Back to Journals » Clinical and Experimental Gastroenterology » Volume 19
Gastric Proton Pumpopathy Associated with Protein Sorting Machinery: A Narrative Review
Authors Niyomugabo AP ![]() , Ntakirutimana L
, Ntakirutimana L ![]() , Habineza JC, Alagbonsi AI
, Habineza JC, Alagbonsi AI ![]()
Received 9 October 2025
Accepted for publication 30 January 2026
Published 8 February 2026 Volume 2026:19 573016
DOI https://doi.org/10.2147/CEG.S573016
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Vipul Yagnik
Aime Patrick Niyomugabo, Leopold Ntakirutimana, Jean Claude Habineza, Abdullateef Isiaka Alagbonsi
Department of Physiology, School of Medicine and Pharmacy, College of Medicine and Health Sciences, University of Rwanda, Huye, Rwanda
Correspondence: Abdullateef Isiaka Alagbonsi, Department of Physiology, School of Medicine and Pharmacy, College of Medicine and Health Sciences, University of Rwanda, Huye, Rwanda, Email [email protected]
Background: Gastric acid secretion by parietal cells depends on the trafficking and exocytosis of H⁺/K⁺-ATPase-rich tubulovesicles (TVs) to the apical membrane in response to histamine stimulation via cyclic AMP elevation. However, the role of parietal cell’s protein sorting machinery [eg, coat protein (COP) II/COP I vesicle proteins, soluble NSF attachment protein receptors (SNAREs), Rab GTPases, clathrin-adaptor proteins, etc.] in the dysregulation of gastric acid homeostasis remains poorly understood. This review synthesizes available evidence on the role of protein sorting machinery in gastric proton pumpopathy, leading to dysregulation of gastric acid secretion.
Methods: A thorough search was conducted on PubMed, with Wiley and African Journal Online as supplementary sources, and 15 original research articles published between January 1988 and July 2025 in English were included.
Results: Misfolding of α- and β-subunits within the endoplasmic reticulum (ER) results in retention and degradation of the pump, leading to impaired acid secretion. Defective coat protein (COP) II/I vesicular transport disrupts Golgi processing and slows the delivery of proton pumps to tubulovesicles, while impaired Rab GTPase activity interferes with vesicle targeting toward the apical canalicular membrane. Also, dysregulation of SNARE complexes and adaptor proteins prevents effective vesicle docking and fusion, further compromising membrane insertion. In addition, abnormalities in endosomal recycling pathways hinder the retrieval and reinsertion of proton pumps, destabilizing long-term acid secretion.
Conclusion: Defects in sorting machinery lead to gastric proton pumpopathy, causing impaired acid secretion. Thus, protein sorting regulators represent promising therapeutic targets to restore proper pump localization, which is a paradigm shift toward disease-modifying therapies that act upstream of the pump, potentially offering solutions for proton pump inhibitor (PPI)-resistant cases and congenital gastric acid disorders.
Keywords: gastric acid, gastric proton pump, parietal cell, protein sorting machinery, pumpopathy
Introduction
The regulation of gastric acid secretion is crucial for gastrointestinal homeostasis, as it contributes to food digestion, nutrient absorption, microbial defense, and maintaining optimal pH levels within the stomach.1 At the core of this process lies the gastric proton pump, a P-type ATPase also known as the H⁺/K⁺-ATPase, predominantly located in the apical membrane of gastric parietal cells.2 This enzyme is a heterodimer composed of an α-subunit, which carries the catalytic activity and ion transport function, and a β-subunit, which plays a critical role in stabilizing and trafficking the α-subunit.3 Both subunits are synthesized in the rough endoplasmic reticulum (ER) and require proper folding and assembly before being transported to the apical membrane of gastric parietal cells.3 This specialized ion transporter functions by exchanging intracellular H⁺ ions for extracellular K⁺ ions, utilizing energy from ATP hydrolysis to establish the highly acidic environment characteristic of the gastric lumen.4 Overstimulation of this pump can cause several gastrointestinal diseases, including peptic ulcer disease (PUD).
