Back to Journals » Drug Design, Development and Therapy » Volume 20
GABAA Receptor Modulators as Emerging Therapeutic Agents for Depressive Disorders
Received 12 March 2026
Accepted for publication 14 May 2026
Published 8 June 2026 Volume 2026:20 608834
DOI https://doi.org/10.2147/DDDT.S608834
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Bo-Yun Guan,1 Yang Li2
1The Second Norman Bethune of Jilin University, Changchun, Jilin, 130000, People’s Republic of China; 2Department of Obstetrics and Gynecology, The Second Norman Bethune of Jilin University, Changchun, Jilin, 130000, People’s Republic of China
Correspondence: Yang Li, Department of Obstetrics and Gynecology, The Second Norman Bethune of Jilin University, Changchun, Jilin, 130000, People’s Republic of China, Tel +86 18843015077, Email [email protected]
Abstract: Depression is a serious mental disorder that affects individuals across diverse demographic and socioeconomic groups. Its pathophysiology is multifactorial and incompletely understood, and currently available pharmacological treatments remain limited in efficacy and scope. This article examines the involvement of the gamma-aminobutyric acid (GABA)ergic system in the pathogenesis of depression and reviews the pharmacological effects, therapeutic efficacy, and recent research developments related to various GABAA receptor modulators in the treatment of depressive disorders, including benzodiazepines, intravenous anesthetics, and neurosteroids. These agents may exert antidepressant and anxiolytic effects through modulation and restoration of GABAergic neurotransmission. In addition, certain agents, such as etomidate and allopregnanolone, have been reported to enhance synaptic plasticity and attenuate neuroinflammatory processes, thereby contributing to the reduction of depressive symptoms through multiple neurobiological mechanisms.
Keywords: BDNF, GABAA receptor modulators, GABAARs, neurosteroids
Introduction—Medical Research and Advances in the Treatment of Depression
Overview of Depression
Depression is a prevalent chronic psychiatric disorder encountered in clinical practice and may arise from multiple etiological factors. It is primarily characterized by persistent depressed mood and anhedonia and may also present with sleep disturbances, psychomotor retardation, executive dysfunction, and impaired social functioning. In severe cases, suicidal ideation may occur.1 Depression is associated with substantial morbidity. It not only impairs daily functioning and quality of life but is also linked to an increased risk of various comorbid medical conditions. Furthermore, depression imposes a significant socioeconomic burden.
Pathogenesis of Depression
The pathogenesis of depression is complex and multifactorial. Environmental exposures, psychological stressors, and genetic susceptibility each contribute to disease development to varying degrees. The heritability of major depressive disorder has been estimated at approximately 30%.2 Although depression is widely considered to result from interactions between genetic and environmental factors, the precise underlying mechanisms remain incompletely elucidated. At the molecular and neurobiological levels, the GABAergic deficit hypothesis posits that reduced GABA levels result in depression. Such abnormal changes tend to return to normal after effective antidepressant treatment. Alongside the above hypothesis, other theories can also account for depression pathogenesis. These include the monoamine neurotransmitter and receptor hypothesis, dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis, impairments in neuroplasticity and brain-derived neurotrophic factor (BDNF) signaling, and the inflammation and cytokine hypothesis.
Current Treatments for Depression
A stepwise and progressive treatment strategy is generally adopted in the management of depression.3 Therapeutic interventions are implemented according to symptom severity and clinical response and may include psychological interventions, pharmacotherapy, and electroconvulsive therapy (ECT).
Pharmacological treatment options include monoamine oxidase inhibitors (MAOIs), which increase synaptic concentrations of monoamine neurotransmitters by inhibiting monoamine oxidase activity. Iproniazid is a representative non-selective, irreversible MAOI. Owing to its non-selective and irreversible inhibition, it is associated with an increased risk of hypertensive crisis. Tricyclic antidepressants (TCAs) represent another major class of antidepressants. These agents inhibit presynaptic norepinephrine and 5-hydroxytryptamine (5-HT) reuptake transporters and antagonize postsynaptic adrenergic α1 and α2 receptors as well as histamine H1 receptors.4,5 By inhibiting norepinephrine and 5-HT reuptake, TCAs increase synaptic concentrations of these neurotransmitters, thereby alleviating depressive symptoms. However, receptor antagonism contributes to a broad adverse effect profile, which may limit tolerability.6,7
Selective serotonin reuptake inhibitors (SSRIs) were subsequently developed to improve safety and tolerability. These agents selectively inhibit 5-HT reuptake by blocking the 5-HT transporter, thereby increasing synaptic 5-HT concentrations. Compared with TCAs, SSRIs exhibit lower affinity for adrenergic α1 and α2 receptors, histamine H1 receptors, muscarinic receptors, and dopamine D2 receptors, resulting in a reduced incidence of certain adverse effects. Fluoxetine is a representative SSRI that remains widely used in the treatment of depression. Nevertheless, adverse effects such as nausea, insomnia, and sexual dysfunction may occur.7
More recently, esketamine has been introduced into clinical practice as a novel antidepressant agent. Ketamine was originally developed as a dissociative anesthetic; however, clinical trials have demonstrated that intravenous administration of ketamine at sub-anesthetic doses produces rapid and robust antidepressant effects.8
Several mechanistic hypotheses have been proposed. The disinhibition hypothesis posits that ketamine inhibits gamma-aminobutyric acid (GABA)ergic interneurons expressing N-methyl-D-aspartate receptors (NMDARs). This leads to disinhibition of glutamatergic pyramidal neurons and increases in glutamate release. Subsequent activation of postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) enhances BDNF release and downstream protein synthesis. These molecular events contribute to the observed antidepressant effects.9
Conversely, some experimental studies indicate that ketamine may reverse depressive-like behaviors through activation of GABAergic interneurons.10 Despite its therapeutic efficacy, ketamine is associated with potential risks. Subanesthetic doses may induce transient elevations in blood pressure and heart rate, whereas higher doses are associated with an increased risk of serious cardiovascular events.11 The identification of ketamine’s rapid antidepressant effects represents a significant advance in the field, facilitating the exploration of novel molecular targets and accelerating the development of next-generation rapid-acting antidepressants.
Antidepressant Effects of GABAA Receptor Modulators
Stress- and Depression-Induced Neuronal Atrophy and Synapse Loss—Restoration of the GABAergic System as a Potential Novel Approach for Depression
Neuroimaging studies and postmortem analyses have demonstrated that individuals with major depressive disorder exhibit reduced brain volume and decreased neuronal density in the dorsolateral prefrontal cortex (dlPFC), findings that indicate a reduction in synaptic number and functional connectivity within this region.12,13 Chronic stress has been shown to induce synaptic instability, dendritic atrophy characterized by reduced dendritic length and branching, and decreased dendritic spine density and strength.14 In addition, chronic stress impairs hippocampal and prefrontal cortex (PFC) function and reduces the expression of AMPARs and NMDARs.15 These alterations contribute to disruption of the excitatory–inhibitory balance within neural circuits. Consequently, therapeutic strategies for depression have evolved from approaches primarily aimed at modulating neurotransmitter concentrations to interventions focused on restoring synaptic structure and functional plasticity.
Research on ketamine has further emphasized that modulation of excitatory–inhibitory circuit dynamics may represent a promising therapeutic direction. The GABAergic system constitutes the principal inhibitory neurotransmitter system in the human brain. In depressive disorders, reductions in central GABA concentrations have been observed, accompanied by decreased expression of enzymes and proteins involved in GABA synthesis.16 Restoration of GABAergic neurotransmission may therefore represent a viable therapeutic strategy for the treatment of depression.
GABA Neurotransmission and the Synaptic Restorative Role of BDNF
GABA Neurotransmission
GABA is synthesized through a metabolic pathway commonly referred to as the GABA shunt. In this process, GABA is produced from glutamate derived from the tricarboxylic acid cycle. A portion of the synthesized GABA is released into the synaptic cleft to exert inhibitory neurotransmission, whereas the remaining fraction is converted into succinate and re-enters the tricarboxylic acid cycle.17,18 Following synaptic release, GABA binds to GABAARs and GABABRs located on postsynaptic membranes. Activation of GABAARs increases chloride ion conductance, leading to membrane hyperpolarization and reduced neuronal excitability.18 GABABR activation mediates slower inhibitory signaling through G-protein–coupled mechanisms. Extracellular GABA is subsequently metabolized by GABA transaminase (GABA-T) to succinic semialdehyde, which re-enters the tricarboxylic acid cycle. In addition, GABA undergoes reuptake via specific transporters for reuse. A proportion is taken up by glial cells, where it is metabolized or recycled and subsequently transported back to neurons, thereby re-entering the GABA shunt for resynthesis (Figure 1).19,20 This tightly regulated cycle maintains inhibitory neurotransmission and excitatory–inhibitory balance within neural circuits.
 |
Figure 1 Synaptic release, receptor activation, reuptake, and metabolic recycling of GABA. |
Role of BDNF in Stress and Depression
BDNF-Mediated Neuronal Plasticity and Therapeutic Effects
Neuronal plasticity refers to the capacity of the nervous system to reorganize its structural and functional properties in response to internal or external stimuli.21 Depression has been associated with structural and functional abnormalities in neural networks. Structural remodeling and restoration of synaptic connectivity are therefore considered critical for the recovery of normal neural circuit function.22 BDNF exerts its biological effects through two principal receptor systems: the low-affinity p75 neurotrophin receptor and the high-affinity tropomyosin receptor kinase B (TrkB). TrkB receptors exist in multiple isoforms, including the full-length TrkB-FL and the truncated TrkB-T1. Binding of BDNF to TrkB-FL induces receptor phosphorylation and activates three major intracellular signaling cascades: the MAPK/ERK pathway, the PI3K-Akt pathway, and the PLCγ–Ca2⁺ pathway.23 These pathways collectively regulate synaptic plasticity and excitatory–inhibitory balance (Figure 2).
