Back to Journals » Drug Design, Development and Therapy » Volume 20
Fused Indole-Diazepines with a Bridgehead Nitrogen Atom: Synthesis and Pharmaceutical Significance
Authors Zhou M, Yang X, Wang S ![]()
Received 4 December 2025
Accepted for publication 22 March 2026
Published 8 April 2026 Volume 2026:20 554857
DOI https://doi.org/10.2147/DDDT.S554857
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Solomon Tadesse Zeleke
Mengyang Zhou,1 Xiaohong Yang,2 Shutao Wang3
1Department of Pharmacy, China-Japan Union Hospital of Jilin University, Changchun, 130033, People’s Republic of China; 2School of Pharmaceutical Sciences, Jilin University, Changchun, 130021, People’s Republic of China; 3Department of Pharmacy, The Second Hospital of Jilin University, Changchun, 130041, People’s Republic of China
Correspondence: Shutao Wang, Department of Pharmacy, The Second Hospital of Jilin University, Changchun, 130041, People’s Republic of China, Email [email protected]
Abstract: Indole-fused diazepines with a bridgehead nitrogen atom represent a unique and privileged scaffold in medicinal chemistry, offering considerable potential for the development of novel pharmaceuticals. The bridgehead nitrogen atom acts as a key pharmacophoric element. It rigidifies the molecular conformation, modulates electronic properties, and participates in target binding via hydrogen bonding or electrostatic interactions. These features significantly influence biological activity, target selectivity, and pharmacokinetic profiles. This review comprehensively summarizes the synthesis, biological activities, and therapeutic potential of indole-fused diazepines, with emphasis on their structural diversity and pharmacological properties. We highlight recent advances in synthetic methodologies as well as important historical studies. We also discuss their implications for drug discovery, provide insights into structure-activity relationships (SAR), and propose future research directions for this class of compounds.
Keywords: indole-fused diazepines, synthetic methodologies, biological activities, structure-activity relationships
Introduction
In the vast landscape of medicinal compounds, certain molecular frameworks have emerged as privileged structures due to their exceptional pharmacological potential.1 One such privileged scaffold arises from the fusion of a benzene ring with a 1,4-diazepine moiety. This framework has shown remarkable efficacy in the treatment of central nervous system (CNS) disorders. Prominent examples include diazepam, estazolam, and lorazepam, which remain indispensable in modern clinical practice.2 Modification of these structures through the incorporation of heterocycles such as pyrrole, furan, and indole has yielded novel scaffolds with enhanced therapeutic potential. Furthermore, the strategic repositioning of nitrogen atoms within the diazepine ring has facilitated the exploration of various isomers, including 1,2-diazepines, 1,3-diazepines, and 1,5-diazepines (Figure 1). These modifications diversify the chemical space and generate diverse isomers, encompassing linear, angular, and peri-fused forms, each with distinct structural and functional characteristics. Each of these isomers possesses distinct structural and functional properties, thereby enriching the medicinal chemistry toolkit.
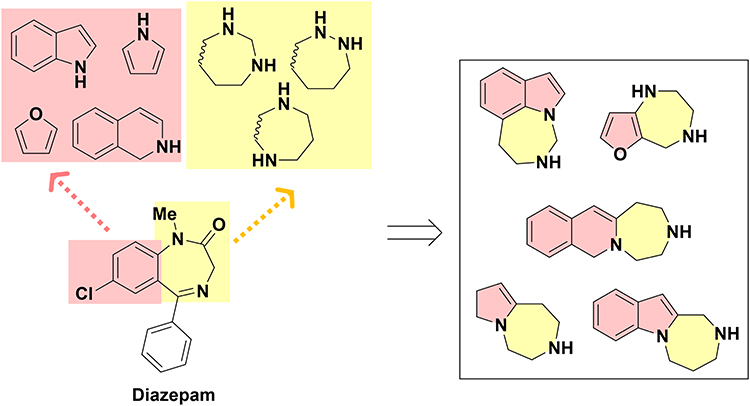 |
Figure 1 Molecular modification of diazepam. |
Indole and diazepine are two of the most impactful heterocyclic structures in drug design.3–6 Indole, in particular, is distinguished for its unique biological activities and widespread occurrence in natural products, bioactive compounds, and pharmaceuticals.7,8 The fusion of indole with diazepine through a bridgehead nitrogen atom creates a privileged scaffold with significant potential for drug discovery.9,10 The bridgehead nitrogen atom is not merely a structural linker but a pivotal pharmacophore. Its fixed position in the tricyclic core restricts conformational flexibility, ensuring optimal alignment with biological targets. Additionally, its electron density enables specific interactions with active site residues, directly influencing target selectivity and potency. Furthermore, the increasing interest in seven-membered heterocycles, exemplified by diazepine, can be attributed not only to their diverse bioactivities but also to their prevalence across a broad range of bioactive compounds.11–14 Synthesizing seven-membered rings is more challenging than preparing five- or six-membered counterparts due to significant entropic costs and transannular interactions during ring closure.15 Bridgehead nitrogen-fused tricyclic indoles present even greater complexity. Their unique biological activities and inherent synthetic challenges have drawn significant interest and driven advances in new organic synthetic methodologies.16,17 These intricate structures hold great promise for the development of novel therapeutic agents and the innovation of synthetic strategies.
For medicinal chemists, synthesizing hybrid molecules represents one of the most promising strategies in the design of novel pharmaceutical agents.18,19 This approach integrates two bioactive heterocyclic components into a single framework, potentially producing compounds with diverse pharmacological properties. Notably, indole-fused diazepines have emerged as a particularly appealing structural motif in drug discovery. These molecules incorporate both an indole moiety and a seven-membered ring system, both of which exhibit significant biological activities (Figure 2). For example, derivatives bearing an indole-diazepine fused core have been identified as inhibitors of myeloid cell leukemia 1 (Mcl-1),20 and ribosomal S6 kinase (RSK), with representatives such as BIX 02565.21 Furthermore, they demonstrate selectivity for the 5-HT2C receptor, suggesting their potential as anxiolytics.22 Additionally, they inhibit glycogen synthase kinase-3 (GSK-3)23,24 and c-Met25 and have been recognized for their anti-anxiety properties,26 among other functions.
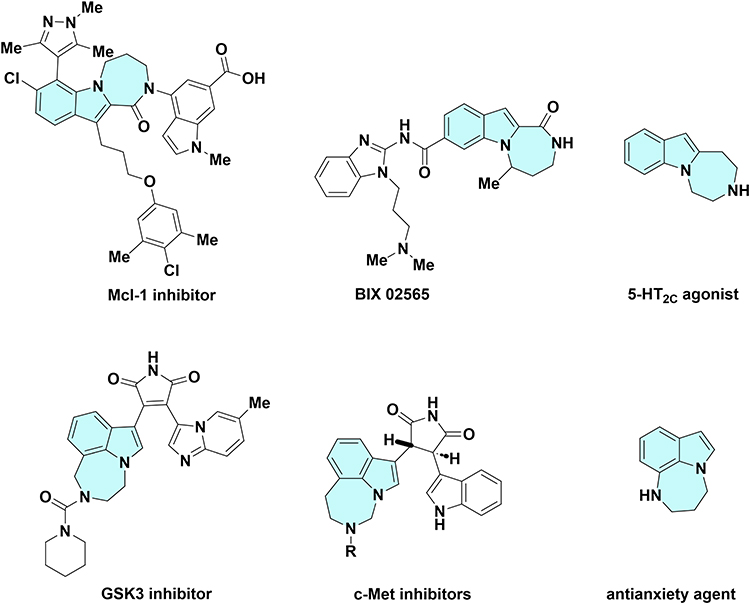 |
Figure 2 Bioactive molecules containing indole-diazepine frameworks. |
Indole-diazepine frameworks are tricyclic systems formed by fusing a seven-membered nitrogen-containing ring with a bicyclic indole structure. The fusion yields intriguing scaffolds that offer significant potential for the exploration of novel biologically active compounds. The fusion of these three rings generates numerous isomers, thereby broadening the chemical space for drug discovery. Motivated by this concept, we developed a series of indole/pyrrole-fused 1,4-diazepanone scaffolds.27 These structures are accessible through the fusion of 1H-indole/pyrrole-2-carboxylic acids with Morita-Baylis-Hillman (MBH)-derived allylamines. MBH adducts are known to enhance bioactivity, suggesting their potential applications for these scaffolds.28–33 We recently expanded the scope of these scaffolds through Lewis base-catalyzed [4 + 3] annulation of indole-2-carboxamides with MBH carbonates.34 This method offers efficient access to densely substituted indole-fused diazepanones for applications in drug discovery.
Several recent reviews have addressed the synthesis of 3, n-fused tricyclic indole frameworks featuring a functionalized medium-sized ring at the C–C bond position of the indole.35,36 However, a considerable gap remains in the literature. To date, no comprehensive study has reviewed synthetic advancements in annulated indole-diazepine structures, particularly those containing a bridgehead nitrogen atom. This structural feature theoretically allows for the construction of four linear annelations and four peri-fused isomers, all with the empirical formula C12H14N2. However, not all theoretically possible structures have been synthesized; only the realized variants are discussed herein (Figure 3). This article elucidates foundational principles of this structural class and guides future development of indole-fused diazepines, emphasizing the role of the bridgehead nitrogen atom in drug discovery. We focus exclusively on compounds containing this bridgehead nitrogen atom, as it is crucial to the integrity of both rings. We delineate synthetic methodologies for these structures and review the biological properties of their functionalized derivatives.
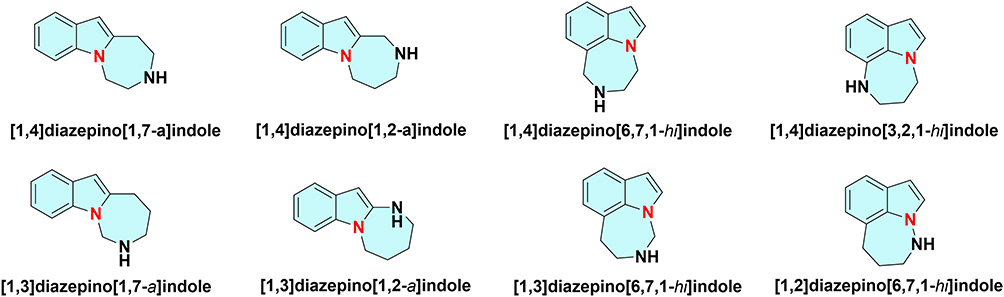 |
Figure 3 Indole-fused diazepines with a bridgehead N atom. |
Synthetic Strategies
Various synthetic strategies have been employed to construct these complex, biologically relevant scaffolds (Figure 3), including base-mediated, metal-catalyzed, radical, and phosphine-catalyzed cyclizations, as well as miscellaneous transformations. These methodologies form the carbon-nitrogen bonds required for ring formation and enable precise control over the configuration and electronic environment of the bridgehead nitrogen atom. The stereoelectronic properties of this nitrogen, such as its hybridization state, lone pair orientation, and spatial relationship to the indole nucleus, directly govern the three-dimensional architecture of the fused ring system. This in turn defines the conformational preferences that are critical for biological target recognition. Base-mediated cyclization has emerged as a particularly powerful approach. This strategy offers mechanistic simplicity, using basic catalysts to activate nucleophilic moieties (eg, indole N–H or amino groups) for intramolecular C–N bond formation, even in the presence of diverse functional groups. These features provide robust access to functionalized derivatives. The following section focuses first on the application of base-mediated cyclization in the synthesis of indole-fused diazepines, highlighting its significance in medicinal chemistry.
Base-Mediated Cyclization
Base-mediated cyclization provides a robust, conceptually simple platform for constructing indole-fused diazepines. A judiciously chosen base activates either the indole N–H or a tethered amino group to form the pivotal C–N bond. The key challenge is controlling selectivity: directing cyclization toward a specific fusion pattern while suppressing competing reactions at the C2 or C3 position of the indole nucleus. The following sections therefore examine how the same strategic principle is fine-tuned for each topological subtype. We begin with indole-1,7-fused 1,4-diazepines. Their formation requires the base to deprotonate the nucleophilic site and modulate the electronics and sterics of the forming seven-membered ring, thereby directing cyclization to the N1/C7 position. Subsequent sections extend this analysis to 1,2-, 6,7,1-, and 3,2,1-fusion modes, demonstrating how this mechanistic blueprint adapts to diverse indole-diazepine architectures.
Indole-1,7-Fused with 1,4-Diazepines
Indole-1,7-fused 1,4-diazepines are a key structural subtype in this class. Base-mediated cyclization forms these structures through the selective activation of nucleophilic moieties (eg, indole N–H or amino groups) by basic catalysts. Figure 4 illustrates the synthetic framework for this subtype, showing the core cyclization site, substrate features, and product structures.
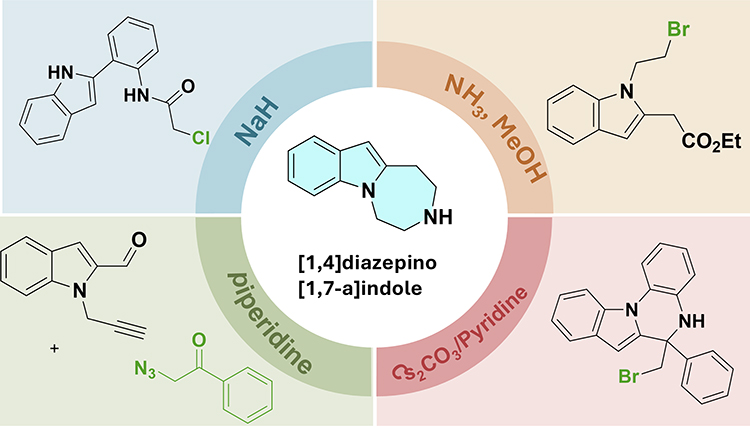 |
Figure 4 Schematic illustration of base-mediated cyclization for indole-1,7-fused 1,4-diazepines. |
Duncan’s group reported the first synthesis of an indole-1,7-fused 1,4-diazepine scaffold (Scheme 1).37 They cyclized chloroacetamidophenylindole 1 to compound 2 using NaH in DMF. These compounds showed analgesic activity in mice, laying the foundation for further exploration of cyclization reactions.38 Sundberg et al applied a similar strategy, treating substrate 3 with NaH in THF to form product 4.39
 |
Scheme 1 Synthesis of indolo[1,2-d][1,4]benzodiazepine-6-ones. Note: Base-mediated intramolecular N-alkylation of chloroacetamidoarylindole 1 furnishes tricyclic lactam 2; similarly, substrate 3 undergoes cyclization to afford tetracyclic product 4 bearing a CO2Me substituent. Selected examples: 2a, 2b and 4a. |
Base-mediated cyclization also constructs the 6,7-dihydro-5H-benzo[5,6][1,4]diazepino[1,7-a]indole scaffold (Scheme 2).40 Starting from compound 5, chloroacetylation and intramolecular cyclization constructed the lactam core, which was reduced to compound 6. Conversion of compound 6 to 7 proceeded via four steps: alkylation with tert-butyl bromoacetate, deprotection, amidation with piperidine, and amide reduction. Hydrolysis of the methyl ester in compound 7 and salt formation afforded the target scaffold 8 in good overall yield. This sequence demonstrates efficient assembly of the fused diazepinoindole skeleton through selective cyclization and functional group manipulation.
 |
Scheme 2 Synthesis of 6,7-dihydro-5H-benzo[5,6][1,4]diazepino[1,7-a]indole scaffold. Note: Chloroacetylation and intramolecular cyclization of 5 furnish the lactam core, which undergoes further reduction to afford 6. Sequential alkylation, deprotection, amidation with piperidine, and amide reduction of 6 provide 7, while hydrolysis of the methyl ester in 7 gives 8. |
Ennis et al reported the synthesis of compound 12 (Scheme 3).22 The route began with conversion of 9 to tricyclic lactam 10. Treatment of compound 9 with ammonia-saturated methanol at 50°C effected this conversion. Lactam 10 was reduced to indoline 11 using BH3-SMe2 in THF. Oxidation of indoline 11 with DDQ in dioxane afforded azepino[1,7-a]indole 12. Related scaffolds include chiral indolo[2,3-a]quinolizidines found in monoterpenoid indole alkaloids. Mirabal-Gallardo et al converted compound 13 to compound 14 through selective acylation with chloroacetyl chloride (K2CO3, DMF).41 This chloroacetyl group enabled subsequent cyclization to the indoloquinolizidine core.
 |
Scheme 3 Synthesis of compound 12 and 14. Note: Intramolecular cyclization of 9 affords tricyclic lactam 10, which is reduced to indoline 11; subsequent oxidative aromatization yields compound 12. Selective N-acylation of 13 followed by intramolecular cyclization furnishes indoloquinolizidine 14, with the bridgehead stereochemistry illustrated by bold and hashed wedges. |
Thikekar et al developed a divergent route to indole-fused diazepines 18 and 19 from common precursor 15 (Scheme 4).42 Reaction conditions dictated the product distribution. The transformation of compound 15 to compounds 18 and 19 involved distinct reaction pathways influenced by reaction conditions and substituents. To form compound 18, compound 15 was treated with KI and Cs2CO3 in refluxing MeCN for 3 hours. Intramolecular N-alkylation generated aziridine 16, which underwent hydrolysis during workup to give compound 18 bearing a hydroxyl group at the quaternary carbon. For compound 19, compound 15 was treated with pyridine in a sealed tube at 140°C for 3 hours. This condition induced skeletal rearrangement of aziridine 16 (formed in situ) to afford 19. Both electron-donating and electron-withdrawing substituents were tolerated, giving satisfactory yields. This divergent approach leverages aziridine reactivity to access different indole-fused diazepines from a common precursor.
 |
Scheme 4 Synthesis and a plausible pathway for transformation of compound 15 to indole-fused diazepines 18 and 19. Note: Intramolecular N-alkylation of 15 affords aziridine intermediate 16; ring-opening of 17 with water during aqueous workup gives 18, while pyridine-mediated rearrangement of 17 furnishes 19. Selected examples: 18a, 18b and 19a. |
Gour et al developed a route to 1,2,3-triazole-fused indolo[1,4]diazepines via sequential Knoevenagel condensation and intramolecular azide-alkyne cycloaddition (Scheme 5).43 Treatment of azide 21 with N-propargyl indole 20 in the presence of catalytic piperidine in methanol afforded compound 22 in good yield. This one-pot process initiates with Knoevenagel condensation between 20 and 21, facilitated by piperidine, to form intermediate III, which then undergoes intramolecular [3+2] azide-alkyne cycloaddition to yield the final product.
 |
Scheme 5 Synthesis and proposed mechanism for transformation of N-propargyl indole 20 and azide 21 to 1,2,3-triazole-fused indolo[1,4]diazepine 22. Note: Knoevenagel condensation of piperidine with 20 forms iminium ion intermediate I, whose deprotonation generates enolate II; nucleophilic attack on enamine 21a affords intermediate III, followed by dehydration to IV and intramolecular [3+2] cycloaddition to final product 22. |
Indole-1,2-Fused with 1,4-Diazepines
Base-mediated cyclization constructs indole-1,2-fused 1,4-diazepines with excellent regioselectivity and functional group compatibility. Figure 5 summarizes representative substrate structures and product frameworks for this structural subtype.
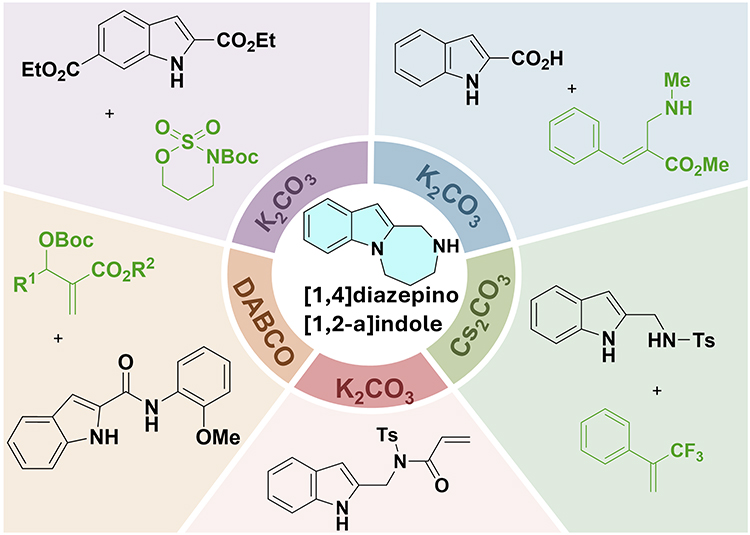 |
Figure 5 Schematic illustration of base-mediated cyclization for indole-1,2-fused 1,4-diazepines. |
Boyer et al reported a series of 1-oxo-2,3,4,5-tetrahydro-1H-[1,4]diazepino[1,2-a]indole-8-carboxamides 28 (Scheme 6).44 Their synthetic strategy involved a sequential ring-opening, deprotection and cyclization cascade starting from indole diester 23 and sulfamidates 24–26, which afforded the key intermediate 27. Base-mediated hydrolysis and coupling with primary amines then afforded products 28. Shaw et al cyclized functionalized 2-indole amides 29 with 1,3-dibromopropane under basic conditions to give diazepinones 30.45
 |
Scheme 6 Synthesis of tricyclic indole diazepinones 28 and 30. Note: Compound 23 reacts with sulfamidates 24–26 via a base-mediated ring-opening/deprotection/cyclization cascade to afford 27, which furnishes 28 upon treatment with primary amines; selected example 28a features a benzimidazole-2-yl amide and dimethylaminopropyl side chain. Intramolecular cyclization of 29 with 1,3-dibromopropane followed by LiOH-mediated hydrolysis yields 30. |
In 2021, we reported a one-pot synthesis of indole-fused 1,4-diazepanones 34 via amide coupling and intramolecular aza-Michael addition of 1H-indole-2-carboxylic acids 31 with MBH-derived allylamines 32 (Scheme 7).27 Amide intermediate 33 formed in 98% yield under standard conditions. Cyclization with K2CO3 or K3PO4 in DMSO gave single isomer 34a. In the one-pot procedure, elevated temperature and increased base loading improved yields: K2CO3 gave 34a in 83% yield at 80°C. Allylamines 32 tolerated various substituents, though steric hindrance reduced yields. Electron-donating groups on the phenyl ring (R1) improved yields and stereoselectivity. Aliphatic aldehyde-derived allylamines gave moderate to good yields. Indole-2-carboxylic acid derivatives 31 accommodated most substituents without significant effect.
 |
Scheme 7 One-pot synthesis of highly substituted indole-fused 1,4-diazepanones via sequential amide coupling and intramolecular aza-Michael addition. Note: Stepwise: Amide intermediate 33 is formed from 31a and 32a, then cyclized to (±)-trans-34a. One-pot: Amide coupling of 31 with MBH-derived allylamines 32 followed by intramolecular aza-Michael addition; elevated temperature and increased base loading improve yields. Selected examples: 34a, 34b, 34c and 34d. Trans-stereochemistry is indicated by hashed wedges. |
In 2022, Hao et al reported acid-base switchable cyclization to seven-membered nitrogen heterocycles (Scheme 8).46 When substrates 35 were reacted with a Lewis base in 1,4-dioxane at 40°C, chemoselective N1 cyclization occurred preferentially over the C3 acylation process. These conditions rapidly afforded indole-fused [1,4]diazepines 36 in good yields. Zeng et al developed an ipso-defluorinative functionalization of diamine 37 with (trifluoromethyl)alkene 38 to give monofluoroalkene 39.47 For example, unsymmetric tethered 1,2-diamines reacted with Cs2CO3 in DMF at room temperature to give indole-fused medium-sized rings efficiently.
 |
Scheme 8 Synthesis of compound 36 and 39. Note: Base-mediated chemoselective N1 cyclization of 35 (favored over C3 acylation) affords 36; selected examples: 36a, 36b and 36c. Ipso-defluorinative functionalization of diamine 37 with (trifluoromethyl)alkene 38 yields monofluoroalkene 39. |
More recently, we reported a Lewis base-catalyzed [4+3] annulation of indole-2-carboxamides 40 with MBH carbonates 41 (Scheme 9).34 A removable o-methoxyphenyl directing group controlled regioselectivity, giving highly substituted diazepanones 42. The reaction proceeds via Lewis base-catalyzed N-allylic alkylation followed by intramolecular Michael cyclization. Optimized conditions (20 mol% DABCO, MeCN, r.t.) gave high regio- and stereoselectivity. A removable o-methoxyphenyl directing group on the carboxamide forms an intramolecular hydrogen bond with the amide NH, reducing its nucleophilicity and directing attack to the indole NH. MBH carbonates tolerated diverse substituents: electron-donating (–Me, –MeO) or withdrawing (–Cl, –CO2Me) groups on aromatic rings, bulky naphthyl groups, and various esters (Me, t-Bu, Bn). Aliphatic MBH carbonates reacted with lower efficiency. Indole-2-carboxamides with diverse ring substituents and N-alkyl derivatives also gave products.
 |
Scheme 9 Synthesis and proposed mechanism for transformation of indole-2-carboxamide 40 and MBH carbonate 41 to indole-1,2-fused diazepinone 42. Note: Indole-2-carboxamide 40a reacts with MBH carbonate 41 to afford diazepinone 42 with high regio/stereoselectivity. DABCO reacts with 41a to form allylic ylide I and tBuO−, which deprotonates indole N–H of 40a to give II; SN2′ substitution with I yields III, deprotonation of which gives IV, followed by intramolecular Michael cyclization via V to trans-42a. Selected examples: 42a, 42b, 42c and 42d. |
The mechanism begins with nucleophilic attack of the Lewis base on MBH carbonate 41a to give allylic nitrogen ylide I. The in situ generated t-BuO− selectively deprotonates the indole N–H (the amide N–H is less acidic due to hydrogen bonding with the OMP group). Intermediate II undergoes SN2′ substitution to give intermediate III. Deprotonation at the amide nitrogen gives intermediate IV, which cyclizes via intramolecular Michael addition. Proton transfer gives the thermodynamically favored trans-isomer. This method offers broad substrate scope, high selectivity, and potential for drug discovery applications.