Globally, the incidence and prevalence of PUD have consistently increased from 1990 to 2021 by 11.05% and 8.77%, respectively, though deaths and disability-adjusted life years (DALYs) related to PUD have reduced by 15.94% and 27.8%, respectively, within this period. Furthermore, the global age-standardized rates (ASRs) have also reduced within this period for PUD-associated incidence (40.3%), prevalence (41.1%), death (61.5%), and DALYs (63.1%), with men exhibiting a higher number and ASRs of these PUD-associated metrics than women across most age cohorts.5
Traditionally, research has focused on the biochemical and electrophysiological properties of this proton pump, with an emphasis on its catalytic mechanism and pharmacological inhibition, particularly through proton pump inhibitors (PPIs).6 However, recent discoveries in molecular cell biology have shifted attention toward the subcellular trafficking and localization mechanisms that govern the correct functioning of this pump, implicating protein sorting machinery as key players in the maintenance of gastric physiology.7 The proper biosynthesis, folding, post-translational modification, and apical targeting of the H⁺/K⁺-ATPase depend on the integrity of protein sorting pathways, which involve a complex network of chaperones, coat proteins, small GTPases (eg, Rabs), SNAREs, and adaptor complexes.8 This set of sorting machinery ensures that the α-subunit (ATP4A) and β-subunit (ATP4B) of the proton pump are correctly assembled in the endoplasmic reticulum, processed in the Golgi apparatus and trafficked via vesicular pathways to the canalicular membrane of parietal cells.9 Any defect in these trafficking processes, whether genetic, acquired, or environmentally induced, can result in misfolding, mistargeting, or retention of the proton pump, thereby compromising its function.10 These molecular abnormalities are referred to as channelopathies, a term traditionally used for ion channel dysfunction but increasingly extended to include transporters and pumps whose behavior is influenced by intracellular trafficking machinery, though recent studies have suggested that pumpopathy is more appropriate for disorders of a pump like the proton pump.11,12
Disruption of the gastric proton pump’s normal trafficking can manifest clinically in several ways. Loss-of-function pumpopathy, due to improper delivery of the H⁺/K⁺-ATPase to the apical membrane, may contribute to hypochlorhydria or achlorhydria, which are associated with iron and vitamin B12 deficiency, gastrointestinal infections, autoimmune gastritis, and pernicious anemia.2 Conversely, gain-of-function mutations or trafficking dysregulation resulting in constitutive activation or overexpression of the proton pump may exacerbate acid-related diseases such as gastroesophageal reflux disease (GERD), Barrett’s esophagus, or peptic ulcer disease.13 Notably, mutations in genes encoding trafficking regulators such as Rab11, clathrin adaptors, and SNARE complexes have been shown to impair acid pump trafficking in experimental models, directly linking protein sorting machinery dysfunction to pathogenesis.8
Despite the widespread clinical use of PPIs and histamine (H2) receptor antagonists to manage hyperacidity, these drugs target the luminal activity of the proton pump and do not address underlying intracellular defects.14 Moreover, long-term acid suppression has been linked with adverse events, including those related to acid inhibition (eg, ischemic cardiac diseases, cerebral ischemic disease, dementia, acute intestinal nephritis, chronic kidney diseases) and those not related to acid inhibition (pneumonia, gastric carcinoid tumor, gastrointestinal infection, changes in gastric microbiome, iron and vitamin B12 deficiency, bone fracture, etc).15–18 These disadvantages of the current PPI treatment underscore the need for novel approaches that target upstream mechanisms in acid regulation. The investigation of protein sorting machinery as a regulatory checkpoint in acid secretion presents an innovative paradigm for therapeutic intervention, one that offers the possibility of correcting proton pump mislocalization, restoring membrane integrity, and ultimately rebalancing acid secretion at the cellular level.19
Currently, there is no review comprehensively evaluating the role of the protein sorting machinery in gastric proton pumpopathy. Therefore, this review aims to comprehensively explore the pathophysiological link between protein sorting machinery dysfunction and gastric proton pumpopathy, shedding light on how intracellular trafficking defects can give rise to or exacerbate gastric acid disorders. This understanding may not only improve diagnostic precision but also facilitate the development of targeted therapies that go beyond symptomatic relief and address the cellular origins of gastric proton pumpopathy.
Methodology
Study Design and Search Strategy