 |
Figure 2 Intracellular signaling pathways activated by BDNF–TrkB receptor interaction. |
(1) PI3K-Akt pathway: Activation of PI3K-Akt signaling interacts with mammalian target of rapamycin and downstream effectors to promote protein synthesis, cellular growth, and survival.24 Antidepressant-like effects of liquiritin have been associated with modulation of this pathway.25 Furthermore, BDNF-induced PI3K-Akt activation enhances dendritic translocation of postsynaptic density protein 95 following NMDA receptor activation, linking this pathway to activity-dependent synaptic potentiation.26,27
(2) MAPK pathway: TrkB activation initiates a kinase cascade that stimulates the MAPK signaling pathway and produces ERK, which plays a regulatory role in cell proliferation and differentiation.28 BDNF-mediated MAPK signaling has also been shown to regulate AMPA receptor trafficking and dendritic spine delivery in a bidirectional manner.29,30
(3) PLC pathway: The PLCγ pathway contributes to multiple BDNF-mediated processes, including neuronal differentiation and survival, neurite outgrowth of dopaminergic neurons, phosphorylation of NMDARs and AMPARs, transcriptional regulation of GABA receptors, and facilitation of hippocampal integration and synaptic plasticity.31–36
TrkB-T1 functions primarily as a dominant-negative receptor by forming heterodimers with TrkB-FL, thereby attenuating canonical BDNF signaling.37 In addition to this inhibitory role, TrkB-T1 has been implicated in BDNF sequestration and intracellular trafficking, neurite outgrowth, cytoskeletal regulation in astrocytes and glioma cells, modulation of Rho GTPase activity, and potential activation of PLCγ and MAPK pathways.38,39
BDNF exerts neuroprotective and pro-survival effects, including inhibition of apoptosis and promotion of cell survival.40 It also plays a central role in synaptic maintenance and function. BDNF enhances excitatory transmission by increasing the amplitude of AMPA receptor-mediated miniature excitatory postsynaptic currents and strengthens inhibitory transmission by increasing the frequency of miniature inhibitory postsynaptic currents and enlarging GABAergic presynaptic terminals.41 In addition, BDNF promotes dendritic growth and branching, increases synapse number and density, and facilitates synaptic plasticity through modulation of intracellular calcium dynamics, regulation of AMPAR trafficking, facilitation of retrograde signaling, and enhancement of long-term potentiation in the hippocampus and dentate gyrus.29,42–46 Collectively, these mechanisms support the role of BDNF as a critical mediator of synaptic restoration and plasticity in depressive disorders and highlight its potential as a disease-modifying therapeutic target.
Antidepressant Effects of GABAA Receptor Modulators
Pharmacological Actions of GABAA Receptor Modulators
GABAA Receptors
GABAA receptors (GABAARs) are ligand-gated ion channel receptors activated by GABA. Structurally, GABAARs are pentameric ligand-gated ion channels composed of five subunits arranged around a central chloride-conducting pore. These receptors are encoded by a family of 19 homologous genes, categorized into subunit classes based on sequence homology: α1–6, β1–3, γ1–3, δ, ε, θ, π, and ρ1–3. Most native GABAARs are assembled from two α subunits, two β subunits, and either one γ or one δ subunit, forming heteropentameric receptor subtypes.17,47 The regional distribution, synaptic versus extrasynaptic localization, pharmacological profile, and physiological properties of GABAARs are determined by their specific subunit composition (see Table 1).48–65 Variations in subunit assembly contribute to functional heterogeneity across brain regions.
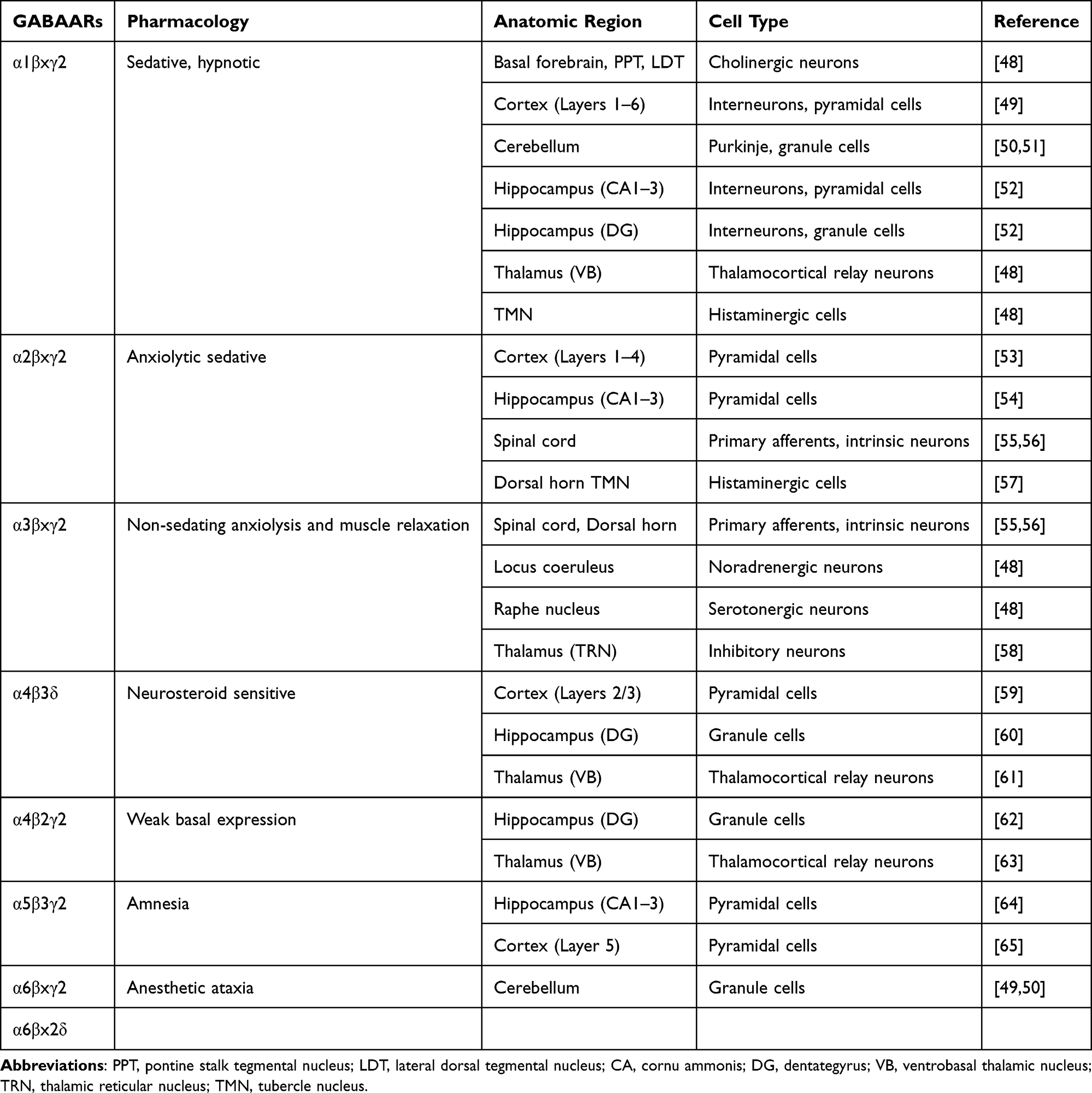 |
Table 1 Subunit Composition, Functional Diversity, and Regional Distribution of GABAAR Subtypes |
Function of GABAARs and Their Relationship with Depression
The GABAergic deficit hypothesis of depression posits that reduced inhibitory neurotransmission contributes to the pathophysiology of depressive disorders and that restoration of GABAergic function is associated with therapeutic improvement.
