Back to Journals » Clinical and Experimental Gastroenterology » Volume 15
Functional, Diagnostic and Therapeutic Aspects of Bile
Authors Ahmed M ![]()
Received 30 January 2022
Accepted for publication 3 July 2022
Published 20 July 2022 Volume 2022:15 Pages 105—120
DOI https://doi.org/10.2147/CEG.S360563
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Koulaouzidis
Monjur Ahmed
Division of Gastroenterology and Hepatology, Thomas Jefferson University Hospital, Philadelphia, PA, USA
Correspondence: Monjur Ahmed, Division of Gastroenterology and Hepatology, Thomas Jefferson University Hospital, 132 South 10th Street, Main Building, Suite 468, Philadelphia, PA, 19107, USA, Tel +304-633-1354, Fax +215-755-1850, Email [email protected]
Abstract: Bile is a unique body fluid synthesized in our liver. Enterohepatic circulation preserves bile in our body through its efficient synthesis, transport, absorption, and reuptake. Bile is the main excretory route for bile salts, bilirubin, and potentially harmful exogenous lipophilic substances. The primary way of eliminating cholesterol is bile. Although bile has many organic and inorganic contents, bile acid is the most physiologically active component. Bile acids have a multitude of critical physiologic functions in our body. These include emulsification of dietary fat, absorption of fat and fat-soluble vitamins, maintaining glucose, lipid, and energy homeostasis, sustenance of intestinal epithelial integrity and epithelial cell proliferation, reducing inflammation in the intestine, and prevention of enteric infection due to its antimicrobial properties. But bile acids can be harmful in certain altered conditions like cholecystectomy, terminal ileal disease or resection, cholestasis, duodenogastric bile reflux, duodenogastroesophageal bile reflux, and bile acid diarrhea. Bile acids can have malignant potentials as well. There are also important diagnostic and therapeutic roles of bile acid and bile acid modulation.
Keywords: bile, function of bile acids, diagnostic and therapeutic roles of bile acids, cholestasis, bile acid diarrhea
Introduction
Bile (also called gall) is a unique dark green to yellowish-green, thick, sticky, and slightly alkaline fluid produced by the liver, stored and concentrated in the gallbladder, and then released into the second part of the duodenum. It subsequently travels to the terminal ileum, where it gets absorbed by the ileal enterocytes and returns back to the liver via the portal venous system in a process known as enterohepatic circulation. Bile has a unique composition and many physiologic functions. Utilizing the functional aspects of bile, bile acid, and its modulation are used therapeutically and in different diagnostic tests. We will discuss the various compositions of bile, physiology of bile, functions of bile, variation of serum levels of bile acids, lithogenic bile, bile acid metabolism in altered conditions, duodenogastric bile reflux, duodenogastroesohageal bile reflux, cholestasis, bile acid diarrhea, bile acid and malignancy, utilization of bile acid and its modulations in diagnostic tests and treatments.
Composition of Bile
It reflects the functional status of hepatocytes (which constitute 65% of the liver cell population) and cholangiocytes (which include 5% of the liver cell population). Approximately 95% of bile is water, and 5% is a complex mixture of organic and inorganic solutes that include (1) electrolytes (sodium, potassium, calcium, chloride, and bicarbonate), (2) endogenous constituents like bile acid and bile salts, bilirubin, cholesterol, phospholipid, carbohydrates, proteins, enzymes, steroids, amino acids, vitamins, immunoglobulin A (IgA), immunoglobulin G (IgG) cytokines, porphyrins, mineral salts, trace elements, and heavy metals, and (3) exogenous substances like xenobiotics, drugs and environmental toxins.1 Bile acids are wedge-shaped steroid acids formed by removing the last three carbon atoms of cholesterol’s terminal aliphatic side chain.2 They are amphipathic molecules having a hydrophilic side and a hydrophobic side (Figure 1). Cholic acid and chenodeoxycholic acid (CDCA) are the primary bile acids synthesized by hepatocytes in the liver either by classic or alternative pathways. 95% of bile acid synthesis occurs in the classical pathway that involves cytochrome P450-mediated oxidation of cholesterol and 14 enzymatic steps; the most important one is the rate-limiting enzyme cholesterol 7 alpha-hydroxylase (CYP7B1) which causes hydroxylation of the 7th steroid nucleus of cholesterol. The cholic acid synthesis also requires sterol 12α-hydroxylase (CYP8B1). 5% of bile acid synthesis occurs via an alternate pathway or acidic pathway, which involves the inner mitochondrial membrane enzyme 27-hydroxylase (CYP27A1) widely present in all tissues in the body.3 Transport of cholesterol to mitochondrial CYP27A1 by steroidogenic acute regulatory protein (StarD1), hydroxylation of cholesterol to 3 oxysterols (26-hydroxycholesterol, 25-hydroxycholesterol, and 24-hydroxycholesterol), and subsequent 7α-hydroxylation of these oxysterols by CYP7B1 are the main steps of the alternate pathway. The ultimate controlling enzyme of cellular oxysterol levels is CYP7B1.
 |
Figure 1 Bile acid. |
Oxysterols are subsequently transported back to the hepatocytes and are converted to bile acids.4 The primary bile acids are lipid-soluble and typically get conjugated to taurine or glycine in hepatocytes to form water-soluble conjugated bile acids (taurocholic acid/taurochenodeoxycholic acid or glycocholic acid/glycochenodeoxycholic acid), also called bile salts before secretion into the biliary canaliculi. Deoxycholic acid, lithocholic acid (ie, dehydroxylated chenodeoxycholic acid), and ursodeoxycholic acid are the secondary bile acids formed in the colon by bacterial deconjugation and dehydroxylation of primary bile acids. Usually, both primary and secondary bile acids are conjugated to taurine or glycine.5 In humans, the total amount of bile acids is about 2 to 3 grams. Cholic acid, chenodeoxycholic acid, and deoxycholic acid constitute 95% of the bile acid pool, whereas lithocholic acid, ursodeoxycholic acid, and other minor bile acids contribute only 5% of the bile acid pool. The most concentrated organic solutes in bile are bile salts. Bile becomes concentrated when the gallbladder absorbs water and electrolytes. Cholesterol and phospholipids (lecithin) are secreted from the hepatocytes into the bile in the form of small spherical membranous vesicles, also called unilamellar vesicles. Cholesterol and lecithins are virtually water-insoluble, and bile acids make them soluble because of their hydrophilic and hydrophobic sides. Initially, the bile acids aggregate to form simple micelles when the concentration exceeds the critical micellar concentration (CMC), ie, ~ 2 mmol/L. Each simple micelle appears like a disc with a diameter of about 3 nm, with its hydrophobic side facing inward and a hydrophilic side facing outward. In the gallbladder, cholesterol becomes solubilized within the inner hydrophobic portion, and phospholipid becomes incorporated into the outer hydrophilic part of the simple micelle to form a mixed micellar solution. Each mixed micelle (Figure 2) is about 4 to 8 nm in diameter and can solubilize cholesterol three times more than a simple micelle. One mole of bile salt can solubilize two moles lecithin.6 The cholesterol solubility is maximum when the molar ratio of phospholipid to bile acids varies between 0.2 to 0.3.7
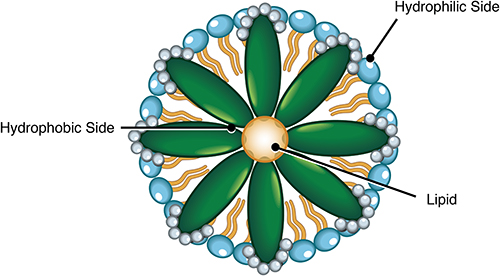 |
Figure 2 Mixed micelle. |
Physiology of Bile
Bile is a lipid-rich complex iso-osmotic fluid primarily synthesized and secreted by hepatocytes into the biliary canaliculi at a rate of about 500 mg per day. The intercellular junctions (tight, gap, and adherens junctions) attach hepatocytes to form liver plates. The tight junctions also called the blood-biliary barrier, keep biliary canalicular bile away from blood circulation.8 Transport proteins are at the hepatocytes’ basolateral (sinusoidal) and apical (canalicular) membranes. Exogenous and endogenous substances seen in the bile are transported from the blood into the hepatocytes through the basolateral membrane, and subsequently, they are secreted from the hepatocytes into the biliary canaliculi (~1 μm in diameter) through the apical membrane. Gap junctions can allow intercellular communication and enable the contraction of biliary canaliculi from the centrilobular zone to the periportal zone.9 Thus, both tight junctions and gap junctions are crucial in biliary secretion, and impairment of the functions of these junctions may result in intrahepatic cholestasis. Hepatocytes produce bile by secreting bile salts, conjugated bilirubin, protein, cholesterol, phospholipid ions, and water into the biliary canaliculi through the canalicular membrane considered as the central secretory apparatus. Transporting bile salts into the biliary canaliculi is an active process against the concentration gradient with the help of transport proteins’ energy and is the primary stimulus for bile formation. Water and inorganic ions follow the osmotic and electrochemical gradient. The canalicular bile flows into the intrahepatic bile ducts (ductal bile), where the cholangiocytes further modify the composition of bile as they secrete bicarbonate-rich fluid. About 50% of the hepatic component of bile flow (approximately 225 mL per day) depends on the osmotic force of bile acids, also called bile acid-dependent fraction (BADF) of bile, and the rest is independent of the osmotic force of bile acids also called bile acid independent fraction (BAIF) of bile.10 Glutathione and bicarbonate prompt BAIF. A certain proportion of bile secreted between meals flows directly through the hepatic ducts and the common bile duct into the duodenum. However, most hepatic bile passes into the gall bladder even during the postprandial period for storage and concentration.11 The gallbladder (GB) concentrates the bile 5 to 18 times the strength of its original secretion under fasting conditions.