Indole-6,7,1-Fused with 1,4-Diazepines
Base-mediated cyclization also constructs indole-6,7,1-fused 1,4-diazepine cores. Manning et al reported diazepinone 44 (Scheme 10).48 DIPEA and T3P promoted cyclization of acid 43 to give compound 44. DeRatt et al prepared oxadiazepanone 47 via basic ring-opening of an oxetane.49 Acid 45 was coupled with amine 46 using HATU, then treated with Cs2CO3 at 70°C to give compound 47 in 85% yield.
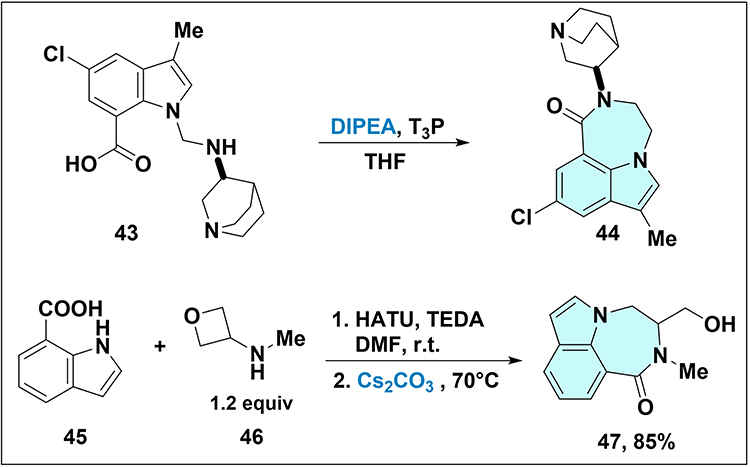 |
Scheme 10 Synthesis of compound 44 and 47. Note: Intramolecular amidation of 43 forms the diazepinone core 44. For the formation of 47: Step 1: Amide coupling of indole-2-carboxylic acid 45 with oxetane-containing amine 46; Step 2: Cs2CO3-mediated intramolecular oxetane ring-opening at 70°C, affording oxadiazepanone 47. |
Indole-3,2,1-Fused with 1,4-Diazepines
Anizon and Moreau cyclized substrates to seven-membered compounds 49 and 50 using dibromoalkanes under basic conditions (Scheme 11).50
 |
Scheme 11 Intramolecular N-alkylation of pyrrolo[2,3-a]carbazole with 1,3-dibromopropane. Note: Intramolecular N-alkylation of 48 with 1,3-dibromopropane gives 49. Compound 50, a derivative with a formyl group at pyrrole C1 and a 3-(piperidin-1-yl)propoxy side chain at C5, displays selective cytotoxicity against AML cells. |
Indole-1,7-Fused with 1,3-Diazepines
Indole-1,7-fused 1,3-diazepines exhibit diverse biological activities. Tabernine B (Figure 6), a carboline indole alkaloid from Tabernaemontana elegans, represents a notable example.51 This compound has demonstrated significant biological potential, including the ability to induce necrosis in human hepatoma HuH-7 cells and circumvent drug resistance in mouse lymphoma cell lines, making it a valuable candidate for cancer therapy.51,52 The discovery of such natural products has spurred interest in exploring the synthetic potential of indole-1,7-fused 1,3-diazepines, with the aim of developing novel compounds with enhanced therapeutic properties.
 |
Figure 6 Structure of Tabernine B. |
McCombie and Vice reported the synthesis of the indole-1,7-fused 1,3-diazepine skeleton (Scheme 12).53 They used 2,2′-indole dimer 51 as the starting material. Urea 52 was obtained in 81% yield via successive C–C and N–C bond formation in the presence of N-chlorocarbonyl isocyanate. The use of 2,6-di-tert-butylpyridine as a base was crucial for the reaction to proceed efficiently and selectively toward the desired product.
 |
Scheme 12 Synthesis of urea 52 from 2,2′-indole dimer and N-chlorocarbonyl isocyanate. Note: Reaction of 2,2′-indole dimer 51 with N-chlorocarbonyl isocyanate followed by 2,6-di-tert-butylpyridine affords urea. |
Indole-1,2-Fused with 1,3-Diazepines
Ghosh et al treated compound 53 with LDA and HMPA in THF at −78°C to give 1,3-diazepine-fused indoloquinazolinone 54 (Scheme 13).54 Intramolecular SN2 cyclization occurs via attack of the quinazolinone nitrogen on the bromopropyl side chain. This demonstrates LDA-mediated cyclization as a route to functionalized indole-1,2-fused 1,3-diazepines.
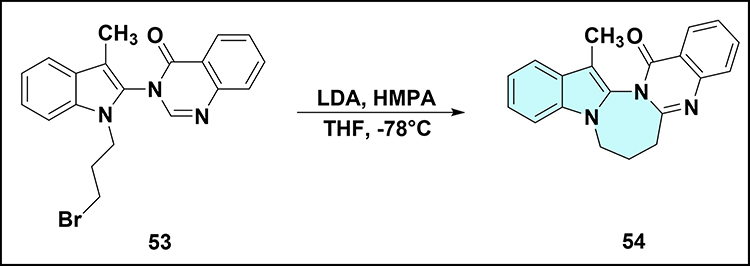 |
Scheme 13 LDA-promoted intramolecular aza-cyclization of compound 53 to 1,3-diazepine-fused indoloquinazolinone 54. Note: The reaction proceeds via intramolecular SN2 cyclization wherein the quinazolinone nitrogen atom attacks the bromopropyl side chain, forming the seven-membered 1,3-diazepine ring. |
These base-mediated cyclization reactions share several notable commonalities. Firstly, they all leverage the reactivity of bases to initiate intramolecular cyclization processes, enabling the construction of complex heterocyclic structures. Whether it is the use of sodium hydride, potassium carbonate, or ammonia in a basic environment, the base plays a crucial role in facilitating bond formation and rearrangement. Secondly, many of these reactions involve the use of chloroacetyl-related reagents. Chloroacetyl chloride, for example, is employed in multiple syntheses to introduce key functional groups that are essential for subsequent cyclization steps. This indicates its importance as a versatile synthetic intermediate in these types of reactions.
The various examples discussed above highlight the broad applicability of base-mediated cyclization in generating scaffolds with diverse structures. This method enables flexible introduction of functional groups and precise construction of indole-fused diazepine cores with distinct substitution patterns. Future work in this area should focus on optimizing reaction conditions, exploring new base systems, and expanding the scope of substrates to access even more complex indole-fused diazepine derivatives. Additionally, the insights gained from these studies can be applied to other cyclization strategies, such as radical and metal-catalyzed methods, to develop more efficient and sustainable synthetic pathways for the construction of these privileged scaffolds.
Metal-Catalyzed Cyclization
Transition-metal-catalyzed C–H functionalization constructs complex heterocyclic systems with high selectivity. Metal catalysts regulate reaction sites and pathways in indole synthesis, enabling cyclizations inaccessible by traditional methods. The following sections detail the research progress in this field, highlighting how different metal catalysts (eg, palladium, rhodium, copper, gold) enable regioselective and efficient formation of C–N/C–C bonds to access these privileged heterocyclic frameworks.
Indole-1,7-Fused with 1,4-Diazepines
In recent years, the field of transition metal-catalyzed C–H functionalization of the indole nucleus has seen remarkable progress.55 Indole-1,7-fused with 1,4-diazepines represent a privileged class of heterocyclic scaffolds with notable biological relevance, particularly in the context of drug design and development. These tricyclic systems have attracted significant attention due to their unique conformational properties and potential for selective molecular recognition. As illustrated in Figure 7, transition metal catalysis has emerged as a powerful strategy for the construction of these complex frameworks, enabling the assembly of diverse substitution patterns around the central indole-diazepine core.
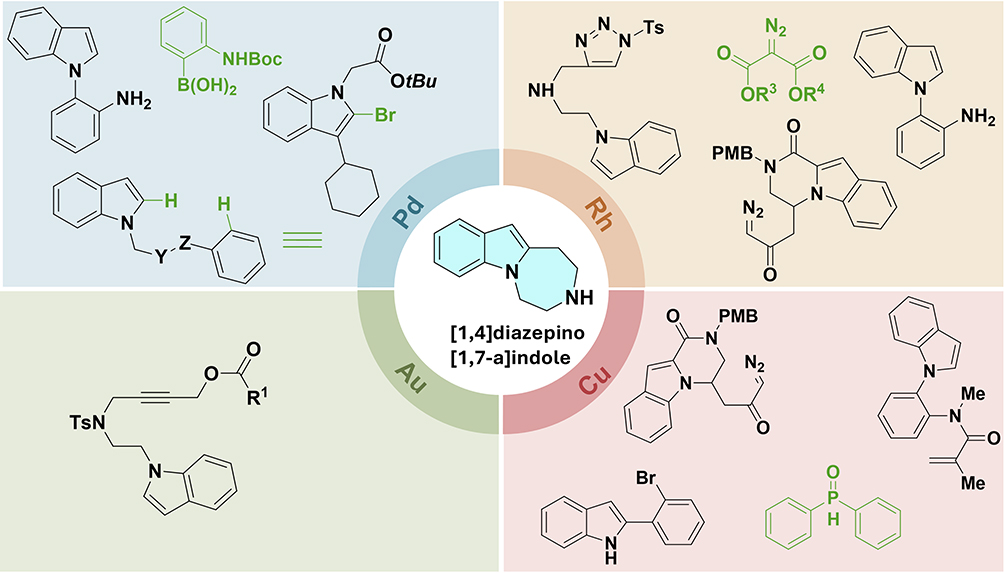 |
Figure 7 Schematic illustration of metal-catalyzed cyclization for indole-1,7-fused 1,4-diazepines. |
This figure provides a concise overview of the key transition metals (Pd, Rh, Cu, and Au) employed in these transformations, along with representative starting materials. The modular nature of these catalytic approaches allows for the introduction of a wide range of functional groups, which is critical for fine-tuning the biological activity of the resulting compounds. Building on this foundation, the following section will detail a specific metal-catalyzed protocol for the synthesis of these fused heterocycles, highlighting the mechanistic insights and synthetic versatility of this methodology.
The heteroatom-assisted, selective C2–H functionalization of indoles combined with cascade annulation represents a powerful strategy for synthesizing diverse fused-indole heterocyclic systems. Pintori and Greaney reported an efficient method for the indole system (Scheme 14).56 They chose to focus on N-alkylated indoles 55 in their study. During the screening process, it was discovered that indole compounds with a strong electron-withdrawing group at the C3 position were identified as suitable substrates for dehydrogenative seven-membered ring formation. Through catalyst optimization, they found that in the presence of Pd(OAc)2 (10 mol%) and excess Cu(OAc)2, with DMA as the solvent, the reaction was highly effective for C–C bond formation, giving compound 56 in good yield. The reaction mechanism begins with indole C2 palladation to form complex I, which can be trapped. Subsequently, a concerted metalation-deprotonation step takes place to generate intermediate III. Finally, reductive elimination gives rise to the products and Pd(0), which is then reoxidized by Cu(II) to maintain the catalytic cycle.
 |
Scheme 14 Synthesis and proposed mechanism for transformation of N-alkylated indole 55 to indole-fused diazepine 56 via intramolecular oxidative C–H coupling. Note: C2 palladation of 55a gives Pd(II) complex I; concerted metalation-deprotonation by K2CO3 affords six-membered palladacycle II. Reductive elimination of II provides III, and Cu(OAc)2-mediated reoxidation of Pd(0) completes the catalytic cycle to yield 56a. Selected examples: 56a and 56b. |
Building on this fundamental research of palladium-catalyzed reactions in indole systems, Zheng et al explored the synthesis of a series of indolobenzodiazepines 60 (Scheme 15).57 The methodology consisted of a Suzuki reaction of the bromoindole derivative 57 to obtain a pendant N-protected 2-aminoaryl group. Boc deprotection allowed spontaneous ring closure and the generation of the intermediate 59. Subsequent hydrolysis resulted in the desired diazepinone 60.
 |
Scheme 15 Synthesis of 2-arylindole-based NS5B inhibitors 60. Note: Suzuki coupling of bromoindole 57 with boronic acid 58 affords 59, which undergoes TFA-mediated deprotection to give 60a. Representative NS5B inhibitors: 60a, 60b and 60c. |
Meanwhile, other researchers focused on different synthetic routes for indole-fused compounds. Watson et al developed the intramolecular arylation of 2-(N-indolyl)alkanamide 61 to indolo-fused benzodiazepinone 62, using a catalyst derived from a 1:1 mixture of Pd(OAc)2 and triphenylphosphine in toluene under microwave irradiation (Scheme 16).58 The reaction proceeded via direct C2 arylation of the indole by the aryl halide, in competition with the base-mediated enolate arylation. The addition of potassium acetate and tetrabutylammonium bromide facilitated direct cyclization to avoid enolate byproducts.
 |
Scheme 16 Palladium-catalyzed synthesis of indolo-fused benzodiazepinones 62 via direct C–H arylation of 2-(N-indolyl)amides 61. Note: The reaction proceeds via indole C2 arylation competing with enolate arylation; Pd(OAc)2 and TABA promote selective cyclization to suppress enolate byproducts. Selected examples: 62a, 62b and 62c. |
In the pursuit of more efficient and regioselective methods, Thikekar and Sun explored an efficient and regioselective palladium(II)-catalyzed [5+2] annulation reaction for the synthesis of an indole-diazepine library (Scheme 17).59 Unprotected o-indoloanilines 63 reacted with internal alkynes 64 under microwave irradiation to form diverse imine-containing 1,2-fused indole[1,7-a]diazepines 65 in moderate to excellent yields. The formation of [5+2] cycloaddition to give a seven-membered heterocyclic ring was achieved by employing palladium (II) as catalyst, without relying on pre-activation of the aromatic fragment with halides or alkynes. The reaction was performed under microwave irradiation, using indolo-substituted aniline, diphenylacetylene, Pd(OAc)2, NaOPiv H2O and PivOH in DMSO at 150°C for 40 min to deliver the target product. The method exhibited broad compatibility with electron-rich and deficient substituents on the indole ring (eg, –Me, –OMe, –Cl, –CN) and the aniline ring, as well as symmetric and unsymmetric internal alkynes (eg, diphenylacetylene, alkyl-aryl alkynes), affording compound 65 in moderate to excellent yields. A plausible catalytic cycle involves the initial formation of [Pd(PivO)]+. This species then coordinates with o-indoloaniline 63a, activating the C2–H bond to form a palladacycle I, followed by coordination with an alkyne to generate intermediate II. Subsequent alkyne insertion gives intermediate III, followed by C–N reductive elimination to form intermediate IV. Tautomerization of intermediate IV yields the product 65a, with regeneration of [Pd(PivO)]+ by pivalic acid and O2.
 |
Scheme 17 Synthesis and proposed mechanism for transformation of o-indoloaniline 63 and alkyne 64 to 1,2-fused indole-diazepine 65 via palladium-catalyzed [5+2] annulation. Note: Pd(OAc)2 and PivOH generate active [Pd(PivO)]+; coordination and C2–H activation of 63a form palladacycle I. Alkyne 64a coordinates to give II, and alkyne insertion affords III. C–N reductive elimination yields IV, and tautomerization of IV furnishes 65a, with [Pd(PivO)]+ regenerated by PivOH and O2. Selected examples: 65a, 65b, 65c and 65d. |
Expanding on these diverse synthetic strategies, Gao et al reported an alternative approach for the synthesis of N-phenyl annulated indoles using a palladium-catalyzed and norbornene-mediated double alkylation process (Scheme 18).60 They chose 5-cyanoindole or 5-nitroindole 66 as substrates and used dibromoalkane 67 as the alkylating reagent. The synthesis strategy involved C2 alkylation of the substrates, followed by intramolecular N–H cyclization to form the annulated indole skeleton 68 in moderate yields. A relatively higher reaction temperature facilitated the transformation.
 |
Scheme 18 Pd-catalyzed reaction of electron-withdrawing substituted indoles 66 with 1,5-dibromopentane 67. Selected examples: 68a and 68b. |
In the field of transition-metal-catalyzed organic synthesis, rhodium-catalyzed reactions have attracted significant attention due to their unique reactivity and selectivity. Particularly, the insertion reactions involving metal-carbene intermediates enable the construction of carbon-carbon and carbon-heteroatom bonds, providing a powerful tool for the synthesis of complex molecules.61 The following section elaborates on the applications of rhodium-catalyzed reactions in different reaction systems, with a notable example being the work by Yang et al, who reported an intramolecular annulation of tethered N-sulfonyl-1,2,3-triazoles with indole ring 69 to afford fused azepine derivative 70 using rhodium(II) catalysts in a one-pot procedure (Scheme 19).62 The optimal conditions were identified as 5 mol% [Rh2(Oct)4] in dry DCE at 80°C, followed by reduction with NaBH3CN, affording products in high yields. A wide range of substrates were tolerated, including indolyl triazoles with various substituents (electron-donating or withdrawing groups on indole cores), affording N-bridgehead azepine derivatives in moderate to good yields. In this study, 1-sulfonyl-1,2,3-triazoles 69 served as a stable precursor of rhodium(II) azavinyl carbenes, enabling a series of novel transformations, such as transannulation with unsaturated compounds, functionalization of X–H bonds (where X = C, N, or O), and 1,2-migration. The reaction proceeded through a rhodium(II) azavinyl carbene intermediate, which initiated the intramolecular C–H transannulation with indolyl rings.
 |
Scheme 19 Synthesis and a plausible pathway for transformation of 1-sulfonyl-1,2,3-triazole 69 to fused azepine derivative 70 via Rh(II)-catalyzed intramolecular annulation. Note: Step 1: Rh2(Oct)4-mediated generation of rhodium(II) azavinyl carbene; Step 2: NaBH3CN reduction to yield the saturated azepine. 69 acts as a stable carbene precursor, forming the rhodium(II) azavinyl carbene; the carbene undergoes intramolecular C–H transannulation with the indolyl ring, and reduction of intermediate II by NaBH3CN gives 70. Selected examples: 70a, 70b and 70c. |
Expanding on the potential of rhodium-catalyzed transformations, Nemoto and Hamada explored the synthesis of nitrogen-bridged bicyclic compounds using rhodium-catalyzed carbenoid insertions (Scheme 20).63 In one example, they used an indole-fused diazo compound 71 as the substrate and 0.4 mol% Rh2(NHCOtBu)4 as the catalyst to obtain the corresponding 6-azabicyclo[3.2.2]nonane product 72 in 69% yield. The mechanism involved the formation of a Rh-associated N-ylide, followed by an acyl group-selective Stevens [1,2]-shift process.
 |
Scheme 20 Synthesis and a plausible pathway for transformation of indole-fused diazo compound 71 to 6-azabicyclo[3.2.2]nonane 72 via Rh(II)-catalyzed formal carbenoid insertion. Note: The reaction involves the formation of a Rh-associated N-ylide intermediate from diazo compound and the rhodium(II) catalyst, followed by an acyl group-selective Stevens [1,2]-shift process to construct the bridged bicyclic framework. |
Building on these advances, Dhole et al reported a Rh(III)-catalyzed annulation strategy for the synthesis of indolo-fused diazepines via reactions between o-indoloanilines 73 and diazo compounds 74 (Scheme 21).64 The optimized conditions included using [RhCp*Cl2]2 as the catalyst, AgSbF6 as the additive, and ethanol as the solvent at 60°C for 3 hours. Under these conditions, the C2-alkylated intermediate was converted to the desired diazepine product 75 with high selectivity. Substrate scope studies demonstrated that various o-indolo anilines (bearing electron-donating or withdrawing groups, halides, or heterocyclic substituents) and diazo compounds (eg, dialkyl diazomalonates) were compatible with the catalytic system, affording the target heterocycles in good to excellent yields. The reaction mechanism involves the initial generation of an active Rh(III) species, which coordinates with o-indolyl aniline 73a and undergoes C2–H cleavage to form a six-membered rhodacycle intermediate II. Subsequent coordination of the diazo ester leads to nitrogen extrusion and formation of a metal carbene species III. Migratory insertion and protonolysis then produce a C2-alkylated intermediate VI, which undergoes nucleophilic attack by the free amine group, assisted by the Lewis acid [RhCp*]2+, to yield the final product 75a and regenerate the catalyst.
 |
Scheme 21 Synthesis and proposed mechanism for transformation of o-indoloaniline 73 and diazo compound 74 to indolo-fused diazepine 75 via Rh(III)-catalyzed annulation. Note: Anion exchange between [RhCp*Cl2]2 and AgSbF6 generates active Rh(III) species [RhCp*X2]+ (I); 73a coordinates to I, undergoes C2–H cleavage to form rhodacycle II. Diazo ester 74a coordinates induce N2 extrusion to form carbene III, which inserts into the Rh–C bond to give IV. HSbF6-mediated protonolysis of IV yields V, and subsequent protonolysis of V (assisted by [RhCp*]2+) forms VI, whose cyclization affords 75a and regenerates the catalyst. Selected examples: 75a, 75b, 75c and 75d. |
Rhodium catalysts have long been lauded for their remarkable efficiency in catalyzing carbenoid reactions. However, their practical utility in large-scale synthesis of complex molecular targets is severely hampered by their scarcity. This limitation has spurred the scientific community to seek alternative catalysts derived from more abundant metals. In this context, copper emerges as a highly promising candidate. Its eco-friendliness, low cost, and wide availability make it an attractive substitute for precious metals like rhodium.65 Harada et al made a significant contribution in this area (Scheme 22).66 They continued to investigate this same substrate 76, in search of more sustainable catalytic options. Their approach specifically utilized a sustainable copper catalyst. It involved the direct treatment of isoindolinone with a diazocarbonyl unit 76. Conducted in chlorobenzene solvent with 0.05 mol% of the copper catalyst, and stirred at 55°C for 24 h, the desired product 77 was obtained in a moderate yield. This work not only demonstrated the potential of copper-catalyzed reactions but also provided a new synthetic route for a specific class of compounds.
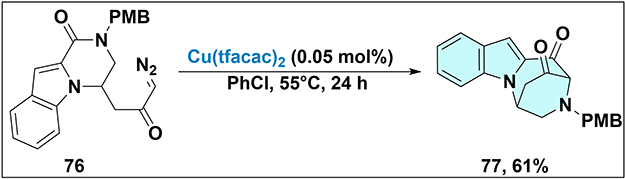 |
Scheme 22 Synthesis of indole-annulated polycyclic compound 77 via copper catalysis. Note: Cu(tfacac)2-catalyzed transformation of 77 from 76. This method employs a sustainable copper catalyst at a very low loading (0.05 mol%) for the synthesis of 77 via formal amide insertion. |
Building on the concept of copper-catalyzed reactions, Pradhan et al further advanced the field by disclosing an efficient one-pot strategy for the synthesis of 6,7-dihydro-5H-benzo[5,6][1,4]diazepino[1,7-a]indoles 80 (Scheme 23).67 Using 2-phenyl-N-tosylaziridine 78 and 2-(2-bromophenyl)-1H-indole 79, the ring-opening reaction was performed at the N site of the indole in the presence of NaH as a strong base, followed by Cu powder-mediated cyclization, yielding the target compound. A variety of structurally different aziridines were reacted with compound 79, yielding the corresponding compound 80 in high yields. Notably, when enantiopure (R)-2-phenyl-N-tosylaziridine was reacted with 2-(2-bromophen-yl)-1H-indole under the same reaction conditions, (R)-80a was obtained in high yield with an outstanding enantiomeric excess (ee). The reaction mechanism is quite intricate. It commences with an SN2-type ring opening of aziridine 78 with 2-(2-bromophen-yl)-1H-indole 79. In the experimental procedure, compound 79 was first treated with NaH in DMF, which enabled the ring-opening reaction with the indolyl N center. Then, Cu powder was added, and the reaction mixture was heated to 125–130°C to promote the C–N bond-forming cyclization, yielding products 80 as the sole product.
 |
Scheme 23 Synthesis and proposed mechanism for transformation of N-tosylaziridine 78 and 2-(2-bromophenyl)-1H-indole 79 to compound 80 via copper-mediated ring-opening cyclization. Note: NaH deprotonates 79 to give indolyl anion I, which opens aziridine 78; the resulting intermediate II coordinates to Cu(I), which then undergoes aryl bromide oxidative addition to Cu(III) species III. Reductive elimination delivers cyclized product 80 and regenerates the catalyst. Selected examples: 80a, 80b, 80c and (R)-80a. |
In 2025, Wang et al reported an efficient radical cascade cyclization strategy to generate indole-fused diazepine derivatives (Scheme 24).68 The synthesis started with the use of N-(2-(1H-indol-1-yl)phenyl)-N-methylmethacrylamide 81a as the starting material. The reaction was catalyzed by Cu(OTf)2 in the presence of K2S2O8 as the oxidant in acetonitrile at 100°C for 10 hours. This strategy leveraged the reactivity of phosphoryl radicals to initiate the cyclization process. The optimized conditions resulted in the formation of the desired product 83a with an isolated yield of 86% and excellent diastereoselectivity (up to >20:1 d.r). The synthesis tolerated a diverse range of substituents, including electron-donating or withdrawing groups on the benzene ring, various substituted indole scaffolds, sterically hindered methyl groups at the indole 3-position, and modified N-amide substituents. Phosphoryl sources bearing aryl or naphthyl groups were also compatible. The reaction mechanism involves the generation of a phosphoryl radical, which adds to the acrylamide double bond, followed by a diastereoselective addition to the indole ring, leading to the formation of the final product.