Preclinical studies provide supporting evidence for this hypothesis. Deletion of the γ2 subunit in mice induces depressive-like phenotypes. Heterozygous (γ2⁺/−) mice exhibit anxiety- and depression-like behaviors in behavioral paradigms, accompanied by hyperactivity of the HPA axis.66 Elevated glucocorticoid levels associated with HPA axis activation further reduce GABAAR expression in forebrain regions, particularly in the frontal cortex and ventral hippocampus.67,68
Chronic stress has been shown to induce loss of hippocampal interneurons, and γ2 subunit deficiency exacerbates this process. This interaction establishes a reciprocal cycle of GABAergic dysfunction and HPA axis hyperactivity, resulting in progressive impairment of hippocampal function. Moreover, increased HPA axis activity may influence responsiveness to antidepressant treatment. γ2⁺/− mice demonstrate a significantly attenuated response to fluoxetine compared with desipramine, indicating that altered GABAAR function may modulate pharmacological sensitivity.66,69
Deletion of the γ2 subunit also markedly reduces hippocampal neurogenesis, neuronal maturation, differentiation, and survival, thereby increasing vulnerability to anxiety- and mood-related phenotypes and reducing resilience under conditions of chronic stress.70,71 Importantly, the γ2 subunit constitutes a critical binding site for benzodiazepines (BZDs). Its deletion substantially decreases benzodiazepine binding site availability, thereby directly influencing the pharmacodynamic efficacy of BZD-related agents.72
Specific α subunits of GABAARs have been implicated in the regulation of depression-related behaviors. The α3 subunit is expressed in monoaminergic neurons and primarily mediates GABAergic inhibition of dopaminergic, serotonergic (5-HT), and noradrenergic systems.73 Mice with complete deletion of the α3 subunit (α3−/−) demonstrate increased active swimming time in the forced swim test (FST), a finding generally interpreted as reflecting reduced behavioral despair.74 GABAARs containing the α2 subunit are considered to play a critical role in affective regulation, as these receptors are highly expressed in limbic regions, including the amygdala, hippocampus, and nucleus accumbens,75,76 which are central to emotional processing and implicated in the pathophysiology of depression.77,78
Studies using α2 heterozygous (α2⁺/−) and homozygous knockout (α2−/−) mice and comparing their behavior with wild-type littermates in the novelty suppressed feeding test, FST, and tail suspension test have demonstrated that α2−/− mice exhibit increased behavioral despair, indicating that the α2 subunit serves as an important regulator of anxiety- and depression-related behaviors within the GABAergic system.77 The α5 subunit primarily contributes to synaptic plasticity, cognition, and memory and has been associated with depression, aging, and cognitive impairment; it is also a key determinant of dendritic inhibition.79 α5-containing GABAARs are localized on the dendrites of pyramidal neurons apposed to somatostatin-expressing interneurons (SST interneurons), predominantly within the middle and deep layers of the hippocampus and frontal cortex.80
Reduced expression of SST and related markers has been reported in postmortem brain tissue from individuals with depression, schizophrenia, bipolar disorder, and Alzheimer’s disease. Notably, the affected regions overlap with those enriched in α5-containing GABAARs.81,82 Experimental evidence further demonstrates that decreased SST expression or impaired SST interneuron function induces anxiety- and depression-like phenotypes as well as cognitive deficits,83–85 whereas enhancement of SST interneuron activity produces antidepressant-like effects.86,87 These findings suggest that the α5 subunit may represent a potential therapeutic target for depression.
Moreover, the α5 subunit interacts with multiple other GABAAR subunits; for example, the β3 subunit frequently co-assembles with α5, and regions exhibiting high β3 expression in the hippocampus also demonstrate elevated α5 expression.88 α5-containing GABAARs may also form heteropentameric assemblies with α1 and α2 subunits. In mixed α subunit-containing receptors, preferential assembly of α5 with γ2 has been observed, generating benzodiazepine binding sites characterized by α5-specific pharmacological properties. Therefore, pharmacological modulation of the α5 subunit may exert antidepressant effects through subunit-specific interactions and the pharmacological modulation of α5-containing GABAARs using selective positive or negative allosteric modulators.79,89
The β subunits play a critical role in the assembly and functional regulation of GABAARs and are essential for conferring receptor biological activity.89 Among these, the β3 subunit is of particular importance.
First, β3 is required for the assembly of functional GABAARs and serves as a key determinant of inhibitory transmission in the hippocampus; deletion of the β3 subunit markedly reduces inhibitory synaptic function, whereas deletion of β1 or β2 produces no significant alterations.
Second, the β3 subunit preferentially co-assembles with α2 and α3 subunits, thereby contributing to the modulation of anxiety- and depression-related behaviors.89–91 Experimental evidence indicates that propofol selectively targets β3-containing GABAARs and attenuates anxiety-like behaviors by inhibiting corticotropin-releasing hormone neuron activity within the paraventricular nucleus of the hypothalamus.92
These findings indicate an association between the β3 subunit and anxiety-related phenotypes and suggest that pharmacological targeting of β3-containing GABAARs to enhance GABAergic transmission may mitigate anxiety symptoms.
GABAARs containing the δ subunit and postpartum depression: GABAARs containing the δ subunit have been implicated in the pathophysiology of postpartum depression. Mice lacking the δ subunit display postpartum depression–like behaviors, whereas wild-type mice exhibit either no significant behavioral alterations or comparatively milder depressive-like phenotypes following parturition.93 It has been proposed that these behavioral changes are associated with fluctuations in central neurosteroid concentrations occurring before and after delivery.94 Collectively, these findings support a significant association between δ subunit–containing GABAARs and vulnerability to postpartum depression.
Positive Allosteric Modulators of GABAARs
Positive allosteric modulators (PAMs) of GABAARs comprise a class of agents that enhance receptor activity in the presence of GABA. This group includes BZDs, barbiturates, neuroactive steroids, and general anesthetics such as etomidate. The selectivity, efficacy, and pharmacodynamic properties of PAMs are closely influenced by the specific GABAAR subunits to which they bind. As presented in Table 2, different GABAAR modulators interact with distinct receptor interfaces, resulting in diverse pharmacological effects.90,95–103 Figure 3 presents the binding sites of commonly used modulators.
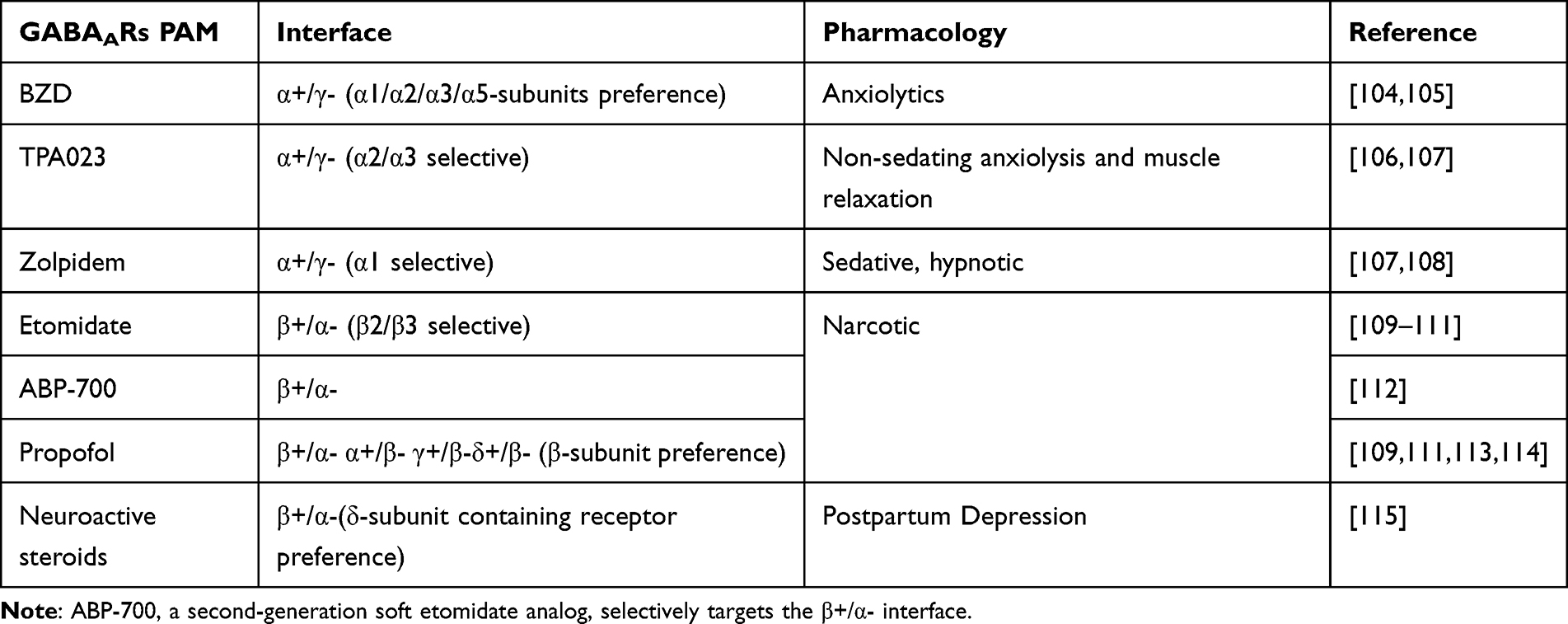 |
Table 2 Receptor Binding Interfaces and Pharmacodynamic Profiles of GABAAR Agonists |
 |
Figure 3 Subunit-specific binding sites and allosteric modulation of GABAAR modulators. |
Effects of GABAARs and Their Modulators in Patients with Depression
The functional properties of GABAARs are highly dependent on receptor subtype composition.104 Distinct subtypes are associated with different therapeutic profiles in depression. Accordingly, GABAAR modulators are categorized in this review based on subtype selectivity, and representative agents from each category are discussed.
Modulators Targeting α Subunits—BZD and Others
BZDs are GABAAR PAMs that bind at the α⁺/γ− subunit interface, with the γ subunit being essential for binding and subtype selectivity.17 As the γ2 subunit is widely expressed across GABAAR subtypes, BZDs generally exhibit limited subtype selectivity. Clinically, BZDs are widely used as anxiolytics but are not typically regarded as primary antidepressant agents.94 However, certain BZDs, such as alprazolam and adinazolam, have demonstrated antidepressant-like effects comparable to or exceeding those of conventional antidepressants, potentially through preferential modulation of α2-containing GABAARs.94
Current evidence indicates that BZDs enhance chloride ion conductance by binding to the α⁺/γ− interface, thereby potentiating GABA-mediated inhibitory neurotransmission.105 Nevertheless, several limitations restrict their use. BZDs require the presence of GABA for activity and are ineffective in its absence. In addition, long-term use is associated with adverse effects, including cognitive impairment tolerance, dependence, and withdrawal symptoms.106,107 Consequently, BZDs are infrequently used as primary antidepressant therapies.
