steroid Disorders. Waltham, MA: Elsevier; 2014:111–123. | |
Costa-Santos M, Kater CE, Auchus RJ; Brazilian Congenital Adrenal Hyperplasia Multicenter Study Group. Two prevalent CYP17 mutations and genotype-phenotype correlations in 24 Brazilian patients with 17-hydroxylase deficiency. J Clin Endocrinol Metab. 2004;89(1):49–60. | |
Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104(4):545–556. | |
Charmandari E, Ichijo T, Jubiz W, et al. A novel point mutation in the amino terminal domain of the human glucocorticoid receptor (hGR) gene enhancing hGR-mediated gene expression. J Clin Endocrinol Metab. 2008;93(12):4963–4498. | |
Liddle GW, Bledsoe T, Coppage WS. A familial renal disorder simulating primary aldosteronism but with negligible aldosterone secretion. Trans Assoc Am Physicians. 1963;76:199–213. | |
Shimkets RA, Warnock DG, Bositis CM, et al. Liddle’s syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79(3):407–414. | |
Abriel H, Loffing J, Rebhun JF, et al. Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle’s syndrome. J Clin Invest. 1999;103(5):667–673. | |
Wilson FH, Disse-Nicodème S, Choate KA, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001; 293(5532):1107–1112. | |
Boyden LM, Choi M, Choate KA, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482(7383):98–102. | |
Choi M, Scholl UI, Yue P, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. 2011;331(6018):768–772. | |
Hiramatsu K, Yamada T, Yukimura Y, et al. A screening test to identify aldosterone-producing adenoma by measuring plasma renin activity. Results in hypertensive patients. Arch Intern Med. 1981;141(12):1589–1593. | |
Funder JW, Carey RM, Fardella C, et al; Endocrine Society. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2008;93(9):3266–3281. | |
Auchus RJ, Wians FH, Anderson ME, et al. What we still do not know about adrenal vein sampling for primary aldosteronism. Horm Metab Res. 2010;42(6):411–445. | |
Rossi GP, Barisa M, Allolio B, et al. The Adrenal Vein Sampling International Study (AVIS) for identifying the major subtypes of primary aldosteronism. J Clin Endocrinol Metab. 2012;97(5):1606–1614. | |
Young WF, Stanson AW. What are the keys to successful adrenal venous sampling (AVS) in patients with primary aldosteronism? Clin Endocrinol (Oxf). 2009;70(1):14–17. | |
Auchus RJ, Chandler DW, Singeetham S, et al. Measurement of 18-hydroxycorticosterone during adrenal vein sampling for primary aldosteronism. J Clin Endocrinol Metab. 2007;92(7):2648–2651. | |
Nakamura Y, Satoh F, Morimoto R, et al. 18-oxocortisol measurement in adrenal vein sampling as a biomarker for subclassifying primary aldosteronism. J Clin Endocrinol Metab. 2011;96(8):E1272–E1278. | |
Auchus RJ, Michaelis C, Wians FH Jr, et al. Rapid cortisol assays improve the success rate of adrenal vein sampling for primary aldosteronism. Ann Surg. 2009;249(2):318–321. | |
Betz MJ, Degenhart C, Fischer E, et al. Adrenal vein sampling using rapid cortisol assays in primary aldosteronism is useful in centers with low success rates. Eur J Endocrinol. 2011;165(2):301–306. | |
Irony I, Kater CE, Biglieri EG, Shackleton CH. Correctable subsets of primary aldosteronism. Primary adrenal hyperplasia and renin responsive adenoma. Am J Hypertens. 1990;3(7):576–582. | |
Jackson B, Valentine R, Wagner G. Primary aldosteronism due to a malignant ovarian tumour. Aust N Z J Med. 1986;16(1):69–71. | |
Todesco S, Terribile V, Borsatti A, Mantero F. Primary aldosteronism due to a malignant ovarian tumor. J Clin Endocrinol Metab. 1975;41(5):809–819. | |
Monticone S, Hattangady NG, Nishimoto K, et al. Effect of KCNJ5 mutations on gene expression in aldosterone-producing adenomas and adrenocortical cells. J Clin Endocrinol Metab. 2012;97(8):E1567–E1572. | |
Oki K, Plonczynski MW, Luis Lam M, Gomez-Sanchez EP, Gomez-Sanchez CE. Potassium channel mutant KCNJ5 T158A expression in HAC-15 cells increases aldosterone synthesis. Endocrinology. 2012;153(4):1774–1782. | |
Scholl UI, Nelson-Williams C, Yue P, et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Natl Acad Sci U S A. 2012;109(7):2533–2538. | |
Beuschlein F, Boulkroun S, Osswald A, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet. 2013;45(4):440–444, 444e1. | |
Neri G, De Toni R, Tortorella C, et al. Ouabain chronic infusion enhances the growth and steroidogenic capacity of rat adrenal zona glomerulosa: the possible involvement of the endothelin system. Int J Mol Med. 2006;18(2):315–319. | |
Hajnóczky G, Csordás G, Hunyady L, et al. Angiotensin-II inhibits Na+/K+ pump in rat adrenal glomerulosa cells: possible contribution to stimulation of aldosterone production. Endocrinology. 1992;130(3):1637–1644. | |
Azizan EA, Poulsen H, Tuluc P, et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet. 2013;45(9):1055–1060. | |
Scholl UI, Goh G, Stölting G, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet. 2013;45(9):1050–1054. | |
Kontak AC, Wang Z, Arbique D, et al. Reversible sympathetic overactivity in hypertensive patients with primary aldosteronism. J Clin Endocrinol Metab. 2010;95(10):4756–4761. | |
Menon DV, Arbique D, Wang Z, Adams-Huet B, Auchus RJ, Vongpatanasin W. Differential effects of chlorthalidone versus spironolactone on muscle sympathetic nerve activity in hypertensive patients. J Clin Endocrinol Metab. 2009;94(4):1361–1366. | |
Raheja P, Price A, Wang Z, et al. Spironolactone prevents chlorthalidone-induced sympathetic activation and insulin resistance in hypertensive patients. Hypertension. 2012;60(2):319–325. | |
Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation. 1991;83(6):1849–1865. | |
Young M, Fullerton M, Dilley R, Funder J. Mineralocorticoids, hypertension, and cardiac fibrosis. J Clin Invest. 1994 Jun;93(6):2578–2583. | |
Rocha R, Rudolph AE, Frierdich GE, et al. Aldosterone induces a vascular inflammatory phenotype in the rat heart. Am J Physiol Heart Circ Physiol. 2002;283(5):H1802–H1810. | |
Sato A, Saruta T. Aldosterone-induced organ damage: plasma aldosterone level and inappropriate salt status. Hypertens Res. 2004;27(5):303–310. | |
Pimenta E, Gordon RD, Ahmed AH, et al. Cardiac dimensions are largely determined by dietary salt in patients with primary aldosteronism: results of a case-control study. J Clin Endocrinol Metab. 2011;96(9):2813–2820. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.