Bile secretion and GB function are under neurohormonal control. Secretin, cholecystokinin (CCK), glucagon, and sulfated gastrin can increase the amount of bile volume and inorganic ion excretion into the bile. Secretin increases bicarbonate-rich fluid secretion by binding to its receptors at the basolateral side of cholangiocytes. This process involves efflux of chloride through activation of chloride channels and bicarbonate secretion via AE2/SLC4A2-mediated CL/HCO exchange.12 Secretin-mediated bile flow is increased by vasoactive intestinal polypeptide (VIP) and decreased by dopamine, non-sulfated gastrin, and somatostatin. CCK can cause GB contraction and sphincter of Oddi relaxation. Acetylcholine through the vagus nerve helps maintain GB muscle tone.13
Catecholamines can increase bile flow by binding to beta receptors in the biliary tract, although the exact mechanism is unknown. Certain bile acids can produce a higher volume of bile, also called choleretic bile acids, and ursodeoxycholic acid is a classic example. The total bile flow is about 800 to 1000 mL of bile every day, ie, 40 to 50 mL/hour before concentrated in the gallbladder. Hepatocytes secrete 75% of bile, and cholangiocytes secrete 25% of bile.14 After a standard meal, the gallbladder contracts to empty the concentrated bile into the duodenum. Bile flows through the duodenum and jejunum into the ileum. An apical sodium-dependent bile acid transporter (ASBT) on the ileal enterocytes in the terminal ileum actively absorbs 95% of luminal bile acids. Bile acid then goes to the basolateral membrane of ileal enterocytes to be transported into the portal venous system by an organic solute transporter α/β heterodimer. Bile acids travel through the portal circulation into the liver. Bile acids and bilirubin are transported into the hepatocytes across the sinusoidal membrane with the help of sodium taurocholate cotransporter (NTCT) and organic anion transporters, thus establishing the enterohepatic circulation. Then, using the bile salt export pump (BSEP), bile acids are secreted across the canalicular membrane in an ATP-dependent fashion into the biliary canaliculi.15
Thus, the enterohepatic circulation (Figure 3) is an anatomical, physiological, and partly vascular process that involves the transport of bile acids, bilirubin, drugs, and substances from the liver into the bile, followed by their passage into the intestinal lumen, absorption across the terminal ileal mucosa into the portal circulation and finally, uptake into the hepatocytes. Terminal ileal absorption of bile acids leads to the accumulation of 2 to 3 gm of bile acid called “bile acid pool.” This cycle happens 10 to 12 times per day and can handle 20 to 30 grams of bile acids per day. Each time, small amounts of bile salt escape terminal ileal absorption (400 to 800 mg daily) and enter into the colon, where some of the secondary bile acids formed by bacterial deconjugation and dehydroxylation are absorbed.16 When secondary bile acids get absorbed through the colon mucosa passively, they can damage the DNA of colonic epithelial cells, and epidemiological studies showed a correlation of increased fecal secondary bile acid concentration and increased incidence of colon cancer. The gut cannot absorb about 5% of bile acids, and hepatic biosynthesis can replenish this loss. Nonsteroidal anti-inflammatory drugs, antibiotics, warfarin, digoxin, opioids, and hormones are secreted from the hepatocytes into the bile and undergo enterohepatic circulation. Many lipophilic xenobiotics can also utilize enterohepatic circulation with repeated damage to the liver. Antibiotics may reduce enterohepatic circulation by suppressing intestinal bacterial deconjugation and dehydroxylation.
 |
Figure 3 Enterohepatic circulation. |
Within the ileal enterocytes, bile acids activate the nuclear receptor farnesoid X (FXR) and membrane-bound G-protein-coupled bile acid receptor 1, also called Gpbar1, or Takeda G protein-coupled receptor or triglyceride receptor 5 (TGR5). The primary bile acids chenodeoxycholic acid and cholic acid are the most potent FXR activator/endogenous ligand, followed by deoxycholic acid and lithocholic acid.17 On the other hand, the secondary bile acids lithocholic acid and deoxycholic acid are the most potent activators of TGR5.18 FXR and TGR5 are co-expressed in the L cells present in the epithelial layer of the ileum and colon.
FXR activation in the terminal ileum induces the production of fibroblast growth factor 19 (FGF19), which is a bile acid-induced ileal hormone that is secreted into the portal venous system and circulates to the hepatocytes in the liver where it binds to the cell surface receptor complex of β Klotho protein and fibroblast growth factor receptor 4 (FGFR4) complex. Activation of this complex reduces bile acid synthesis by downregulating CYP7A1 and CYP8B1 expression.19 This feedback inhibition is the principal mechanism of control of bile acid biosynthesis. FGF19 can also induce protein and glycogen synthesis but inhibits glycolysis, gluconeogenesis, and lipogenesis in the liver. A low FGF19 level has been found in primary bile acid diarrhea.20
Different organs, like the small intestine, colon, liver, gallbladder, kidney, and adrenal cortex, express FXR-α. Activation of intestinal FXR can improve insulin sensitivity and energy metabolism to prevent diabetes, non-alcoholic fatty liver disease (NAFLD), and obesity.