α2 subunit–selective modulator—TPA023: Activates targeting α2-containing GABAARs are of particular interest given the established association between the α2 subunit and affective regulation. Although no agent demonstrates complete subtype specificity, compounds with relative selectivity may offer therapeutic advantages. TPA023 is a selective modulator with preferential activity at α2/α3 subtypes. Its modulation of α2/α3-containing receptors mediates anxiolytic effects, while its low intrinsic activity at α1-containing receptors minimizes sedative effects and reduces the risk of α1-associated adverse outcomes, including dependence liability.102,108,109 The favorable pharmacological profile and relatively mild adverse effect spectrum of TPA023 suggest potential utility as a novel antidepressant candidate.
Bidirectional pharmacological modulation of α5-GABAARs for antidepressant effects: Selective targeting of α5-containing GABAARs has also demonstrated antidepressant potential. α5-selective PAMs, such as GL-II-73, retain anxiolytic properties similar to those of BZDs while additionally exhibiting pro-cognitive effects in models of acute stress, chronic stress, and aging-related memory impairment.79,110 Conversely, α5-selective negative allosteric modulators (NAMs) have also demonstrated antidepressant-like effects. The proposed mechanism appears to involve modulation of glutamatergic neurotransmission, in a manner analogous to that of ketamine.111
Modulators Targeting β Subunits—Etomidate and Propofol
Numerous anesthetic agents enhance GABAAR-mediated inhibitory neurotransmission through direct interaction with receptor subunits, particularly intravenous anesthetics such as etomidate and propofol, as well as neuroactive steroid anesthetics.112–114
Etomidate and propofol function as GABAAR positive allosteric modulators with preferential activity at receptors containing the β3 subunit. Etomidate selectively modulates GABAARs incorporating β2 or β3 subunits, thereby enhancing inhibitory synaptic transmission.47,100 In addition, etomidate exerts suppressive effects on HPA axis activity. The β subunits are essential for GABAAR assembly and stability, and the β3 subunit plays a central role in maintaining inhibitory transmission within the hippocampus, a region closely implicated in the pathophysiology of depression.
Although direct antidepressant effects of etomidate alone have not been extensively reported, propofol has demonstrated antidepressant-like properties in preclinical models. Its mechanism of action is pharmacologically similar to that of etomidate. In mice exposed to chronic unpredictable mild stress, propofol administration reduces depressive-like behaviors, supporting its antidepressant potential.115 Both etomidate and propofol are widely used as anesthetic agents for induction during ECT. Comparative studies indicate that etomidate may provide superior therapeutic outcomes compared with propofol in the context of ECT, potentially due to distinct pharmacodynamic characteristics.100,116–118
Evidence indicates that etomidate may enhance the therapeutic efficacy of ECT through modulation of BDNF. BDNF regulates the expression of nuclear factor erythroid 2–related factor 2 (Nrf2) in astrocytes.119 Experimental findings demonstrate that ECT alleviates depressive-like behaviors in chronic unpredictable mild stress (CUMS) models by activating the BDNF/Nrf2 signaling pathway, thereby reducing ferroptosis in hippocampal neurons. Etomidate has been demonstrated to augment the antidepressant effects of ECT by upregulating BDNF/Nrf2 signaling and inhibiting hippocampal neuronal ferroptosis.117 These observations suggest that etomidate may exert adjunctive antidepressant effects through modulation of BDNF-dependent neuroprotective pathways.
Modulators Targeting Multiple Subunits—Neurosteroids
Neurosteroids are steroids synthesized within the central nervous system that regulate neuronal excitability through rapid, non-genomic mechanisms.120 They are primarily produced locally in the hippocampus and other brain regions. Based on structural characteristics, neurosteroids are broadly classified into pregnane neurosteroids (eg., allopregnanolone, APα) and sulfated neurosteroids (Table 3). These compounds have long been recognized for their sedative, anesthetic, and anticonvulsant properties in both animal models and humans. Their biological effects involve multiple mechanisms, including modulation of GABAARs, regulation of glutamatergic neurotransmission, and influence on synaptic plasticity.121
 |
Table 3 Molecular Targets and Pharmacological Actions of Neurosteroids in the Central Nervous System |
With respect to GABAARs, neurosteroids may function as either positive or negative allosteric modulators, depending on their molecular structure. Pregnane neurosteroids such as APα act as potent positive modulators of GABAARs, whereas sulfated neurosteroids generally function as negative modulators.103,122 The magnitude and direction of neurosteroid effects are influenced by GABAAR subunit composition; specific α and γ subunits modulate efficacy and potency, and receptors containing the δ subunit demonstrate particular sensitivity to neurosteroid-induced potentiation of GABA responses.123–125
Although the δ subunit does not constitute part of the canonical neurosteroid binding site, its presence enhances functional responses following ligand binding. Certain neurosteroids, including pregnenolone sulfate (PS) and dehydroepiandrosterone sulfate (DHEAS), also positively modulate NMDA-type glutamate receptors.126
Neurosteroids have demonstrated significant antidepressant effects through mechanisms involving modulation of the GABAergic and glutamatergic systems, regulation of BDNF signaling, anti-inflammatory activity, and normalization of HPA axis dysregulation. Direct administration of APα alleviates depressive-like behaviors in animal models of depression, and PS and DHEAS have demonstrated antidepressant effects in both preclinical and clinical studies.127–130
Neurosteroids exhibit particularly pronounced therapeutic effects in postpartum depression, likely related to their modulation of δ subunit–containing GABAARs, which are strongly implicated in this condition. Neurosteroid-based therapeutics, including brexanolone and zuranolone, have been developed and approved for the treatment of postpartum depression.131 Although mechanistic investigations have largely focused on GABAAR and NMDAR modulation, increasing evidence indicates that synaptic plasticity is a critical mediator of their antidepressant efficacy.
Neurosteroids regulate BDNF expression and contribute to neuronal protection and regeneration.132 Experimental findings indicate that APα increases BDNF levels in the hippocampus and amygdala, potentially through GABAAR-dependent mechanisms133 (Figure 4). In immature neurons, APα induces depolarization via GABAAR-mediated activation of voltage-gated L-type Ca2⁺ channels, resulting in altered chloride flux and increased intracellular Ca2⁺ signaling. Elevated Ca2⁺ levels may activate and phosphorylate calcium/calmodulin-dependent protein kinase II (CaMKII), thereby promoting transcription factor activation and increasing BDNF expression, ultimately supporting synaptic plasticity.134–136
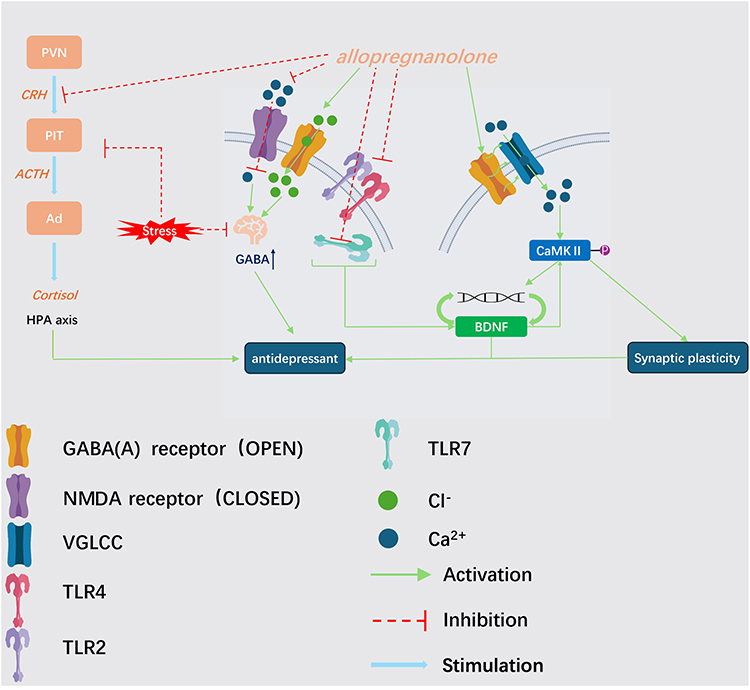 |
Figure 4 Molecular mechanisms underlying the antidepressant effects of allopregnanolone (APα). |
Additional evidence indicates that estradiol enhances BDNF expression in the mouse brain, promoting synaptogenesis and neuronal plasticity.137 Administration of progesterone has also been demonstrated to produce antidepressant-like effects; however, inhibition of its conversion to APα markedly attenuates these effects, indicating that APα mediates the primary antidepressant activity. The anti-inflammatory properties of neurosteroids further contribute to their effects on synaptic function.132
Exogenous APα inhibits lipopolysaccharide (LPS)-induced pro-inflammatory responses by preventing activation of Toll-like receptor 4 (TLR4) and suppressing signaling through MyD88-dependent TLR pathways, including TLR7 and TLR2.138,139 These actions preserve hippocampal neuronal viability and reduce inflammation-associated neural injury.140 Moreover, APα suppresses HPA axis hyperactivity, thereby facilitating regulation of acute stress responses, stabilizing BDNF expression, and protecting hippocampal neurons.121,141 Collectively, the multifaceted mechanisms of neurosteroids underscore their therapeutic potential as novel and broadly applicable antidepressant agents.