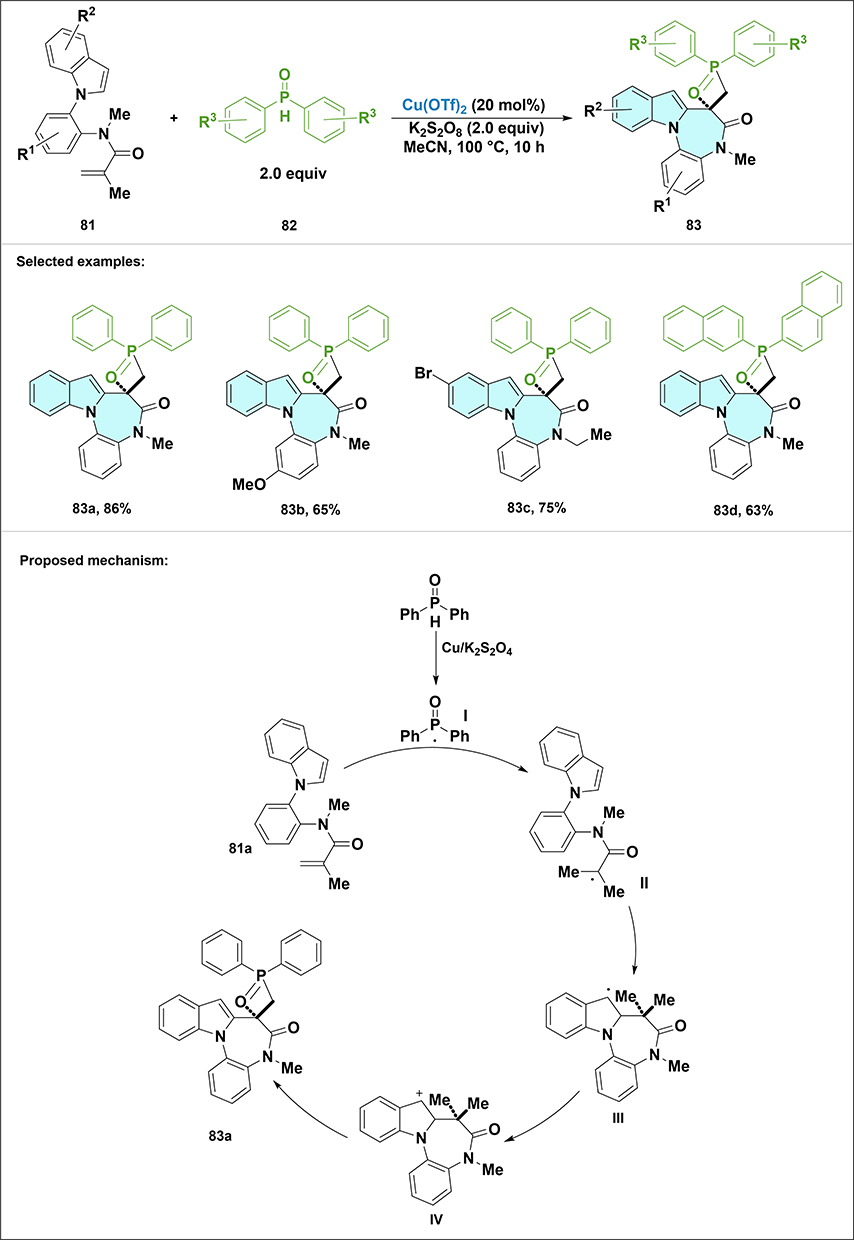 |
Scheme 24 Synthesis and proposed mechanism for transformation of compound 81 to indole-fused diazepine 83 via copper-catalyzed diastereoselective phosphorylation cyclization. Note: Diphenylphosphine oxide 82 undergoes single-electron oxidation by Cu(II)/K2S2O8 to generate phosphoryl radical I, which adds to the acrylamide moiety of 81a to form radical II; diastereoselective intramolecular cyclization onto the indole gives III, which is oxidized by Cu(II) to cation IV. Deprotonation of IV yields 83a. Selected examples: 83a, 83b, 83c and 83d. |
Gold catalysis, a rapidly evolving domain within the realm of homogeneous catalysis, is gradually being unraveled through the concept of carbophilic π-acids.69 These carbophilic π-acids serve as linchpins in the activation of C–C multiple bonds. Among the various gold-catalyzed reactions, the cyclization of indole derivatives has garnered substantial acclaim. This is primarily attributed to its remarkable efficacy in the construction of intricate polyheterocyclic frameworks, which are of great significance in organic synthesis and medicinal chemistry.70
Yang et al presented a significant advancement in this field (Scheme 25).71 They described an efficient gold(I)-catalyzed intramolecular cyclization of indolyl propargylic esters. By employing different gold catalytic systems, they achieved the selective synthesis of two distinct types of polycyclic indoline derivatives with high selectivity. Specifically, treatment of indolyl propargylic ester 84 with 5 mol% of [Au(tBuXPhos)][OTf] in anhydrous DCE at room temperature, polycyclic oxabridged-ring products 85 were formed. In contrast, when [(IPr)Au(MeCN)][SbF6] was used as the catalyst under atmospheric moisture conditions, product 86 was obtained instead of product 85. The proposed mechanism involves gold(I) activating the substrate’s triple bond, followed by 1,2-acyloxy migration to form a gold-vinyl carbene intermediate II. The carbonyl group then nucleophilically attacks this carbene, generating a 1,3-dipolar intermediate III that undergoes [3+2] cycloaddition with the indole’s C2–C3 bond to form oxabridged-ring products 85. In the presence of water and an alternative gold catalyst, these oxabridged products undergo hydrolysis to afford derivatives 86.
Additionally, they explored another reaction pathway for a specific substrate: when using substrate 87 (bearing a 4-Me group on the indole core) and replacing the catalyst with [Au(nBuPAd2)(MeCN)][SbF6], a new product 88 was obtained through a 7-exo-dig cyclization process, which retained the intact indole ring structure due to the blocked formal [3+2] cycloaddition caused by steric hindrance. Moreover, they delved into an asymmetric diastereoselective strategy mediated by chiral auxiliaries to introduce chirality into product 90. The optimal conditions for this asymmetric transformation involved using AgNTf2 as the co-catalyst in DCE at 0°C, which led to good to excellent ee values.
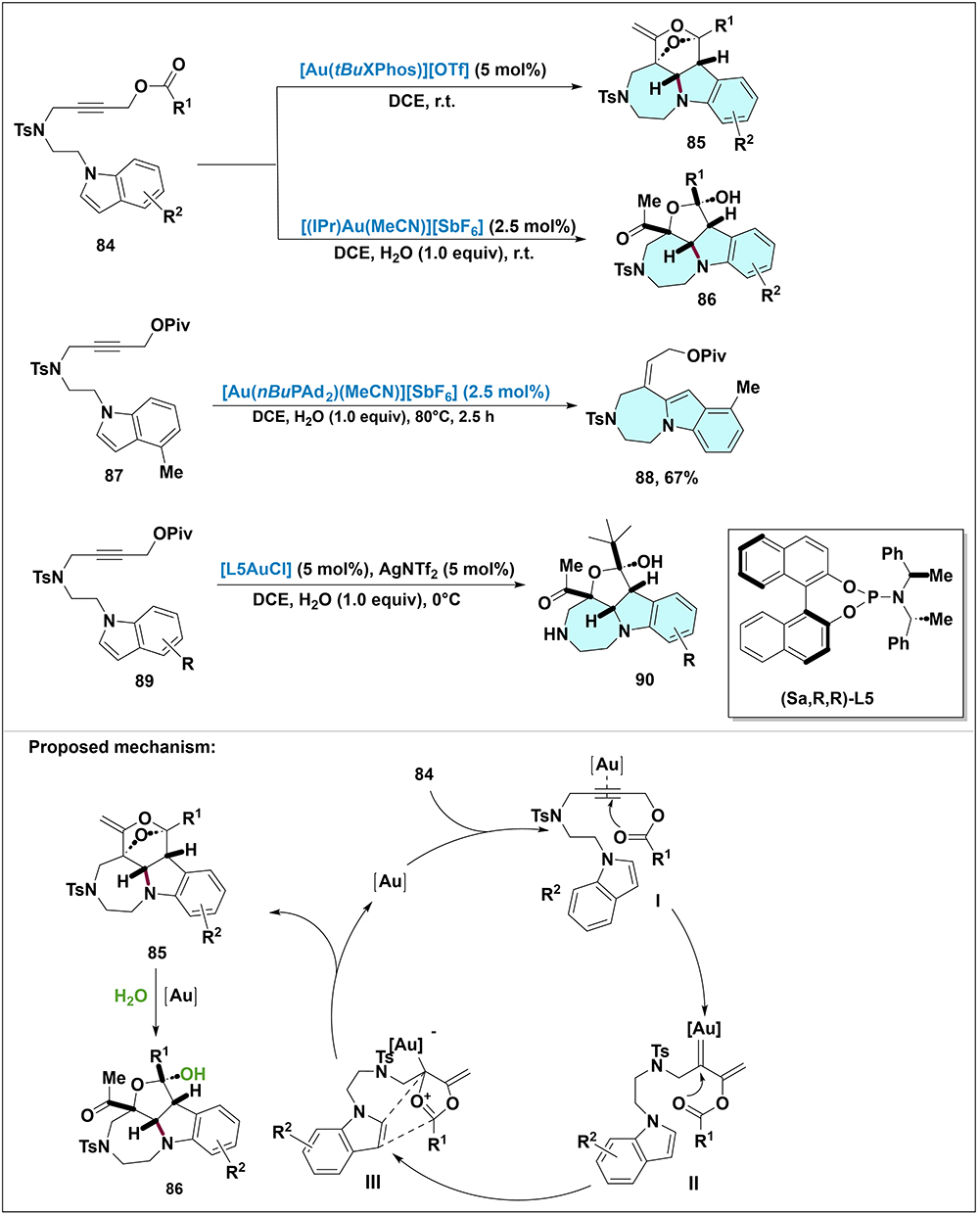 |
Scheme 25 Synthesis and proposed mechanism for transformation of indolyl propargylic ester to polycyclic indoline via gold(I)-catalyzed intramolecular cyclization. Note: Reaction of 84 with [Au(tBuXPhos)][OTf] affords oxabridged product 85, while treatment with [(IPr)Au(MeCN)][SbF6] yields hydrolyzed product 86. 7-exo-dig product 88 is formed from 87 with [Au(nBuPAd2)(MeCN)][SbF6], and asymmetric product 90 is obtained from 89 using [L5AuCl]/AgNTf2 with chiral ligand (Sa, R, R)-L5. Mechanism: Gold(I) activates the alkyne of 84 to form π-complex I; 1,2-acyloxy migration generates gold–vinyl carbene II, and carbonyl oxygen attack affords 1,3-dipolar intermediate III. [3+2] Cycloaddition with the indole C2–C3 bond gives oxabridged product 85, which hydrolyzes to 86 with H2O and alternative gold catalysts. |
Building on the foundation of indole-based gold-catalyzed reactions, Yang et al made another important contribution (Scheme 26).72 They developed a gold(I)-catalyzed method to construct azepines from indole substrates 91 with a remote alkyne connected to the N1 position. Through an optimization process, they determined that using 5 mol% [(IPr)Au(MeCN)][SbF6] as the catalyst and reacting highly functionalized indoles in DCE at a relatively high temperature gave the best results. Significantly, the prepared hydroarylation products 92 could be further transformed into polycyclic carbazoles 93 through an oxidation step. Polycyclic carbazoles are privileged polyheterocyclic units in medicinal chemistry, highlighting the potential applications of this reaction in drug synthesis.
 |
Scheme 26 Gold(I)-catalyzed intramolecular hydroarylation of alkynylindoles for the synthesis of azepino[1,2-a]indoles. Note: Gold(I)-catalyzed intramolecular hydroarylation of 91 gives 92, which undergoes oxidative cyclization to form 93. Selected examples: 92a, 92b, 92c, 93a and 93b. |
Indole-1,2-Fused with 1,4-Diazepines
Indole-1,2-fused with 1,4-diazepines constitute a unique class of fused heterocycles with promising biological potential. Transition metal catalysis has proven to be a powerful tool for the construction of these complex frameworks, enabling the formation of multiple bonds in a single operation and accommodating a wide range of functional groups. As illustrated in Figure 8, two major metal-catalyzed strategies, namely palladium and gold catalysis, have dominated the synthesis of these compounds, each offering distinct advantages in terms of substrate scope and reaction selectivity.
 |
Figure 8 Schematic illustration of metal-catalyzed cyclization for indole-1,2-fused 1,4-diazepines. |
This figure provides a concise snapshot of the key transition metals employed in these transformations, along with representative starting materials. The modular nature of these catalytic approaches allows for the introduction of a wide range of functional groups, which is critical for fine-tuning the biological activity of the resulting compounds. Building on this foundation, the following section will detail a specific metal-catalyzed protocol for the synthesis of these fused heterocycles, highlighting the mechanistic insights and synthetic versatility of this methodology.
Pintori and Greaney reported a Pd(II)-catalyzed oxidative C–H coupling without the need for pre-functionalization to achieve diazepane analogs (Scheme 27).56 A screening of N-alkylated indoles substrates 94 showed that a strong electron-withdrawing group at the indole 3-position was essential for heterobiaryl annulation. The reaction proceeded effectively, mediated by Pd(OAc)2 in the presence of K2CO3 and excess Cu(OAc)2 as additives. Compound 95 was obtained in a high yield from C2 oxidative coupling of the symmetrical precursor. The C–H bonds of benzimidazole and pyrazole could also be utilized to construct highly functionalized biheteroaryls.
 |
Scheme 27 Synthesis of diazepane analogs 95 via intramolecular oxidative coupling of indole and heteroarenes 94. Selected examples: 95a, 95b, 95c and 95d. |
Liu et al reported a Pd-catalyzed domino cyclization for the synthesis of 1,2-fused tricyclic indole 98 (Scheme 28).73 The procedure started with the reaction of compound 96 and 97. The best yield of the target product was obtained when 1,4-dioxane was used as the solvent, K2CO3 as the base, and Pd(OAc)2 and PPh3 were used at 10 and 40 mol%, respectively. Electron-withdrawing groups (eg, fluoro, chloro, cyano) on the benzene rings afforded moderate to excellent yields, whereas electron-donating or strongly electron-withdrawing groups led to decreased yields. Among protected substrates, benzenesulfonyl-protected derivatives with electron-donating groups outperformed those bearing electron-withdrawing groups. The proposed reaction mechanism involves a Pd-catalyzed Sonogashira coupling of compounds 96a and 97a to form intermediate III, followed by indole cyclization and an intramolecular transamidation reaction to afford key intermediate VI via IV (path 1). Subsequent Michael addition yields the final product 98a. Another possible pathway (path 2) is that intermediate III can be converted to intermediate V through an intermolecular transamidation reaction before indole cyclization, and then intermediate VI is formed via indole cyclization of intermediate V. Path 1 is proposed to be the more favorable pathway, although path 2 cannot be completely ruled out.
 |
Scheme 28 Synthesis and proposed mechanism for transformation of 2,2,2-trifluoro-N-(2-iodophenyl)acetamide 96 and N-(prop-2-yn-1-yl)-N-tosylacrylamide 97 to 1,2-fused tricyclic indole scaffold 98 via palladium-catalyzed domino cyclization. Note: Path 1 (favored): Pd-catalyzed Sonogashira coupling of 96a and 97a forms intermediate III, which undergoes indole cyclization to IV, followed by intramolecular transamidation to VI, and finally Michael addition to yield 98a. Path 2 (possible): Intermediate III undergoes intermolecular transamidation to V, which then cyclizes to VI before forming 98a. Selected examples: 98a, 98b, 98c and 98d. |
Gold-catalyzed transition-metal-catalyzed cascade reactions have garnered substantial interest due to their exceptional chemo- and stereoselectivity, as well as their commendable atom economy.74 Among the diverse array of catalysts, silver salts and gold complexes have distinguished themselves as highly desirable options.75 Their unique capacity to serve as potent electrophilic activators is the root of their allure. They can facilitate the reactivity of alkynes with a broad spectrum of nucleophiles under benign reaction conditions, rendering them indispensable in the realm of selective organic synthesis.
For instance, Zhou et al described an intriguing synthetic route to benzo[e]indolo[1,2-a]pyrrolo/pyrido[2,1-c][1,4]diazepine-3,9-diones 101 (Scheme 29).76 The derivatives of (2-aminophenyl)(4-methyl-1H-indol-1yl)methanone 99 and alkynoic acid 100 were used as the starting materials. When the reaction was performed in dry toluene in the presence of cocatalysts, namely 20 mol% of AgSbF6 and 5 mol% of Au catalyst, in a sealed tube at 120°C, moderate to excellent yields were obtained. Electronic variation on the indole (4–Me, 5–F, 6–OMe, 5–CN) or on the aniline ring (5–Me, 5–Cl, 5–F) was well tolerated, except for a strong electron-withdrawing cyano group, which retained the final cyclization. Alkyl chains (n-hexyl) on the alkynoic acid were also accommodated without erosion of yield. The reaction mechanism is as follows: Initially, the catalyst triggers the cyclization of alkynoic acid 100, leading to the formation of the activated enol-lactone intermediate I. Subsequently, the amino group of aromatic primary amine 99 attacks intermediate I, resulting in the generation of ketoamide II. This ketoamide II then undergoes nucleophilic addition and subsequent dehydration (a cascade process) to transform into N-acyl iminium ion III. Finally, the nucleophilic addition of intermediate III to the alkene part in the indole ring of the substrate produces the ring-closure product 101.
 |
Scheme 29 Synthesis and proposed mechanism for transformation of compound 99 and alkynic acid 100 to compound 101 via AgSbF6/Au-catalyzed one-pot cascade cyclization. Note: Under gold catalysis, alkynoic acid 100 cyclizes to enol-lactone I, which is trapped by aromatic primary amine 99 to form ketoamide II. A cascade of nucleophilic addition and dehydration generates N-acyl iminium ion III, which undergoes intramolecular nucleophilic addition to the indole alkene to afford cyclic product 101. Selected examples: 101a, 101b, 101c and 101d. |
Furthermore, gold catalysts have also been utilized by Feng et al for highly selective cascade reactions to generate a single skeleton (Scheme 30).77 In this context, the gold catalyst acts as a Lewis acid catalyst to produce enol lactone I and the iminium ion intermediate III. This approach consists of a two-step one-pot process: First, 2-(1H-indol-1-yl)ethanamines 102, alkynoic acid 103, and an Au(I) catalyst were treated in water under air and irradiated for 30 minutes at 150°C to form keto-amide intermediate. After the reaction mixture was cooled, CF3CO2H was added directly to promote N-acyliminium ion formation and cyclization, and the mixture was irradiated for another 30 minutes at 150°C to yield the target product 104. Additionally, they found that the reaction could alternatively also be carried out in DCE under the catalysis of 5 mol% of Au(PPh3)Cl.78
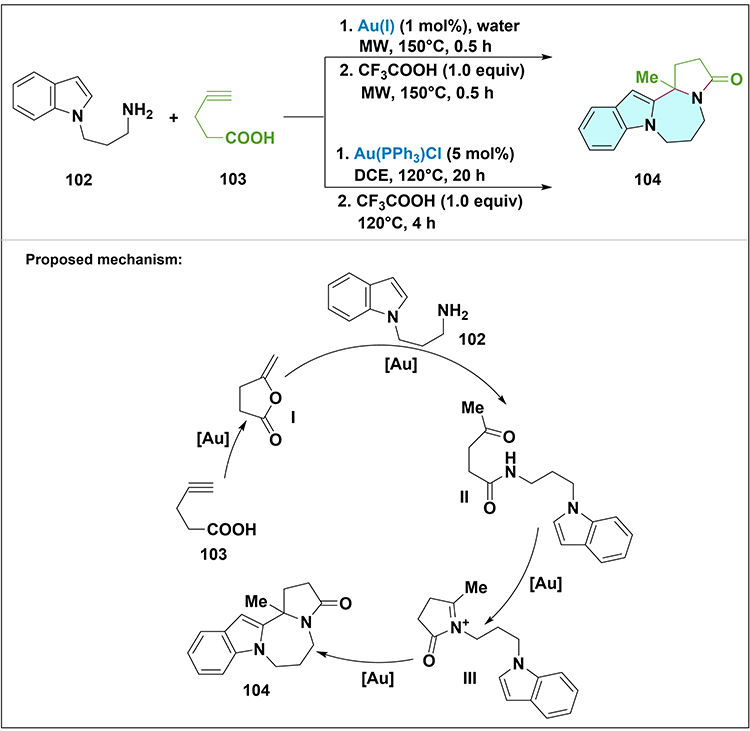 |
Scheme 30 Synthesis and proposed mechanism for transformation of 2-(1H-indol-1-yl)ethanamine 102 and alkynoic acid 103 to compound 104 via gold-catalyzed two-step one-pot cascade cyclization. Note: Gold(I) acts as a Lewis acid: it first activates the alkyne in 103 to form a π-complex that cyclizes to enol lactone I, which then undergoes ammonolysis with 102 to yield keto-amide II; Au-catalyzed cyclization and dehydration of II affords N-acyliminium ion III, and intramolecular nucleophilic attack of the indole C2 on III gives cyclized product 104. |
Basceken et al reported the gold-catalyzed intramolecular cyclization of N-propargyl indole derivatives with pyrazole to afford 7-endo-dig cyclization products 107 (Scheme 31).79 Interestingly, the substituents attached to the triple bond were associated with the 6-exo-dig 106 and 7-endo-dig cyclization 107 processes. For example, the alkyne carbon atoms substituted with phenyl or methyl groups were exclusively attacked by the pyrazole nitrogen atom at the C1 carbon atom because of geometry selectivity. It was the distance between the alkyne carbon atom and the gold atom that determined whether the positive charge is on the internal or terminal alkyne carbon atom.
 |
Scheme 31 Gold-catalyzed intramolecular cyclization of N-propargyl indole derivatives 105 containing pyrazole units for the synthesis of pyrazolodiazepinoindole. Note: When R = H, the reaction afforded 6-exo-dig cyclization product 106; when R = Ph, p-Me-Ph, p-OMe-Ph, m-NO2-Ph, p-F-Ph, or Me, the reaction afforded 7-endo-dig cyclization product 107. |
Indole-6,7,1-Fused with 1,4-Diazepines
Metal-catalyzed strategies have enabled efficient construction of indole-6,7,1-fused 1,4-diazepines by leveraging transition metals to facilitate bond formation and regioselective cyclization. Feng et al reported a method for synthesizing dibenzazepine-based heterocycles via Ni-catalyzed reductive aminocarbonylation of bromonitroarenes (Scheme 32).80 Using CO as the carbonyl source, Mn as the reductant, and TMSCl as an additive, the reaction with bromo-substituted indole 108 afforded the corresponding dibenzodiazepinone 109 in 85% yield. This approach demonstrates the utility of nickel catalysis in constructing fused heterocycles under reductive conditions.
 |
Scheme 32 Synthesis of dibenzazepine-based heterocycle 109 via Ni-catalyzed reductive aminocarbonylation of bromonitroarenes 108. |
In 2024, Ghosh et al developed a free amine-directed C–H bond activation strategy for synthesizing indole-fused benzodiazepines (Scheme 33).6 The heterobiaryl intermediates 110 underwent a Lewis acid-catalyzed double Michael addition with dimethyl acetylenedicarboxylate 111, yielding the target scaffolds 112 in moderate yields. This method showcases the efficiency of Lewis acid-catalyzed C–H functionalization for complex heterocycle assembly.