Hepatic FXR controls the expression of bile acid transporters to keep low levels of intracellular bile acids. It decreases bile acid transport from the portal circulation by inhibiting the expression of NTCT and increases the excretion of intracellular bile acids into the biliary canaliculi by activating the expression of BSEP. Hepatic FXR also increases the expression of an atypical nuclear receptor, which subsequently decreases bile acid synthesis by suppressing the transcription of cholesterol 7a-hydroxylase. Thus, enterohepatic FXR plays a significant role in bile acid synthesis, excretion, and detoxification.21 Hepatic FXR protects the liver and suppresses the development of hepatocellular cancer in multiple ways: preventing hepatic apoptosis and inflammation by modulating NF-κB and cytokine-inducible SH2-containing protein, promoting hepatic repair and regeneration, decreasing hepatic fibrosis, activating the expression of short heterodimer partner (SHP), and regulating bile acid homeostasis.22
Although many human tissues detect TGR5, it is highly expressed in the intestine, liver, and gall bladder epithelial cells and exerts its paracrine effects. In the intestine, bile acid-induced activation of TGR5 protects intestinal barrier function, reduces inflammation, and stimulates glucagon-like peptide-1 (GLP-1) secretion from enteroendocrine L-cells in the ileocolonic mucosa. Usually, rapid activation of L-cells occurs after a meal due to the release of glucose-stimulated insulinotropic peptide (GIP) and gastrin-releasing peptide (GRP) from the proximal small bowel. Activation of TGR5 in pancreatic β cells in pancreatic islets can release insulin directly.23 TGR5 is found close to cystic fibrosis transmembrane conductance regulator (CFTR) and ASBT in epithelial cells in the gall bladder. TGR5 activation stimulates chloride secretion into the bile through CFTR, causes smooth muscle relaxation, and enhances refilling of the gall bladder.24 In the liver, only Kupffer cells, sinusoidal endothelial cells, and cholangiocytes (not hepatocytes) express TGR5. It plays a vital role in hepatic lipid metabolism as it prevents fasting-induced hepatic steatosis by decreasing fatty acid uptake in the liver and increasing beta-oxidation of fatty acids in the liver. It stimulates nitric oxide synthesis in sinusoidal endothelial cells, mediates paracrine vasodilation, and thus connects bile acids with hepatic hemodynamics.25
TGR5 activation stimulates thyroid hormone deiodinase 2 (Dio2) in adipose tissue, converting T4 to T3. Thus, activation of TGR5 regulates glucose and lipid metabolism and homeostasis of energy.26,27 Intestinal bacteria degrade bilirubin to urobilinogen. The gut absorbs 50% of urobilinogen to get excreted in the urine, and the rest of urobilinogen gets converted to stercobilinogen, which gives the stool color brown.28
Because of efficient enterohepatic circulation and 70 to 90% of hepatic first-pass metabolism, the fasting level of serum bile acids in the systemic circulation remains low (2–3 µmol/L) compared to that in the portal circulation (10–18 µmol/L). In the postprandial period (15 to 60 minutes after meals), the level of bile acid increases not only in the portal circulation (37−49 µmol/L) but also in the systemic circulation (5–6 µmol/L) as a small amount of bile acids escape the enterohepatic circulation.29 The serum bile acids bind to serum albumin and get filtered through the glomerular basement membrane (about 100 μmol/day). However, only 1–2 μmol of bile acids get excreted through the urine daily because of efficient proximal tubular reuptake and reabsorption utilizing ASBT and α/β heterodimer transporters in renal epithelial cells.30,31
The Function of Bile in the Liver
Bile helps drain metabolic waste products from the liver into the duodenum. It also acts as a route for cholesterol, bile salts, and bilirubin excretion. Toxins from liver detoxification processes and certain metals such as copper, zinc, and mercury get excreted through bile. Hepatic bile acid FXR inhibits hepatic phosphoenolpyruvate (PEP) carboxykinase activity (which converts oxaloacetate to phosphoenolpyruvate) and stimulates glycogen synthetase activity. Thus, bile acid FXR signaling inhibits gluconeogenesis, stimulates glycogen synthesis, enhances insulin sensitivity, and controls glucose metabolism.32 Hepatic bile acid-TGR5 signaling regulates lipid metabolism and prevents hepatic steatosis.
The Function of Bile in the Intestine
The primary function of bile acids in the intestine is digestion and absorption of fat (cholesterol, triglycerides), fat-soluble vitamins (vitamins A, D, E, K), other lipophilic nutrients, iron, calcium, and provitamin beta-carotene. Bile acids have detergent-like emulsifying power to break dietary large fat globules into tiny emulsion droplets, thus significantly increasing their surface area for lipase action.33 As a result, lipase can digest the emulsified fat quickly. Bile acts as a suitable medium allowing the interaction of fat and fat-splitting enzymes. Bile acid micelles also carry the digested fat to the intestinal brush border, facilitating absorption. Deoxycholic acid may affect intestinal smooth muscle activity, and chenodeoxycholic acid is potent in causing intestinal secretion.34 Bile acids have a role in maintaining epithelial integrity and epithelial cell proliferation. They can also reduce intestinal inflammation and establish normoglycemia through activation of TGR5, as mentioned before. Bile acids can control cholesterol, triglycerides, glucose, energy hemostasis, bile acid synthesis, and enterohepatic circulation by activating different signaling pathways. Bile salts also have bacteriostatic and potent antimicrobial activity against invasive microorganisms in the gut. They can stimulate mucin secretion from gut epithelial cells, inhibit bacterial adhesion to the gut mucosa, and bind enterotoxin in the lumen of the gut. Bile acids can also stimulate innate immunity in the liver and intestine by activating FXR and TGR5 in macrophages, dendritic cells, and natural killer T cells. They play an essential role in maintaining the microbial ecology of the intestine. Bacterial colonization can increase with the change in the concentration of bile salts. In cirrhosis of the liver, small intestinal bacterial overgrowth can occur due to decreased intraluminal bile salts.35
Variation in Serum Bile Acids
A mild increase in serum bile acids occurs following meals. But the marked elevation of serum bile acids can occur in various liver diseases like acute viral hepatitis, cirrhosis of the liver, cholestasis, cholangitis, liver failure, hemochromatosis, portal vein thrombosis, Budd-Chiari syndrome, portosystemic shunt, and Wilson disease. An increased serum bile acid level in bariatric surgery like sleeve gastrectomy improves metabolism and weight loss by activating TGR5 and FXR receptors. In cholestatic liver diseases, the serum bile acid can be as high as 100 times the average concentration, and this increase is mainly due to conjugated bile acids.36 Increased bile acids in hepatocytes, as seen in cholestasis, can cause hepatocyte injury because of its inherent detergent activity and are partly responsible for hepatotoxicity in cholestatic liver diseases.37 Elevated serum bile acids (both fasting and postprandial) have high sensitivity (93%) for the detection of cirrhosis of the liver in the presence of normal transaminases.38 In genetic liver disorders like Gilbert syndrome, Crigler-Najjar syndrome, and Dubin-Johnson syndrome, serum bile acid can be normal.
Diminished serum bile acids can be seen in fasting, delayed gastric emptying, and terminal ileal disease or resection.
Lithogenic Bile
This means that the bile tends to form gallstones. Bile becomes lithogenic when the bile gets supersaturated with cholesterol either due to hypersecretion of biliary cholesterol or decreased bile acid output.39 Cholesterol solubility in the bile depends on two variables: bile salt-lecithin ratio and the total lipid ((bile salts + lecithin + cholesterol) concentration in the bile. The lithogenic index is the ratio of the actual cholesterol present in the bile to the maximal cholesterol dissolvable in that bile sample. The suspicion of “lithogenic bile” comes in the setting of cholesterol gallstone disease, biliary microlithiasis, biliary sludge, and biliary microcrystal disease. Several medical and surgical conditions can be associated with the formation of lithogenic bile. These include obesity, pregnancy, genetic predisposition, high-calorie diet, oral contraceptive pills, certain medications like clofibrate, ileal disease, or resection with bile salt depletion. Lithogenic bile stimulates the gallbladder to hypersecrete mucus that promotes the formation of cholesterol microcrystals and gallstones.
Bile Acid Metabolism in Altered Conditions
Cholecystectomy
Gallbladder cholangiocytes secrete high levels of FGF19 into bile. FGF19 plays a vital role in the negative feedback regulation of bile acid synthesis. As a result, cholecystectomy doubles the production of bile acids.40 Because of the absence of the gallbladder, there is a continuous flow of bile salts into the intestine. Increased entry of primary bile acids into the intestine increases the pool size of secondary bile acids, particularly deoxycholic acid, due to increased exposure of primary bile acids to bacterial deconjugation and dehydroxylation in the colon.41 Deoxycholic acid irritates the colon mucosa, causing water and electrolytes secretion and increasing colon motility, shortening colon transit time.42 Up to 12% of post-cholecystectomy patients develop chronic diarrhea (ie, ≥ 3 liquid stools per day for >4 weeks), usually a few weeks after surgery, ie, post-cholecystectomy diarrhea.43
Terminal Ileal Resection or Terminal Ileal Disease
Terminal ileum is 3 to 4 cm long. It is responsible for the active absorption of 95% of luminal bile acids and vitamin B12 and fat not yet absorbed in the proximal small bowel. Terminal ileal resection may be necessary if there is a fibrotic stricture not amenable to endoscopic intervention, symptomatic internal or enterocutaneous fistula of the terminal ileum due to Crohn’s disease, terminal ileal disease refractory to biologic agents, neoplastic involvement of the terminal ileum due to neuroendocrine tumor or adenocarcinoma, uncontrolled terminal ileal infection and uncontrolled terminal ileal bleeding due to ulcer or injury.44 As mentioned before, short ileal disease or resection of less than 100 cm may cause bile acid-induced diarrhea or choleraic diarrhea by unabsorbed bile salts. Long ileal disease or more than 100 cm resection may cause bile salt deficiency and fat malabsorption, leading to steatorrhea. Colonic bacteria can also secrete lipases that degrade unabsorbed dietary triglycerides and release hydroxy fatty acids, increasing colonic secretion.45 As little as 10 cm of ileal disease or resection can derange bile acid metabolism with an increased propensity to develop cholelithiasis and urolithiasis.46 Cholelithiasis may include both cholesterol and pigment stones. Bile acid malabsorption may cause supersaturation of bile with cholesterol, leading to cholesterol gallstones in many patients. Pigment gallstones can also be formed as malabsorbed bile acids in the colon solubilize unconjugated bilirubin promoting increased absorption of conjugated bilirubin from the colon and ultimately increased rate of bilirubin secretion into the bile.47 Bile acid deficiency may lead to fat malabsorption, and unabsorbed long-chain fatty acid in the colon binds to calcium and magnesium to create soaps. Normally oxalate binds to calcium to form insoluble and unabsorbable calcium oxalate. Calcium is now unavailable; oxalate binds to sodium to form soluble and absorbable sodium oxalate, resulting in enteric hyperoxaluria and calcium oxalate renal stone.48
Duodenogastric Bile Reflux (DGR)
Mild retrograde bile flow from the duodenum into the stomach can be physiological. Although mild bile reflux may not cause any gastric injury, excessive bile reflux may cause alkaline gastritis. Bile acids can directly damage the gastric surface epithelium and disrupt the mucosal barrier leading to back-diffusion of hydrogen ions and histamine-mediated vascular response to hyperemia and edema. Patients can develop chronic gastritis, gastric ulceration, gastric atrophy, and intestinal metaplasia. Intestinal metaplasia can be pronounced in the presence of H. pylori infection.49 Bile acid is toxic to the gastric mucosa and is also considered a risk factor for the development of gastric cancer.50 Primary DGR can occur due to antroduodenal motility disorder. Secondary DGR generally follows surgical alteration of gastroduodenal anatomy (pyloroplasty, distal gastric resection with Billroth I or II reconstruction) or cholecystectomy.51 Patients with DGR may present with dyspepsia, nausea, and bilious emesis. Upper endoscopy may show the presence of bile in the stomach, and biopsy may reveal the presence of chemical gastritis. DGR can be quantified by diisopropyl iminodiacetic acid (DISIDA) scan, hydroxy iminodiacetic acid (HIDA) scan, or 24-hour intragastric bile monitoring with the Bilitec device.52 Excessive DGR can also promote the development of duodenogastroesophageal (DGER) non-acid reflux.