This study employed a narrative synthesis. An initial literature review was performed using databases such as PubMed, African Journal Online, and Wiley Online Library, focusing on studies published from January 1988 to July 2025. The search targeted publications on gastric H⁺/K⁺-ATPase, commonly referred to as the gastric proton pump, with particular attention to pumpopathy and disorders associated with its dysfunction. Key search terms included “gastric proton pump”, “H⁺/K⁺-ATPase”, “channelopathies”, “pumpopathy”, and “proton pump disorders”. To explore the molecular mechanisms underlying these conditions, the review incorporated terms such as “protein trafficking”, “vesicular transport”, “protein sorting machinery”, “apical membrane targeting”, and “parietal cells”. Because the correct localization and function of the gastric proton pump depend on precise intracellular transport pathways, the review also included related cellular processes and structures such as “endosomes”, “SNARE proteins”, “Rab GTPases”, and “cytoskeletal elements” (Supplementary File S1). Both human and experimental model studies (including “mice”, “rats”, “cell lines”, and “in vitro” systems) were included. Articles were selected based on inclusion and exclusion criteria focused on the functional relevance of protein sorting defects in gastric proton pump-related pathologies.
Eligibility Criteria
The inclusion and exclusion criteria for the selection of the articles are summarized in Table 1.
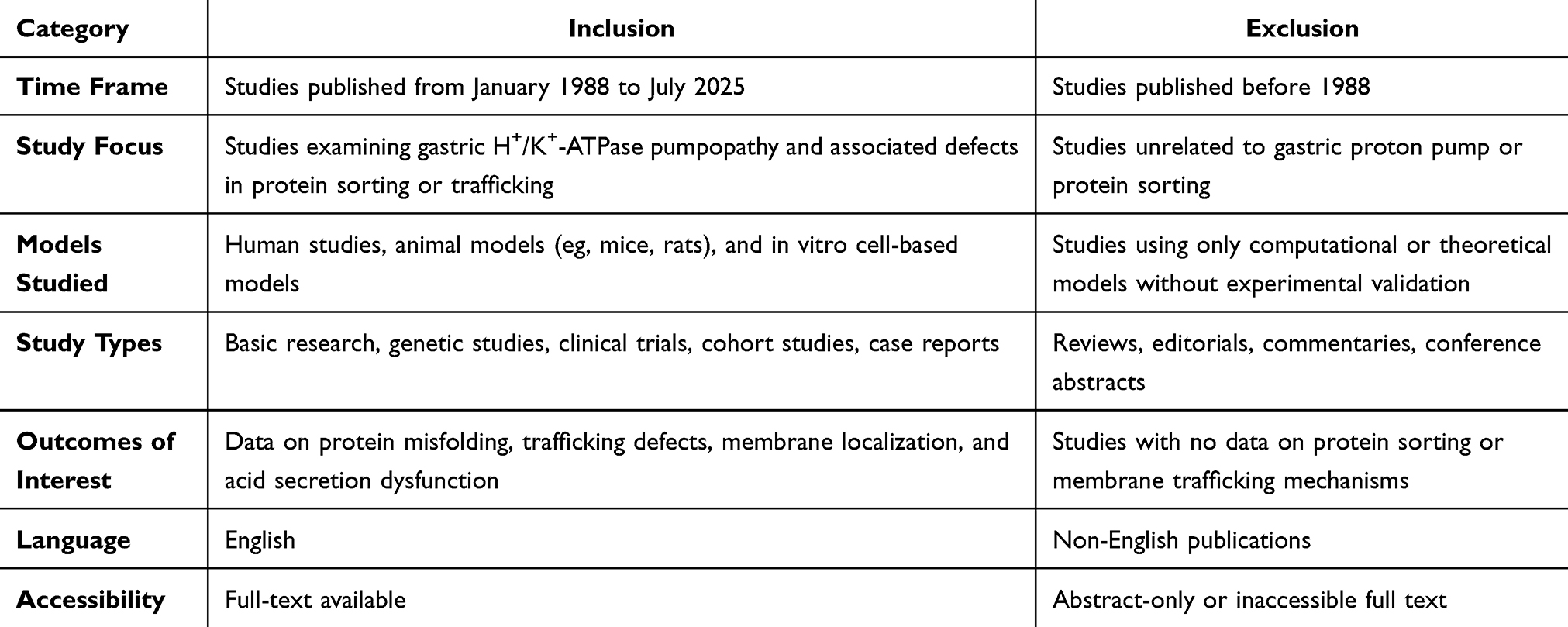 |
Table 1 Inclusion and Exclusion Criteria for Articles |
Data Extraction and Analysis
From each included study, the following data were extracted from the full text: (i) first author and year of publication, (ii) type of study (human and animal); (iii) model details (eg, transgenic mice, parietal cells, gastric mucosa biopsies, patient-derived samples, etc.); (iv) type of intervention (eg, genetic knockout or mutation of sorting machinery proteins, pharmacological inhibition, overexpression, CRISPR/Cas9 gene editing, etc.); (v) target proteins or pathways (eg, H⁺/K⁺-ATPase, coat protein (COP) II/COP I vesicle proteins, SNAREs, Rab GTPases, clathrin-adaptor proteins, etc.); (vi) Key outcome measures (eg, acid secretion levels, intracellular trafficking efficiency, proton pump localization, endoplasmic reticulum stress markers, protein expression or degradation, etc.); and (vii) mechanisms of dysfunction (eg, defective vesicle budding, misfolding and ER retention, impaired membrane targeting, autophagy, or proteasomal degradation pathways). All data were extracted manually by all reviewers using a pre-designed and piloted standardized data collection form20–28 (Supplementary File S2). Discrepancies in interpretation or categorization were resolved by mutual consensus after discussion among the review team.
Mechanistic Categorization and Thematic Analysis
The synthesis of findings has been structured as follows. First, the mechanistic categorization would group results based on the cellular location of the defect, distinguishing between sorting machinery defects (issues with protein synthesis and trafficking within the cell) and true pumpopathy (problems with the pump’s function at the membrane). This is further categorized by clinical context, contrasting normal acid secretion with pathological states like congenital hypochlorhydria and autoimmune gastritis. The review would then map mechanisms to specific cellular pathways, such as ER/Golgi trafficking and vesicular transport involving Rab GTPases, which guide the proton pump to the parietal cell’s apical membrane. For the thematic analysis, findings were synthesized into functional categories, including proton pump synthesis and folding, vesicle trafficking and docking, and final apical membrane insertion and function. The review highlighted contradictory findings, such as how mutations in the same sorting protein might lead to different clinical outcomes. Finally, it addressed significant knowledge gaps, emphasizing the need for more research into receptor-specific functions of different sorting proteins and the circadian influence on gastric acid secretion.
Results
Initially, we identified 165 articles (94 from Wiley Online Library, 52 from the African Journal Online, and 19 from PubMed). Among the 165 articles, 35 were duplicates from all search databases, and the duplicates were removed before screening, leaving 130 for titles and abstracts screening. Thereafter, some 60 articles that were not relevant to the study were removed, leaving 70 articles that underwent full-text screening for inclusion eligibility. Following the full-text screening, 55 articles were excluded, while only 15 articles that fully met our inclusion criteria were selected for the review process (Figure 1).
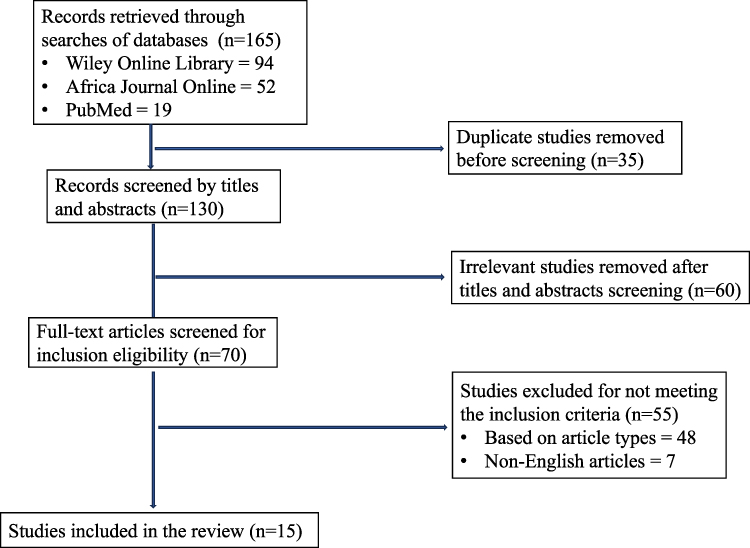 |
Figure 1 PRISMA flow chart of the article selection process. |
From the 15 studies reviewed, various targets for the effects of gastric proton pumpopathy associated with protein sorting machinery were noted, including misfolding of H⁺/K⁺-ATPase Subunits (n = 2 study), Defective COPII/COPI vesicular transport (n = 2 studies), impaired Rab GTPase-mediated targeting (n = 2 studies), SNARE complex dysregulation (n = 3 studies), faulty adaptor proteins and sorting signals (n = 3 studies), and the disruption in endosomal recycling pathways (n = 3 studies).