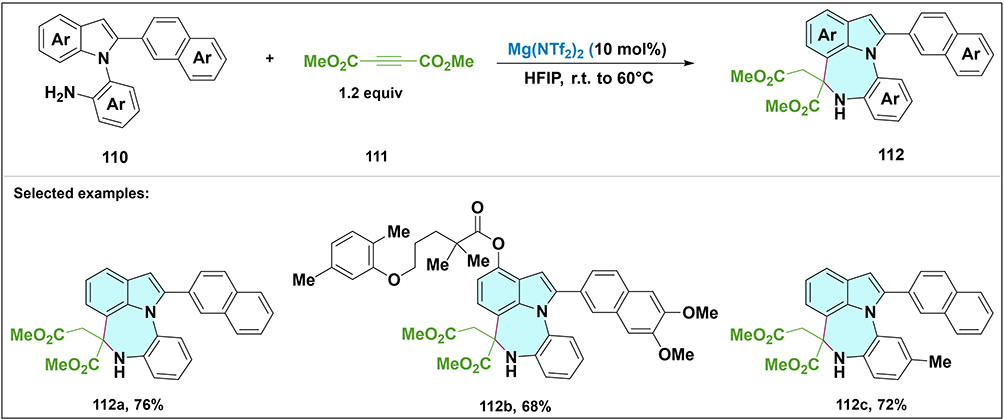 |
Scheme 33 Mg(NTf2)2-catalyzed double Michael addition of free amine-containing heterobiaryls 110 with 111 for the synthesis of indole-fused benzodiazepines 112. Selected examples: 112a, 112b and 112c. |
Indole-3,2,1-Fused with 1,4-Diazepines
In 2022, Li et al reported the synthesis of benzoimidazo[1,4]diazepinoindoles 115 via double Ullmann cross-coupling cyclization (Scheme 34).81 The conversion of simple 2-bromophenyl-imidazoles 113 and 7-bromo-indole 114 into [1,4]diazepino[3,2,1-hi]indoles 115 was achieved using DMSO in the presence of CuI/o-phen and K2CO3 at 90°C. A wide range of aryl and heteroaryl substituents on both the imidazole and bromoaryl moieties were well-tolerated, affording the target diazepinoindoles in good yields, regardless of electronic or steric effects.
A plausible reaction mechanism for the double Ullmann cross-coupling cyclization was proposed: Initially, copper interacts with o-phen to form copper complex I. Then, intermediate II is produced via an intermolecular oxidative addition reaction of compound 113a with copper complex I. Subsequently, intermediate II reacts with 114 to form intermediate III, which undergoes an addition/intramolecular C(sp2)–N coupling/elimination process to give the target structure 115a.
 |
Scheme 34 Synthesis and proposed mechanism for transformation of 2-bromophenyl-imidazole 113 and 7-bromo-indole 114 to benzoimidazo[1,4]diazepinoindole 115 via copper-catalyzed double Ullmann cross-coupling cyclization. Note: Copper forms complex I with o-phen, which undergoes oxidative addition with 113a to give intermediate II. Reaction of II with 114 affords III, which proceeds via addition/intramolecular C(sp2)–N coupling/elimination to form IV. Further intramolecular C(sp2)–N coupling of IV generates V, and subsequent reductive elimination delivers product 115a and regenerates the copper catalyst. Selected examples: 115a, 115b, 115c and 115d. |
Indole-1,7-Fused with 1,3-Diazepines
Transition-metal-catalyzed C–H bond functionalization is an appealing strategy for the construction of significant synthetic units. Xie et al successfully employed metal catalysis to convert brominated indole structure 116 into the chiral N-fused polycyclic 117 (Scheme 35).82 The reaction was carried out in DMA at 110°C, using Pd(OAc)2 as the catalyst, along with PivOH and K2CO3 as additives. The yield of the desired product 117 was 68%, and its ee was 96%.
 |
Scheme 35 Palladium-catalyzed intramolecular C–H functionalization of brominated N-alkylated indole derivative 116 for the synthesis of N-fused polycyclic compound 117. |
Xu et al developed a rhodium(III)-catalyzed tandem strategy for synthesizing diazepine-1,3-diones 120 (Scheme 36).83 The reaction involved C–H alkylation and intramolecular amination of indole formamides 118 with 3-bromo-3,3-difluoropropene 119, yielding the target compounds in moderate yields. Conducted in HFIP at 120°C under an argon atmosphere with a rhodium complex as the catalyst, the mechanism starts with anion exchange between rhodium and AgOAc, triggering ortho C–H activation to form a cyclic rhodium intermediate I. Olefin insertion generates intermediate II, which undergoes protonation with acetic acid to release alkylation intermediate III. Intramolecular cyclization of III via intramolecular amination furnishes intermediate IV. This intermediate then experiences sequential dehydrohalogenation and hydrolysis, wherein water and acetic acid facilitate the transformation, ultimately yielding product 120a. This approach highlights the efficiency of transition-metal catalysis in constructing fluorinated heterocyclic skeletons via sequential C–H functionalization and ring closure.
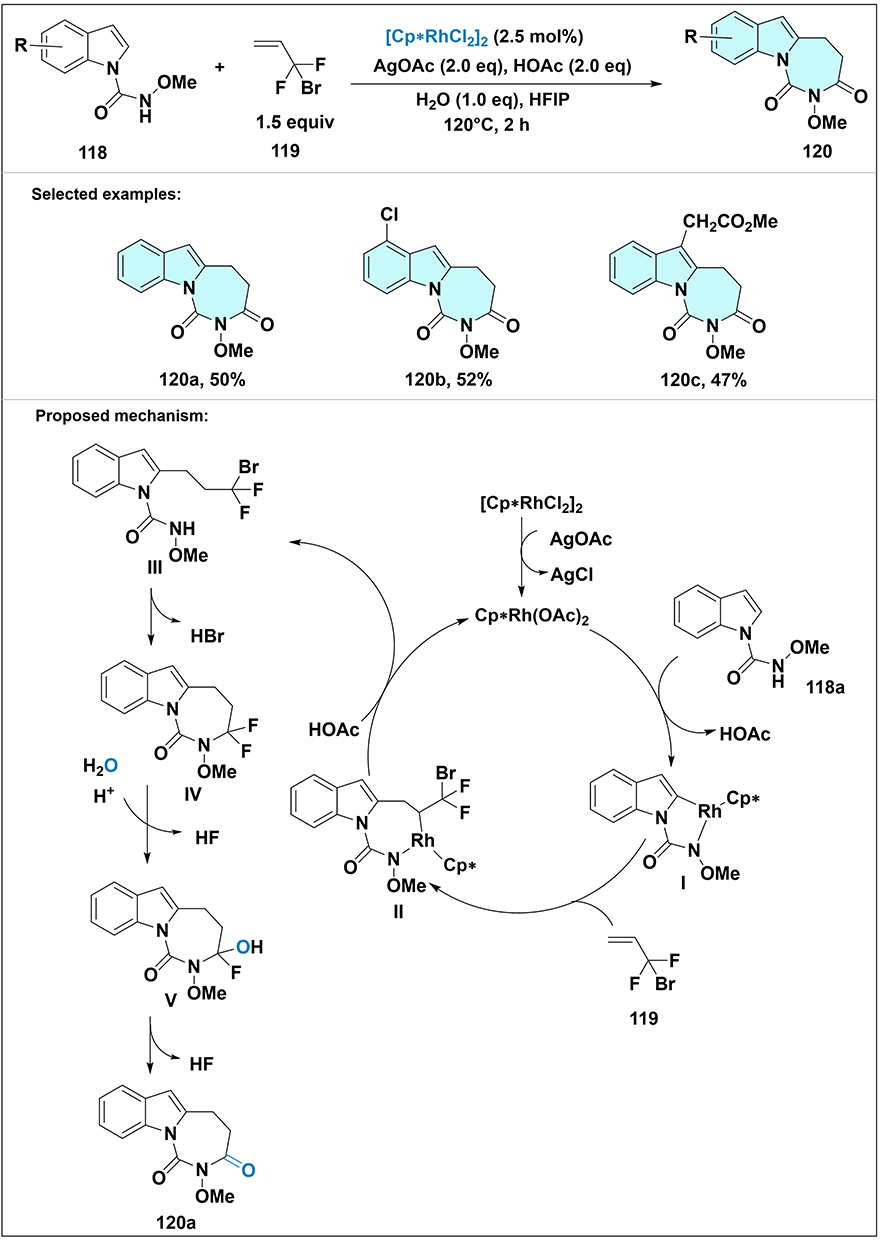 |
Scheme 36 Synthesis and proposed mechanism for transformation of indole formamide 118 and 3-bromo-3,3-difluoropropene 119 to diazepine-1,3-dione 120 via Rh(III)-catalyzed tandem C–H alkylation/intramolecular amination. Note: The Rh catalyst undergoes anion exchange with AgOAc, triggers ortho C–H activation to form rhodium intermediate I; olefin insertion gives II, protonation by HOAc yields III; intramolecular amination forms IV, which undergoes sequential dehydrohalogenation and hydrolysis to deliver final product 120a. Selected examples: 120a, 120b and 120c. |
Indole-1,2-Fused with 1,3-Diazepines
Kiruthika and Perumal further advanced the field by developing a copper-catalyzed intermolecular approach involving ynamide cyclization and intramolecular N-arylation to construct the indole fused 1,3-diazepine ring 122 (Scheme 37).84 The most favorable outcome was achieved when the reaction was conducted in the presence of CuI, L2, and Cs2CO3 in THF at 80°C, yielding the desired product 122 in 70% yield.
 |
Scheme 37 Synthesis and proposed mechanism for transformation of ynamide 121 to indole-fused 1,3-diazepine 122 via copper-catalyzed intramolecular N-arylation. Note: Base-mediated intramolecular hydroamidation of 121 and deacetylation generate 2-amidoindole II. CuI activates the aryl halide to form copper–aryl intermediate III, which undergoes nucleophilic addition to yield IV; reductive elimination delivers product 122 and regenerates the copper catalyst. |
The reaction mechanism commences with base-promoted intramolecular hydroamidation of ynamide, followed by deacetylation of intermediate I, culminating in the formation of 2-amidoindole II. In the presence of a proximal aryl halide, the 2-amidoindole II then undergoes intramolecular N-arylation. The CuI catalyst coordinates with the aryl halide, forming an active copper-aryl intermediate III. This intermediate reacts with the nitrogen atom of 2-amidoindole, facilitating the formation of a new C–N bond through a nucleophilic addition process. This step completes the construction of the indole-fused 1,3-diazepine ring, resulting in the final product. This study demonstrates the potential of combining base-mediated cyclization with metal-catalyzed reactions to achieve more efficient and selective synthesis of indole-1,2-fused 1,3-diazepines.
Indole-6,7,1-Fused with 1,2-Diazepines
Only one example of constructing an indole fused 1,2-diazepine ring has been reported by Melnyk et al (Scheme 38).85 The palladium-catalyzed intramolecular cyclization of amide 123 afforded a new class of tetracyclic product 124 in 41% yield. Optimization of reaction conditions revealed that an excess of P(Ph)3 was essential to promote the desired Heck-type cyclization and suppress competing side reactions.
 |
Scheme 38 Synthesis of indole-6,7,1-fused 1,2-diazepine 124 via Pd(OAc)2-mediated transformation of 123. |
Despite the broad medicinal potential of indole-fused diazepines highlighted in Section 2, indole-fused 1,2-diazepines remain an underdeveloped subclass, with only one synthetic example reported to date. This striking scarcity can be attributed to three key challenges inherent to the 1,2-diazepine scaffold and its fusion with indole:
Thermodynamic Instability of the 1,2-Diazepine Core
The 1,2-diazepine ring features adjacent nitrogen atoms in a seven-membered heterocycle, leading to significant electron density delocalization and ring strain. Compared to the more stable 1,3- or 1,4-diazepine isomers, 1,2-diazepines exhibit an intrinsic tendency toward rearrangement (eg, ring contraction to five-membered pyrazoles) or decomposition under standard synthetic conditions (eg, acid/base catalysis, metal activation). The indole ring is a π-conjugated system. Its fusion with the 1,2-diazepine core further increases structural instability. This is caused by steric clash between the benzene moiety of indole and the adjacent nitrogen atoms of 1,2-diazepine, which limits the formation of the desired tricyclic scaffold.
Regioselectivity Challenges in Cyclization
The synthesis of indole-fused 1,2-diazepines requires precise intramolecular bond formation between the indole and 1,2-diazepine fragments. In Melnyk et al’s work,85 palladium-catalyzed intramolecular cyclization of amide precursors relied on strict control of ligand stoichiometry (excess PPh3) to avoid off-target reactions– specifically, competitive C–C bond formation at the indole C3 position instead of the desired C–N bond for 1,2-diazepine fusion. For most other potential precursors (eg, indole-alkyne or indole-azide derivatives), the 1,2-diazepine’s adjacent nitrogens act as competing nucleophilic sites, leading to the formation of undesired isomers (eg, indole-fused 1,3-diazepines) or linear byproducts, which discourages further synthetic exploration.
Limited Biological Incentive and Synthetic Priority
Historically, medicinal chemistry research has prioritized 1,4-diazepines (eg, diazepam analogs) and 1,3-diazepines (eg, Tabernine B) due to their well-documented biological activities (eg, CNS modulation, anticancer effects). In contrast, 1,2-diazepines have fewer validated therapeutic targets–no indole-fused 1,2-diazepine has been reported to bind to key enzymes (eg, Mcl-1, GSK-3) or receptors (eg, 5-HT2C). This lack of biological validation reduces the incentive for synthetic chemists to overcome the scaffold’s inherent challenges, creating a “low-priority” cycle that perpetuates the scarcity of reports.
Looking forward, advances in stabilization strategies (eg, introducing electron-withdrawing groups onto the 1,2-diazepine ring to mitigate rearrangement) or bioinformatics-guided target prediction (eg, AI-driven screening for potential 1,2-diazepine-binding proteins) could rekindle interest in this understudied subclass, bridging the gap between synthetic feasibility and medicinal potential.
Radical Cyclization
After exploring the potential of base-mediated cyclization and metal-catalyzed cyclization in constructing indole-fused diazepine scaffolds, we now turn our attention to another powerful synthetic strategy: radical cyclization. Radical cyclization has emerged as a highly promising approach for synthesizing complex heterocyclic structures, owing to its unique reaction mechanisms and mild reaction conditions. Recent advancements in photochemical and radical chemistry have significantly expanded the scope of these reactions, making them essential tools for the synthesis of nitrogen-containing heterocycles. The high efficiency, straightforward operation, and good functional group compatibility render radical cyclization an attractive method for constructing indole-fused diazepines with promising biological activities.86,87
Figure 9 provides a visual summary of the diverse radical cyclization strategies employed for the construction of indole-1,7-fused 1,4-diazepines, including photoredox-catalyzed and thermal radical processes. As illustrated, these methodologies leverage various radical precursors, such as α-acetoxy ketones, selenosulfonates, acrylamides, and halogenated amides, to construct the target scaffold through distinct mechanistic pathways. The following sections detail representative examples of these radical cyclization strategies, ranging from early photochemical approaches to modern photoredox-catalyzed methods.
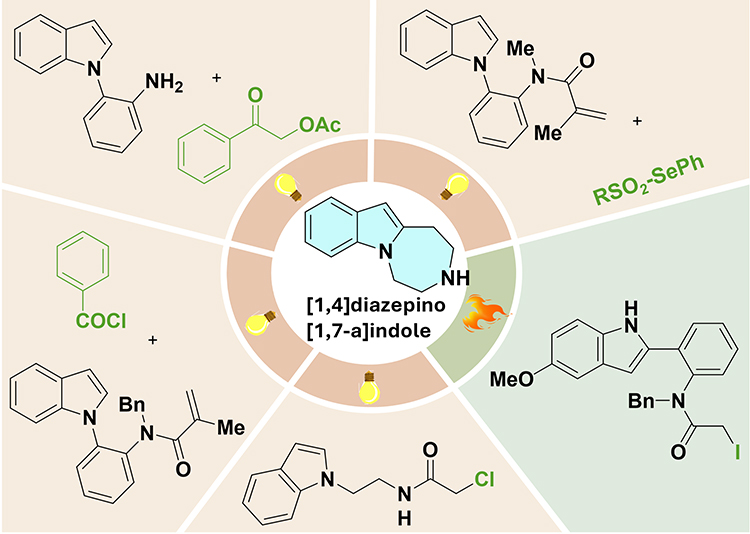 |
Figure 9 Schematic illustration of radical cyclization for indole-1,7-fused 1,4-diazepines. |
Naruto and Yonemitsu were among the early researchers to explore the potential of photochemical reactions in the synthesis of nitrogen-containing heterocycles (Scheme 39).88 They described a photocyclization process aimed at accessing azepinoindoles. When N-chloroacetyl-1-indolylethylamine 125 was irradiated with a 400 W high-pressure mercury lamp in a 50% aqueous ethanol solution containing potassium carbonate, it underwent ortho-photocyclization. However, the yield of the target product 126 was disappointingly low, only 9.9%. This low yield indicated the challenges in optimizing such photochemical reactions at that time. Subsequently, Sundberg et al also investigated the photochemical synthesis of heterocyclic compounds.89 They reported the formation of an iboga alkaloid analog through a Diels–Alder reaction photolyzed with a 450 W hanovia mercury lamp under nitrogen protection. In their experiment, a solution of chloroacetamide 127 in a mixture of NaHCO3, methanol, and water was used. Despite the efforts, the yield of product 128 was only 14%, further highlighting the difficulties in achieving high-efficiency photochemical reactions in these early studies.
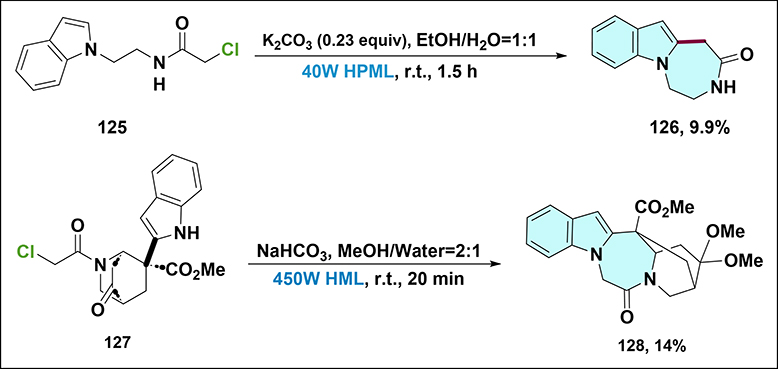 |
Scheme 39 Synthesis of compound 126 and 128. Note: Compound 126 is obtained from 125 using K2CO3 under 40 W HPML irradiation, while compound 128 is prepared from 127 with NaHCO3 under 450 W HML irradiation. |
In an attempt to overcome these limitations and improve reaction efficiency, Lian et al developed a more advanced strategy (Scheme 40).90 They reported a photoredox-catalyzed cascade reaction for the synthesis of fluorinated indolo[1,2-d]benzodiazepine derivatives. Using indole-substituted anilines 129 and fluoro-substituted acetate 130 as model substrates, using fac-Ir(ppy)3 as the photocatalyst and Et3N as the base under the irradiation of a 23 W spiral compact fluorescent lamp bulb, they first achieved a substitution reaction at the indole C2 position, followed by intramolecular amidation to afford the fluorinated product 131 in moderate yields. The reaction exhibited good tolerance toward various functional groups, including methyl, methoxy, and chloro substituents. This work not only demonstrated a new synthetic route but also provided insights into the reaction mechanism. The proposed mechanism involves the excitation of fac-Ir(ppy)3, which then undergoes single-electron transfer (SET) with BrCF2CO2Et to form radical I. Radical I adds to substrate 129a to form intermediate II, which is then oxidized via SET to intermediate III with concomitant regeneration of the photocatalyst. Intermediate III is deprotonated to IV, and finally, intramolecular amidation of intermediate IV affords the product.
 |
Scheme 40 Synthesis and proposed mechanism for the transformation of indole-substituted aniline 129 and fluorinated acetate 130 to fluorinated indolo[1,2-d]benzodiazepine 131 via photoredox-catalyzed radical cascade cyclization. Note: Under 23 W CFL irradiation, fac-Ir(ppy)3 is excited to Ir*(III), which undergoes SET with 130 to form radical I and Ir(IV). Radical I adds to substrate 129a to generate intermediate II, which is oxidized via SET to III while regenerating the photocatalyst. Deprotonation of III yields IV, and intramolecular amidation of IV affords product 131a. Selected examples: 131a, 131b, 131c and 131d. |
Building on these studies, Brambilla et al further expanded the application of visible-light photoredox catalysis to access a series of novel indole-fused 1,4-diazepinones (Scheme 41).3 The design focused on constructing the diazepinone scaffold via a photoredox-catalyzed cascade reaction, leveraging N-indolyl phenylacrylamides 132 as substrates and aroyl chlorides 133 as radical precursors. The key was to combine radical addition to the C=C double bond with intramolecular cyclization at the indole C2 position to form the fused ring system. Optimal conditions involved 1 mol% Ir(ppy)3 as the photocatalyst, 2,6-lutidine as the base, and MeCN as the solvent, under 40 W blue LED irradiation at room temperature for 20 h. This process typically afforded two diastereomeric diazepinones, arising from an axial chirality center at the N–C(aryl) bond. Notably, when sterically hindered 2,6-dimethylphenyl substituents were present at the C3 position of the starting indole, the reaction gave a single product 134a, albeit with a reduced yield of 65%. Substrate scope encompassed aroyl chlorides with electron-donating or withdrawing groups (eg, 4–OMe, 4–F) and indoles with various substituents (eg, 5–OMe, 6–CF3).
 |
Scheme 41 Synthesis and proposed mechanism for transformation of N-indolyl phenylacrylamide 132 and aroyl chloride 133 to indole-fused 1,4-diazepinone 134 via photoredox-catalyzed cyclization. Note: Photoexcitation of Ir3+ to *Ir3+, SET reduction of aroyl chloride 133a to generate aroyl radical I and Ir4+; radical I adds to the acryloyl double bond of 132a to form intermediate II, followed by cyclization at the indole C2 position to give III; reduction of III by Ir4+ yields cationic intermediate IV, which undergoes aromatization to afford 134a. Selected examples: 134a;134b/134b′; 134c/134c′ and 134d/134d′. |
The proposed mechanism for the formation of indole-fused 1,4-diazepinones is also depicted. Initially, excitation of *Ir(ppy)3 under blue LED irradiation generates an excited *IrIII species, which undergoes SET to reduce 4-methylbenzoyl chloride 133a to its radical anion, concomitant with oxidation to *IrIV. This anion then fragments, losing chlorine to form aroyl radical I. Subsequent addition of radical I to the acryloyl double bond of the acryloyl group on indole 132a forms intermediate II. Cyclization at the C2 position of indole results in intermediate III, which, upon reduction of *IrIV, yields cationic intermediate IV. Finally, aromatization of intermediate IV delivers the final products.