DGER alone may not cause damage to the esophageal mucosa, but in combination with acid reflux, it may cause significant esophageal mucosal injury, including the development of Barrett’s esophagus.53 Bile reflux into the esophagus is not uncommon in patients with Barrett’s esophagus and early esophageal carcinoma (EAC). Conjugated bile acids (taurocholic acid and glycocholic acids) can increase the invasiveness and metastasis of EAC by activating sphingosine 1-phosphate receptor 2 (S1PR2).54 Bilitec is the investigation of choice for diagnosing DGER, and it should always be done in combination with a 24 hour esophageal pH study.55 The treatment of DGR is nonspecific, challenging, and unsatisfactory. Prokinetic agents can decrease DGR by improving gastric emptying. PPI can reduce the potentially harmful effect of DGER by reducing gastroesophageal acid reflux. In the case of cholecystectomy, bile acid binders like cholestyramine can be helpful. A duodenal switch operation should be considered in patients with transpyloric DGR not responding to medical therapy.56 DGER can be treated by Nissen Fundoplication if lifestyle modification, PPI, and baclofen fail.57
Cholestasis
Is a condition in which there is diminished bile flow either due to impairment of bile formation or intra-hepatic or extra-hepatic biliary obstruction. As a result, there is the retention of bile acids, bilirubin, and other substances normally get excreted in the bile. In cholestasis, serum bile acids are generally greater than 10 µmol/L. Because of their detergent-like activity, retained bile acids in the liver can cause membrane dysfunction and membrane injury to hepatocytes and cholangiocytes. Retained bile acids can injure all biological membranes throughout the body. Retained cholesterol in the liver can cause dysfunction of integral membrane proteins due to increased cholesterol deposition in the membranes. In cholestasis, conjugation of unconjugated bilirubin to conjugated bilirubin continues, and as a result, conjugated hyperbilirubinemia is one of the principal signs of cholestasis. Chronic cholestasis can lead to hepatic fibrosis, cirrhosis, hepatocellular carcinoma (HCC), and cholangiocarcinoma.58 High concentrations of intrahepatic bile acids can damage the hepatocyte DNA leading to increased mutation of tumor suppressor genes and oncogenes. In a large prospective study, Stepien et al found an association of HCC and increased concentrations of major circulating bile acids with a higher proportion of taurine-conjugated bile acids from a few years to the development of HCC. This study may indicate that an altered bile acid metabolism may have a role in developing HCC.59 Under cholestatic conditions, conjugated bile acids can activate the S1PR2 and the AKT and ERK1/2 signaling pathways and play a critical role in promoting invasive growth of cholangiocarcinoma, as shown in the mouse model.60 Primary sclerosing cholangitis (PSC) combined with inflammatory bowel disease (IBD) has a high risk of developing colon cancer with a cumulative risk of 50% after 25 years of illness, ie, 5-fold higher than in IBD without PSC, possibly due to exposure of the colon to toxic bile.
Bile Acid Diarrhea (BAD) or Choleraic Diarrhea
Bile acids can cause diarrhea by increasing colonic mucosal permeability, stimulating the colonic mucosa to secrete water and electrolytes, and accelerating colon motility by enhancing propulsive high amplitude colonic contractions.61 Patients generally present with chronic or intermittent watery diarrhea, nocturnal bowel movements, fecal incontinence, abdominal pain, and flatulence.62 The prevalence of BAD is over 1% in the general population of the western world.63 BAD is classified as type 1, type 2, and type 3 BAD.
Type 1 BAD is due to ileal resection or ileal dysfunction. >90% of the patients have ileal resection due to Crohn’s disease, and many patients have unresected ileal inflammatory Crohn’s disease or radiation enteritis.
Type 2 BAD is idiopathic. However, recent work suggests that this condition results from excessive bile acid production due to defective negative feedback inhibition of bile acid synthesis due to deficiency of FGF19. 33% of diarrhea-predominant irritable bowel syndrome (IBS-D) patients have type 2 BAD.64
Type 3 BAD is due to gastrointestinal diseases that affect absorption but do not cause ileal dysfunction. These include small intestinal bacterial overgrowth (SIBO), post-cholecystectomy, post-vagotomy, chronic pancreatitis, celiac disease, and microscopic colitis.65 Metformin can cause BAD by inhibiting bile acid absorption from the terminal ileum and increasing hepatic bile acid synthesis.66
Diagnosis of BAD
The following tests are currently used to diagnose BAD.
- (75) Selenium homocholic acid taurine (75SeHCAT) 7-day retention test: 75SeHCAT is a gamma radiolabeled synthetic bile acid resistant to bacterial degradation and utilizes the same enterohepatic circulation as natural bile acids. The patient swallows a capsule of 75SeHCAT, which gets distributed in the gut in an hour. After 1 hour, a baseline abdominal and pelvic gamma camera scan is done, representing 100% retention. A follow-up gamma camera scan is done on day 7, and its radioactivity is divided by the radioactivity detected at the baseline scan. This value indicates retention of 75SeHCAT in the body. In healthy subjects, there should be a retention of ≥15%. 10 −15% retention indicates mild BAD, 5–10% retention moderate BAD, and <5% retention severe BAD.67 Although the 75SeHCAT retention test is considered the gold standard test to diagnose BAD, it is not available in the United States but is used in many European countries.
- Fasting serum 7αC4 (7 α-hydroxy-4-cholesten-3-one) level: 7αC4 is an intermediate product in bile acid synthesis secondary to the activity of CYP7A1 and correlates well with bile acid synthesis.68 In patients with chronic diarrhea, fasting serum 7αC4 of ≥52.5 ng/mL may suggest BAD.69
- Fecal bile acid test: The patient is instructed to eat a diet containing 100 grams of fat for 2 days. Then stools are collected in a kit for 48 hours and kept frozen to maintain bile acid stability. The stool sample is then mailed to the reference laboratory, where fecal bile acid is measured by high-performance liquid chromatography and mass spectrometry. The total and individual primary and secondary bile acids can be measured. Currently, BAD is diagnosed if (a) the total fecal bile acid is >2337µmol/48hours, or (b) the primary fecal bile acid level is more than 10% (without elevated total fecal BAs), or (c) total fecal bile acid is more than 1000µmol/48hours plus primary fecal bile acids is more than 4%.70
- Serum Fibroblast Growth Factor 19 (FGF19): Serum FGF-19 is low in a prospective clinical study.71 It is an Enzyme-Linked Immunosorbent Assay (ELISA) and can be used as a screening test in patients with BAD. Fasting serum FGF19 levels of ≤61.7pg/mL can be diagnostic for bile acid diarrhea.72
- Therapeutic trial: At present, a therapeutic trial with bile acid binders is most commonly used to diagnose BAD. They bind to bile acids in the intestinal lumen preventing exposure of the colon to free bile acids.