Summary and Perspectives
With the continued advancement of research on depression, therapeutic approaches have progressively shifted toward precision medicine. This review has examined the therapeutic potential of GABAAR modulators in depressive disorders. Owing to their complex pentameric structure and diverse subunit composition, GABAARs exhibit substantial functional heterogeneity. Conventional GABAAR modulators generally lack subunit selectivity, which may contribute to a broad spectrum of adverse effects. Accordingly, the development of subunit-selective GABAAR modulators represents a promising strategy for next-generation antidepressant therapies. Several representative selective GABAAR modulators have been discussed, and their pharmacological mechanisms and therapeutic profiles have been summarized.
Although neurosteroid-based agents have already been used in the treatment of postpartum depression, the broader clinical application of GABAAR-targeting compounds in depressive disorders remains an area of considerable potential. Furthermore, investigation of GABAAR modulators should extend beyond receptor-level mechanisms alone. Multiple agents, including etomidate and neurosteroid-based compounds, have been demonstrated to regulate BDNF expression.117 Given the functional interplay between the GABAergic system and BDNF signaling, pharmacological agents with multimodal mechanisms may offer a rational and promising direction for the development of future rapid-acting antidepressants.
Abbreviations
5-HT (5-hydroxytryptamine); AMPARs (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors); APα (allopregnanolone); BDNF (brain-derived neurotrophic factor); BZDs (benzodiazepines); CaMKII (calcium/calmodulin-dependent protein kinase II); CRH (corticotropin-releasing hormone); CUMS (chronic unpredictable mild stress); DHEAS (dehydroepiandrosterone sulfate); dlPFC (dorsolateral prefrontal cortex); ECT (electroconvulsive therapy); FST (forced swim test); GABA (gamma-aminobutyric acid); GABAARs (gamma-aminobutyric acid type A receptors); GABA-T (GABA transaminase); GTPase (guanosine triphosphatase); HPA (hypothalamic-pituitary-adrenal); IP3R (inositol trisphosphate receptor); LPS (lipopolysaccharide); MAOIs (monoamine oxidase inhibitors); MAPK/ERK (mitogen-activated protein kinase/extracellular signal-regulated kinase); mTOR (mammalian target of rapamycin); MyD88 (myeloid differentiation primary response 88); NAMs (negative allosteric modulators); NMDARs (N-methyl-D-aspartate receptors); Nrf2 (nuclear factor erythroid 2-related factor 2); PAMs (positive allosteric modulators); PFC (prefrontal cortex); PI3K-Akt (phosphatidylinositol 3-kinase-protein kinase B); PLCγ (phospholipase C gamma); PS (pregnenolone sulfate); PSD-95 (postsynaptic density protein 95); PVN (paraventricular nucleus); SST (somatostatin); SSRIs (selective serotonin reuptake inhibitors); TCA (tricarboxylic acid); TCAs (tricyclic antidepressants); TLR4 (Toll-like receptor 4); TRPC (transient receptor potential canonical); TrkB (tropomyosin receptor kinase B).
Data Sharing Statement
All data generated or analysed during this study are included in this article. Further enquiries can be directed to the corresponding author.
Ethical Statement
This narrative review is based exclusively on published literature and does not involve any original research with human participants, human tissue, or animal subjects. Therefore, ethical approval from an institutional review board and informed consent were not required.
Acknowledgments
We are particularly grateful to all the people who have given us help on our article.
Funding
No external funding received to conduct this study.
Disclosure
The authors declare that they have no competing interests.
References
1. Guo WF, Cao XL, Sheng L, Li JX, Zhang LK, Ma YZ. Expert consensus on diagnosis and treatment of major depressive disorder by integrated chinese and western medicine. Chin J Integr Tradit West Med. 2020;40(2):141–17.
2. Kendall KM, Van Assche E, Andlauer TFM, et al. The genetic basis of major depression. Psychol Med. 2021;51(13):2217–2230. doi:10.1017/S0033291721000441
3. Guidi J, Tomba E, Cosci F, et al. The role of staging in planning psychotherapeutic interventions in depression. J Clin Psychiatry. 2017;78(4):456–463. doi:10.4088/JCP.16r10736
4. Olubodun-Obadun TG, Ishola IO, Adesokan TP, et al. Antidepressant- and anxiolytic-like actions of Cajanus cajan seed extract mediated through monoaminergic, nitric oxide-cyclic GMP and GABAergic pathways. J Ethnopharmacol. 2023;306:116142. doi:10.1016/j.jep.2023.116142
5. Mundula T, Baldi S, Gerace E, et al. Role of the intestinal microbiota in the genesis of major depression and the response to antidepressant drug therapy: a narrative review. Biomedicines. 2023;11(2):550. doi:10.3390/biomedicines11020550
6. Hillhouse TM, Porter JH. A brief history of the development of antidepressant drugs: from monoamines to glutamate. Exp Clin Psychopharmacol. 2015;23(1):1–21. doi:10.1037/a0038550
7. Papakostas GI. Tolerability of modern antidepressants. J Clin Psychiatry. 2008;69:8–13.
8. Basso L, Bönke L, Aust S, et al. Antidepressant and neurocognitive effects of serial ketamine administration versus ECT in depressed patients. J Psychiatr Res. 2020;123:1–8. doi:10.1016/j.jpsychires.2020.01.002
9. Zanos P, Gould TD. Mechanisms of ketamine action as an antidepressant. Mol Psychiatry. 2018;23(4):801–811. doi:10.1038/mp.2017.255
10. Ren Z, Pribiag H, Jefferson SJ, et al. Bidirectional homeostatic regulation of a depression-related brain state by gamma-aminobutyric acidergic deficits and ketamine treatment. Biol Psychiatry. 2016;80(6):457–468. doi:10.1016/j.biopsych.2016.02.009
11. Zhou YL, Liu W-J, Wang C-Y, et al. Cardiovascular effects of repeated subanaesthetic ketamine infusion in depression. J Psychopharmacol. 2021;35(2):159–167. doi:10.1177/0269881120936909
12. Buchholz F, Meffert M, Bazin P-L, et al. Highfield imaging of the subgenual anterior cingulate cortex in uni- and bipolar depression. Front Psychiatry. 2024;15:1462919. doi:10.3389/fpsyt.2024.1462919
13. Rigucci S, Serafini G, Pompili M, et al. Anatomical and functional correlates in major depressive disorder: the contribution of neuroimaging studies. World J Biol Psychiatry. 2010;11(2 Pt 2):165–180. doi:10.3109/15622970903131571
14. Duman RS, Voleti B. Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents. Trends Neurosci. 2012;35(1):47–56. doi:10.1016/j.tins.2011.11.004
15. Mayegowda SB, Thomas C. Glial pathology in neuropsychiatric disorders: a brief review. J Basic Clin Physiol Pharmacol. 2019;30(4). doi:10.1515/jbcpp-2018-0120
16. Godfrey KEM, Gardner AC, Kwon S, et al. Differences in excitatory and inhibitory neurotransmitter levels between depressed patients and healthy controls: a systematic review and meta-analysis. J Psychiatr Res. 2018;105:33–44. doi:10.1016/j.jpsychires.2018.08.015
17. Ghit A, Assal D, Al-Shami AS, et al. GABA(A) receptors: structure, function, pharmacology, and related disorders. J Genet Eng Biotechnol. 2021;19(1):123. doi:10.1186/s43141-021-00224-0
18. Zhang Q, Zhu L, Li H, et al. Insights and progress on the biosynthesis, metabolism, and physiological functions of gamma-aminobutyric acid (GABA): a review. PeerJ. 2024;12:e18712. doi:10.7717/peerj.18712
19. Pehrson AL, Sanchez C. Altered γ-aminobutyric acid neurotransmission in major depressive disorder: a critical review of the supporting evidence and the influence of serotonergic antidepressants. Drug Des Devel Ther. 2015;9:603–624. doi:10.2147/DDDT.S62912
20. Rowley NM, Madsen KK, Schousboe A, et al. Glutamate and GABA synthesis, release, transport and metabolism as targets for seizure control. Neurochem Int. 2012;61(4):546–558. doi:10.1016/j.neuint.2012.02.013
21. Wolpaw JR. Harnessing neuroplasticity for clinical applications. Brain. 2012;135(Pt 4):e215–e216. doi:10.1093/brain/aws017
22. Castrén E. Neuronal network plasticity and recovery from depression. JAMA Psychiatry. 2013;70(9):983–989. doi:10.1001/jamapsychiatry.2013.1
23. Gupta VK, You Y, Gupta V, et al. TrkB receptor signalling: implications in neurodegenerative, psychiatric and proliferative disorders. Int J Mol Sci. 2013;14(5):10122–10142. doi:10.3390/ijms140510122
24. Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012;4(9):a011189. doi:10.1101/cshperspect.a011189
25. Tao W, Dong Y, Su Q, et al. Liquiritigenin reverses depression-like behavior in unpredictable chronic mild stress-induced mice by regulating PI3K/Akt/mTOR mediated BDNF/TrkB pathway. Behav Brain Res. 2016;308:177–186. doi:10.1016/j.bbr.2016.04.039
26. Leal G, Comprido D, Duarte CB. BDNF-induced local protein synthesis and synaptic plasticity. Neuropharmacology. 2014;76:639–656. doi:10.1016/j.neuropharm.2013.04.005
27. Yoshii A, Constantine-Paton M. BDNF induces transport of PSD-95 to dendrites through PI3K-AKT signaling after NMDA receptor activation. Nat Neurosci. 2007;10(6):702–711. doi:10.1038/nn1903
28. Numakawa T, Kajihara R. Roles of MAPKs, including those activated by BDNF/TrkB, and their contribution in neurodegenerative diseases. Int J Mol Sci. 2026;27(2):984. doi:10.3390/ijms27020984
29. Keifer J. Regulation of AMPAR trafficking in synaptic plasticity by BDNF and the impact of neurodegenerative disease. J Neurosci Res. 2022;100(4):979–991. doi:10.1002/jnr.25022
30. Chuang HW, Wang T-Y, Huang -C-C, et al. Echinacoside exhibits antidepressant-like effects through AMPAR-Akt/ERK-mTOR pathway stimulation and BDNF expression in mice. Chin Med. 2022;17(1):9. doi:10.1186/s13020-021-00549-5
31. Hellmann J, Rommelspacher H, Wernicke C. Long-term ethanol exposure impairs neuronal differentiation of human neuroblastoma cells involving neurotrophin-mediated intracellular signaling and in particular protein kinase C. Alcohol Clin Exp Res. 2009;33(3):538–550.