In a recent study, Oishi et al reported an efficient photocatalyzed cascade reaction for the synthesis of indole-fused benzodiazepines (Scheme 42).91 The key design involved the generation of a phenacyl radical from α-acetoxy acetophenone 135a under photoredox conditions, followed by its addition to 2-(3-methyl-1H-indol-1-yl)aniline 136a and subsequent cyclodehydration to afford the fused heterocyclic structure. The optimal system was identified using 10-phenylphenothiazine (PTH) as the photocatalyst, Na2CO3 as the base, and MeCN as the solvent, under irradiation with a 370 nm LED. This combination delivered the target product 137a in 80% yield. The reaction exhibited good tolerance toward a broad range of substrates. α-acetoxy aryl ketones bearing electron-donating or electron-withdrawing groups on the aryl ring reacted smoothly to afford corresponding indole-fused benzodiazepines in moderate to good yields. Indole derivatives bearing methyl, methoxy, bromo, or chloro substituents at the C5 position, as well as 3-aryl-substituted indoles were also suitable substrates, with p-methoxyphenyl-substituted indole yielding product 137c in 89% yield. Notably, substituents on the N-aryl ring were compatible. The reaction initiates with the single-electron reduction of α-acetoxy acetophenone 135a by photoexcited PTH, leading to the generation of a phenacyl radical. This radical adds to the indole moiety of compound 136a to form radical adduct I, which undergoes single-electron oxidation by PTH+. Subsequent deprotonation and cyclodehydration afford the indole-fused benzodiazepine product.
 |
Scheme 42 Synthesis and proposed mechanism for transformation of α-acetoxy acetophenone 135 and 2-(3-methyl-1H-indol-1-yl)aniline 136 to indole-fused benzodiazepine 137 via photocatalyzed cascade reaction. Note: 370 nm LED excitation of PTH generates excited-state PTH*, which undergoes single-electron reduction of α-acetoxy acetophenone 135a to afford phenacyl radical with release of acetate and formation of PTH•+; the phenacyl radical adds to the indole C3 position of 136a to form radical adduct I, which is oxidized by PTH•+ to generate cationic intermediate II; subsequent deprotonation and cyclodehydration of II furnishes the final product 137a. Selected examples: 137a, 137b and 137c. |
The synthesis of indole-fused diazepinones was achieved by Shi et al via a visible-light-induced sulfonylation cyclization, designed to leverage radical chemistry for efficient medium-ring formation (Scheme 43).92 The key design involved the homolytic cleavage of the S–Se bond in bench-stable selenosulfonates 139 under visible-light irradiation to generate sulfonyl radicals, which initiated cascade cyclization with N-(2-(1H-indol-1-yl)phenyl)acetamides 138. The optimal system utilized 30 W blue LEDs as the light source and biomass-derived 2-Me-THF as the solvent and proceeded under air at room temperature for 2 h, without external photocatalysts, additives, or bases. This setup delivered the target product 140a in 82% yield, outperforming other solvents (eg, THF gave 80%, DMF gave 65%) and confirming the necessity of continuous light irradiation (no product formed in the dark). A broad substrate scope was observed. Indoles bearing electron-donating (eg, –Me, –OMe) or electron-withdrawing groups (eg, –Cl, –CN, –CO2Me) at the 3-position reacted smoothly to form the corresponding diazepinones in good yields. Substituents on the indole benzene ring (eg, –F, –Br, –CF3) and arylamine moiety (eg, –Me, –OMe, –Cl) were also compatible. Selenosulfonates with diverse substituents (eg, aryl, naphthyl, thiophene, alkyl) further expanded the scope. The reaction proceeds through a radical pathway, in which the S–Se bond in selenosulfonates is cleaved under visible-light irradiation to generate sulfonyl radicals. These radicals react with the double bond of indole substrate, forming radical intermediates that undergo intramolecular cyclization to produce the desired indole-fused diazepinones.
 |
Scheme 43 Synthesis and proposed mechanism for transformation of compound 138 and selenosulfonate 139 to indole-fused diazepinone 140 via visible-light-induced sulfonylation cyclization. Note: Under blue light irradiation, homolytic cleavage of the S–Se bond in selenosulfonate 139a generates electrophilic sulfonyl radical (Ts•) and phenylseleno radical (PhSe•); addition of Ts• to the indole C3=C2 double bond of 138a forms carbon-centered radical intermediate I, which undergoes intramolecular cyclization to give II; oxidation of II by PhSe• followed by deprotonation generates intermediate III, which eliminates PhSeH to afford the final product 140a. Selected examples: 140a, 140b, 140c and 140d. |
Wang et al reported an efficient method to synthesize indole-fused 1,4-diazepine derivatives 143 through a photocatalyzed sulfonation–cyclization reaction (Scheme 44).68 Under optimized conditions, using Ru(bpy)3Cl2 (2 mol%) as the photocatalyst, Na2CO3 as the base, and blue LEDs irradiation in MeCN at room temperature, the reaction of N-(2-(1H-indol-1-yl)phenyl)-N-methylmethacrylamide 141a with tosyl chloride 142a delivered the target product 143a in 91% isolated yield. This strategy exhibited broad substrate compatibility, tolerating various substituted phenylsulfonyl chlorides, heteroarylsulfonyl chlorides, and substituted indoles, with the corresponding products obtained in good yields. Mechanistic studies revealed that the reaction proceeds via a radical pathway, starting from the sulfonyl radical, which adds to the acrylamide alkene moiety, followed by diastereoselective cyclization with the indole double bond, oxidation, and deprotonation to form the final product.
 |
Scheme 44 Synthesis and proposed mechanism for transformation of compound 141 and tosyl chloride 142 to indole-fused 1,4-diazepine 143 via Ru-photocatalyzed sulfonation–cyclization. Note: Blue light excitation of Ru(bpy)3Cl2 generates the excited photocatalyst, which reduces 142a via SET to generate sulfonyl radical. Radical addition to 141a forms I, followed by diastereoselective cyclization at the indole C2 position to give II. Oxidation of II to III and subsequent deprotonation affords 143a. Selected examples: 143a, 143b, 143c and 143d. |
Shi et al developed a novel visible-light-induced sulfonylation–cyclization–selenylation reaction to rapidly construct highly functionalized indole-fused diazepinones 146 (Scheme 45).93 The synthesis employed 2-Me-THF, derived from biomass feedstocks, as the solvent, aligning with green chemistry principles. The process was carried out under additive-, base-, and external photosensitizer-free conditions, highlighting its atom economy and environmental sustainability. The synthetic pathway involves the use of indole derivatives 144 and selenosulfonates 145 as starting materials. Under blue LED irradiation, the selenosulfonate undergoes homolysis to generate an electrophilic sulfonyl radical and a seleno radical. The sulfonyl radical adds to the double bond of the indole derivative, forming a carbon-centered radical intermediate I, which then undergoes intramolecular cyclization. Subsequent oxidation by a seleno radical and deprotonation yield the sulfonylation–cyclization intermediate IV. Finally, electrophilic addition of the PhSe+ species to the C3 position of the indole, followed by deprotonation, affords the desired product 146.
 |
Scheme 45 Synthesis and proposed mechanism for transformation of indole derivative 144 and selenosulfonate 145 to indole-fused diazepinone 146 via visible-light-induced sulfonylation–cyclization–selenylation cascade. Note: Under blue light irradiation, homolytic cleavage of the S–Se bond in selenosulfonate 145a generates sulfonyl radical (Ts•) and phenylseleno radical (PhSe•); addition of Ts• to the acrylamide double bond of 144a forms carbon-centered radical I, which undergoes intramolecular cyclization to give II; oxidation of II by O2 generates carbocation III, which upon deprotonation affords IV; finally, electrophilic selenylation of IV at the indole C3 position by PhSe+, followed by deprotonation, furnishes 146a. Selected examples: 146a, 146b, 146c and 146d. |
The synthetic method for compound 146 exhibited a broad substrate scope. Indoles with various substituents, including electron-withdrawing groups and electron-donating groups at different positions on the indole ring, were compatible with this transformation, affording the corresponding products in moderate to good yields. Selenosulfonates with different substituents on the benzene ring (eg, hydrogen, electron-withdrawing groups, and various heterocyclic substituents) could also be employed to successfully synthesize the desired indole-fused medium-sized diazepinones.
Looking back at the development of this field, it is also important to mention the work of Bremner et al (Scheme 46).94 They reported a radical cyclization process to access the pharmacologically important paullone scaffold from indolyl iodoacetamide derivatives. For instance, the synthesis of indolo[1,2-d][1,4]benzodiazepin-6-one 148 from compound 147 was achieved via a nucleophilic cyclization process at the indolic nitrogen, which was generated in situ by treatment with Bu3SnH and AIBN in refluxing mesitylene. This work further demonstrates the utility of radical processes in constructing biologically relevant heterocyclic frameworks.
 |
Scheme 46 Radical cyclization of indolyl iodoacetamide 147 for the synthesis of indolo[1,2-d][1,4]benzodiazepin-6-one 148. |
The studies summarized above demonstrate the remarkable versatility of radical reactions in the synthesis of complex indole-fused heterocyclic compounds. While most of these reactions employ visible-light photoredox catalysis to generate radical intermediates via SET processes, it is important to note that radical chemistry extends beyond photoredox-mediated mechanisms. For example, the radical cyclization reported by Bremner et al employs thermal radical initiation via Bu3SnH and AIBN,94 highlighting that both photochemical and thermal pathways can effectively mediate radical transformations. A notable commonality among these reactions is the presence of halogen substituents in the substrates. Halogens (eg, chlorine and iodine) play a crucial role in initiating radical processes, either by facilitating SET from the excited photocatalyst or via thermal initiation. These halogenated substrates serve as efficient radical precursors, enabling the formation of reactive intermediates that mediate the subsequent cyclization or functionalization steps. This shared feature underscores the importance of halogens in radical chemistry as versatile handles for generating radical species under mild conditions. Future endeavors may focus on developing more efficient photocatalytic systems, exploring synergies between photoredox and thermal radical processes, and integrating radical chemistry with other synthetic strategies to achieve more sustainable and efficient pathways for the synthesis of complex molecules.
Radical cyclization has proven to be a highly effective and versatile strategy for the synthesis of indole-fused diazepine structures. Its ability to operate under mild conditions and the compatibility with a wide range of functional groups render this approach particularly attractive for the construction of complex heterocyclic scaffolds with potential biological applications. Future work in this area may focus on further optimizing reaction conditions, exploring novel photocatalytic systems, and integrating radical chemistry with other synthetic methodologies to develop more sustainable and efficient pathways for the assembly of complex molecules. The insights gained from these studies will undoubtedly continue to drive the development of new and innovative synthetic methods in the field of heterocyclic chemistry.
Following the discussion on radical cyclization, while this approach offers mild reaction conditions and compatibility with diverse functional groups for the synthesis of complex indole-fused diazepine structures, it still faces notable limitations. For instance, some reactions rely on photocatalysis or elevated temperatures, and there is a strong dependence on halogen-containing substrates, which restricts its applicability to a broader substrate scope and the sustainability of synthetic routes.
To address the limitations of radical cyclization, including reliance on photocatalysis, elevated temperatures, and halogen-containing substrates, researchers have turned to alternative synthetic strategies that offer enhanced operational flexibility, substrate generality, and sustainability for the construction of indole-fused diazepines. Among these, phosphine-catalyzed cyclization has emerged as a particularly powerful approach, especially for the synthesis of indole-1,2-fused 1,4-diazepines. Phosphine catalysts, as versatile Lewis bases, enable unique reaction pathways (eg, annulation reactions involving MBH derivatives or redox-neutral cascade processes) that eliminate the need for harsh reaction conditions or halogenated starting materials. Their ability to modulate nucleophilicity and regioselectivity also enables the efficient construction of the seven-membered diazepine ring while preserving sensitive functional groups on the indole scaffold. The next section focuses on the development and application of phosphine-catalyzed cyclization strategies for accessing indole-1,2-fused 1,4-diazepines, highlighting their advantages in overcoming the drawbacks of radical cyclization.
Phosphine-Catalyzed Cyclization
Wang et al detailed the development of an N-alkylation-initiated, redox-neutral [5 + 2] annulation reaction between 3-alkylindoles and o-aminobenzaldehydes, culminating in the efficient synthesis of indole-1,2-fused 1,4-benzodiazepines 151 (Scheme 47).95 The use of substituted indoles 149 and 2-(pyrrolidin-1-yl)benzaldehydes 150 in the presence of 20 mol% of 1,1′-binaphthyl-2,2′-diyl hydrogen phosphate (PA) in DCM at 80°C was identified as the optimal reaction conditions. This annulation reaction exhibited a broad substrate scope. Both 3-alkylindoles bearing electron-donating or electron-withdrawing substituents, and o-aminobenzaldehydes with diverse substituents at various ring positions, reacted smoothly under the optimized conditions. The corresponding products were obtained in moderate to good yields. A cascade mechanism was proposed: PA-catalyzed N-alkylation of 3-methylindole 149a with and 2-(pyrrolidin-1-yl)benzaldehyde 150a forms 1-indolylmethanol I. Protonic acid-catalyzed dehydration of intermediate I affords carbocation intermediate II, which then undergoes intramolecular [1,5]-hydride transfer to produce iminium ion intermediate III. Finally, intramolecular Friedel–Crafts alkylation of the carbocation in intermediate III with the indole ring yields product 151a.
 |
Scheme 47 Synthesis and proposed mechanism for transformation of 3-alkylindole 149 and 2-(pyrrolidin-1-yl)benzaldehyde 150 to indole-1,2-fused 1,4-benzodiazepine 151 via PA-catalyzed N-alkylation-initiated [5 + 2] annulation. Note: PA-catalyzed N-alkylation of 3-methylindole 149a with 150a forms intermediate I; protonic acid-catalyzed dehydration of I affords carbocation II; [1,5]-hydride transfer of II generates iminium ion III; finally, intramolecular Friedel-Crafts alkylation of III (attack at indole C2 position) yields 151a. Selected examples: 151a, 151b, 151c and 151d. |
Similarly, annulation of tosyl-protected tryptamine 152 with o-aminobenzaldehyde 153 also delivered the scaffolds in high yields (Scheme 48).4 This method provided a straightforward route to indole-1,2-fused 1,4-benzodiazepines 154 with high efficiency and selectivity, highlighting the versatility and utility of the PA-catalyzed [5+2] annulation strategy.
 |
Scheme 48 PA-catalyzed synthesis of indole-1,2-fused 1,4-benzodiazepines 154 from tosyl-protected tryptamine 152 and N-alkyl o-aminobenzaldehydes 153. Selected examples: 154a, 154b, 154c and 154d. |
Zhu et al developed a straightforward synthetic method for indole-1,2-fused 1,4-diazepinones 157 via phosphine-catalyzed [4+3] annulations between dinucleophilic indoles 155 and MBH carbonates 156 (Scheme 49).5 Here, the indoles acted as innovative four-atom building blocks. The optimal conditions for this reaction were established as 65°C in MeCN, employing 20 mol% (p-MeC6H4)3P as the catalyst. Electron-withdrawing substituents at the indole C3 position were found to significantly enhance the nucleophilicity of the N atom, thereby facilitating the annulation transformation. Consequently, a diverse array of indole-1,2-fused 1,4-diazepinones were efficiently synthesized in one step, delivering products in good to high yields.
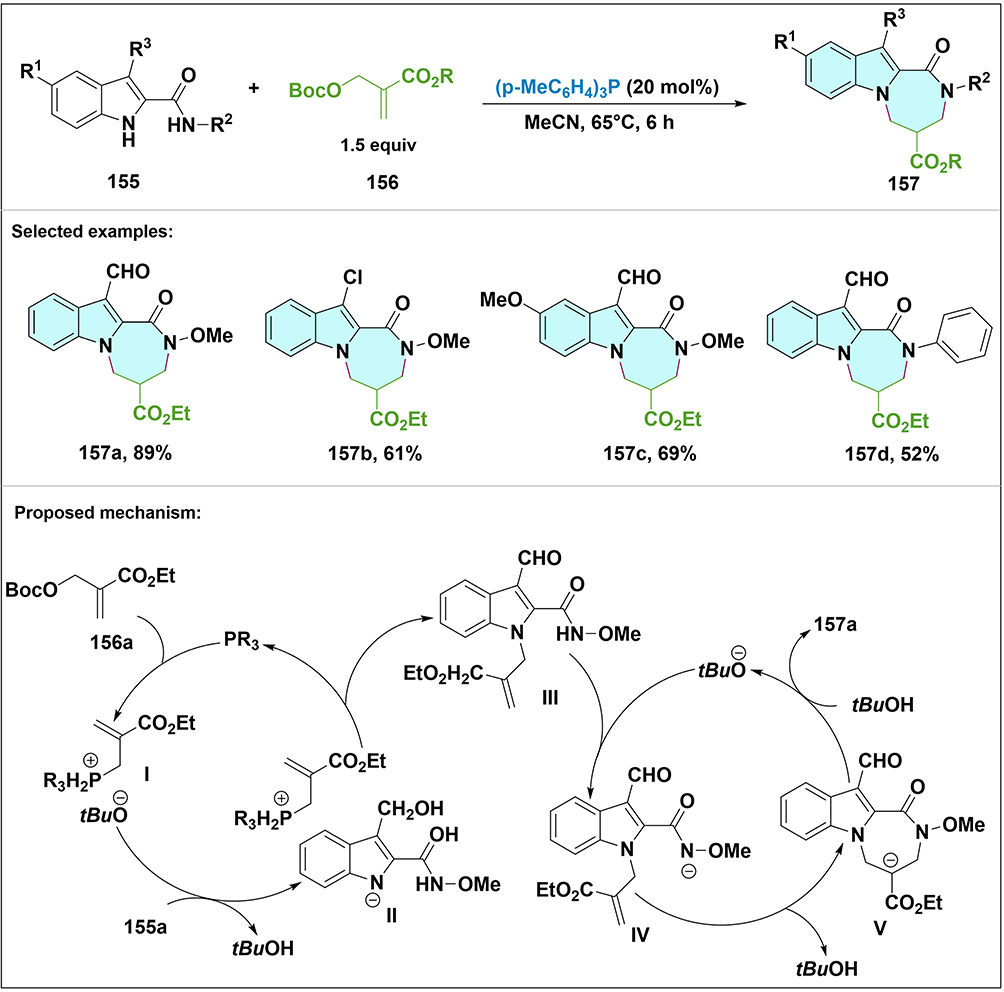 |
Scheme 49 Synthesis and proposed mechanism for transformation of dinucleophilic indole 155 and MBH carbonate 156 to indole-1,2-fused 1,4-diazepinone 157 via phosphine-catalyzed [4+3] annulation. Note: Nucleophilic addition of phosphine to MBH carbonate 156a with concomitant decarboxylation generates allylic phosphonium ylide I and tBuO−; selective deprotonation of indole N1-H by tBuO− gives II; SN2′ substitution of II with I affords allylic alkylation intermediate III with regeneration of phosphine catalyst; deprotonation of amide N-H in III by tBuO− generates IV; intramolecular Michael cyclization of IV forms cyclic intermediate V; proton transfer from tBuOH to V yields 157a. Selected examples: 157a, 157b, 157c and 157d. |
A proposed mechanism for the formation of these diazepinones is outlined as follows. First, the phosphine catalyst undergoes nucleophilic addition to the MBH carbonate 156a with concomitant decarboxylation, yielding intermediate I. Concurrently, the tert-butoxide anion, generated in situ, serves as a base, abstracting a proton from the N1 atom of the indole ring and leading to intermediate II. Subsequently, ion pair II undergoes SN2′ substitution to afford allylic alkylation intermediate III, with regeneration of the phosphine catalyst. Next, the N4′ atom in the amide moiety of intermediate III is deprotonated by the in situ generated tert-butoxide anion, owing to its relatively high acidity, forming intermediate IV. Finally, intermediate IV undergoes intramolecular Michael cyclization to furnish the final product 157a as the final cyclic product.
Miscellaneous Transformations
After surveying the mainstream strategies: base-mediated, metal-catalyzed, radical, and phosphine-catalyzed cyclization, we now turn to a heterogeneous collection of “miscellaneous transformations” that resist clean classification. These reactions do not hinge on a single catalyst or unified mechanism; instead, they capitalize on tailored chemistries such as multicomponent couplings, oxidation-triggered cyclization, or functional-group-specific rearrangements. By finely tuning substrate reactivity, they deliver rapid access to intricate indole-fused diazepine cores, often in scenarios where classical methods falter. Their inherent modularity accommodates every fusion topology (1,7-, 1,2-, 6,7,1-, etc.) and offers a practical gateway to structural outliers or heavily decorated derivatives.
Indole-1,7-Fused with 1,4-Diazepines
Hutait et al presented a convenient synthesis of a lactam-fused β-carboline library using a Ugi four-center three-component reaction, a type of isonitrile-based multicomponent reaction (Scheme 50).96 Optimization began with β-carboline aldehyde 158, amine 159, and 4-tolylsulfonylmethyl isocyanide 160 in methanol at room temperature, delivering the target lactam-annulated carboline 161 in moderate to good yields. Parallel reactions employing diverse amines and isonitriles afforded a library of products. Anilines and benzylamine proved optimal. This one-pot approach efficiently constructs complex heterocycles by leveraging the synergistic reactivity of multiple functional groups, enabling flexible diversification of amine and isocyanide substituents.
 |
Scheme 50 Ugi four-center three-component reaction of β-carboline aldehyde 158, amine 159 and isocyanide 160 for the synthesis of lactam-annulated β-carbolines 161. Selected examples: 161a, 161b and 161c. |
In a distinct approach, Danieli and Passarella employed pyridine sulfur trioxide complex, the key reagent in Parikh–Doering Oxidation, to catalyze C–N bond formation for the synthesis of the indole polyheterocycle 163 (Scheme 51).97 Intramolecular oxidative cyclization of substrate 162 proceeded in the presence of triethylamine, affording unique indole polyheterocycle derivatives as two isomers in 34% and 27% yield, respectively. This method highlights the utility of oxidative activation in driving cyclization reactions, even with sensitive functional groups present.
 |
Scheme 51 Synthesis of compound 163 from 162 via Parikh–Doering Oxidation. |
Indole-1,2-Fused with 1,4-Diazepines
The synthesis of indole-fused lactam rings has been achieved through multiple strategies. One route involves bis-mesylation of diol 164, subsequent azide displacement, Me3P-mediated reduction, and in situ cyclization to form lactam 165, which was further transformed to primary amine 166 (Scheme 52).98 Separately, Putey et al reported the preparation of tetrahydro[1,4]diazepino[1,2-a]indol-1-ones 168 via lactone-to-lactam rearrangement of compound 167 in MeOH/THF.99
 |
Scheme 52 Two synthetic strategies for indole-fused lactam scaffolds. Note: Bis-mesylation of diol 164, subsequent azide displacement, trimethylphosphine-mediated reduction, and in situ cyclization to form lactam 165, which is further transformed to primary amines 166a (X = 3-pyridyl) or 166b (X = 2-thiazolyl); lactone-to-lactam rearrangement of lactone 167 upon treatment with ammonia gas to afford 168. |
Gour et al reported a catalyst-free approach for constructing polycyclic indole-substituted 1,2,3-triazole 170 via an intermolecular [3+2] azide-alkene cycloaddition reaction (Scheme 53).100 Employing N-bromoalkyl 2-indolyl α, β-unsaturated ketones as substrates, extensive optimization identified a water/acetone (1:1) solvent system at room temperature as the optimal conditions, with no external catalyst required.
 |
Scheme 53 Intramolecular azide-alkene cascade reaction of 169 for the synthesis of polycyclic indole-substituted 1,2,3-triazoles 170. Selected examples: 170a, 170b and 170c. |
Indole-6,7,1-Fused with 1,4-Diazepines
Acid-mediated cyclization emerges as a pivotal strategy due to its ability to efficiently activate electrophilic sites and facilitate the formation of nitrogen-containing heterocycles under mild conditions. Specifically, acid catalysts can protonate reactive centers to promote intramolecular cyclization involving the reactive carbon sites on the indole ring and the carbon intermediates from the diazepine precursor, a process particularly suited for constructing the conjugated diazepine ring. One notable example is the synthesis of indole-6,7,1-fused 1,4-diazepine core (Scheme 54).101 The authors utilized indoline derivative 171 as starting material. It underwent ring closure in the presence of formalin, acetic acid, and catalytic sulfuric acid at 70°C, affording the target product 172.
 |
Scheme 54 Synthesis of indole-6,7,1-fused 1,4-diazepine 172 by acid-mediated cyclization of 5-fluoroindoline derivative 171 with formalin; selected example 173 is a glycogen synthase kinase-3 inhibitor. |
Alternative synthetic approaches outside traditional catalysis have been developed to access indole-6,7,1-fused 1,4-diazepines, leveraging unique reaction chemistries for complex scaffold construction. Azepinoindole-fused ring systems were synthesized via oxidative dehydrogenation and subsequent functionalization (Scheme 55).102 Treatment of compound 174 with DDQ in refluxing benzene induced dehydrogenation to form compound 175. The reaction was followed by slow addition of POCl3 in DMF at 0°C, stirring at room temperature for 17 hours to introduce an aldehyde group, which was then condensed with hydroxylamine to afford oxime 177.
 |
Scheme 55 Synthesis of compound 177. Note: Oxidation of 174 with DDQ in refluxing benzene to afford unsaturated lactam 175; Vilsmeier–Haack formylation of 175 with POCl3/DMF to give aldehyde 176; Condensation of 176 with hydroxylamine hydrochloride/NaOH to furnish oxime 177. |
Indole-3,2,1-Fused with 1,4-Diazepines
Triazidochlorosilane (TACS), generated in situ as triazidochlorosilane (from SiCl4 and 3NaN3), serves as a versatile nitrogen source for constructing nitrogen-rich heterocycles.103 El-Ahl et al utilized this reagent to develop a synthetic route to ring-expanded azepinoindole-triazole 180, starting from azepinoindolone 178 (Scheme 56).104 The TACS-mediated process proceeds via siloxy azide I and gem-diazidoalkane II intermediates, followed by a Beckmann-like rearrangement of gem-diazidoalkane to form imidoyl azide III, which cyclizes to the target product 180. This strategy highlights the utility of azide chemistry in assembling complex fused rings with precise nitrogen incorporation.
 |
Scheme 56 Synthesis and proposed mechanism for transformation of azepinoindolone 178 to azepinoindole-triazole 180 via TACS-mediated [3+2] cycloaddition. Note: Reaction of TACS 179 with 178 generates siloxy azide intermediate I, which is converted to gem-diazidoalkane II; II undergoes Beckmann-like rearrangement to form imidoyl azide III, which cyclizes to afford 180. |
Indole-1,7-Fused with 1,3-Diazepines
Liu et al developed a method for synthesizing polycyclic-fused indoles from 2-indolyl aryl carbinols through a Lewis acid-catalyzed consecutive [1,5]-hydrogen migration and 7-cyclization process (Scheme 57).105 The highest yield was achieved with this protocol at 80°C in toluene with a catalytic amount of BF3·Et2O. In this methodology, indolodiazepine 182 was obtained via C–N connection rather than C–C cyclization due to the low nucleophilicity of the indole C3 position and the steric hindrance from the methyl group at its C1 position. 2-indolyl aryl carbinol derivatives with various electron-donating and electron-withdrawing substituents on the indole or benzene ring attached to the amino group, as well as a tetrahydroisoquinoline-derived substrate 181c, underwent the reaction smoothly to afford the target indolodiazepine products. A plausible mechanism for the formation of product 182a involves with the dehydration of substrate 181a to form an iminium ion I under acid catalysis. This intermediate then undergoes a [1,5]-hydride shift, followed by a C–N cyclization at the indole nitrogen atom, which is favored by steric hindrance from the methyl group, yielding indolodiazepine 182a.
 |
Scheme 57 Synthesis and proposed mechanism for transformation of 3-methyl-2-indolylmethanol 181 to indolodiazepine 182 via BF3·Et2O-catalyzed dehydration/[1,5]-hydride shift/7-cyclization sequence. Note: BF3·Et2O-promoted dehydration of 181a generates iminium ion I, which undergoes [1,5]-hydride transfer to afford II; subsequent [1,7] C–N cyclization furnishes 182a. Selected examples: 182a, 182b and 182c. |
Qu et al described the synthesis of benzodiazepino-indoles 184 from the indoloquinolines 183 (Scheme 58).106 The reaction was conducted using phosphoryl chloride in MeCN, resulting in the formation of the chloro-iminium intermediate I. The subsequent rearrangement involves the migration of the N-benzyl group from the indole C3 position to C2, leading to the cleavage of the indole C2-N bond and formation of the imine nitrogen atom in intermediate II. Finally, the corresponding [1,3]benzodiazepino[1,7-a]indoles 184 were obtained with yields ranging from 40% to 50%. It was found that the activation of the two methoxy groups appears to be crucial in facilitating the rearrangements at C3 and C2, enabling the subsequent cyclization onto the indole nitrogen atom.