Management of BAD
Bile acid binders or bile acid sequestrants like cholestyramine, colestipol, or colesevelam are the mainstays of BAD treatment. They are effective irrespective of the type of BAD. If the patient is intolerant to one bile acid binder, it should be switched to another bile acid binder. As bile acid binders can bind other medications, patients should take those medications 1 hour before or 4–6 hours after the intake of bile acid binders. If bile acid binders fail, obeticholic acid, liraglutide, or octreotide can be tried to control BAD.73,74 Physicians should treat the underlying cause or associated diseases. Patients with Crohn’s ileitis should be treated with steroids which can induce the expression of ASBT.75 Patients with SIBO should be treated with antibiotics. Microscopic colitis is generally treated with budesonide. Patients with diarrhea and steatorrhea due to ileal resection should also be treated with a low-fat diet, medium-chain triglyceride, and fat-soluble vitamins (A, D, E, and K). Patients with pancreatic insufficiency should be treated with pancreatic enzyme supplementation.
Bile Acid and Malignancy
The role of bile acid in the development and progression of esophageal, gastric, hepatocellular, and cholangiocarcinoma has been discussed. Bile acids, particularly secondary bile acids, play a role in the initiation and progression of colon cancer. The possible mechanisms include colonic epithelial damage followed by inflammation and hyperproliferation of colonic epithelial cells, stimulation of reactive oxygen species with oxidative damage to the DNA and genomic instability, damage to the p53 gene, resistance to apoptosis, stimulation of β-catenin and multiple oncogenic signaling, and enhancement of angiogenesis.76
Diagnostic Roles of Bile Acid and Bile Acid Modulation
Intrahepatic Cholestasis of Pregnancy
In the late 2nd trimester and 3rd trimester of pregnancy, serum bile acid measurement can be very useful when patients present with pruritus. It can give a clue to the early diagnosis of intrahepatic cholestasis of pregnancy when other liver parameters can be normal.77
Hepatobiliary Diseases
As bile acids escape the enterohepatic circulation in hepatobiliary diseases, elevated serum bile acids can also be a sensitive diagnostic indicator of these diseases.78 If this test is performed 2 hours postprandially, the sensitivity of this test can be further increased.79
Cirrhosis of Liver
Total and individual bile acids, particularly primary conjugated bile acids, can be used as noninvasive markers in the diagnosis and prognosis of cirrhosis of the liver as well as potential indicators of the development of hepatocellular carcinoma (HCC) in early cirrhosis of the liver.80 The risk of HCC is increased when there is an elevated serum concentration of primary bile acids and taurine-conjugated bile acid but a reduced concentration of secondary bile acids.81
BAD
The 14C-glycocholate (14C-BA) breath and stool test was used in the past for detecting SIBO and BAD but is not used nowadays because of its radiation exposure and availability of better tests.82 Instead, 75SeHCAT 7 day retention test is used in diagnosing BAD.
Liver Transplantation
After orthotopic liver transplantation, serum bile acid reflects the grafted liver function. Serum bile acid monitoring can be helpful in the early recognition of graft dysfunction after orthotopic liver transplantation.83
Therapeutic Roles of Bile Acid and Bile Acid Modulation
Ursodeoxycholic Acid (UDCA) or Ursodiol
It is a hydrophilic secondary bile acid found in human and other mammalian bile, although it was first discovered in bear bile, deriving its name from Ursus, ie, bears. It is an epimer of chenodeoxycholic acid. It is widely used as the drug of first choice in different cholestatic liver diseases, where it works on multiple pathogenic mechanisms. It protects the injured cholangiocytes from the toxic effects of bile acids. It detoxifies hydrophobic bile acids. It stimulates impaired hepatobiliary secretion. It protects hepatocytes from bile acid-induced apoptosis. In primary biliary cholangitis (PBC), UDCA improves the liver function and delays the progression of the disease to develop severe fibrosis or cirrhosis of the liver, leading to prolongation of transplant-free survival.84 It also suppresses immunity by interfering with T cell and B cell function. UDCA improves liver chemistry in primary sclerosing cholangitis but does not improve survival. UDCA has also been used in other cholestatic liver diseases like intrahepatic cholestasis of pregnancy, graft vs host disease, cystic fibrosis, and progressive familial intrahepatic cholestasis (PFIC).85
UDCA decreases cholesterol absorption from the gut, biliary cholesterol saturation, and biliary lithogenic index (a measure of cholesterol micellar solubility). UDCA is a safe and effective treatment in dissolving small (≤5 mm in diameter) cholesterol-rich and radiolucent stones in a functioning gallbladder.86
Obeticholic Acid (OCA)
It is a synthetic derivative of chenodeoxycholic acid. It acts as a selective and potent agonist of FXR, mainly present in enterocytes and hepatocytes. Its binding potency to FXR is more than 100 fold higher than natural bile acids.87 OCA was approved for the treatment of PBC a few years ago. OCA can decrease bile acid production by increasing the synthesis of FGF19. OCA can also improve hepatic bile acid excretion by stimulating BSEP. Thus, OCA can reduce toxic bile build-up in PBC.88
Bile Acid Sequestrants (BAS)
These include cholestyramine, colestipol, and colesevelam. They have been used in various clinical conditions:
- Hyperlipidemia. BAS can bind bile acids and bile salts in the small bowel. As a result, the enterohepatic circulation of bile acids is interrupted, and there is increased conversion of cholesterol into bile acid, depleting intrahepatic cholesterol. This effect leads to increased low-density lipoprotein (LDL) receptor formation. Upregulation of LDL receptors causes catabolism of LDL and decreases serum LDL levels. BAS is mainly used in patients with hyperlipidemia where the primary abnormality is increased LDL-C. BAS can also reduce coronary artery disease progression and outcome.89
- Cholestatic liver diseases. BAS is used as an anti-pruritic agent in cholestatic liver diseases.
- BAS is considered the first-line BAD agent.90
Ileal Bile Acid Transport Inhibitors (IBATis)
IBATis prevent bile acid reuptake from the ileal lumen into the terminal ileal enterocytes by inhibiting ASBT. This leads to (a) increased bile acid delivery into the colon where bile acid increases colonic secretory and motor activity, (b) increased bile acid synthesis in the liver because of decreased ileal FXR activation by bile acids and reduced synthesis of FGF19 This is evidenced by increased serum bile acid precursor C4 (7α-hydroxy-4-cholesten-3-one), (c) decreased serum total cholesterol and LDL cholesterol as increased bile acid synthesis consumes cholesterol stores in the liver and depletion of cholesterol stores promotes expression of cholesterol receptors on the hepatocytes, (d) improvement of liver histology in mouse models of cholestatic liver diseases and non-alcoholic steatohepatitis (NASH) due to interruption of enterohepatic circulation and decreased bile acid pool,91,92 (e) increased serum level of glucagon-like peptide-1 (GLP-1) probably due to the effect of bile acids on enteroendocrine L cells93 and (f) decreased severity and incidence of necrotizing enterocolitis in premature rats.94 As a result, IBATis have the potential of treating chronic constipation, cholestatic liver disease, non-alcoholic fatty liver disease (NAFLD), NASH, atherosclerosis, type II diabetes mellitus, and necrotizing enterocolitis.95 IBATi blocks ASBT not only on ileal enterocytes but also on the renal tubules and thus increases bile acid output both by fecal and renal excretion. Several IBATis are being investigated in clinical trials for different indications. In September 2021, Food and Drug Administration (FDA) in the United States approved Maralixibat (Livmarli) oral solution as the first and only medication for treating cholestatic pruritus in patients with Alagille syndrome one year of age and older. In July 2021, FDA approved Odevixibat (Bylvay) as the first treatment for pruritus in patients with PFIC. In 2018, Elobixibat (Goofice) was approved in Japan to treat chronic idiopathic constipation.