32. Sotogaku N, Tully SE, Gama CI, et al. Activation of phospholipase C pathways by a synthetic chondroitin sulfate-E tetrasaccharide promotes neurite outgrowth of dopaminergic neurons. J Neurochem. 2007;103(2):749–760. doi:10.1111/j.1471-4159.2007.04849.x
33. Liu M, Kay JC, Shen S, et al. Endogenous BDNF augments NMDA receptor phosphorylation in the spinal cord via PLCγ, PKC, and PI3K/Akt pathways during colitis. J Neuroinflammation. 2015;12:151. doi:10.1186/s12974-015-0371-z
34. Tao W, Chen Q, Zhou W, et al. Persistent inflammation-induced up-regulation of brain-derived neurotrophic factor (BDNF) promotes synaptic delivery of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor GluA1 subunits in descending pain modulatory circuits. J Biol Chem. 2014;289(32):22196–22204.
35. Roberts DS, Hu Y, Lund IV, et al. Brain-derived neurotrophic factor (BDNF)-induced synthesis of early growth response factor 3 (Egr3) controls the levels of type A GABA receptor alpha 4 subunits in hippocampal neurons. J Biol Chem. 2006;281(40):29431–29435.
36. Colgan LA, Hu M, Misler JA, et al. PKCα integrates spatiotemporally distinct Ca(2+) and autocrine BDNF signaling to facilitate synaptic plasticity. Nat Neurosci. 2018;21(8):1027–1037.
37. Ishii T, Warabi E, Mann GE. Circadian control of p75 neurotrophin receptor leads to alternate activation of Nrf2 and c-Rel to reset energy metabolism in astrocytes via brain-derived neurotrophic factor. Free Radic Biol Med. 2018;119:34–44. doi:10.1016/j.freeradbiomed.2018.01.026
38. Holt LM, Hernandez RD, Pacheco NL, et al. Astrocyte morphogenesis is dependent on BDNF signaling via astrocytic TrkB.T1. Elife. 2019;8: e44667
39. Tessarollo L, Yanpallewar S. TrkB truncated isoform receptors as transducers and determinants of bdnf functions. Front Neurosci. 2022;16:847572. doi:10.3389/fnins.2022.847572
40. Bazzari AH, Bazzari FH. BDNF therapeutic mechanisms in neuropsychiatric disorders. Int J Mol Sci. 2022;23(15).
41. Lu VB, Colmers WF, Smith PA. Long-term actions of BDNF on inhibitory synaptic transmission in identified neurons of the rat substantia gelatinosa. J Neurophysiol. 2012;108(2):441–452. doi:10.1152/jn.00457.2011
42. Lazo OM, Gonzalez A, Ascaño M, et al. BDNF regulates Rab11-mediated recycling endosome dynamics to induce dendritic branching. J Neurosci. 2013;33(14):6112–6122. doi:10.1523/JNEUROSCI.4630-12.2013
43. Kowiański P, Lietzau G, Czuba E, et al. BDNF: a key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol Neurobiol. 2018;38(3):579–593.
44. Nakata H, Nakamura S. Brain-derived neurotrophic factor regulates AMPA receptor trafficking to post-synaptic densities via IP3R and TRPC calcium signaling. FEBS Lett. 2007;581(10):2047–2054.
45. Gangarossa G, Perez S, Dembitskaya Y, et al. BDNF controls bidirectional endocannabinoid plasticity at corticostriatal synapses. Cereb Cortex. 2020;30(1):197–214. doi:10.1093/cercor/bhz081
46. Toader C, Serban M, Munteanu O, et al. From synaptic plasticity to neurodegeneration: bdnf as a transformative target in medicine. Int J Mol Sci. 2025;26(9):4271.
47. Olsen RW. GABA(A) receptor: positive and negative allosteric modulators. Neuropharmacology. 2018;136(Pt A):10–22. doi:10.1016/j.neuropharm.2018.01.036
48. Winsky-Sommerer R. Role of GABAA receptors in the physiology and pharmacology of sleep. Eur J Neurosci. 2009;29(9):1779–1794. doi:10.1111/j.1460-9568.2009.06716.x
49. Wisden W, Laurie DJ, Monyer H, et al. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J Neurosci. 1992;12(3):1040–1062. doi:10.1523/JNEUROSCI.12-03-01040.1992
50. Jechlinger M, Pelz R, Tretter V, et al. Subunit composition and quantitative importance of hetero-oligomeric receptors: gaba a receptors containing α 6 subunits. J Neurosci. 1998;18(7):2449–2457. doi:10.1523/JNEUROSCI.18-07-02449.1998
51. Nusser Z, Sieghart W, Somogyi P. Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci. 1998;18(5):1693–1703. doi:10.1523/JNEUROSCI.18-05-01693.1998
52. Gao B, Fritschy JM. Selective allocation of GABAA receptors containing the alpha 1 subunit to neurochemically distinct subpopulations of rat hippocampal interneurons. Eur J Neurosci. 1994;6(5):837–853. doi:10.1111/j.1460-9568.1994.tb00994.x
53. Nusser Z, Sieghart W, Benke D, et al. Differential synaptic localization of two major gamma-aminobutyric acid type A receptor alpha subunits on hippocampal pyramidal cells. Proc Natl Acad Sci U S A. 1996;93(21):11939–11944. doi:10.1073/pnas.93.21.11939
54. Sperk G, Schwarzer C, Tsunashima K, et al. GABA(A) receptor subunits in the rat hippocampus I: immunocytochemical distribution of 13 subunits. Neuroscience. 1997;80(4):987–1000. doi:10.1016/S0306-4522(97)00146-2
55. Bohlhalter S, Weinmann O, Mohler H, et al. Laminar compartmentalization of GABAA-receptor subtypes in the spinal cord: an immunohistochemical study. J Neurosci. 1996;16(1):283–297. doi:10.1523/JNEUROSCI.16-01-00283.1996
56. Paul J, Zeilhofer HU, Fritschy JM. Selective distribution of GABA(A) receptor subtypes in mouse spinal dorsal horn neurons and primary afferents. J Comp Neurol. 2012;520(17):3895–3911. doi:10.1002/cne.23129
57. May AC, Fleischer W, Kletke O, et al. Benzodiazepine-site pharmacology on GABAA receptors in histaminergic neurons. Br J Pharmacol. 2013;170(1):222–232. doi:10.1111/bph.12280
58. Browne SH, Kang J, Akk G, et al. Kinetic and pharmacological properties of gaba a receptors in single thalamic neurons and gaba a subunit expression. J Neurophysiol. 2001;86(5):2312–2322. doi:10.1152/jn.2001.86.5.2312
59. Huang S, Hokenson K, Bandyopadhyay S, et al. Brief dark exposure reduces tonic inhibition in visual cortex. J Neurosci. 2015;35(48):15916–15920. doi:10.1523/JNEUROSCI.1813-15.2015
60. Mortensen M, Smart TG. Extrasynaptic alphabeta subunit GABAA receptors on rat hippocampal pyramidal neurons. J Physiol. 2006;577(Pt 3):841–856. doi:10.1113/jphysiol.2006.117952
61. Jia F, Pignataro L, Schofield CM, et al. An extrasynaptic gaba a receptor mediates tonic inhibition in thalamic vb neurons. J Neurophysiol. 2005;94(6):4491–4501. doi:10.1152/jn.00421.2005
62. Liang J, Suryanarayanan A, Abriam A, et al. Mechanisms of reversible gaba a receptor plasticity after ethanol intoxication. J Neurosci. 2007;27(45):12367–12377. doi:10.1523/JNEUROSCI.2786-07.2007
63. Sur C, Farrar SJ, Kerby J, et al. Preferential coassembly of α4 and δ subunits of the γ-aminobutyric acid a receptor in rat thalamus. Mol Pharmacol. 1999;56(1):110–115. doi:10.1124/mol.56.1.110
64. Caraiscos VB, Elliott EM, You-Ten KE, et al. Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by alpha5 subunit-containing gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A. 2004;101(10):3662–3667. doi:10.1073/pnas.0307231101
65. Yamada J, Furukawa T, Ueno S, et al. Molecular basis for the GABAA receptor-mediated tonic inhibition in rat somatosensory cortex. Cereb Cortex. 2007;17(8):1782–1787. doi:10.1093/cercor/bhl087
66. Shen Q, Lal R, Luellen BA, et al. gamma-Aminobutyric acid-type A receptor deficits cause hypothalamic-pituitary-adrenal axis hyperactivity and antidepressant drug sensitivity reminiscent of melancholic forms of depression. Biol Psychiatry. 2010;68(6):512–520. doi:10.1016/j.biopsych.2010.04.024
67. Hooper A, Paracha R, Maguire J. Seizure-induced activation of the HPA axis increases seizure frequency and comorbid depression-like behaviors. Epilepsy Behav. 2018;78:124–133. doi:10.1016/j.yebeh.2017.10.025
68. Volkova EP, Rozov AV, Nadareishvili GG, et al. Corticosterone induces rapid increase in the amplitude of inhibitory response in hippocampal synapses with asynchronous gaba release. Bull Exp Biol Med. 2016;160(5):628–631. doi:10.1007/s10517-016-3234-4