 |
Scheme 58 Synthesis and proposed mechanism for transformation of indoloquinoline 183 to benzodiazepino-indole 184 via POCl3-mediated ring-expansion reaction. Note: Activation of 183 by POCl3 forms chloro-iminium intermediate I; I undergoes benzyl migration from C3 to C2 with concomitant cleavage of the indole C2 bond to generate iminium ion II; final cyclization onto the indole nitrogen affords 184. Selected examples: 184a, 184b and 184c. |
Indole-1,2-Fused with 1,3-Diazepines
de la Mora et al published the initial synthesis of 1,3-diazepino[1,2-a]indole 186 (Scheme 59).107 Heating the azidoindole 185 in a solution of bromobenzene at 180°C in a sealed metal reactor led to the formation of 1,3-diazepino[1,2-a]indole 186 in 60% yield. The absence of an electron-withdrawing substituent at the indole C3 position precluded the formation of tricyclic compounds. It has been proposed that the synthesis of compound 186 occurs through an intramolecular 1,3-dipolar cycloaddition between the azido group and the indole C2=C3 double bond, resulting in the formation of an intermediate triazoline I. Subsequent nitrogen extrusion from intermediate I, potentially involving an intermediate aziridine, ultimately yields the tricyclic product 186.
 |
Scheme 59 Intramolecular 1,3-dipolar cycloaddition of 1-α-azidoalkylindoles 185 for the synthesis of tricyclic 1,3-diazepino[1,2-a]indoles 186. Note: Thermally induced intramolecular 1,3-dipolar cycloaddition of 185 forms triazoline intermediate I via reaction of the azide with the indole C2=C3 bond; nitrogen extrusion from I (potentially involving an aziridine intermediate) affords 186. |
In a subsequent study, Yang et al reported the synthesis of compound 189 via 1,4-Michael addition and cyclocondensation reactions involving 1,4-benzoquinone 187 and heterocyclic ketene aminals 188 (Scheme 60).108 The optimal reaction conditions were identified as acetic acid as the catalyst and refluxing 1,4-dioxane as the solvent. The heterocyclic ketene aminals undergo a potential aza-ene addition mechanism with 1,4-benzoquinone, leading to the formation of adduct II. Subsequent imine–enamine tautomerization results in the formation of compound III, followed by cyclocondensation and dehydration to afford the desired product 189.
 |
Scheme 60 Synthesis and proposed mechanism for the transformation of heterocyclic ketene aminal 188 and 1,4-benzoquinone 187 to 1,3-diazepino[1,2-a]indole 189 via acetic acid-catalyzed cyclocondensation. Note: Protonation of 187 generates electrophilic quinone I, which undergoes an aza-ene reaction with 188a to afford adduct II; imine-enamine tautomerization of II gives III; final cyclocondensation of III (-H2O) furnishes 189a. Selected examples: 189a, 189b and 189c. |
Indole-6,7,1-Fused 1,3-Diazepines
A Chinese patent, published as CN102757435A, describes the synthesis of a novel class of trans-3-indole-based 1,3-diazepine compounds 192, which have shown significant anti-tumor activity in tumor-bearing mouse models (Scheme 61).25 The key step in the synthetic procedure for the ring closure involved heating secondary amine 190 with paraformaldehyde in dry toluene, which was followed by six additional steps to yield indole-6,7,1-fused 1,3-diazepines 192 as a class of c-Met inhibitors for cancer treatment.
 |
Scheme 61 Synthesis of indole-6,7,1-fused 1,3-diazepines. Note: Pictet-Spengler-type cyclization of secondary amine 190 with paraformaldehyde to afford tetracyclic intermediate 191; subsequent transformation of 191 furnishes 192 as a class of c-Met inhibitors for cancer therapy. |
These miscellaneous transformations highlight the diversity and creativity in synthetic chemistry, demonstrating that even less conventional methods can play a crucial role in the construction of complex heterocyclic systems. By exploring these alternative pathways, researchers can access novel scaffolds and functional groups that may offer unique biological activities and therapeutic potential. Future work in this area may focus on further optimizing these methods and exploring their applications in the synthesis of other complex natural products and pharmaceuticals.
As summarized in the preceding sections, the construction of indole-fused diazepines with a bridgehead nitrogen atom has been achieved through a rich arsenal of synthetic methodologies, including base-mediated cyclization, transition-metal-catalyzed C–H activation, radical cascade cyclization, phosphine-catalyzed annulation, and various miscellaneous transformations such as Ugi reactions, oxidative cyclization, and rearrangement-based ring expansions. Each strategy offers unique advantages in terms of regioselectivity, functional group tolerance, scalability, and structural diversity, enabling access to all major fusion modes, including 1,7-, 1,2-, 6,7,1-, and 3,2,1-indole-diazepine architectures.
Notably, base-mediated and phosphine-catalyzed protocols have emerged as particularly robust and modular platforms for late-stage diversification, while metal-catalyzed C–H activations and photoredox radical cyclizations continue to push the boundaries of step and atom economy. A comprehensive comparison of these mainstream synthetic strategies, along with miscellaneous transformations, highlights their distinct advantages, limitations, and applicability across key metrics critical for medicinal chemistry and scale-up synthesis (Table 1).
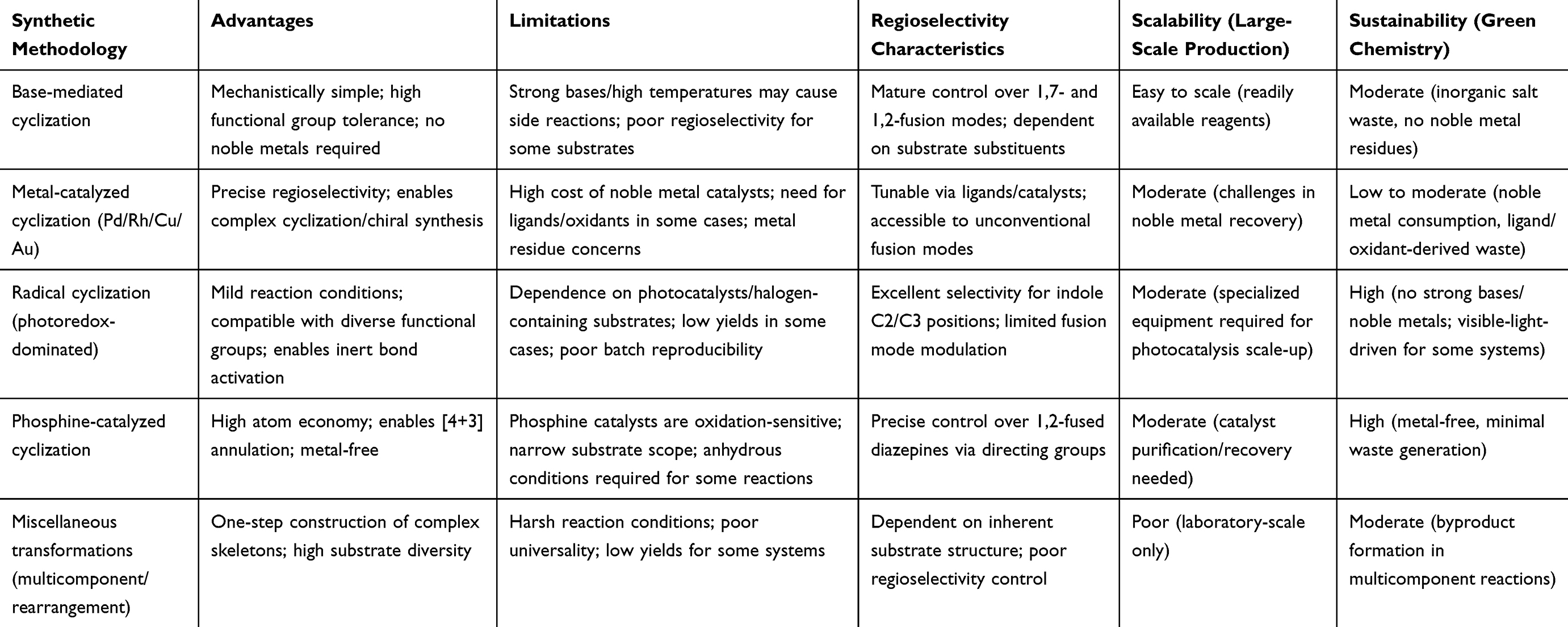 |
Table 1 Comparative Analysis of Synthetic Methodologies for Indole-Fused Diazepines with a Bridgehead Nitrogen Atom |
As reflected in the table, each strategy offers unique trade-offs: base-mediated and phosphine-catalyzed methods stand out for their modularity and sustainability, making them preferred for late-stage derivatization, while metal-catalyzed and radical approaches excel in accessing complex or unconventional fusion patterns through precise regiocontrol and step economy. Despite these advances, several challenges remain, such as the underexplored indole-1,2-fused 1,3-diazepine and indole-6,7,1-fused 1,2-diazepine scaffolds, in which thermodynamic instability and regioselectivity issues limit synthetic accessibility. Future efforts are expected to focus on asymmetric variants, adaptation to flow chemistry, and AI-guided route optimization to further enhance synthetic efficiency and enable library generation for biological screening, thereby addressing current limitations such as catalyst cost, substrate scope restrictions, and scalability barriers in these methodologies.
With these powerful synthetic tools in hand, the stage is now set to systematically explore the biological landscape of indole-fused diazepines. The following sections critically survey their therapeutic relevance across antiviral, anticancer, cardiovascular, and CNS-related disorders, with a particular emphasis on SAR that underpins their target selectivity and drug-like potential.
Biological Significance Along with SAR Study
Indole-fused diazepines with a bridgehead nitrogen atom exhibit diverse biological activities that are tightly governed by three core structural variables: (1) the fusion mode between indole and diazepine rings (eg, 1,7-fused vs. 1,2-fused), (2) the positional arrangement of nitrogen atoms in the diazepine ring (1,3-, 1,4-diazepines), and (3) the substitution patterns on the indole/diazepine scaffolds (eg, electron-donating/withdrawing groups, chiral substituents). These structural features directly modulate the molecule’s spatial conformation, electron density distribution, and binding affinity to biological targets (eg, enzymes, receptors, DNA). Below, we systematically link these structural characteristics to specific biological targets and pharmacological outcomes, with a focus on mechanistic insights and SAR trends.
Antiviral Activity
A series of conformationally constrained tetracyclic compounds based on a 6,7-dihydro-5H-benzo[5,6][1,4]diazepino[7,1-a]indole scaffold were designed and synthesized as potent Hepatitis C Virus (HCV) nonstructural protein 5B (NS5B) RNA polymerase inhibitors (Figure 10).40 The tetracyclic core, constructed by bridging the indole moiety (a 6,5-bicyclic system) and the C2-phenyl ring through a seven-membered ring, is essential for maintaining an optimal dihedral angle (~46°), which facilitates optimal binding to the allosteric thumb domain of NS5B. The bridgehead nitrogen atom, anchored in the tricyclic indole core, plays a pivotal role in constraining the dihedral angle to the optimal ~46°. This structural confinement avoids over-planarization or excessive twisting of the scaffold, both of which are associated with reduced NS5B inhibitory potency. Among ring sizes evaluated, the seven-membered ring, with oxygen or NH as the bridging atom (X), demonstrated superior potency, while smaller or larger rings led to reduced activity due to over-planarization or excessive twisting, respectively.
 |
Figure 10 Structure and SAR of 6,7-dihydro-5H-benzo[5,6][1,4]diazepino[1,7-a]indole 8. |
Key to the enhanced efficacy of compound 8 was the introduction of a piperidine-containing basic substituent at the N5-position of the diazepine ring. This modification significantly improved both biochemical and cellular potency (NS5B IC50 = 9 nM; replicon EC50 = 35 nM in 10% FCS) and, importantly, minimized the potency shift in the presence of human serum albumin (EC50 = 84 nM with 4% HSA). This low HSA binding was clinically advantageous, as it reduced protein binding losses in vivo and maintained free drug concentrations in the liver—the primary site of HCV infection. Furthermore, compound 8 exhibited no significant cytotoxicity (CC50 > 20 μM), resulting in a favorable selectivity index (SI > 238), underscoring its potential as a promising lead for anti-HCV drug development.
Compound 60a represented a key indolo-fused heterocyclic inhibitor of HCV NS5B polymerase (IC50 = 0.07 μM) with notable PK profiles (Figure 11).57 Structurally, it features a conformationally constrained indolobenzodiazepinone core stabilized by a seven-membered lactam bridge (linking the indole N1 and the ortho position of the 2-aryl moiety), which is critical for its inhibitory activity. The bridgehead nitrogen atom serves as a critical structural anchor within this seven-membered lactam bridge, constraining the dihedral angle to ~43° between the indole and pendant phenyl rings. This conformational preorganization positions the inhibitor optimally within the NS5B thumb domain, complementing the hydrogen-bonding network formed by the pendant side chain with Arg503 and Arg498, a configuration that further enhances preorganized binding to the NS5B thumb domain—distinct from less constrained analogs or those with alternative bridges.
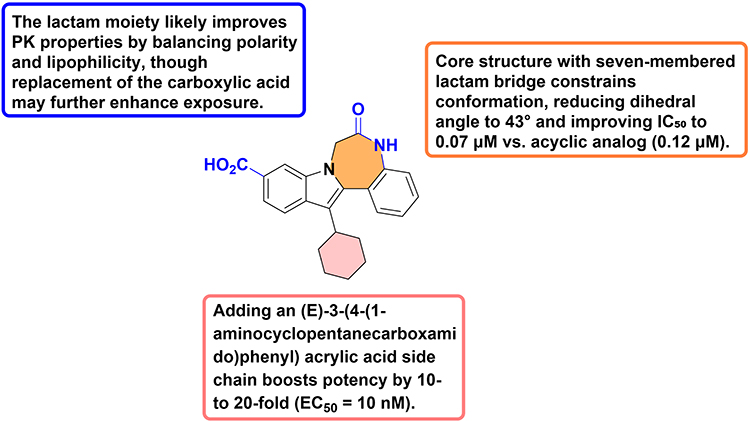 |
Figure 11 Structure and SAR of 13-cyclohexyl-6-oxo-6,7-dihydro-5H-indolo[2,1-d][1,4]benzodiazepine-10-carboxylic acid 60a. |
In terms of PK properties in rats, compound 60a outperformed other truncated analogs: it displayed an oral bioavailability of 17.6%, an area under the curve (AUC) of 2.2 μM·h, and a clearance of 35 mL/min/kg, surpassing the hydrocarbon-bridged (F = 6%, AUC = 1.3 μM·h, clearance = 53 mL/min/kg) and oxa-bridged (F = 5%, clearance = 170 mL/min/kg) analogs. The superior PK profile of compound 60a was attributed to the polar lactam bridge (with C=O and NH groups), which improved aqueous solubility and reduced metabolic clearance, while the cyclohexyl substituent optimized hydrophobic interactions without inducing excessive steric hindrance. Collectively, these features positioned compound 60a as a promising truncated lead for further optimization, particularly through modification of the carboxylic acid moiety to improve systemic exposure while preserving target potency.
Compound 60b was a heteroaryl-fused tetracyclic indole-diamide compound that functions as a potent allosteric inhibitor of the HCV NS5B polymerase (Figure 12).109 It bound to the “thumb” domain of NS5B, as supported by its reduced activity against the P495L mutant NS5B enzyme (genotype 1b, IC50 > 0.6 μM) compared to the wild-type enzyme (genotype 1b, IC50 = 0.024 μM). It exhibited excellent anti-HCV efficacy across key models: it inhibited HCV genotype 1a and 1b replicons with EC50 values of 0.028 μM and 0.021 μM, respectively, and showed low cytotoxicity with a CC50 of 45 μM in the replicon system, yielding a calculated therapeutic index >2140 against genotype 1b. Its potency was attributed to structural features including a pyridine-fused tetracyclic core (which matched the spatial conformation of the NS5B “thumb” domain pocket better than five-membered heterocycle-fused analogs) and a conformationally constrained bridging element that optimized alignment with the enzyme’s active site.
 |
Figure 12 Structure and SAR of heteroaryl-fused tetracyclic indole-diamide 60b. |
However, its pharmacokinetic profile in rats was suboptimal, with oral bioavailability of less than 1% and undetectable oral AUC, likely due to poor solubility and membrane permeability. Despite this limitation, compound 60b was valuable for HCV drug development: it expanded the SAR of NS5B inhibitors (validating pyridine as a viable bioisostere of phenyl for sustaining potency) and highlighted future optimization directions, such as introducing polar groups to improve solubility or designing prodrugs to enhance membrane permeability.
Anticancer Activity
Indole-fused diazepines exhibit anticancer activity by targeting key drivers of tumorigenesis, including anti-apoptotic proteins (eg, Mcl-1), oncogenic kinases (eg, proviral insertion site in moloney murine leukemia virus (Pim), RSK), and DNA-intercalating pathways. Their tricyclic structure enables selective binding to protein active sites or DNA grooves.
Compound 30a, a representative tricyclic indole diazepinone derivative, emerged as a potent and selective inhibitor of Mcl-1, a critical antiapoptotic protein in cancer progression (Figure 13).45 Its tricyclic core was derived from the cyclization of linear indole amides using 1,3-dibromopropane under basic conditions, a modification that proved pivotal for both Mcl-1 binding affinity and pharmaceutical properties. This cyclization constructed a bridgehead nitrogen atom that rigidified the molecular conformation to lock in the optimal binding pose with Mcl-1 and eliminated two hydrogen bond donors and rotatable bonds. These changes addressed the poor permeability of earlier linear analogs by boosting compound 30a’s passive permeability to 1.3×10−5 cm/s. Three key substitutions on the tricyclic core contributed to its exceptional Mcl-1 inhibitory activity. First, the 7-(1,3,5-trimethyl-1H-pyrazol-4-yl) group was critical for occupying the hydrophobic P2 pocket of Mcl-1, as it enhanced binding through hydrophobic interactions with surrounding residues. This is supported by SAR trends showing that installation of this group resulted in a 4-fold potency enhancement compared to analogs lacking it. Second, the 6-chloro atom further stabilized binding by forming weak electrostatic interactions with the backbone carbonyl of A227 in Mcl-1. Third, the indole-6-carboxylic acid headgroup acted as a polar anchor, compensating for the activity loss caused by replacing the acidic acylsulfonamide with a neutral amide linkage. This headgroup was far more effective than monocyclic alternatives—for instance, replacing it with a para-benzyl carboxylic acid reduced affinity by over 20-fold.
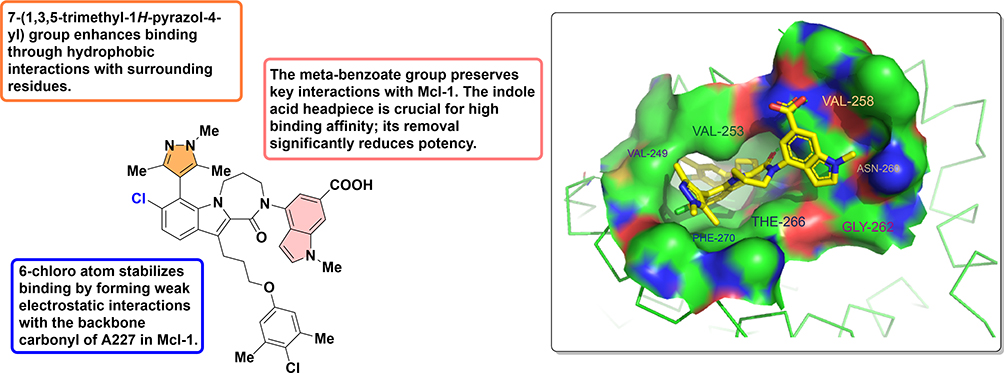 |
Figure 13 Structure and SAR of tricyclic indole diazepinone Mcl-1 inhibitor 30a. |
In biological evaluations, compound 30a exhibited robust activity with specificity for Mcl-1. In Mcl-1-sensitive multiple myeloma H929 cells, it inhibited proliferation with a GI50 of 2.0±0.3 μM and induced caspase 3/7 activation (with an EC50 of 0.76 μM)—both hallmark events of Mcl-1-mediated apoptosis. In contrast, it showed minimal activity against Mcl-1-resistant K562 cells (with a GI50 of 11.5±1.5 μM), which confirmed its dependence on Mcl-1 for cytotoxicity. Its selectivity was equally notable: it exhibited negligible affinity for other Bcl-2 family proteins (with a Ki of >10 μM for Bcl-xL and >36 μM for Bcl-2), a critical advantage over non-selective inhibitors like ABT-263, which caused thrombocytopenia via Bcl-xL inhibition. Even in the presence of 1% fetal bovine serum (a condition that mimicked in vivo environments), compound 30a retained strong binding (with a Ki of 2.8±1.7 nM), highlighting its potential for in vivo efficacy.
Compound 50 was a key indolopyrrolobenzodiazepine derivative with selective cytotoxicity against acute myeloid leukemia (AML) cells (Figure 14).110 Its core structure comprised a rigid, fused tricyclic scaffold (indole-pyrrole-benzodiazepine), which was essential for binding to Pim kinases, and it had two critical structural modifications: a 1-formyl group and a 5-oxyether side chain. The 1-formyl group was indispensable for Pim kinase inhibitory activity, as all active analogs in the series retained this moiety; meanwhile, the 5-position carried a 3-(piperidin-1-yl)propoxy chain, which consisted of a 3-methylene linker, a piperidine ring, and an ether oxygen bridge that balanced the molecule’s lipophilicity and flexibility.
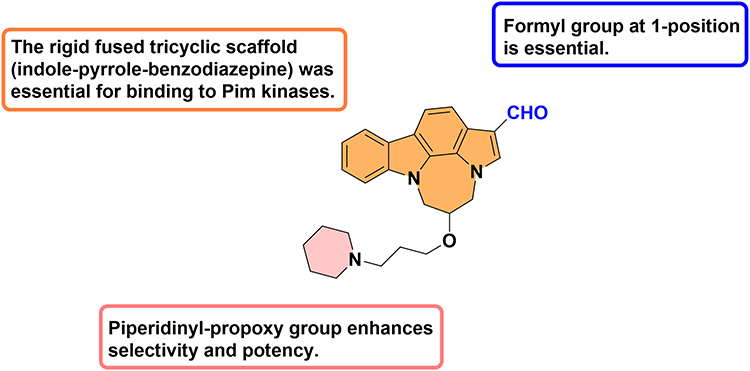 |
Figure 14 Structure and SAR of indolopyrrolobenzodiazepine derivative 50. |
It exhibited modest inhibition of Pim-1 (IC50 = 42 nM) and Pim-3 (IC50 = 50 nM) and showed superior selectivity toward AML cells compared to normal fibroblasts (NRK cells). Unlike its analogs with shorter linkers or polar terminal groups, the 3-methylene-piperidine side chain of compound 50 enhanced membrane permeability and compatibility with AML cell membranes. Additionally, the piperidine ring prevented excessive polarity–an issue seen in morpholine-containing compounds that impaired cell penetration. Notably, it induced apoptosis in AML cells with chemo-resistant phenotypes (including those with Bcl-2 overexpression or p53 knockdown). This selective cytotoxicity may be partially attributed to the favorable physicochemical properties conferred by the piperidine side chain, potentially facilitating intracellular accumulation or off-target effects beyond Pim kinase inhibition, although further mechanistic studies are warranted.
Compound 22 was a 1,2,3-triazole-fused indolo[1,4]diazepine derivative with prominent anticancer activity (Figure 15).43 Its core structure featured a rigid, planar framework formed by the fusion of an indole ring, a 1,4-diazepine ring, and a 1,2,3-triazole ring, with an unsubstituted benzoyl group attached at the 12-position. This planar tricyclic scaffold was critical for its DNA-intercalating ability, as confirmed by molecular docking studies, which showed that the triazole-fused diazepine moiety inserted between the dC5-dG6 (chain A) and dC7-dG8-dA9 (chain B) base pairs of DNA, while the benzoyl group interacted with the dG-B8 nucleotide in the DNA groove through hydrogen bonding and hydrophobic interactions. In vitro screening against the NCI-60 cell panel revealed broad-spectrum anticancer activity for compound 22 at 10 μM, with the highest growth inhibition (94.02%) observed in MDA-MB-468 breast cancer cells, significantly outperforming analogs with other substituted benzoyl groups.