Misfolding of H⁺/K⁺-ATPase Subunits
Protein folding within the ER is tightly regulated by a network of molecular chaperones, such as BiP (GRP78), calnexin, and calreticulin, which ensure that newly synthesized proteins acquire their correct three-dimensional structure.29 If the α- or β-subunits of the H⁺/K⁺-ATPase are misfolded due to genetic mutations, environmental stress, or disruptions in ER homeostasis, they are recognized by the ER quality control system and are retained within the ER.30 Genetic mutations in the ATP4A (α-subunit) or ATP4B (β-subunit) genes can result in amino acid substitutions or truncations that prevent proper folding or assembly,31 thereby rendering the proton pump non-functional.32 Environmental or cellular stressors, such as oxidative stress, glucose deprivation, calcium imbalance, or viral infections, can impair the function of ER chaperones, thereby increasing the likelihood of protein misfolding.33 Additionally, overproduction of either subunit may overwhelm the ER’s capacity to manage protein folding, contributing to misfolding and aggregation.33
Once identified as misfolded or unassembled, the defective H⁺/K⁺-ATPase subunits are directed toward ER-associated degradation (ERAD).29 This process involves tagging proteins with ubiquitin, retro-translocating them from the ER into the cytosol via translocon channels, such as Sec61, and then degrading them in the 26S proteasome.3 The functional consequence of misfolded H⁺/K⁺-ATPase subunits is a loss of gastric acid secretion, which clinically manifests as hypochlorhydria or achlorhydria.34 These conditions are associated with impaired digestion and nutrient absorption, especially of iron and vitamin B12, leading to anemia.34 In severe cases, such as in individuals with congenital mutations in ATP4A, lifelong achlorhydria occurs.33 Autoimmune atrophic gastritis may also target the subunits, leading to their misfolding or degradation via immune-mediated mechanisms.35
Defective COPII/COPI Vesicular Transport
Defective COPII and COPI vesicular transport can severely impair the intracellular trafficking and functional expression of the gastric H⁺/K⁺-ATPase in parietal cells.36 Normally, the COPII complex, comprising key proteins such as Sec23/Sec24 and Sec13/Sec31, mediates the anterograde transport of newly synthesized H⁺/K⁺-ATPase α- and β-subunits from the ER to the Golgi apparatus for maturation and glycosylation.4 Disruption in COPII function, whether due to mutations, protein misfolding, or ER stress, leads to retention of the pump subunits within the ER, triggering ERAD and preventing their delivery to the apical canalicular membrane.4 Conversely, the COPI complex is responsible for retrograde transport from the Golgi to the ER, ensuring retrieval of mislocalized or unassembled subunits, recycling of vesicle components, and maintenance of ER-Golgi homeostasis.37 Defects in COPI function result in mis-sorting, loss of essential trafficking machinery, and reduced recycling efficiency, which ultimately diminishes the pool of functional proton pumps available for acid secretion.38 Together, impaired COPII/COPI trafficking disrupts the precise vesicular cycling required for the recruitment and retrieval of H⁺/K⁺-ATPase in response to secretory stimuli, contributing to hypochlorhydria and related gastric pathologies.35,39
Impaired Rab GTPase-Mediated Targeting
Impaired Rab GTPase-mediated targeting disrupts the precise delivery and recycling of the gastric H⁺/K⁺-ATPase to the apical canalicular membrane of parietal cells, which is essential for regulated acid secretion.4 Rab GTPases function as molecular switches, cycling between an active GTP-bound state and an inactive GDP-bound state, thereby orchestrating vesicle docking, tethering, and fusion with specific membrane compartments.4 In gastric parietal cells, Rab11 and Rab27 are particularly important for targeting tubulovesicles containing H⁺/K⁺-ATPase to the canalicular membrane during stimulation. At the same time, Rab2, Rab6, and Rab8 contribute to Golgi-to-plasma membrane trafficking and recycling.40 Defects in Rab protein expression, mutations impairing their GTP-binding/hydrolysis cycle, or disruptions in their interaction with effectors (eg, motor proteins, tethering factors, SNAREs) cause mistargeting or retention of the proton pump in cytoplasmic vesicles or incorrect compartments.33 This mislocalization reduces the number of functional pumps at the apical membrane, leading to diminished gastric acid secretion and contributing to conditions such as hypochlorhydria or achlorhydria.41 Additionally, impaired Rab-mediated recycling can disturb the rapid retrieval of the pumps during the resting phase, altering vesicle homeostasis and potentially triggering cellular stress responses.38,42
SNARE Complex Dysregulation
SNARE complex dysregulation interferes with the final step of vesicular fusion.3 Under normal conditions, vesicle-associated v-SNAREs (such as VAMP2 or VAMP8) interact with target membrane t-SNAREs (such as syntaxin 3/4 and SNAP-23) to form a tight four-helix bundle that brings the vesicle and plasma membranes into proximity, enabling membrane fusion and insertion of the proton pump into the membrane.34 This process is tightly regulated by accessory proteins like Munc18 and synaptotagmin, which ensure that fusion occurs only upon appropriate secretory stimuli (eg, histamine, gastrin, acetylcholine).34 Dysregulation of the SNARE machinery,43 whether due to altered expression, post-translational modifications, mutations, or defects in regulatory proteins, can disrupt complex assembly or disassembly.3 Such impairments prevent efficient fusion, causing the H⁺/K⁺-ATPase to remain trapped in the cytoplasmic vesicle pool instead of being deployed to the functional site.41 Over time, this not only reduces gastric acid secretion (hypochlorhydria) but can also disturb vesicle turnover, leading to abnormal parietal cell morphology and triggering compensatory or pathological cellular responses.44
Faulty Adaptor Proteins and Sorting Signals