TGR5 Agonist
6g (2-thio-imidazole derivative) is a selective and potent oral TGR5 agonist that stimulates GLP-1 secretion. It has a robust glucose-lowering effect in the mouse model.96 It can also reduce obesity and hepatic steatosis by controlling mice’s blood glucose. In recent years, TGR5 agonist has been a therapeutic target in different metabolic diseases, including type II diabetes mellitus, NAFLD, obesity, atherosclerosis, and malignancy.97
In the mouse model, engineered FGF19 was found to have anti-steatotic, anti-inflammatory, and anti-fibrotic properties with a resolution of histologic features of NASH.98
Future Role of Bile Acids as Therapeutic Agents in Cancer
Synthetic derivatives of CDCA (HS-1199 and HS-1200) and UDCA (HS-1183) can inhibit cell growth and induce apoptosis in cancer cells. These agents have anti-tumor activity in gastric cancer, HCC, breast cancer, cervical cancer, prostate cancer, and T-cell leukemia.99–104
Future Role of Bile Acids as Drug Absorption Enhancer
Because of the unique amphipathic structure and physicochemical properties, bile acids or modified bile acids can solubilize and enhance absorption of many drugs from the terminal ileum, buccal mucosa, nasal mucosa, cornea, skin and rectum, and thus improve bioavailability of many drugs as shown in many experimental animals and humans.105–107
Summary
Bile is an essential body fluid produced in our liver. It has a unique composition of fluid, organic and inorganic materials. The enterohepatic circulation is an efficient mechanism of preserving its contents. Bile production and bile flow are under neurohormonal control. The primary function of bile is the excretion of cholesterol and bilirubin from the liver and digestion and absorption of fat and fat-soluble vitamins from the small bowel. Bile acids can control their synthesis in the liver and have many endocrine and metabolic functions. Bile acids work through the nuclear receptor FXR and membrane receptor TGR5. By activating FXR, bile acids (1) controls hepatic bile acid synthesis via the formation of FGF-19, (2) keeps intracellular bile acid level low by inhibiting NTCT and activating BSEP and thus helps in detoxification in the liver, and (3) controls glucose metabolism by stimulating glycogen synthesis, inhibiting gluconeogenesis and increasing insulin sensitivity. Through the activation of TGR5, bile acids (1) lower blood glucose by stimulating GLP-1 secretion from L cells in the ileocolonic mucosa and by releasing insulin directly from pancreatic β cells (2) stimulate chloride secretion from the gallbladder, (3) enhance gallbladder refilling by smooth muscle relaxation, (4) protect intestinal epithelial barrier and (5) reduce intestinal inflammation. Bile acids also have potent antimicrobial activity in the intestine. They also help maintain the intestinal microbiome. Serum bile acid is markedly increased in cholestatic conditions and other liver diseases, including acute viral hepatitis, cirrhosis, HCC, liver failure, hemochromatosis, portal vein thrombosis, Budd-Chiari syndrome, portosystemic shunts, and Wilson disease. Lithogenic bile has an increased propensity to develop gallstones. It can be found in pregnant women, obese individuals, persons taking a high-calorie diet, patients with ileal disease or resection with bile salt depletion, and patients on certain medications like nicotinic acid and clofibrate. After cholecystectomy, hepatic bile synthesis increases, and the colon’s exposure to secondary bile acids causes diarrhea. Terminal ileal disease or resection of less than 100 cm may cause BAD, whereas terminal ileal disease or resection of more than 100 cm may cause steatorrhea. DGR can cause gastric ulceration, chronic gastritis, gastric atrophy, and intestinal metaplasia. DGR is also considered a risk factor for developing gastric cancer. DGR can be seen during endoscopy, but a more objective diagnosis can be made by DISIDA scan, HIDA scan, or Bilitec device. DGER, along with acid reflux, may injure the esophageal mucosa, including the formation of Barrett’s esophagus, and Bilitec in combination with 24 hours esophageal pH should be done to diagnose it. Cholestasis is characterized by the retention of bile acids, bilirubin, and other bile contents that would normally be excreted in bile. Retained bile acids are toxic to hepatocytes and cholangiocytes and may cause hepatic fibrosis, cirrhosis, hepatoma, and cholangiocarcinoma in long-standing cases. BAD is due to the secretagogue and colonic prokinetic property of bile acids. BAD is diagnosed by therapeutic trials, elevated fasting serum 7αC4, low serum FGF19, elevated fecal bile acids and elevated 75SeHCAT 7-day retention test. BAD is managed by BAS and treatment of the underlying cause. Serum bile acid can be a valuable marker of intrahepatic cholestasis of pregnancy, cirrhosis of the liver, and hepatocellular carcinoma. UDCA is used in different cholestatic liver diseases and dissolves small cholesterol gallstones. Obeticholic acid delays the progression of PBC. BAS is used in BAD and hyperlipidemia, and cholestatic liver diseases. IBATi is used to treat constipation and cholestatic pruritus in Alagille syndrome and PFIC. However, they have the potential of treating NAFL, NASH, type 2 diabetes mellitus, atherosclerosis, and necrotizing enterocolitis. TGR5 agonists can control blood glucose and reduce obesity and NAFLD. Synthetic derivatives of CDCA and UDCA have anti-tumor activity and can be used as potential therapeutic agents against different malignancies in the future.
Author Contributions
Monjur Ahmed solely made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; has agreed on the journal to which the article has been submitted; and agrees to be accountable for all aspects of the work.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Boyer JL. Bile Formation and Cholestasis. In: Schiff ER, Sorrell MF, Maddrey WC, editors. Schiff’s Diseases of the Liver. Philadelphia: Lippincott, Williams & Wilkins; 2002:135–165.
2. Hofmann AF, Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. 2008;65(16):2461–2483. PMID: 18488143. doi:10.1007/s00018-008-7568-6
3. Chen I, Cassaro S. Physiology, bile acids. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2021. PMID: 31747172.
4. Chiang JY. Negative feedback regulation of bile acid metabolism: impact on liver metabolism and diseases. Hepatology. 2015;62(4):1315–1317. PMID: 26122550; PMCID: PMC4589461. doi:10.1002/hep.27964
5. Nagana Gowda GA, Shanaiah N, Cooper A, Maluccio M, Raftery D. Bile acids conjugation in human bile is not random: new insights from (1) H-NMRspectroscopy at 800 MHz. Lipids. 2009;44(6):527–535. PMID: 19373503; PMCID: PMC5459358. doi:10.1007/s11745-009-3296-4
6. Carey MC, Small DM. Micelle formation by bile salts. Physical-chemical and thermodynamic considerations. Arch Intern Med. 1972;130(4):506–527. PMID: 4562149. doi:10.1001/archinte.1972.03650040040005
7. Di Ciaula A, Garruti G, Lunardi Baccetto R, et al. Bile acid physiology. Ann Hepatol. 2017;16:s4–s14. PMID: 29080336. doi:10.5604/01.3001.0010.5493
8. Kojima T, Yamamoto T, Murata M, Chiba H, Kokai Y, Sawada N. Regulation of the blood-biliary barrier: interaction between gap and tight junctions in hepatocytes. Med Electron Microsc. 2003;36(3):157–164. PMID: 14505059. doi:10.1007/s00795-003-0220-5
9. Reshetnyak VI. Physiological and molecular biochemical mechanisms of bile formation. World J Gastroenterol. 2013;19(42):7341–7360. PMID: 24259965; PMCID: PMC3831216. doi:10.3748/wjg.v19.i42.7341
10. Almajid AN, Sugumar K. Physiology, bile. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2021.
11. Jazrawi RP, Pazzi P, Petroni ML, et al. Postprandial gallbladder motor function: refilling and turnover of bile in health and in cholelithiasis. Gastroenterology. 1995;109(2):582–591. PMID: 7615209. doi:10.1016/0016-5085(95)90348-8
12. Banales JM, Prieto J, Medina JF. Cholangiocyte anion exchange and biliary bicarbonate excretion. World J Gastroenterol. 2006;12(22):3496–3511. PMID: 16773707; PMCID: PMC4087566. doi:10.3748/wjg.v12.i22.3496
13. Kaminski DL, Nahrwold DL. Neurohormonal control of biliary secretion and gallbladder function. World J Surg. 1979;3(4):449–456. PMID: 516757. doi:10.1007/BF01556105
14. Hundt M, Basit H, John S. physiology, bile secretion. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2021. PMID: 29262229.