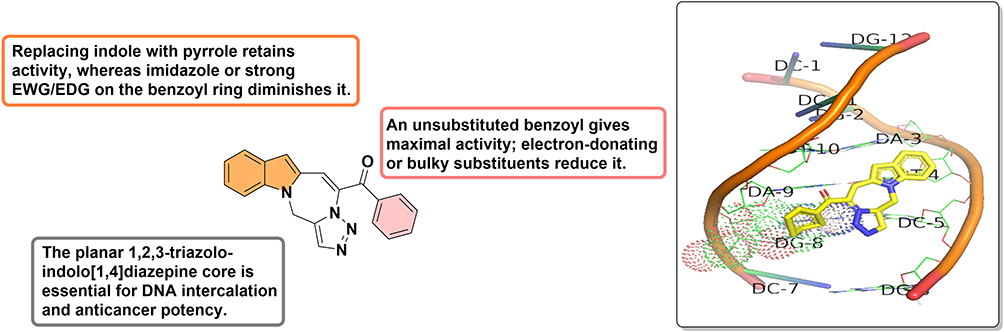 |
Figure 15 Structure and SAR of 1,2,3-triazole-fused indolo[1,4]diazepine derivative 22. |
Mechanistically, compound 22 exerted its anticancer effects by inducing DNA damage and subsequent apoptosis. It caused a dose-dependent accumulation of MDA-MB-468 cells in the sub-G1 phase (30.04% at 2.5 μM vs. 4.12% in the control) and increased the proportion of early apoptotic cells from 0.02% to 66.70% at 2.5 μM, accompanied by mitochondrial membrane potential loss (as indicated by JC-1 staining). Biophysical studies, including DNA nanodrop and viscosity assays, demonstrated that compound 22 intercalated into DNA, leading to increased absorbance at 260 nm and higher relative viscosity compared to ethidium bromide and doxorubicin, confirming its strong DNA-binding affinity. Notably, it exhibited low cytotoxicity toward normal human keratinocytes (HaCaT cells, 35.23% growth inhibition), highlighting its tumor selectivity and potential as a lead compound for further development.
Anti-Heart Failure
Compound 28a (BIX 02565), a RSK inhibitor derived from the 1-oxo-2,3,4,5-tetrahydro-1H-[1,4]diazepino[1,2-a]indole-8-carboxamide scaffold, was optimized for cellular potency, kinase selectivity, and aqueous solubility to serve as a candidate for investigating RSK as a therapeutic target in heart failure (Figure 16).111 It exhibited exceptional activity, with an RSK2 IC50 of 1 nM and a cellular HLR-CREB IC50 of 20 nM. Notably, the clinical application of indole-fused diazepines as cardiovascular agents requires attention to potential drug-drug interactions, especially with concurrent use of anticoagulants (eg, warfarin) commonly prescribed for heart failure patients.112 Structurally, it featured an (R)-1-methyl-substituted indole lactam core and a 2-(dimethylaminopropyl)benzimidazole amide moiety. The benzimidazole was critical for high binding affinity: it maintained coplanarity with the amide bond via 2-imino tautomerism, formed a hydrogen bond with Leu150 of RSK2, and engaged in π-stacking with Phe357—advantages not observed with isoxazole or pyrazole analogs. The (R)-methyl substitution on the lactam core conferred a 14-fold enantiomeric potency advantage over the (S)-enantiomer, as the (R)-methyl fitted precisely into a lipophilic indentation in the RSK2 binding pocket, while gem-dimethylation impaired activity due to steric clash. Additionally, its aqueous solubility of 26 μg/mL, enabled by the dimethylaminopropyl group, balanced potency and druggability, making it suitable for both in vitro and in vivo studies.
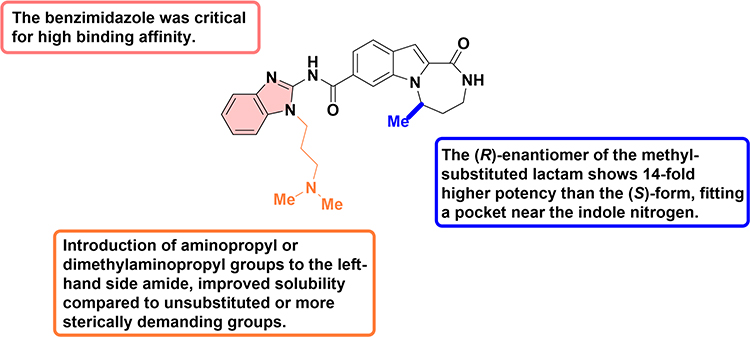 |
Figure 16 Structure and SAR of indole diazepinone RSK inhibitor 28a. |
CNS Activity
Indole-fused diazepines modulate CNS function by targeting serotonin receptors (eg, 5-HT2C) for anxiety disorders and GSK-3 for neurodegenerative diseases (eg, Alzheimer’s disease, AD). The 2,3,4,5-tetrahydro-1H-[1,4]diazepino[1,7-a]indole core 12 bound to human 5-HT2C with a Ki of 4.8 nM and displayed 4-fold selectivity over 5-HT2A (Ki = 18 nM) (Figure 17).22 Functional FLIPR assays showed 80% intrinsic efficacy. Loss of the indole aromaticity in the corresponding indoline derivative reduced affinity by more than 150-fold (5-HT2C Ki = 721 nM; 5-HT2A Ki = 805 nM), underscoring the importance of the fully fused tricyclic scaffold. N-Alkylation on the azepine nitrogen progressively decreased 5-HT2C affinity and efficacy without improving 2A/2C selectivity. Substitution at C11 of the indole also failed to improve the binding profile. In the mouse shock-induced aggression model, compound 12 increased latency to fight in a dose-dependent manner (3 mg kg−¹: 12.8 ± 2.3 s; 30 mg kg−¹: 27.0 ± 4.5 s), comparable to alprazolam yet without motor impairment on the rotarod. These data identified compound 12 as a potent, well-tolerated 5-HT2C agonist template for anxiolytic development.
 |
Figure 17 Structure and SAR of 5-HT2C agonist 12. |
Compound 44, a diazepinone-based partial agonist developed for irritable bowel syndrome therapy, exhibited exceptional subnanomolar binding affinity for the human 5-HT3A receptor with a Ki of 0.2 ± 0.03 nM, comparable to the antagonist alosetron (Ki = 0.50 ± 0.05 nM) (Figure 18).48 Despite its high affinity, compound 44 displayed low intrinsic activity (~7% of 5-HT Emax), consistent with a low-efficacy partial agonist profile. Its structure featured an indole core with a chlorine at the 5-position and a methyl at the 3-position, coupled with an (S)-3-aminoquinuclidine moiety—stereochemistry critical for potency. The –Cl/Me substitution combination on the indole ring enhanced affinity by leveraging hydrophobic interactions and potential halogen bonding with the receptor, while the (S)-quinuclidine enabled optimal fitting into the binding pocket.
 |
Figure 18 Structure and SAR of 5-HT3 partial agonist 44. |
In terms of ADME properties, compound 44 exhibited a favorable profile: it inhibited only CYP1A2, with an IC50 of 8.8 μM, while showing no significant inhibition of other CYP isoforms (IC50 > 50 μM). Additionally, its metabolic clearance in human liver microsomes was 2.3 μL/min/mg, comparable to alosetron (h-CLint = 3 μL/min/mg), suggesting a low potential for drug-drug interactions.
Compound 168a held notable potential for treating cyclin-dependent kinases (CDK)- and GSK-mediated diseases, particularly cancers and neurodegenerative disorders (Figure 19).99 As a key CDK inhibitor in the tetrahydro[1,4]diazepino[1,2-a]indol-1-one series, it targeted CDK1 (a core cell cycle regulator dysregulated in cancer) and CDK5 (abnormally upregulated in Alzheimer’s and Parkinson’s diseases, as well as in ischemic brain injury); critically, it also exhibited inhibitory activity against GSK-3α/β, a kinase implicated in diabetes, Alzheimer’s disease, and inflammatory disorders, thereby expanding its therapeutic scope beyond CDKs alone. Structurally, it featured a fused 2,3,4,5-tetrahydro[1,4]diazepino[1,2-a]indol-1-one core, with three functionally critical substituents: a hydroxyl group at the 4-position of the diazepine ring, a hydroxyl group at the 8-position of the indole moiety, and an iodine atom at the 11-position of the indole ring—all essential for its kinase inhibitory activity and subsequent disease-targeting potential.
 |
Figure 19 Structure and SAR of CDK/GSK inhibitor 168a. |
It exhibited potent submicromolar inhibitory activity against these disease-relevant kinases: it had an IC50 of 0.4 μM against CDK5/p25, 0.4 μM against CDK1/cyclin B, and 1.1 μM against GSK-3α/β—a potency that, while slightly lower than its CDK inhibition, still positioned it as a multi-kinase modulator capable of addressing overlapping pathological pathways (eg, CDK5 and GSK-3 cross-talk in Alzheimer’s disease). Importantly, the 8-hydroxy group and 11-iodine atom were indispensable for this activity—larger substituents (eg, 8-benzyloxy) or non-iodine groups at the 11-position (eg, hydrogen, methyl) led to complete loss of inhibitory potency (IC50 > 10 μM), emphasizing that precise structural optimization was key to retaining its ability to target kinases linked to cancer, neurodegeneration, and GSK-3-associated disorders. While the aforementioned heterocyclic scaffolds target specific receptors or kinases, natural product-derived compounds such as DL-3-n-butylphthalide have shown neuroprotective effects in ischemic stroke models through modulation of oxidative stress and neuroinflammatory pathways,113 suggesting that diverse chemical frameworks may converge on common therapeutic endpoints for CNS disorders.
The SAR of indole-fused diazepines is not only shaped by substituent modifications but also fundamentally governed by two core structural features: the fusion pattern between the indole and diazepine rings, and the positional arrangement of nitrogen atoms within the seven-membered diazepine heterocycle. These structural variables directly dictate the spatial conformation of the molecule, its ability to interact with target proteins (eg, enzyme active sites, receptor pockets), and ultimately its biological activity spectrum. By systematically dissecting the impact of these two features, we can better rationalize the diverse therapeutic potentials of different subclasses of indole-fused diazepines, as exemplified by the distinct biological roles of indole-fused 1,4-diazepines and indole-fused 1,3-diazepines below.
Nitrogen-containing heterocycles exhibit diverse biological activities across therapeutic areas, including anticancer, antiviral, cardiovascular, and CNS disorders, as well as emerging applications in other fields.114 Indole-fused diazepines exhibit structurally diverse, biology-driven activities, with their therapeutic potential tightly linked to the fusion pattern between the indole and diazepine rings, as well as the position of nitrogen atoms in the seven-membered heterocycle. For indole-fused 1,4-diazepines, distinct fusion modes enable selective targeting of key disease-related pathways: Indole-1,7-fused 1,4-diazepines, for instance, act as NS5B antagonists against HCV,109 5-HT2C-selective anxiolytics,22 and DNA-intercalating anticancer agents,43 while their indole-1,2-fused counterparts (eg, tetrahydro[1,4]diazepino[1,2-a]indole derivatives) show broad biomedical potential. Natural products like Streptocarbazole A (from Streptomyces sp. FMA) exhibit cytotoxicity against A-549 and HL-60 cells and arrest HeLa cells at the G2/M phase,115 and evodiagenine (from Evodia rutaecarpa) functions as a photosensitizer for metastatic breast cancer,116,117 while synthetic derivatives target oncogenic kinases including CDK,99 Mcl-120 and RSK.21 Indole-6,7,1-fused 1,4-diazepines exert their effects by modulating metabolic and neurodegenerative disease-related enzymes (GSK-3;23 poly(ADP-ribose) polymerase-1102), whereas the compact tetracyclic core of indole-3,2,1-fused 1,4-diazepines facilitates interactions with oncogenic kinases,110 thereby expanding their utility in cancer therapy. In contrast, indole-fused 1,3-diazepines, distinguished by the position of nitrogen atoms in the diazepine ring, offer complementary activities: Indole-1,7-fused 1,3-diazepines (eg, natural alkaloid Tabernines B from Tabernaemontana elegans) induce necrosis in HuH-7 hepatoma cells and overcome drug resistance in mouse lymphoma cell lines,51,52 while indole-1,2-fused 1,3-diazepines leverage unique 1,2-fusion-derived electronic properties to potentially unlock novel pharmacological profiles. This underscores how structural variation expands the therapeutic scope of this privileged scaffold.
To systematically consolidate the structure-target-pharmacology relationships elaborated above, we summarize the key structural subtypes of indole-fused diazepines, their corresponding biological targets, pharmacological effects and core structural determinants (Figure 20). As reflected in the figure, the medicinal potential of this scaffold is inherently dictated by two interconnected structural features: the fusion mode between the indole and diazepine rings, and the positional arrangement of nitrogen atoms within the seven-membered heterocycle. For instance, indole-1,7-fused 1,4-diazepines are predisposed to targeting HCV NS5B polymerase and 5-HT2C receptors due to their angle-optimized seven-membered ring and aromatic indole core, while indole-1,2-fused 1,4-diazepines rely on directing groups and cyclization-induced rigidity to engage kinases (RSK, Mcl-1) and CNS-related enzymes (CDK/GSK-3). Notably, common structural determinants, such as the indispensability of the bridgehead nitrogen, the role of planar tricyclic frameworks in DNA/protein binding, and the modulation of lipophilicity-permeability balance via side-chain substitutions, emerge across subtypes, providing modular design principles for future derivative development.
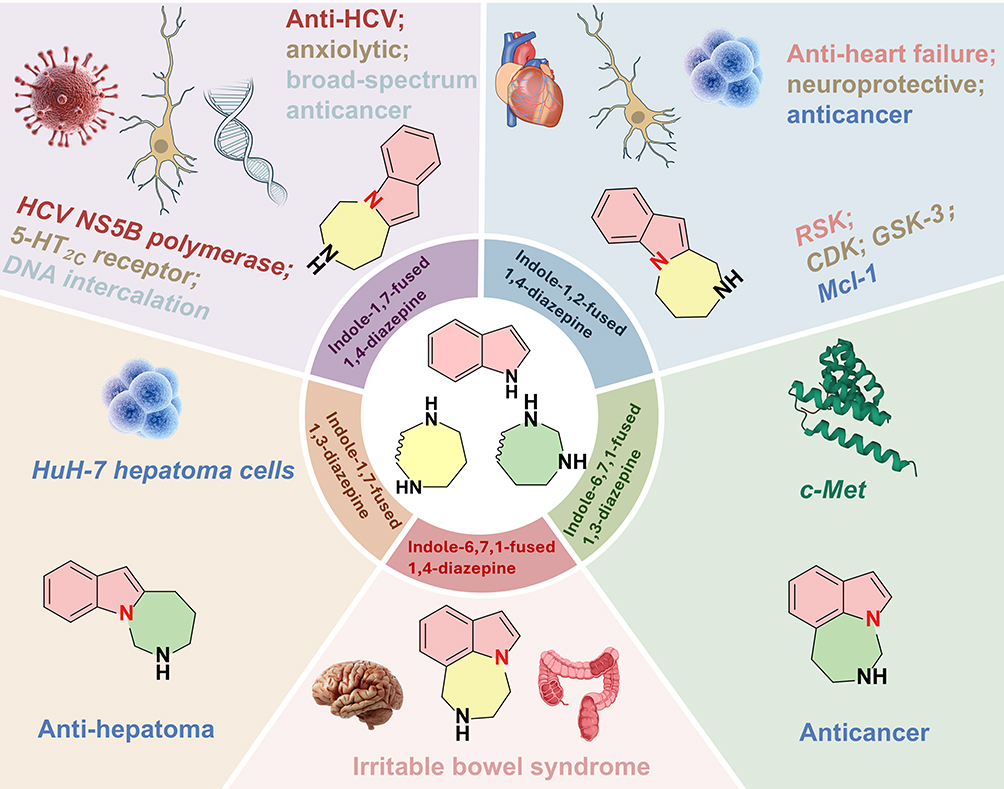 |
Figure 20 Graphical overview of structure–target–activity relationships for indole-fused diazepines. |
The integrated overview provided in Figure 20 not only reinforces the scaffold’s privileged status in medicinal chemistry but also identifies unmet needs for future research, such as the underexplored indole-1,2-fused 1,3-diazepine and indole-6,7,1-fused 1,2-diazepine subtypes, where thermodynamic instability and regioselectivity challenges limit synthetic accessibility and biological validation. By leveraging the structural design principles summarized herein (eg, fusion mode-target matching, substituent optimization for pocket binding), future efforts can rationally design derivatives with enhanced potency, selectivity, and pharmacokinetic properties, accelerating the translation of this scaffold from preclinical research to clinical applications.
Conclusion and Future Prospects
In conclusion, indole-fused diazepines, with their unique tricyclic structures centered around a bridgehead nitrogen atom, have firmly established themselves as a class of compounds with immense medicinal potential. Throughout this review, the bridgehead nitrogen atom emerges as a defining pharmacophore rather than a mere structural motif, constraining molecular conformation to optimize target binding and mediating key intermolecular interactions that enhance potency. We have systematically elaborated on diverse synthetic methodologies for these compounds, including base-mediated cyclization, metal-catalyzed C–H activation, radical cyclization, and phosphine-catalyzed annulation, which have enabled efficient access to key subclasses such as indole-1,7-fused and 1,2-fused 1,4-diazepines. These two subclasses are extensively studied in the literature, benefiting from robust, generalizable synthetic strategies and validated biological targets (eg, NS5B, Mcl-1, RSK). In contrast, indole-fused 1,2-diazepines and certain peri-fused systems remain underdeveloped, primarily due to intrinsic thermodynamic instability of the 1,2-diazepine core, regioselectivity challenges in forming strained bridgehead fusions, and a lack of biological screening to drive synthetic investment. This differential development of indole-diazepine subclasses reflects the inherent characteristics of the field rather than editorial selection, highlighting both the maturity of well-explored scaffolds and the untapped potential of understudied systems.
Looking forward, future research should prioritize several targeted directions to fully unlock the potential of this scaffold: 1. Asymmetric synthesis of chiral indole-fused diazepines, leveraging chiral catalysts or directing groups to address the lack of enantioselective methods for pharmacologically relevant derivatives; 2. Greener catalytic systems (eg, low-loading metal catalysts, solid acids, or photocatalysts without halogenated substrates) to improve sustainability and scalability; 3. Late-stage functionalization of core scaffolds to rapidly diversify derivatives for biological screening, particularly for underexplored fusion patterns; 4. Exploration of unexplored subtypes (eg, indole-1,2-fused 1,3-diazepines) by developing stabilization strategies (eg, electron-withdrawing substituents) to overcome thermodynamic barriers; 5. Computational and artificial intelligence-guided approaches for target identification (eg, predicting binding to Tau kinases or PPARs) and SAR optimization, accelerating the translation of these heterocycles from synthetic scaffolds to clinical candidates.
In summary, indole-fused diazepines offer a rich landscape for future research. By pursuing these research directions, we can unlock their full potential in drug discovery and development for a wide range of diseases.
Abbreviations
AI, artificial intelligence; AML, acute myeloid leukemia; AUC, Area under the curve; PA, 1,1′-binaphthyl-2,2′-diyl hydrogen phosphate; CDK, cyclin-dependent kinases; CNS, central nervous system; GSK-3, glycogen synthase kinase-3; HCV, Hepatitis C Virus; HSA, human serum albumin; MBH, Morita-Baylis-Hillman; Mcl-1, myeloid cell leukemia 1; NS5B, Nonstructural protein 5B; Pim, proviral insertion site in moloney murine leukemia virus; PTH, 10-phenylphenothiazine; RSK, ribosomal S6 kinase; SAR, structure-activity relationships; SET, single-electron transfer.
Acknowledgments
This work is supported by the Jilin Provincial Natural Science Foundation (No. YDZJ202501ZYTS059). The authors would like to thank Home for Researchers (home-for-researchers.com) for English language editing.
Disclosure
The author(s) report no conflicts of interest in this work.
References
1. Tomašič T. Privileged structures in drug discovery. ChemMedChem. 2020;15(8):726. doi:10.1002/cmdc.202000095
2. Fernandes H, Novais C, Sousa-Pinto B, Soares-da-Silva P, Azevedo LF. Comparative efficacy and safety of benzodiazepines in the treatment of patients with generalized anxiety disorder: a systematic review and network meta-analysis. Psychother Psychosom. 2025;94(5):373–71. doi:10.1159/000546269
3. Brambilla E, Meraviglia S, Moneta E, et al. Synthesis of indole-fused 1,4-diazepinones via photoredox-catalyzed cascade cyclization reaction. Adv Synth Catal. 2023;365(22):3958–3966. doi:10.1002/adsc.202300708
4. Wang L, Song X, Guo F, et al. Diversity-oriented synthesis of indole-fused scaffolds and bis(indolyl)methane from tosyl-protected tryptamine. Org Biomol Chem. 2024;22(14):2824–2834. doi:10.1039/D4OB00099D
5. Zhu Y, Jiang H, Wang Y-N. Phosphine-catalyzed [4+3] annulation reaction of indole derivatives with MBH carbonates: a facile access to indole-1,2-fused 1,4-diazepinones and azepines. Chinese J Chem. 2024;42(3):271–275. doi:10.1002/cjoc.202300505
6. Ghosh S, Koner M, Kunhiraman AA, Baidya M. Free amine-directed redox neutral ruthenium(II) catalysis toward regioselective synthesis of heterobiaryls. Org Lett. 2024;26(15):2987–2992. doi:10.1021/acs.orglett.4c00568
7. Zeng W, Han C, Mohammed S, et al. Indole-containing pharmaceuticals: targets, pharmacological activities, and SAR studies. RSC Med Chem. 2024;15(3):788–808. doi:10.1039/D3MD00677H
8. Bendi A, Versha, Rajni, Singh L, Taruna. Insight into indole-based heterocyclic scaffolds: a medicinal chemistry perspective. ChemistrySelect. 2023;8(48):e202303872. doi:10.1002/slct.202303872
9. Stempel E, Gaich T. Cyclohepta[b]indoles: a privileged structure motif in natural products and drug design. Acc Chem Res. 2016;49(11):2390–2402. doi:10.1021/acs.accounts.6b00265
10. Chadha N, Silakari O. Indoles as therapeutics of interest in medicinal chemistry: bird’s eye view. Eur J Med Chem. 2017;134:159–184. doi:10.1016/j.ejmech.2017.04.003
11. Kaur N. 10 - Synthesis of azepines, diazepines, triazepines, oxepines, oxazepines, and thiapines. In: Kaur N editor. Synthesis of 6- and 7-Membered Heterocycles. Elsevier; 2024:281–323. doi:10.1016/B978-0-443-16118-6.00012-2
12. Gilles P, De Borggraeve WM. 13.05 - 1,3-Diazepines. In: Black DS, Cossy J, Stevens CV editors. Comprehensive Heterocyclic Chemistry IV. Elsevier; 2022:204–242. doi:10.1016/B978-0-12-818655-8.00075-5
13. Stanovnik B. 13.04 - 1,2-Diazepines. In: Black DS, Cossy J, Stevens CV editors. Comprehensive Heterocyclic Chemistry IV. Elsevier; 2022:181–203. doi:10.1016/B978-0-12-818655-8.00099-8
14. Boa AN, McPhillie MJ. 13.06 - 1,4-Diazepines. In: Black DS, Cossy J, Stevens CV editors. Comprehensive Heterocyclic Chemistry IV. Elsevier; 2022:243–268. doi:10.1016/B978-0-12-818655-8.00110-4
15. Illuminati G, Mandolini L. Ring closure reactions of bifunctional chain molecules. Acc Chem Res. 1981;14(4):95–102. doi:10.1021/ar00064a001
16. Pecnard S, Hamze A, Pozzo J-L, Alami M, Provot O. Synthesis of oxazino[4,3-a]indoles and biological applications. Eur J Med Chem. 2021;224:113728. doi:10.1016/j.ejmech.2021.113728
17. Shelke YG, Hande PE, Gharpure SJ. Recent advances in the synthesis of pyrrolo[1,2-a]indoles and their derivatives. Org Biomol Chem. 2021;19(35):7544–7574. doi:10.1039/D1OB01103K
18. Yan Z, Yu B, Lan X, et al. Synthesis, bioactivity evaluation and theoretical study of nicotinamide derivatives containing diphenyl ether fragments as potential succinate dehydrogenase inhibitors. J Mol Struct. 2024;1308:138331. doi:10.1016/j.molstruc.2024.138331
19. Kang L, Gao X-H, Liu H-R, et al. Structure–activity relationship investigation of coumarin–chalcone hybrids with diverse side-chains as acetylcholinesterase and butyrylcholinesterase inhibitors. Mol Divers. 2018;22(4):893–906. doi:10.1007/s11030-018-9839-y
20. Lee T, Bian Z, Belmar J, et al; inventors; Vanderbilt University, USA. assignee. Substituted indoles Mcl-1 inhibitors and their preparation. patent WO2015148854A1. 2015.
21. Boyer SJ, Gao DA, Guo X, et al; inventors; Boehringer Ingelheim International GmbH, Germany. assignee. Preparation of heteroaryloxotetrahydrodiazepinoindolecarboxamide derivatives and analogs for use as RSK inhibitors. patent WO2011071716A1. 2011.
22. Ennis MD, Hoffman RL, Ghazal NB, et al. 2,3,4,5-Tetrahydro- and 2,3,4,5,11,11a-hexahydro-1H-[1,4]diazepino[1,7-a]indoles: new templates for 5-HT2C agonists. Bioorg Med Chem Lett. 2003;13(14):2369–2372. doi:10.1016/S0960-894X(03)00403-7
23. Gunosewoyo H, Midzak A, Gaisina IN, et al. Characterization of maleimide-based glycogen synthase kinase-3 (GSK-3) inhibitors as stimulators of steroidogenesis. J Med Chem. 2013;56(12):5115–5129. doi:10.1021/jm400511s
24. Engler TA, Malhotra S, Burkholder TP, et al. The development of potent and selective bisarylmaleimide GSK3 inhibitors. Bioorg Med Chem Lett. 2005;15(4):899–903. doi:10.1016/j.bmcl.2004.12.063
25. Wang Y, Yan G, Zhang Y; inventors; Chengdu Easton Pharmaceutical Co. Ltd. assignee. Preparation of trans-3-indyl-4-indolodiazepinecycloheptyl-2,5-pyrrolidin-2,5-dione derivatives as antitumor agents. patent CN102757435. 2012.