Faulty adaptor proteins and sorting signals compromise the accurate trafficking of H⁺/K⁺-ATPase subunits through the secretory pathway in gastric parietal cells, ultimately impairing acid secretion.44 Under normal circumstances, adaptor protein complexes such as AP-1, AP-2, and AP-3 recognize specific sorting motifs within the cytoplasmic tails of the proton pump’s α- and β-subunits.32 These motifs (eg, dileucine [LL] signals) direct the recruitment of clathrin coats and guide vesicles toward the correct intracellular compartments, such as tubulovesicles in the cytoplasm during the resting state or the apical canalicular membrane upon stimulation.44 Mutations in the sorting motifs or structural defects in adaptor proteins can disrupt this recognition, leading to the mistargeting of the proton pump to incorrect membranes (eg, basolateral instead of apical) or its retention in the trans-Golgi network or endosomal compartments. Additionally, failure of adaptor proteins to interact with accessory trafficking machinery can hinder clathrin-mediated vesicle formation, reducing the pool of correctly packaged H⁺/K⁺-ATPase available for stimulus-induced exocytosis.45 This misrouting reduces the number of functional pumps at the site of acid secretion, causing hypochlorhydria and potentially contributing to gastric mucosal dysfunction.46
Disruption in Endosomal Recycling Pathways
Disruption in endosomal recycling pathways hinders the retrieval and re-targeting of H⁺/K⁺-ATPase-containing membranes from the canalicular surface back to the intracellular tubulovesicle pool in gastric parietal cells, impairing the dynamic regulation of acid secretion.34 Normally, during the resting phase, endocytic retrieval pathways internalize excess proton pumps from the apical canalicular membrane via clathrin-mediated or clathrin-independent endocytosis.33 These vesicles enter early endosomes, from which the pumps are sorted either for degradation in lysosomes or, more importantly, for recycling through Rab11- and Rab4-regulated recycling endosomes back to the cytoplasmic tubulovesicle reserve. Disruption in this pathway, due to defects in key regulators (eg, Rab GTPases, retromer complex, ESCRT machinery) or cytoskeletal transport systems, can cause pumps to be misrouted to late endosomes and lysosomes, resulting in their degradation instead of reuse.8 This reduces the available pool of functional pumps for rapid redeployment upon stimulation, leading to blunted acid secretory responses.47 Chronic impairment in recycling not only diminishes gastric acid output but can also alter endosomal morphology, disrupt membrane homeostasis, and trigger stress signals within the parietal cell.36
Discussion
Functional Consequences of Sorting Defects
Sorting defects in the trafficking of the gastric proton pump result in loss of parietal cell polarity and equilibrium,29 mislocalization to incorrect cellular compartments or retention within the trans-Golgi network, endosomes, or lysosomes,4 leading to significant functional consequences that reduce functional parietal cell membrane pumps and impair gastric acid secretion.36 Clinically, this manifests as hypochlorhydria or achlorhydria, conditions associated with digestive inefficiency, bacterial overgrowth, and increased susceptibility to infections such as Helicobacter pylori.37 Impaired recycling and degradation pathways cause accumulation of misfolded or mistargeted pumps, triggering cellular responses like ER stress or autophagy, which contribute to parietal cell dysfunction or death,38 impairment of gastric mucosal integrity, and increased risk of gastric diseases like chronic gastritis and gastric atrophy.4 Furthermore, the resulting hypochlorhydria or achlorhydria causes nutrient malabsorption (iron, calcium, vitamin B12), anemia, osteoporosis, and neurological problems. Consequently, the enterochromaffin-like cell hyperplasia increases the chance of polyps and neoplasia, hypochlorhydria causes hypergastrinemia, and damaged cells emit damage-associated molecular patterns (DAMPs), which intensify inflammation.29
Future Perspectives for Diagnostics and Therapeutics
Emerging insights into the molecular mechanisms underlying defective trafficking and sorting of the gastric H⁺/K⁺-ATPase have important diagnostic and therapeutic implications.29 On the diagnostic front, identifying specific mutations or dysregulations in adaptor proteins, Rab GTPases, or SNARE components through genetic screening or biomarker assays could enable early detection of parietal cell dysfunction and related hypochlorhydria.34,48 Advances in molecular imaging techniques may also allow for visualization of proton pump mislocalization in gastric tissue biopsies, thereby improving diagnostic precision.7,33 The current therapeutic landscape for acid-related disorders relies heavily on PPIs and H2 receptor antagonists, which act downstream by suppressing the pump’s luminal activity.3 However, these approaches do not address the underlying intracellular defects in protein sorting machinery that may drive disease progression. The evidence synthesized in this review indicates that modulation of the activities of protein sorting machinery represents promising therapeutic targets,16 as they can restore proper pump localization and normalize gastric acid secretion without complete suppression, potentially offering solutions for PPI-resistant cases and congenital gastric acid disorders.29,34
Hypothetically, gene therapy approaches might correct underlying genetic defects causing trafficking impairments.40 Such targeted interventions hold promise for treating refractory gastric acid disorders, reducing side effects associated with long-term acid suppression, and potentially preventing progression to more severe gastric diseases.4 Continued research into the trafficking machinery will thus pave the way for personalized medicine strategies tailored to specific molecular defects in gastric acid regulation.37 Nanomedicine-targeted therapy, including nanoparticles loaded with peptides and RNA or CRISPR reagents as drugs around primary parietal cells to correct proton pump trafficking defect without side effects from knockdown/long-term PPIs for rebound acid hypersecretion, is another hypothetical but hopeful direction.49 The use of these combinations will evolve, especially as we develop more personalized medicine strategies that combine genetic, proteomic, and imaging information to refine therapy further.