69. Luscher B, Shen Q, Sahir N. The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry. 2011;16(4):383–406.
70. Earnheart JC, Schweizer C, Crestani F, et al. GABAergic control of adult hippocampal neurogenesis in relation to behavior indicative of trait anxiety and depression states. J Neurosci. 2007;27(14):3845–3854. doi:10.1523/JNEUROSCI.3609-06.2007
71. Li R, Wang Y, Yang Y, et al. The α1 and γ2 subunit-containing GABA(A) receptor-mediated inhibitory transmission in the anteroventral bed nucleus of stria terminalis is involved in the regulation of anxiety in rats with substantia nigra lesions. Neuropharmacology. 2023;237:109645. doi:10.1016/j.neuropharm.2023.109645
72. Vinkers CH, Olivier B. Mechanisms underlying tolerance after long-term benzodiazepine use: a future for subtype-selective gaba(a) receptor modulators? Adv Pharmacol Sci. 2012;2012:416864. doi:10.1155/2012/416864
73. Boccalaro IL, Cristiá-Lara L, Schwerdel C, et al. Cell type-specific distribution of GABA A receptor subtypes in the mouse dorsal striatum. J Comp Neurol. 2019;527(12):2030–2046. doi:10.1002/cne.24665
74. Bernardo A, Lee P, Marcotte M, et al. Symptomatic and neurotrophic effects of GABAA receptor positive allosteric modulation in a mouse model of chronic stress. Neuropsychopharmacology. 2022;47(9):1608–1619. doi:10.1038/s41386-022-01360-y
75. Kaufmann WA, Humpel C, Alheid GF, et al. Compartmentation of alpha 1 and alpha 2 GABA(A) receptor subunits within rat extended amygdala: implications for benzodiazepine action. Brain Res. 2003;964(1):91–99. doi:10.1016/S0006-8993(02)04082-9
76. Olsen RW, Wallner M, Rogawski MA, et al. GABA(A) Receptors, Seizures, and Epilepsy, in Jasper’s Basic Mechanisms of the Epilepsies. Noebels JL, Editors.. New York:Oxford University Press; 2024:1025–1046.
77. Vollenweider I, Smith KS, Keist R, et al. Antidepressant-like properties of α2-containing GABA(A) receptors. Behav Brain Res. 2011;217(1):77–80. doi:10.1016/j.bbr.2010.10.009
78. Alizadeh Pahlavani H. Possible role of exercise therapy on depression: effector neurotransmitters as key players. Behav Brain Res. 2024;459:114791. doi:10.1016/j.bbr.2023.114791
79. Prevot TD, Li G, Vidojevic A, et al. Novel benzodiazepine-like ligands with various anxiolytic, antidepressant, or pro-cognitive profiles. Mol Neuropsychiatry. 2019;5(2):84–97. doi:10.1159/000496086
80. Jacob TC. Neurobiology and therapeutic potential of α5-gaba type a receptors. Front Mol Neurosci. 2019;12:179. doi:10.3389/fnmol.2019.00179
81. Song YH, Yoon J, Lee SH. The role of neuropeptide somatostatin in the brain and its application in treating neurological disorders. Exp Mol Med. 2021;53(3):328–338. doi:10.1038/s12276-021-00580-4
82. Lin CW, Chang L-C, Ma T, et al. Older molecular brain age in severe mental illness. Mol Psychiatry. 2021;26(7):3646–3656. doi:10.1038/s41380-020-0834-1
83. Lin LC, Sibille E. Somatostatin, neuronal vulnerability and behavioral emotionality. Mol Psychiatry. 2015;20(3):377–387. doi:10.1038/mp.2014.184
84. Abbas AI, Sundiang MJ, Henoch B, et al. Somatostatin interneurons facilitate hippocampal-prefrontal synchrony and prefrontal spatial encoding. Neuron. 2018;100(4):926–939.e3.
85. Fee C, Prevot TD, Misquitta K, et al. Behavioral deficits induced by somatostatin-positive gaba neuron silencing are rescued by alpha 5 gaba-a receptor potentiation. Int J Neuropsychopharmacol. 2021;24(6):505–518. doi:10.1093/ijnp/pyab002
86. Fuchs T, Jefferson SJ, Hooper A, et al. Disinhibition of somatostatin-positive GABAergic interneurons results in an anxiolytic and antidepressant-like brain state. Mol Psychiatry. 2017;22(6):920–930. doi:10.1038/mp.2016.188
87. Jefferson SJ, Feng M, Chon U, et al. Disinhibition of somatostatin interneurons confers resilience to stress in male but not female mice. Neurobiol Stress. 2020;13:100238. doi:10.1016/j.ynstr.2020.100238
88. Ju YH, Guzzo A, Chiu MW, et al. Distinct properties of murine alpha 5 gamma-aminobutyric acid type a receptors revealed by biochemical fractionation and mass spectroscopy. J Neurosci Res. 2009;87(8):1737–1747. doi:10.1002/jnr.21991
89. Fischell J, Van Dyke AM, Kvarta MD, et al. Rapid antidepressant action and restoration of excitatory synaptic strength after chronic stress by negative modulators of alpha5-containing gabaa receptors. Neuropsychopharmacology. 2015;40(11):2499–2509. doi:10.1038/npp.2015.112
90. Sieghart W, Ramerstorfer J, Sarto-Jackson I, et al. A novel GABA A receptor pharmacology: drugs interacting with the α + β - interface. Br J Pharmacol. 2012;166(2):476–485. doi:10.1111/j.1476-5381.2011.01779.x
91. Sigel E, Steinmann ME. Structure, function, and modulation of GABA(A) receptors. J Biol Chem. 2012;287(48):40224–40231. doi:10.1074/jbc.R112.386664
92. Yu L, Zhu X, Peng K, et al. Propofol alleviates anxiety-like behaviors associated with pain by inhibiting the hyperactivity of pvn crh neurons via gaba a receptor β 3 subunits. Adv Sci. 2024;11(28):e2309059. doi:10.1002/advs.202309059
93. Ferando I, Mody I. Altered gamma oscillations during pregnancy through loss of δ subunit-containing GABA(A) receptors on parvalbumin interneurons. Front Neural Circuits. 2013;7:144. doi:10.3389/fncir.2013.00144
94. Smith KS, Rudolph U. Anxiety and depression: mouse genetics and pharmacological approaches to the role of GABA(A) receptor subtypes. Neuropharmacology. 2012;62(1):54–62. doi:10.1016/j.neuropharm.2011.07.026
95. Atack JR. GABAA receptor alpha2/alpha3 subtype-selective modulators as potential nonsedating anxiolytics. Curr Top Behav Neurosci. 2010;2:331–360. doi:10.1007/7854_2009_30
96. Speigel I, Bichler EK, García PS. The influence of regional distribution and pharmacologic specificity of gaba(a)r subtype expression on anesthesia and emergence. Front Syst Neurosci. 2017;11:58. doi:10.3389/fnsys.2017.00058
97. Chen X, van Gerven J, Cohen A, et al. Human pharmacology of positive GABA-A subtype-selective receptor modulators for the treatment of anxiety. Acta Pharmacol Sin. 2019;40(5):571–582. doi:10.1038/s41401-018-0185-5
98. Philip AB, Brohan J, Goudra B. The role of gaba receptors in anesthesia and sedation: an updated review. CNS Drugs. 2025;39(1):39–54. doi:10.1007/s40263-024-01128-6
99. McGrath M, Yu Z, Jayakar SS, et al. Etomidate and etomidate analog binding and positive modulation of γ-aminobutyric acid type a receptors: evidence for a state-dependent cutoff effect. Anesthesiology. 2018;129(5):959–969. doi:10.1097/ALN.0000000000002356
100. Valk BI, Struys M. Etomidate and its analogs: a review of pharmacokinetics and pharmacodynamics. Clin Pharmacokinet. 2021;60(10):1253–1269. doi:10.1007/s40262-021-01038-6
101. Jayakar SS, Zhou X, Chiara DC, et al. Multiple propofol-binding sites in a γ-aminobutyric acid type A receptor (GABAAR) identified using a photoreactive propofol analog. J Biol Chem. 2014;289(40):27456–27468. doi:10.1074/jbc.M114.581728
102. Li GD, Chiara DC, Cohen JB, et al. Numerous classes of general anesthetics inhibit etomidate binding to gamma-aminobutyric acid type A (GABAA) receptors. J Biol Chem. 2010;285(12):8615–8620.