26. Hester JB Jr, Rudzik AD, Veldkamp W. Pyrrolo[3,2,1-jk][1,4]benzodiazepines and pyrrolo[1,2,3-ef][1,5]benzodiazepines which have central nervous system activity. J Med Chem. 1970;13(5):827–835. doi:10.1021/jm00299a008
27. Wang S, Wang S, Song S, et al. Modular and stereoselective approach to highly substituted indole/pyrrole-fused diazepanones. J Org Chem. 2021;86(9):6458–6466. doi:10.1021/acs.joc.1c00303
28. Xie P, Huang Y. Morita–Baylis–Hillman adduct derivatives (MBHADs): versatile reactivity in Lewis base-promoted annulation. Org Biomol Chem. 2015;13(32):8578–8595. doi:10.1039/C5OB00865D
29. Ashraf R, Zahoor AF, Ahmad S, et al. Recent advances in the chemistry of Morita–Baylis–Hillman (MBH) carbonates: a review. J Heterocycl Chem. 2025;62(10):1171–1217. doi:10.1002/jhet.70058
30. Devi N, Pathania AS, Singh V, Sharma S. Synthesis, biological activities, and structure–activity relationships of morita–baylis–hillman adducts: an update. Arch Pharm. 2024;357(10):e2400372. doi:10.1002/ardp.202400372
31. Larissa Adilis Maria Paiva F, Louise Mangueira de L, Laercia Karla Diega Paiva F, et al. Biological activities of Morita-Baylis-Hillman adducts (MBHA). Mini Rev Med Chem. 2023;23(17):1691–1710. doi:10.2174/1389557523666230202103719
32. Kumar CNSSP, Jayathirtha VR. Bioactive molecules via Morita–Baylis–Hillman chemistry. Synlett. 2025;36(17):2772–2790. doi:10.1055/a-2535-1306
33. Porto RS, Porto VA. Morita–Baylis–Hillman adducts and their derivatives: a patent-based exploration of diverse biological activities. Pharm Pat Anal. 2023;12(3):127–141. doi:10.4155/ppa-2023-0021
34. Wang S, Zhang W, Zhang Z, Xiang J, Zheng L. Directing group-controlled regioselective [4 + 3] annulation of indole-2-carboxamides with MBH carbonates toward highly substituted indole-1,2-fused diazepanones. J Org Chem. 2025;90(16):5651–5661. doi:10.1021/acs.joc.5c00335
35. Nemoto T, Harada S, Nakajima M. Synthetic methods for 3,4-fused tricyclic indoles via Indole ring formation. Asian J Org Chem. 2018;7(9):1730–1742. doi:10.1002/ajoc.201800336
36. Connon R, Guiry PJ. Recent advances in the development of one-pot/multistep syntheses of 3,4-annulated indoles. Tetrahedron Lett. 2020;61(14):151696. doi:10.1016/j.tetlet.2020.151696
37. Duncan RL, Helsley GC, Boswell RF. Synthesis of Indolo and benzimidazoquinazolines and benzodiazepines. J Heterocycl Chem. 1973;10(1):65–70. doi:10.1002/jhet.5570100115
38. Tegeler JJ, Gardenhire EM, Helsley GC; inventors; Hoechst-Roussel Pharmaceuticals, Inc. assignee. 4H-Indolo(1,2-d)(1,2,4)triazolo(4,3-a)(1,4)benzodiazepines as analgesics and their preparation. patent US4897392. 1990.
39. Sundberg RJ, Hong J, Smith SQ, Sabat M, Tabakovic I. Synthesis and oxidative fragmentation of catharanthine analogs. Comparison to the fragmentation — coupling of catharanthine and vindoline. Tetrahedron. 1998;54(23):6259–6292. doi:10.1016/S0040-4020(98)00289-0
40. Ikegashira K, Oka T, Hirashima S, et al. Discovery of conformationally constrained tetracyclic compounds as potent hepatitis C virus NS5B RNA polymerase inhibitors. J Med Chem. 2006;49(24):6950–6953. doi:10.1021/jm0610245
41. Mirabal-Gallardo Y, Soriano M, Caballero J, Alzate-Morales J, Simirgiotis MJ, Santos LS. Synthesis of the indolo 2,3-a quinolizidine ring through the addition of 2-siloxyfurans to imines and intrinsic reaction coordinate calculations. Synthesis-Stuttgart. 2012;44(1):144–150. doi:10.1055/s-0031-1289631
42. Thikekar TU, Selvaraju M, Sun C-M. Skeletally diverse synthesis of indole-fused diazocine and diazepine frameworks by one-pot, two-component cascade reaction. Org Lett. 2016;18(2):316–319. doi:10.1021/acs.orglett.5b03481
43. Gour J, Gatadi S, Pooladanda V, et al. Facile synthesis of 1,2,3-triazole-fused indolo- and pyrrolo[1,4]diazepines, DNA-binding and evaluation of their anticancer activity. Bioorg Chem. 2019;93:103306. doi:10.1016/j.bioorg.2019.103306
44. Boyer SJ, Burke J, Guo X, et al. Indole RSK inhibitors. Part 1: discovery and initial SAR. Bioorg Med Chem Lett. 2012;22(1):733–737. doi:10.1016/j.bmcl.2011.10.030
45. Shaw S, Bian Z, Zhao B, et al. Optimization of potent and selective tricyclic indole diazepinone myeloid cell leukemia-1 inhibitors using structure-based design. J Med Chem. 2018;61(6):2410–2421. doi:10.1021/acs.jmedchem.7b01155
46. Hao Y, Zhou P, Niu K, et al. Synthesis of indole- and pyrrole-fused seven-membered nitrogen heterocycles via acid-base switchable cyclization involving cleavage of amide C−N bonds. Adv Syn Catal. 2022;364(2):281–285. doi:10.1002/adsc.202101126
47. Zeng H, Li H, Jiang H, Zhu C. Steric-switched defluorofunctionalization selectivity: controlled synthesis of monofluoroalkene-masked medium-sized heterocyclic lactams and lactones. Sci China Chem. 2022;65(3):554–562. doi:10.1007/s11426-021-1135-8
48. Manning DD, Guo C, Zhang Z, et al. The discovery of diazepinone-based 5-HT3 receptor partial agonists. Bioorg Med Chem Lett. 2014;24(11):2578–2581. doi:10.1016/j.bmcl.2014.03.074
49. DeRatt LG, Lawson EC, Wang C-Y, Kuduk SD. Mild intramolecular ring opening of oxetanes. Org Lett. 2019;21(23):9642–9645. doi:10.1021/acs.orglett.9b03810
50. Giraud F, Bourhis M, Nauton L, et al. New N-1,N-10-bridged pyrrolo[2,3-a]carbazole-3-carbaldehydes: synthesis and biological activities. Bioorg Chem. 2014;57:108–115. doi:10.1016/j.bioorg.2014.09.004
51. Mansoor TA, Ramalhete C, Molnár J, Mulhovo S, Ferreira MJU. Tabernines A−C, β-carbolines from the leaves of Tabernaemontana elegans. J Nat Prod. 2009;72(6):1147–1150. doi:10.1021/np9001477
52. Mansoor TA, Ramalho RM, Mulhovo S, Rodrigues CMP, Ferreira MJU. Induction of apoptosis in HuH-7 cancer cells by monoterpene and β-carboline indole alkaloids isolated from the leaves of Tabernaemontana elegans. Bioorg Med Chem Lett. 2009;19(15):4255–4258. doi:10.1016/j.bmcl.2009.05.104
53. McCombie SW, Vice SF. Oxidation reactions of some indolocarbazoles. J Org Chem. 1996;61(1):413–415. doi:10.1021/jo951570y
54. Ghosh SK, Nagarajan R. NIS-mediated regioselective amidation of indole with quinazolinone and pyrimidone. RSC Adv. 2014;4(39):20136–20144. doi:10.1039/C4RA02417F
55. Zhang X, Lu X, Zhang P, Dai M, Liang T. Recent advances in the multicomponent reactions of indoles. Eur J Org Chem. 2025;28(15):e202401446. doi:10.1002/ejoc.202401446
56. Pintori DG, Greaney MF. Intramolecular oxidative C−H coupling for medium-ring synthesis. J Am Chem Soc. 2011;133(5):1209–1211. doi:10.1021/ja1090854
57. Zheng X, Hudyma TW, Martin SW, et al. Syntheses and initial evaluation of a series of indolo-fused heterocyclic inhibitors of the polymerase enzyme (NS5B) of the hepatitis C virus. Bioorg Med Chem Lett. 2011;21(10):2925–2929. doi:10.1016/j.bmcl.2011.03.067
58. Watson EL, Ball A, Raw SA, Marsden SP. Synthetic studies on psychotrimine: palladium-catalysed arylation of 2-(N-Indolyl) amides. Synlett. 2016;27(1):146–150. doi:10.1055/s-0035-1560519
59. Thikekar TU, Sun C-M. Palladium-catalyzed regioselective synthesis of 1,2-fused indole-diazepines via 5+2 annulation of o-indoloanilines with alkynes. Adv Synth Catal. 2017;359(19):3388–3396. doi:10.1002/adsc.201700741
60. Gao Y, Li J, Bai S, et al. Direct synthesis of annulated indoles through palladium-catalyzed double alkylations. Org Chem Front. 2020;7(9):1149–1157. doi:10.1039/D0QO00135J
61. Chen D, Dong D-Q, Qiu Z-X, et al. Research progress of rhodium(III)-catalyzed C(sp2)−H bond functionalization. Eur J Org Chem. 2025;28(4):e202401177. doi:10.1002/ejoc.202401177
62. Yang J-M, Zhu C-Z, Tang X-Y, Shi M. Rhodium(II)-catalyzed intramolecular annulation of 1-sulfonyl-1,2,3-triazoles with pyrrole and indole rings: facile synthesis of N-bridgehead azepine skeletons. Angew Chem Int Edit. 2014;53(20):5142–5146. doi:10.1002/anie.201400881
63. Harada S, Kono M, Nozaki T, Menjo Y, Nemoto T, Hamada Y. General approach to nitrogen-bridged bicyclic frameworks by Rh-catalyzed formal carbenoid insertion into an amide C–N bond. J Org Chem. 2015;80(20):10317–10333. doi:10.1021/acs.joc.5b01954
64. Dhole S, Chiu W-J, Sun C-M. Catalyst-controlled chemodivergent annulation to indolo/pyrrolo-fused diazepine and quinoxaline. Adv Syn Catal. 2019;361(12):2916–2925. doi:10.1002/adsc.201900088
65. Soleiman-Beigi M, Mohammadi M, Kohzadi H. An overview on copper in industrial chemistry: from ancient pigment to modern catalysis. Coordin Chem Rev. 2025;529:216438. doi:10.1016/j.ccr.2025.216438
66. Harada S, Kato R, Nemoto T. Construction of functionalized azapolycyclic architectures via formal amide insertion at a low catalyst loading of copper trifluoroacetylacetonate. Adv Syn Catal. 2016;358(19):3123–3129. doi:10.1002/adsc.201600603
67. Pradhan S, Shahi CK, Bhattacharyya A, Chauhan N, Ghorai MK. Syntheses of tetrahydrobenzoazepinoindoles and dihydrobenzodiazepinoindoles via ring-opening cyclization of activated aziridines with 2-(2-Bromophenyl)-1H-indoles. Org Lett. 2017;19(13):3438–3441. doi:10.1021/acs.orglett.7b01397
68. Wang Y, Huang X, Chen J, Xu J, Song Q. Diastereoselective radical cascade cyclization to access indole-fused diazepine derivatives. Org Biomol Chem. 2025;23(18):4349–4354. doi:10.1039/D5OB00219B
69. Stylianakis I, Kolocouris A. Comprehensive overview of homogeneous gold-catalyzed transformations of π-systems for application scientists. Catalysts. 2023;13(6):921. doi:10.3390/catal13060921
70. Reyes RL, Iwai T, Sawamura M. Construction of medium-sized rings by gold catalysis. Chem Rev. 2021;121(14):8926–8947. doi:10.1021/acs.chemrev.0c00793
71. Yang JM, Li PH, Wei Y, Tang XY, Shi M. Gold(I)-catalyzed highly stereoselective synthesis of polycyclic indolines: the construction of four contiguous stereocenters. Chem Commun. 2016;52(2):346–349. doi:10.1039/c5cc08381h
72. Yang J-M, Yao M-L, Li J-C, Liu J-K, Wu B. Access to azepino-annulated benzo[c]carbazoles enabled by gold-catalyzed hydroarylation of alkynylindoles and subsequent oxidative cyclization. Org Lett. 2022;24(36):6505–6509. doi:10.1021/acs.orglett.2c02293
73. Liu YX, Huang YQ, Song HJ, Liu YX, Wang QM. Regio- and chemoselective N-1 acylation of indoles: pd-catalyzed domino cyclization to afford 1,2-fused tricyclic indole scaffolds. Chem Eur J. 2015;21(14):5337–5340. doi:10.1002/chem.201406617
74. Baccalini A, Faita G, Zanoni G, Maiti D. Transition metal promoted cascade heterocycle synthesis through C−H functionalization. Chem Eur J. 2020;26(44):9749–9783. doi:10.1002/chem.202001832
75. Das A, Patil NT. Enantioselective C−H functionalization reactions under gold catalysis. Chem Eur J. 2022;28(20):e202104371. doi:10.1002/chem.202104371
76. Zhou Y, Li J, Ji X, et al. Silver- and gold-mediated domino transformation: a strategy for synthesizing benzo[e]indolo[1,2-a]pyrrolo/pyrido[2,1-c][1,4]diazepine-3,9-diones. J Org Chem. 2011;76(5):1239–1249. doi:10.1021/jo101727r
77. Feng E, Zhou Y, Zhao F, et al. Gold-catalyzed tandem reaction in water: an efficient and convenient synthesis of fused polycyclic indoles. Green Chem. 2012;14(7):1888–1895. doi:10.1039/C2GC35293A
78. Qiao J, Jia XW, Li PY, et al. Gold-catalyzed rapid construction of nitrogen-containing heterocyclic compound library with scaffold diversity and molecular complexity. Adv Synth Catal. 2019;361(6):1419–1440. doi:10.1002/adsc.201801494
79. Basceken S, Kaya S, Balci M. Intramolecular gold-catalyzed and NaH-supported cyclization reactions of N-propargyl indole derivatives with pyrazole and pyrrole rings: synthesis of pyrazolodiazepinoindole, pyrazolopyrazinoindole, and pyrrolopyrazinoindole. J Org Chem. 2015;80(24):12552–12561. doi:10.1021/acs.joc.5b02419
80. Feng Z, Ma J-A, Cheung CW. Ni-catalysed intramolecular reductive aminocarbonylation of 2-haloaryl-tethered nitroarenes for the synthesis of dibenzazepine-based heterocycles. Org Chem Front. 2022;9(14):3869–3875. doi:10.1039/D2QO00699E
81. Li Z-H, Chen H, Liu J-Q, Wang X-S. CuI-catalyzed synthesis of benzoimidazo[1,4]diazepinoindoles/indazoles via double Ullmann cross-coupling reaction. Tetrahedron. 2022;121:132835. doi:10.1016/j.tet.2022.132835
82. Xie Y, Zhao Y, Qian B, Yang L, Xia C, Huang H. Enantioselective N–H functionalization of indoles with α,β-unsaturated γ-lactams catalyzed by chiral Brønsted acids. Angew Chem Int Edit. 2011;50(25):5682–5686. doi:10.1002/anie.201102046
83. Xu X, Zhou G, Ju G, Wang D, Li B, Zhao Y. Rhodium(III)-catalyzed benzo[c]azepine-1,3(2H)-dione synthesis via tandem C–H alkylation and intermolecular amination of N-methoxylbenzamide with 3-bromo-3,3-difluoropropene. Chinese Chem Lett. 2022;33(2):847–850. doi:10.1016/j.cclet.2021.07.070
84. Kiruthika SE, Perumal PT. CuI-catalyzed coupling of gem-dibromovinylanilides and sulfonamides: an efficient method for the synthesis of 2-amidoindoles and indolo[1,2-a]quinazolines. Org Lett. 2014;16(2):484–487. doi:10.1021/ol403365t
85. Melnyk P, Legrand B, Gasche J, Ducrot P, Thal C. Synthesis of new annelated indole systems: new entry in the E-azaeburnane series. Tetrahedron. 1995;51(7):1941–1952. doi:10.1016/0040-4020(94)01075-B
86. Bellotti P, Huang H-M, Faber T, Glorius F. Photocatalytic late-stage C–H functionalization. Chem Rev. 2023;123(8):4237–4352. doi:10.1021/acs.chemrev.2c00478
87. Pitre SP, Overman LE. Strategic use of visible-light photoredox catalysis in natural product synthesis. Chem Rev. 2022;122(2):1717–1751. doi:10.1021/acs.chemrev.1c00247
88. Naruto S, Yonemitsu O. Photochemical synthesis of azepinoindoles and azocinoindoles from N-chloroacetylindolylethylamines, and a mechanistic study based on the correlation between quantum yields and calculated frontier electron densities of indole radicals. Chem Pharm Bull. 1980;28(3):900–909. doi:10.1248/cpb.28.900
89. Sundberg RJ, Amat M, Fernando AM. Analogs of the iboga alkaloids. Synthesis and reactions of (.+-.)-15-oxo-20-deethylcoronaridine derivatives. J Org Chem. 1987;52(14):3151–3159. doi:10.1021/jo00390a034
90. Lian G, Li J, Liu P, Sun P. Photoredox-catalyzed radical cascade reaction to synthesize fluorinated pyrrolo[1,2-d]benzodiazepine derivatives. J Org Chem. 2019;84(14):9322–9329. doi:10.1021/acs.joc.9b00937
91. Oishi T, Uchikura T, Akiyama T. Synthesis of indole-fused benzodiazepine derivatives by photocatalyzed cascade reaction. Chem Commun. 2025;61(12):2576–2579. doi:10.1039/D4CC05755D
92. Shi T, Tian M, Zhu Y, et al. Visible-light-induced sulfonylation cyclization to generate indole-fused medium-sized N-heterocycles in 2-Me-THF. J Org Chem. 2025;90(9):3435–3443. doi:10.1021/acs.joc.4c03187
93. Shi T, Tian M, Sun Z, et al. Photochemical aerobic sulfonylation–cyclization–selenylation to indole-fused medium-sized N-heterocycles in 2-Me-THF. Chem Commun. 2025;61(20):4066–4069. doi:10.1039/D4CC06686C
94. Bremner JB, Sengpracha W. An iodoacetamide-based free radical cyclisation approach to the 7,12-dihydro-indolo 3,2-d 1 benzazepin-6(5H)-one (paullone) system. Tetrahedron. 2005;61(23):5489–5498. doi:10.1016/j.tet.2005.03.133
95. Wang S, Shen Y-B, Li L-F, et al. N-alkylation-initiated redox-neutral [5 + 2] annulation of 3-alkylindoles with o-Aminobenzaldehydes: access to indole-1,2-fused 1,4-benzodiazepines. Org Lett. 2019;21(22):8904–8908. doi:10.1021/acs.orglett.9b03011
96. Hutait S, Nayak M, Penta A, Batra S. Convenient synthesis of a library of lactam-fused β-carbolines via the Ugi reaction. Synthesis. 2011;2011(03):419–430. doi:10.1055/s-0030-1258374
97. Danieli B, Lesma G, Martinelli M, Passarella D, Silvani A. Diastereoselective synthesis of 3-oxo-14,15-dihydroandranginine. J Org Chem. 1997;62(19):6519–6523. doi:10.1021/jo970370e
98. Xiong Z, Gao DA, Cogan DA, et al. Synthesis and SAR studies of indole-based MK2 inhibitors. Bioorg Med Chem Lett. 2008;18(6):1994–1999. doi:10.1016/j.bmcl.2008.01.119
99. Putey A, Fournet G, Lozach O, Perrin L, Meijer L, Joseph B. Synthesis and biological evaluation of tetrahydro[1,4]diazepino[1,2-a]indol-1-ones as cyclin-dependent kinase inhibitors. Eur J Med Chem. 2014;83:617–629. doi:10.1016/j.ejmech.2014.06.063
100. Gour J, Gatadi S, Akunuri R, et al. Catalyst-free facile synthesis of polycyclic indole/pyrrole substituted-1,2,3-triazoles. Org Biomol Chem. 2019;17(35):8153–8165. doi:10.1039/C9OB01560D
101. Magnus NA, Ley CP, Pollock PM, Wepsiec JP. Pictet−Spengler based synthesis of a bisarylmaleimide glycogen synthase kinase-3 Inhibitor. Org Lett. 2010;12(16):3700–3703. doi:10.1021/ol101405g
102. Tikhe JG, Webber SE, Hostomsky Z, et al. Design, Synthesis, and evaluation of 3,4-dihydro-2H-[1,4]diazepino[6,7,1-hi]indol-1-ones as inhibitors of poly(ADP-Ribose) polymerase. J Med Chem. 2004;47(22):5467–5481. doi:10.1021/jm030513r
103. Elmorsy SS, El-Ahl -A-AS, Soliman H, Amer FA. Synthesis of triazidochlorosilane (TACS). A novel silicon mediated one pot conversion of aldehydes to nitriles. Tetrahedron Lett. 1995;36(15):2639–2640. doi:10.1016/0040-4039(95)00302-S
104. El-Ahl -A-AS, Elmorsy SS, Soliman H, Amer FA. A facile and convenient synthesis of substituted tetrazole derivatives from ketones or α,β-unsaturated ketones. Tetrahedron Lett. 1995;36(40):7337–7340. doi:10.1016/0040-4039(95)01513-H
105. Liu S, Qu J, Wang B. Substrate-controlled divergent synthesis of polycyclic indoloazepines and indolodiazepines via 1,5-hydride shift/7-cyclization cascades. Chem Commun. 2018;54(57):7928–7931. doi:10.1039/C8CC03804J
106. Qu J, Bhadbhade M, Dobrowolski JC, Kumar N, Black DS. Unusual rearrangements in activated indoles: synthesis of indoloquinolines, dibenzonaphthyridines and indolobenzodiazepines. Tetrahedron. 2020;76(33):131375. doi:10.1016/j.tet.2020.131375
107. de la Mora MA, Cuevas E, Muchowski JM, Cruz-Almanza R. Synthesis of tricyclic-2-aminoindoles by intramolecular 1,3-dipolar cycloaddition of 1-ω-azidoalkylindoles. Tetrahedron Lett. 2001;42(32):5351–5353. doi:10.1016/S0040-4039(01)01006-1
108. Yang L-J, Yan S-J, Chen W, Lin J. A facile route to 1,3-diazaheterocycle-fused [1,2-a]indole derivatives via acetic acid catalyzed cyclocondensation reactions. Synthesis. 2010;2010(20):3536–3544. doi:10.1055/s-0030-1258195
109. Ding M, He F, Hudyma TW, et al. Synthesis and SAR studies of novel heteroaryl fused tetracyclic indole-diamide compounds: potent allosteric inhibitors of the hepatitis C virus NS5B polymerase. Bioorg Med Chem Lett. 2012;22(8):2866–2871. doi:10.1016/j.bmcl.2012.02.063
110. Giraud F, Bourhis M, Ebrahimi E, et al. Synthesis and activities of new indolopyrrolobenzodiazepine derivatives toward acute myeloid leukemia cells. Bioorg Med Chem. 2015;23(22):7313–7323. doi:10.1016/j.bmc.2015.10.031
111. Kirrane TM, Boyer SJ, Burke J, et al. Indole RSK inhibitors. Part 2: optimization of cell potency and kinase selectivity. Bioorg Med Chem Lett. 2012;22(1):738–742. doi:10.1016/j.bmcl.2011.10.029
112. Li HG, Wang Y, Fan R, et al. The effects of ferulic acid on the pharmacokinetics of warfarin in rats after biliary drainage. Drug Des Dev Ther. 2016;10:2173–2180. doi:10.2147/dddt.S107917
113. Zhang H, Wang L, Yang Y, et al. DL-3-n-butylphthalide (NBP) alleviates poststroke cognitive impairment (PSCI) by suppressing neuroinflammation and oxidative stress. Original Research. Front Pharmacol. 2023;13:987293. doi:10.3389/fphar.2022.987293
114. Wang L, Wang X, Deng L, et al. Pexidartinib (PLX3397) through restoring hippocampal synaptic plasticity ameliorates social isolation-induced mood disorders. Int Immunopharmacol. 2022;113:109436. doi:10.1016/j.intimp.2022.109436
115. Fu P, Yang C, Wang Y, et al. Streptocarbazoles A and B, Two novel indolocarbazoles from the marine-derived actinomycete strain Streptomyces sp. FMA. Org Lett. 2012;14(9):2422–2425. doi:10.1021/ol3008638
116. Wang QZ, Liang JY, Feng X. Evodiagenine and dievodiamine, two new indole alkaloids from Evodia rutaecarpa. Chinese Chem Lett. 2010;21(5):596–599. doi:10.1016/j.cclet.2009.12.002
117. Xu C, Wang Q, Feng X, Bo Y. Effect of evodiagenine mediates photocytotoxicity on human breast cancer cells MDA-MB-231 through inhibition of PI3K/AKT/mTOR and activation of p38 pathways. Fitoterapia. 2014;99:292–299. doi:10.1016/j.fitote.2014.10.010
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.