Strengths and Limitations
One of the major strengths of this review lies in the mechanistic depth of its analysis: rather than restricting focus to the catalytic activity of the proton pump, the review highlights the role of protein sorting machinery, providing a novel perspective that expands the definition of proton pumpopathy. The inclusion of diverse study models, ranging from human clinical samples to animal models and in vitro systems, adds robustness to the findings by integrating molecular, genetic, and pathophysiological evidence. Finally, the thematic categorization of results into synthesis, trafficking, docking, and apical insertion creates a structured framework that not only clarifies mechanisms but also identifies where therapeutic intervention may be most effective.
Despite these strengths, certain limitations must be acknowledged. First, the number of studies directly investigating protein sorting machinery in gastric proton pump dysfunction remains limited, which constrains the ability to draw definitive mechanistic conclusions. The heterogeneity of included studies spanning genetic, pharmacological, and cellular approaches introduces variability that complicates direct comparison of outcomes. Another limitation is the reliance on experimental models, which may not fully replicate human gastric physiology or disease progression. Additionally, the exclusion of non-English publications and inaccessible full texts may have introduced language and publication bias, potentially overlooking important contributions from other regions. Lastly, while the review proposes protein sorting machinery as a therapeutic target, clinical evidence supporting this approach is currently lacking, meaning that translational application remains largely speculative at this stage.
New Findings
Expansion of Pumpopathy Concept
Traditionally, gastric proton pumpopathy has been defined in terms of mutations directly affecting the H⁺/K⁺-ATPase subunits or their catalytic properties.50 However, this review highlights a conceptual expansion of the term, demonstrating that dysfunction in protein sorting machinery can equally disrupt gastric proton pump function by impairing its correct localization.14,38 This finding suggests that gastric proton pumpopathy should be considered not only in the context of the proton pump dysfunction but also as “sortopathies”, where intracellular transport defects phenocopy classic ion channel or pump dysfunction.
Dual Clinical Outcomes from Sorting Defects
A striking insight from this review is the recognition that defects in sorting pathways can lead to diametrically opposite clinical outcomes. On one hand, the misfolding or retention of H⁺/K⁺-ATPase in the ER contributes to hypochlorhydria or achlorhydria, which can manifest as iron and vitamin B12 deficiency, recurrent gastrointestinal infections, or pernicious anemia.44 On the other hand, trafficking dysregulation that enhances constitutive apical localization may promote hyperacidic states such as GERD or PUD.44 This duality highlights the importance of evaluating protein sorting defects not as unidirectional errors, but as context-dependent events that can either suppress or exacerbate gastric acid secretion.32
Molecular Convergence Across Disorders
Another novel observation is the convergence of gastric proton pump trafficking defects with broader protein misfolding and degradation pathways seen in systemic diseases such as neurodegeneration.46 Mislocalization of ATP4A or ATP4B subunits can activate ER stress responses and proteasome-mediated degradation, echoing mechanisms described in conditions like Parkinson’s or Alzheimer’s disease.45 This overlap suggests that gastric acid disorders may not be isolated pathologies but part of a wider spectrum of protein misfolding syndromes.33 Such convergence opens the possibility of cross-disciplinary therapeutic strategies, where interventions developed for neurodegenerative diseases could be repurposed to restore gastric acid homeostasis.
Unresolved Contradictions
The review also highlights contradictions in how mutations of the same protein sorting regulators can yield variable outcomes across studies.37 For instance, alterations in Rab GTPases or adaptor proteins sometimes lead to hypochlorhydria in one model but exacerbate hyperacidity in another.50 These discrepancies suggest a strong dependence on contextual factors such as circadian rhythm, hormonal regulators like ghrelin and somatostatin, or the metabolic state of the parietal cell.2 This variability emphasizes the need for more nuanced research into the conditional roles of sorting proteins, as their effects may be shaped by dynamic interactions rather than static pathways.
Conclusions and Recommendations
Conclusions
This review underscores the critical role of protein sorting machinery in the (dys)function of the gastric proton pump. By broadening the concept of gastric proton pumpopathy to include defects in intracellular trafficking pathways, it highlights that mislocalization and misfolding of H⁺/K⁺-ATPase is as clinically significant as its catalytic dysfunction.