15. Dawson PA, Lan T, Rao A. Bile acid transporters. J Lipid Res. 2009;50(12):2340–2357. PMID: 19498215; PMCID: PMC2781307. doi:10.1194/jlr.R900012-JLR200
16. Cai JS, Chen JH. The mechanism of enterohepatic circulation in the formation of gallstone disease. J Membr Biol. 2014;247(11):1067–1082. PMID: 25107305; PMCID: PMC4207937. doi:10.1007/s00232-014-9715-3
17. Fujino T, Une M, Imanaka T, Inoue K, Nishimaki-Mogami T. Structure-activity relationship of bile acids and bile acid analogs in regard to FXR activation. J Lipid Res. 2004;45(1):132–138. PMID: 13130122. doi:10.1194/jlr.M300215-JLR200
18. Beuers U, Trauner M, Jansen P, Poupon R. New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond. J Hepatol. 2015;62(1Suppl):S25–37. PMID: 25920087. doi:10.1016/j.jhep.2015.02.023
19. Ticho AL, Malhotra P, Dudeja PK, Gill RK, Alrefai WA. Intestinal absorption of bile acids in health and disease. Compr Physiol. 2019;10(1):21–56. PMID: 31853951; PMCID: PMC7171925. doi:10.1002/cphy.c190007
20. Lee JM, Ong JR, Vergnes L, et al. Diet1, bile acid diarrhea, and FGF15/19: mouse model and human genetic variants. J Lipid Res. 2018;59(3):429–438. PMID: 29295820; PMCID: PMC5832924. doi:10.1194/jlr.M078279
21. Mencarelli A, Fiorucci S. FXR an emerging therapeutic target for the treatment of atherosclerosis. J Cell Mol Med. 2010;14(1–2):79–92. PMID: 20041971; PMCID: PMC3837604. doi:10.1111/j.1582-4934.2009.00997.x
22. Wang X, Fu X, Van Ness C, Meng Z, Ma X, Huang W. Bile acid receptors and liver cancer. Curr Pathobiol Rep. 2013;1(1):29–35. PMID: 23420103; PMCID: PMC3571718. doi:10.1007/s40139-012-0003-6
23. Kumar DP, Rajagopal S, Mahavadi S, et al. Activation of transmembrane bile acid receptor TGR5 stimulates insulin secretion in pancreatic β cells. Biochem Biophys Res Commun. 2012;427(3):600–605. PMID: 23022524; PMCID: PMC3498511. doi:10.1016/j.bbrc.2012.09.104
24. Lavoie B, Balemba OB, Godfrey C, et al. Hydrophobic bile salts inhibit gallbladder smooth muscle function via stimulation of GPBAR1 receptors and activation of KATP channels. J Physiol. 2010;588(Pt17):3295–3305. PMID: 20624794; PMCID: PMC2976023. doi:10.1113/jphysiol.2010.192146
25. Keitel V, Reinehr R, Gatsios P, et al. The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology. 2007;45(3):695–704. PMID: 17326144. doi:10.1002/hep.21458
26. Reich M, Klindt C, Deutschmann K, Spomer L, Häussinger D, Keitel V. Role of the G Protein-coupled bile acid receptor TGR5 in liver damage. Dig Dis. 2017;35(3):235–240. PMID: 28249265. doi:10.1159/000450917
27. Donepudi AC, Boehme S, Li F, Chiang JY. G-protein-coupled bile acid receptor plays a key role in bile acid metabolism and fasting-induced hepatic steatosis in mice. Hepatology. 2017;65(3):813–827. PMID: 27351453; PMCID: PMC5195921. doi:10.1002/hep.28707
28. Tripathi N, Jialal I. Conjugated hyperbilirubinemia. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2021. PMID: 32965843.
29. Angelin B, Björkhem I, Einarsson K, Ewerth S. Hepatic uptake of bile acids in man. fasting and postprandial concentrations of individual bile acids in portal venous and systemic blood serum. J Clin Invest. 1982;70(4):724–731. PMID: 7119112; PMCID: PMC370280. doi:10.1172/jci110668
30. Craddock AL, Love MW, Daniel RW, et al. Expression and transport properties of the human ileal and renal sodium-dependent bile acid transporter. Am J Physiol. 1998;274(1):G157–69. PMID: 9458785. doi:10.1152/ajpgi.1998.274.1.G157
31. Ballatori N, Christian WV, Lee JY, et al. OSTα-OSTβ: a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology. 2005;42(6):1270–1279. doi:10.1002/hep.20961
32. Cao R, Cronk ZX, Zha W, et al. Bile acids regulate hepatic gluconeogenic genes and farnesoid X receptor via G (alpha) i-protein-coupled receptors and the AKT pathway. J Lipid Res. 2010;51(8):2234–2244. PMID: 20305288; PMCID: PMC2903791. doi:10.1194/jlr.M004929
33. Hoffmann AF, Borgstroem B. The intraluminal phase of fat digestion in man: the lipid content of the micellar and oil phases of intestinal content obtained during fat digestion and absorption. J Clin Invest. 1964;43(2):247–257. PMID: 14162533; PMCID: PMC289518. doi:10.1172/JCI104909
34. Bajor A, Gillberg PG, Abrahamsson H. Bile acids: short and long term effects in the intestine. Scand J Gastroenterol. 2010;45(6):645–664. PMID: 20334475. doi:10.3109/00365521003702734
35. Sannasiddappa TH, Lund PA, Clarke SR. In vitro antibacterial activity of unconjugated and conjugated bile salts on Staphylococcus aureus. Front Microbiol. 2017;8:1581. PMID: 28878747; PMCID: PMC5572772. doi:10.3389/fmicb.2017.01581
36. Neale G, Lewis B, Weaver V, Panveliwalla D. Serum bile acids in liver disease. Gut. 1971;12(2):145–152. PMID: 5548561; PMCID: PMC1411536. doi:10.1136/gut.12.2.145
37. Attili AF, Angelico M, Cantafora A, Alvaro D, Capocaccia L. Bile acid-induced liver toxicity: relation to the hydrophobic-hydrophilic balance of bile acids. Med Hypotheses. 1986;19(1):57–69. PMID: 2871479. doi:10.1016/0306-9877(86)90137-4
38. Mannes GA, Stellaard F, Paumgartner G. Increased serum bile acids in cirrhosis with normal transaminases. Digestion. 1982;25(4):217–221. PMID: 7166204. doi:10.1159/000198835
39. Grundy SM, Metzger AL, Adler RD. Mechanisms of lithogenic bile formation in American Indian women with cholesterol gallstones. J Clin Invest. 1972;51(12):3026–3043. PMID: 4640946; PMCID: PMC332985. doi:10.1172/JCI107130
40. Barrera F, Azócar L, Molina H, et al. Effect of cholecystectomy on bile acid synthesis and circulating levels of fibroblast growth factor 19. Ann Hepatol. 2015;14(5):710–721. PMID: 26256900. doi:10.1016/S1665-2681(19)30766-5
41. Berr F, Stellaard F, Pratschke E, Paumgartner G. Effects of cholecystectomy on the kinetics of primary and secondary bile acids. J Clin Invest. 1989;83(5):1541–1550. PMID: 2708522; PMCID: PMC303859. doi:10.1172/JCI114050
42. Fort JM, Azpiroz F, Casellas F, Andreu J, Malagelada JR. Bowel habit after cholecystectomy: physiological changes and clinical implications. Gastroenterology. 1996;111(3):617–622. PMID: 8780565. doi:10.1053/gast.1996.v111.pm8780565
43. O’Donnell LJ. Post-cholecystectomy diarrhoea: a running commentary. Gut. 1999;45(6):796–797. PMID: 10562574; PMCID: PMC1727755. doi:10.1136/gut.45.6.796
44. Bemelman WA, Warusavitarne J, Sampietro GM, et al. ECCO-ESCP consensus on surgery for Crohn’s disease. J Crohns Colitis. 2018;12(1):1–16. PMID: 28498901. doi:10.1093/ecco-jcc/jjx061
45. Kouker G, Jaeger KE. Specific and sensitive plate assay for bacterial lipases. Appl Environ Microbiol. 1987;53(1):211–213. PMID: 3103532; PMCID: PMC203632. doi:10.1128/aem.53.1.211-213.1987
46. Steiner MS, Morton RA. Nutritional and gastrointestinal complications of the use of bowel segments in the lower urinary tract. Urol Clin North Am. 1991;18(4):743–754. PMID: 1949406. doi:10.1016/S0094-0143(21)00375-X
47. Lapidus A, Akerlund JE, Einarsson C. Gallbladder bile composition in patients with Crohn ‘s disease. World J Gastroenterol. 2006;12(1):70–74. PMID: 16440420; PMCID: PMC4077498. doi:10.3748/wjg.v12.i1.70
48. Gregory JG, Park KY, Schoenberg HW. Oxalate stone disease after intestinal resection. J Urol. 1977;117(5):631–634. PMID: 870715. doi:10.1016/s0022-5347(17)58564-x
49. Sobala GM, O’Connor HJ, Dewar EP, King RF, Axon AT, Dixon MF. Bile reflux and intestinal metaplasia in gastric mucosa. J Clin Pathol. 1993;46(3):235–240. PMID: 8463417; PMCID: PMC501177. doi:10.1136/jcp.46.3.235
50. Fracchia M, Pellegrino S, Secreto P, et al. Biliary bile acid composition in gastric cancer. Int J Clin Lab Res. 1999;29(1):46–48. PMID: 10356664. doi:10.1007/s005990050062
51. Mabrut JY, Collard JM, Baulieux J. Le reflux biliaire duodéno-gastrique et gastro-oesophagien [Duodenogastric and gastroesophageal bile reflux]. J Chir. 2006. 143(6):355–365. French. PMID: 17285081. doi:10.1016/s0021-7697(06)73717-6
52. Vaezi MF, Lacamera RG, Richter JE. Validation studies of Bilitec 2000: an ambulatory duodenogastric reflux monitoring system. Am J Physiol. 1994;267(6 Pt 1):G1050–7. PMID: 7810652. doi:10.1152/ajpgi.1994.267.6.G1050
53. Gillen P, Keeling P, Byrne PJ, Healy M, O’Moore RR, Hennessy TP. Implication of duodenogastric reflux in the pathogenesis of Barrett’s oesophagus. Br J Surg. 1988;75(6):540–543. PMID: 3395818. doi:10.1002/bjs.1800750612
54. Liu R, Li X, Hylemon PB, Zhou H. Conjugated bile acids promote invasive growth of esophageal adenocarcinoma cells and cancer stem cell expansion via sphingosine 1-phosphate receptor 2-mediated yes-associated protein activation. Am J Pathol. 2018;188(9):2042–2058. PMID: 29963993; PMCID: PMC6105923. doi:10.1016/j.ajpath.2018.05.015
55. Vaezi MF, Richter JE. Importance of duodeno-gastro-esophageal reflux in the medical outpatient practice. Hepatogastroenterology. 1999;46(25):40–47. PMID: 10228763.