103. Sugasawa Y. Neurosteroid Binding and Actions on GABA(A) Receptors. Juntendo Iji Zasshi. 2024;70(3):239–244. doi:10.14789/jmj.JMJ24-0002-R
104. Vithlani M, Terunuma M, Moss SJ. The dynamic modulation of GABA(A) receptor trafficking and its role in regulating the plasticity of inhibitory synapses. Physiol Rev. 2011;91(3):1009–1022. doi:10.1152/physrev.00015.2010
105. Sigel E, Lüscher BP. A closer look at the high affinity benzodiazepine binding site on GABAA receptors. Curr Top Med Chem. 2011;11(2):241–246. doi:10.2174/156802611794863562
106. Zetsen SPG, Schellekens AFA, Paling EP, et al. Cognitive functioning in long-term benzodiazepine users. Eur Addict Res. 2022;28(5):377–381. doi:10.1159/000525988
107. Fond G, et al. Benzodiazepine long-term administration is associated with impaired attention/working memory in schizophrenia: results from the national multicentre FACE-SZ data set. Eur Arch Psychiatry Clin Neurosci. 2018;268(1):17–26.
108. Atack JR. GABAA receptor subtype-selective modulators. I. α2/α3-selective agonists as non-sedating anxiolytics. Curr Top Med Chem. 2011;11(9):1176–1202. doi:10.2174/156802611795371350
109. Cerne R, Chrzanowska A, Lippa A, et al. Nonsedating anxiolytics. Pharmacol Biochem Behav. 2024;245:173895.
110. Engin E, Benham RS, Rudolph U. An Emerging Circuit Pharmacology of GABA(A) Receptors. Trends Pharmacol Sci. 2018;39(8):710–732. doi:10.1016/j.tips.2018.04.003
111. Luscher B, Maguire JL, Rudolph U, et al. GABA(A) receptors as targets for treating affective and cognitive symptoms of depression. Trends Pharmacol Sci. 2023;44(9):586–600. doi:10.1016/j.tips.2023.06.009
112. Echeverria-Villalobos M, Fabian CA, Mitchell JG, et al. Cannabinoids and general anesthetics: revisiting molecular mechanisms of their pharmacological interactions. Anesth Analg. 2025;140(6):1401–1413. doi:10.1213/ANE.0000000000007313
113. Kim JJ, Gharpure A, Teng J, et al. Shared structural mechanisms of general anaesthetics and benzodiazepines. Nature. 2020;585(7824):303–308. doi:10.1038/s41586-020-2654-5
114. Hansen SB. Mechanisms of General Anesthesia. Annu Rev Biochem. 2025;94(1):503–530. doi:10.1146/annurev-biochem-030222-121430
115. Yan Y, Ma H, Zhao J. Propofol brings on the light for depression therapy. Neurosci Bull. 2023;39(12):1891–1894. doi:10.1007/s12264-023-01133-7
116. Lihua P, Su M, Ke W, et al. Different regimens of intravenous sedatives or hypnotics for electroconvulsive therapy (ECT) in adult patients with depression. Cochrane Database Syst Rev. 2014;2014(4):Cd009763. doi:10.1002/14651858.CD009763.pub2
117. Li X, Hu J, Zang X, et al. Etomidate improves the antidepressant effect of electroconvulsive therapy by suppressing hippocampal neuronal ferroptosis via upregulating BDNF/Nrf2. Mol Neurobiol. 2023;60(11):6584–6597. doi:10.1007/s12035-023-03499-1
118. Gurel SC, Ozden HC, Karahan S, et al. The superiority of ketofol and etomidate against propofol or thiopental anesthesia for ECT. Asian J Psychiatr. 2022;72:103090. doi:10.1016/j.ajp.2022.103090
119. Ishii T, Warabi E, Mann GE. Circadian control of BDNF-mediated Nrf2 activation in astrocytes protects dopaminergic neurons from ferroptosis. Free Radic Biol Med. 2019;133:169–178. doi:10.1016/j.freeradbiomed.2018.09.002
120. Reddy DS. Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog Brain Res. 2010;186:113–137.
121. Bali A, Jaggi AS. Multifunctional aspects of allopregnanolone in stress and related disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2014;48:64–78. doi:10.1016/j.pnpbp.2013.09.005
122. Akk G, Covey DF, Evers AS, et al. The influence of the membrane on neurosteroid actions at GABA(A) receptors. Psychoneuroendocrinology. 2009;34(1):S59–66. doi:10.1016/j.psyneuen.2009.05.020
123. Feng HJ, Forman SA. Comparison of αβδ and αβγ GABA(A) receptors: allosteric modulation and identification of subunit arrangement by site-selective general anesthetics. Pharmacol Res. 2018;133:289–300. doi:10.1016/j.phrs.2017.12.031
124. Sun MY, Shu H-J, Benz A, et al. Chemogenetic isolation reveals synaptic contribution of δ gaba a receptors in mouse dentate granule neurons. J Neurosci. 2018;38(38):8128–8145. doi:10.1523/JNEUROSCI.0799-18.2018
125. Spigelman I, Li Z, Liang J, et al. Reduced inhibition and sensitivity to neurosteroids in hippocampus of mice lacking the gaba a receptor δ subunit. J Neurophysiol. 2003;90(2):903–910. doi:10.1152/jn.01022.2002
126. Wang C, Marx CE, Morrow AL, et al. Neurosteroid modulation of GABAergic neurotransmission in the central amygdala: a role for NMDA receptors. Neurosci Lett. 2007;415(2):118–123. doi:10.1016/j.neulet.2007.01.004
127. Iwata S, Wakita M, Shin M-C, et al. Modulation of allopregnanolone on excitatory transmitters release from single glutamatergic terminal. Brain Res Bull. 2013;93:39–46. doi:10.1016/j.brainresbull.2012.11.002
128. Khisti RT, Chopde CT. Serotonergic agents modulate antidepressant-like effect of the neurosteroid 3alpha-hydroxy-5alpha-pregnan-20-one in mice. Brain Res. 2000;865(2):291–300. doi:10.1016/S0006-8993(00)02373-8
129. Espallergues J, Mamiya T, Vallée M, et al. The antidepressant-like effects of the 3β-hydroxysteroid dehydrogenase inhibitor trilostane in mice is related to changes in neuroactive steroid and monoamine levels. Neuropharmacology. 2012;62(1):492–502. doi:10.1016/j.neuropharm.2011.09.005
130. Khisti RT, Chopde CT, Jain SP. Antidepressant-like effect of the neurosteroid 3alpha-hydroxy-5alpha-pregnan-20-one in mice forced swim test. Pharmacol Biochem Behav. 2000;67(1):137–143. doi:10.1016/S0091-3057(00)00300-2
131. Kargbo RB. Neurosteroids and postpartum depression: the mechanism, efficacy, and approval of brexanolone and zurzuvae. ACS Med Chem Lett. 2023;14(10):1326–1328. doi:10.1021/acsmedchemlett.3c00388
132. Frye CA, Cleveland DM, Sadarangani A, et al. Progesterone promotes anti-anxiety/depressant-like behavior and trophic actions of bdnf in the hippocampus of female nuclear progesterone receptor, but not 5α-reductase, knockout mice. Int J Mol Sci. 2025;26(3):1173. doi:10.3390/ijms26031173
133. Naert G, Maurice T, Tapia-Arancibia L, et al. Neuroactive steroids modulate HPA axis activity and cerebral brain-derived neurotrophic factor (BDNF) protein levels in adult male rats. Psychoneuroendocrinology. 2007;32(8–10):1062–1078. doi:10.1016/j.psyneuen.2007.09.002
134. Wang JM, Brinton RD. Allopregnanolone-induced rise in intracellular calcium in embryonic hippocampal neurons parallels their proliferative potential. BMC Neurosci. 2008;9(Suppl 2):S11. doi:10.1186/1471-2202-9-S2-S11
135. Jagasia R, Steib K, Englberger E, et al. GABA-cAMP response element-binding protein signaling regulates maturation and survival of newly generated neurons in the adult hippocampus. J Neurosci. 2009;29(25):7966–7977. doi:10.1523/JNEUROSCI.1054-09.2009
136. Tsutsui K. Neurosteroid biosynthesis and action during cerebellar development. Cerebellum. 2012;11(2):414–415. doi:10.1007/s12311-011-0341-7
137. Haraguchi S, Sasahara K, Shikimi H, et al. Estradiol promotes purkinje dendritic growth, spinogenesis, and synaptogenesis during neonatal life by inducing the expression of BDNF. Cerebellum. 2012;11(2):416–417. doi:10.1007/s12311-011-0342-6
138. Balan I, Aurelian L, Schleicher R, et al. Neurosteroid allopregnanolone (3α,5α-THP) inhibits inflammatory signals induced by activated MyD88-dependent toll-like receptors. Transl Psychiatry. 2021;11(1):145. doi:10.1038/s41398-021-01266-1
139. Balan I, Beattie MC, O’Buckley TK, et al. Endogenous neurosteroid (3α,5α)3-hydroxypregnan-20-one inhibits toll-like-4 receptor activation and pro-inflammatory signaling in macrophages and brain. Sci Rep. 2019;9(1):1220. doi:10.1038/s41598-018-37409-6
140. Izumi Y, O’Dell KA, Cashikar AG, et al. Neurosteroids mediate and modulate the effects of pro-inflammatory stimulation and toll-like receptors on hippocampal plasticity and learning. PLoS One. 2024;19(6):e0304481. doi:10.1371/journal.pone.0304481
141. Misztal T, Młotkowska P, Marciniak E, et al. Effects of stress and allopregnanolone on the expression of neurotrophins and trkb receptor in the sheep hippocampus. Int J Mol Sci. 2025;26(13):6190. doi:10.3390/ijms26136190
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.