Recommendations
Future research should prioritize detailed mechanistic studies to clarify how specific sorting proteins influence proton pump trafficking under physiological and pathological conditions. Greater emphasis should also be placed on identifying biomarkers of pump mislocalization to improve early diagnosis of gastric acid disorders. Clinically, translational efforts are needed to explore therapeutic strategies targeting protein sorting pathways, which may provide disease-modifying alternatives to conventional acid suppression. Longitudinal human studies that integrate molecular biology with clinical outcomes will be especially valuable for bridging the gap between bench and bedside. Finally, expanding research into underexplored areas, such as circadian regulation of sorting machinery and its impact on gastric secretion, may reveal additional therapeutic windows and enhance patient-specific management approaches.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kiela PR, Ghishan FK. Physiology of intestinal absorption and secretion. Best Pract Res Clin Gastroenterol. 2016;30(2):145–11. doi:10.1016/j.bpg.2016.02.007
2. Yao X, Forte JG. Cell biology of acid secretion by the parietal cell. Annu Rev Physiol. 2003;65(1):103–131. doi:10.1146/annurev.physiol.65.072302.114200
3. Young VC, Nakanishi H, Meyer DJ, et al. Structure and function of H+/K+ pump mutants reveal Na+/K+ pump mechanisms. Nat Commun. 2022;13(1):5270. doi:10.1038/s41467-022-32793-0
4. Zhang W, Zheng SB, Zhuang Y, et al. H + /K + ATPase expression in human parietal cells and gastric acid secretion in elderly individuals. J Dig Dis. 2013;14(7):366–372. doi:10.1111/1751-2980.12055
5. Hao W, Zheng C, Wang Z, Ma H. Global burden and risk factors of peptic ulcer disease between 1990 and 2021: an analysis from the global burden of disease study 2021. PLoS One. 2025;20(7):e0325821. doi:10.1371/journal.pone.0325821
6. Shin JM, Sachs G. Pharmacology of proton pump inhibitors. Curr Gastroenterol Rep. 2008;10(6):528–534. doi:10.1007/s11894-008-0098-4
7. Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116(2):153–166. doi:10.1016/S0092-8674(03)01079-1
8. Ray P, Nancarrow DJ, Ferrer-Torres D, et al. UBCH5 family members differentially impact stabilization of mutant p53 via RNF128 Iso1 during Barrett’s progression to esophageal adenocarcinoma. Cell Mol Gastroenterol Hepatol. 2022;13(1):129–149. doi:10.1016/j.jcmgh.2021.08.003
9. Sagia GM, Georgiou X, Chamilos G, Diallinas G, Dimou S. Distinct trafficking routes of polarized and non-polarized membrane cargoes in Aspergillus nidulans. Elife. 2024;13. doi:10.7554/eLife.103355
10. Whitsett JA. Genetic disorders influencing lung formation and function at birth. Hum Mol Genet. 2004;13(suppl_2):R207–R215. doi:10.1093/hmg/ddh252
11. Hubner CA. Ion channel diseases. Hum Mol Genet. 2002;11(20):2435–2445. doi:10.1093/hmg/11.20.2435
12. Litan A, Langhans SA. Cancer as a channelopathy: ion channels and pumps in tumor development and progression. Front Cell Neurosci. 2015;9:9. doi:10.3389/fncel.2015.00086
13. Freston JW. The pathophysiological and pharmacological basis of peptic ulcer therapy. Toxicol Pathol. 1988;16(2):260–266. doi:10.1177/019262338801600219
14. Shin JM, Kim N. Pharmacokinetics and pharmacodynamics of the proton pump inhibitors. J Neurogastroenterol Motil. 2013;19(1):25–35. doi:10.5056/jnm.2013.19.1.25
15. Kinoshita Y, Ishimura N, Ishihara S. Advantages and disadvantages of long-term proton pump inhibitor use. J Neurogastroenterol Motil. 2018;24(2):182–196. doi:10.5056/jnm18001
16. Freedberg DE, Kim LS, Yang YX. The risks and benefits of long-term use of proton pump inhibitors: expert review and best practice advice from the American Gastroenterological Association. Gastroenterology. 2017;152(4):706–715. doi:10.1053/j.gastro.2017.01.031
17. Zhan Z, Li J, Zhang W, Huang S, Fang X. Multidimensional assessment of psychiatric adverse events related to proton pump inhibitors: a real‐world, pharmacovigilance study. CNS Neurosci Ther. 2025;31(5). doi:10.1111/cns.70436
18. Alagbonsi A, Manishimwe C, Serge M, Agahozo D, Tito S. Varying psychological stress among Rwandan patients with chronic diseases. Diabetes Metabolic Syndrom Obesity. 2025;18:3249–3258. doi:10.2147/DMSO.S539308
19. López-Hernández T, Ichiro TK, Mori Y, et al. Clathrin-independent endocytic retrieval of SV proteins mediated by the clathrin adaptor AP-2 at mammalian central synapses. Elife. 2022:11. doi:10.7554/eLife.71198
20. Adepoju AA, Muhammad MA, Adamson MM, et al. Genetic and environmental factors associated with alteration of filtration slit proteins and their functions: a scoping review. Front Nephrol. 2025:5. doi:10.3389/fneph.2025.1678502
21. Ajayi JA, Ananias EN, Issa-Lawal M, et al. Mechanisms involved in aminoacidurias: impacts of genetic and environmental factors. Curr Res Physiol. 2025;8:100168. doi:10.1016/j.crphys.2025.100168
22. Hakizimana JC, Alagbonsi AI. Modulation of lactose synthesis and orexinergic‐glucose pathway by sex steroid hormones. Physiol Rep. 2025;13(22). doi:10.14814/phy2.70661
23. Hakizimana JC, Alagbonsi AI. Genetic and environmental factors associated with lactose digestion in African populations. Physiol Genomics. 2025;58(1):32–41. doi:10.1152/physiolgenomics.00268.2025
24. Kampire M, Hakizimana J, Mucumbitsi J, Alagbonsi A. Pathophysiological consequences associated with hormonal contraceptives use in Sub-Saharan Africa: a scoping review. Open Access J Contracept. 2025;16:171–187. doi:10.2147/OAJC.S563680
25. Izabayo P, Hakizimana JC, Uwineza A, Alagbonsi AI. Environmental factors associated with microcephaly in Africa: a systematic review. Neurotoxicol Teratol. 2026;113:107577. doi:10.1016/j.ntt.2025.107577
26. Hakizimana O, Hitayezu J, Uyisenga JP, et al. Genetic etiology of autism spectrum disorder in the African population: a scoping review. Front Genet. 2024:15. doi:10.3389/fgene.2024.1431093
27. Hakizimana JC, Izabayo P, Izukwizabigenza Z, Alagbonsi AI. Orexinergic pathway as a potential therapeutic candidate for the modulation of glucose homeostasis. Front Physiol. 2025;16. doi:10.3389/fphys.2025.1659753
28. Ndinganire G, Ntamukunzi G, Alagbonsi AI. Physiological relevance of autocrine melatonin signaling in pineal and extra-pineal sites: a systematic review. Function. 2026. doi:10.1152/function.101.2025
29. Engevik AC, Kaji I, Goldenring JR. The Physiology of the Gastric Parietal Cell. Physiol Rev. 2020;100(2):573–602. doi:10.1152/physrev.00016.2019
30. Lutfiah U, Jusman SWA, Sadikin M. H,K-ATPase and carbonic anhydrase response to chronic systemic rat gastric hypoxia. Med J Indonesia. 2015;24(3):133–138. doi:10.13181/mji.v24i3.1066
31. Kawasaki-Nishi S, Nishi T, Forgac M. Arg-735 of the 100-kDa subunit a of the yeast V-ATPase is essential for proton translocation. Proc Natl Acad Sci. 2001;98(22):12397–12402. doi:10.1073/pnas.221291798
32. Kimura T, Yoshida A, Tabuchi Y, Ikari A, Takeguchi N, Asano S. Stable expression of gastric proton pump activity at the cell surface. J Biochem. 2002;131(6):923–932. doi:10.1093/oxfordjournals.jbchem.a003183
33. Sahoo N, Gu M, Zhang X, et al. Gastric acid secretion from parietal cells is mediated by a Ca2+ efflux channel in the tubulovesicle. Dev Cell. 2017;41(3):262–273.e6. doi:10.1016/j.devcel.2017.04.003
34. Fährmann M, Kaufhold M, Pfeiffer AF, Seidler U. Protein kinase C‐ α attenuates cholinergically stimulated gastric acid secretion of rabbit parietal cells. Br J Pharmacol. 2003;139(3):545–554. doi:10.1038/sj.bjp.0705211
35. Sachs G, Wallmark B. The gastric H, K ATPase. In: Proton Pump Inhibitors. Birkhäuser Basel; 1999:23–45. doi:10.1007/978-3-0348-8795-3_2
36. Afaa TJ, Etwire VK, Dame JA, Adja-Sai C, Amegan-Aho KH. Prolapse gastropathy: a rare cause of gastrointestinal bleeding in a child. Health Sci Investig J. 2022;3(1):341–344. doi:10.46829/hsijournal.2022.6.3.1.341-344
37. Jones CM, Toh B, Pettitt JM, et al. Monoclonal antibodies specific for the core protein of the β‐subunit of the gastric proton pump (H + /K + ATPase). Eur J Biochem. 1991;197(1):49–59. doi:10.1111/j.1432-1033.1991.tb15881.x
38. Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev. 2007;87(4):1377–1408. doi:10.1152/physrev.00050.2006
39. Adolf F, Rhiel M, Hessling B, et al. Proteomic profiling of mammalian COPII and COPI Vesicles. Cell Rep. 2019;26(1):250–265.e5. doi:10.1016/j.celrep.2018.12.041
40. Derby MC, Gleeson PA. New insights into membrane trafficking and protein sorting. Int Rev Cytol. 2007;7:47–116. doi:10.1016/S0074-7696(07)61002-X
41. Appenzeller-Herzog C, Hauri HP. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci. 2006;119(11):2173–2183. doi:10.1242/jcs.03019
42. Liu H, Xu J, Yao Q, Zhang Z, Guo Q, Lin J. Rab7 is associated with poor prognosis of gastric cancer and promotes proliferation, invasion, and migration of gastric cancer cells. Med Sci Monit. 2020;26. doi:10.12659/MSM.922217
43. Mashima H, Suzuki J, Hirayama T, et al. Involvement of vesicle-associated membrane protein 7 in human gastric epithelial cell vacuolation induced by Helicobacter pylori -produced VacA. Infect Immun. 2008;76(6):2296–2303. doi:10.1128/IAI.01573-07
44. Okamoto CT, Li R, Zhang Z, Jeng YY, Chew CS. Regulation of protein and vesicle trafficking at the apical membrane of epithelial cells. J Control Release. 2002;78(1–3):35–41. doi:10.1016/S0168-3659(01)00479-5
45. Elias AF, Lin BC, Piggott BJ. Ion channels in gliomas—from molecular basis to treatment. Int J Mol Sci. 2023;24(3):2530. doi:10.3390/ijms24032530
46. Sung WJ, Kim D, Zhu A, et al. The lysosome as a novel therapeutic target of EGFR-mediated tumor inflammation. Front Pharmacol. 2022:13. doi:10.3389/fphar.2022.1050758
47. Asano S, Yoshida A, Yashiro H, et al. The cavity structure for docking the K+-competitive inhibitors in the gastric proton pump. J Biol Chem. 2004;279(14):13968–13975. doi:10.1074/jbc.M308934200
48. Heitzmann D, Warth R. No Potassium, No Acid: k + channels and gastric acid secretion. Physiology. 2007;22(5):335–341. doi:10.1152/physiol.00016.2007
49. Lerner M, Lemke D, Bertram H, et al. An extracellular loop of the human non-gastric H,K-ATPase a-subunit is involved in apical plasma membrane polarization. Cell Physiol Biochem. 2006;18(1–3):75–84. doi:10.1159/000095169
50. Abe K, Yamamoto K, Irie K, Nishizawa T, Oshima A. Gastric proton pump with two occluded K+ engineered with sodium pump-mimetic mutations. Nat Commun. 2021;12(1):5709. doi:10.1038/s41467-021-26024-1
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.