56. Strignano P, Collard JM, Michel JM, et al. Duodenal switch operation for pathologic transpyloric duodenogastric reflux. Ann Surg. 2007;245(2):247–253. PMID: 17245178; PMCID: PMC1876986. doi:10.1097/01.sla.0000242714.59254.0e
57. Sifrim D. Management of bile reflux. Gastroenterol Hepatol. 2013;9(3):179–180. PMID: 23961269; PMCID: PMC3745208.
58. Li T, Apte U. Bile acid metabolism and signaling in cholestasis, inflammation, and cancer. Adv Pharmacol. 2015;74:263–302. PMID: 26233910; PMCID: PMC4615692. doi:10.1016/bs.apha.2015.04.003
59. Stepien M, Lopez-Nogueroles M, Lahoz A, et al. Prediagnostic alterations in circulating bile acid profiles in the development of hepatocellular carcinoma. Int J Cancer. 2021;148:609–625. PMID: 34843121. doi:10.1002/ijc.33885
60. Wang Y, Aoki H, Yang J, et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology. 2017;65(6):2005–2018. PMID: 28120434; PMCID: PMC5444993. doi:10.1002/hep.29076
61. Camilleri M. Bile Acid diarrhea: prevalence, pathogenesis, and therapy. Gut Liver. 2015;9(3):332–339. PMID: 25918262; PMCID: PMC4413966. doi:10.5009/gnl14397
62. Walters JRF, Arasaradnam R, Andreyev HJN; UK Bile Acid Related Diarrhoea Network. Diagnosis and management of bile acid diarrhoea: a survey of UK expert opinion and practice. Frontline Gastroenterol. 2019;11(5):358–363. PMID: 32879719; PMCID: PMC7447276. doi:10.1136/flgastro-2019-101301
63. Walters JR, Pattni SS. Managing bile acid diarrhoea. Therap Adv Gastroenterol. 2010;3(6):349–357. PMID: 21180614; PMCID: PMC3002596. doi:10.1177/1756283X10377126
64. Barkun AN, Love J, Gould M, Pluta H, Steinhart H. Bile acid malabsorption in chronic diarrhea: pathophysiology and treatment. Can J Gastroenterol. 2013;27(11):653–659. PMID: 24199211; PMCID: PMC3816948. doi:10.1155/2013/485631
65. Wildt S, Nørby Rasmussen S, Lysgård Madsen J, Rumessen JJ. Bile acid malabsorption in patients with chronic diarrhoea: clinical value of SeHCAT test. Scand J Gastroenterol. 2003;38(8):826–830. PMID: 12940434. doi:10.1080/00365520310004461
66. Scarpello JH, Hodgson E, Howlett HC. Effect of metformin on bile salt circulation and intestinal motility in type 2 diabetes mellitus. Diabet Med. 1998;15(8):651–656. PMID: 9702467. doi:10.1002/(SICI)1096-9136(199808)15:8<651::AID-DIA628>3.0.CO;2-A
67. Vijayvargiya P, Camilleri M, Shin A, Saenger A. Methods for diagnosis of bile acid malabsorption in clinical practice. Clin Gastroenterol Hepatol. 2013;11(10):1232–1239. PMID: 23644387; PMCID: PMC3783593. doi:10.1016/j.cgh.2013.04.029
68. Gälman C, Arvidsson I, Angelin B, Rudling M. Monitoring hepatic cholesterol 7alpha-hydroxylase activity by assay of the stable bile acid intermediate 7alpha-hydroxy-4-cholesten-3-one in peripheral blood. J Lipid Res. 2003;44(4):859–866. PMID: 12562858. doi:10.1194/jlr.D200043-JLR200
69. Camilleri M, Nadeau A, Tremaine WJ, et al. Measurement of serum 7alpha-hydroxy-4-cholesten-3-one (or 7alphaC4), a surrogate test for bile acid malabsorption in health, ileal disease and irritable bowel syndrome using liquid chromatography-tandem mass spectrometry. Neurogastroenterol Motil. 2009;21(7):734–e43. PMID: 19368662; PMCID: PMC2705747. doi:10.1111/j.1365-2982.2009.01288.x
70. Vijayvargiya P, Camilleri M, Chedid V, et al. Analysis of fecal primary bile acids detects increased stool weight and colonic transit in patients with chronic functional diarrhea. Clin Gastroenterol Hepatol. 2019;17(5):922–929.e2. PMID: 29902647; PMCID: PMC6291372. doi:10.1016/j.cgh.2018.05.050
71. Walters JR. Bile acid diarrhoea and FGF19: new views on diagnosis, pathogenesis and therapy. Nat Rev Gastroenterol Hepatol. 2014;11(7):426–434. PMID: 24662279. doi:10.1038/nrgastro.2014.32
72. Vijayvargiya P, Camilleri M, Carlson P, et al. Performance characteristics of serum C4 and FGF19 measurements to exclude the diagnosis of bile acid diarrhoea in IBS-diarrhoea and functional diarrhoea. Aliment Pharmacol Ther. 2017;46(6):581–588. PMID: 28691284; PMCID: PMC5555810. doi:10.1111/apt.14214
73. Hvas CL, Ott P, Paine P, Lal S, Jørgensen SP, Dahlerup JF. Obeticholic acid for severe bile acid diarrhea with intestinal failure: a case report and review of the literature. World J Gastroenterol. 2018;24(21):2320–2326. PMID: 29881241; PMCID: PMC5989246. doi:10.3748/wjg.v24.i21.2320
74. Kårhus ML, Brønden A, Røder ME, Leotta S, Sonne DP, Knop FK. Remission of bile acid malabsorption symptoms following treatment with the glucagon-like peptide 1 receptor agonist liraglutide. Gastroenterology. 2019;157(2):569–571. PMID: 30965026. doi:10.1053/j.gastro.2019.04.002
75. Jung D, Fantin AC, Scheurer U, Fried M, Kullak-Ublick GA. Human ileal bile acid transporter gene ASBT (SLC10A2) is transactivated by the glucocorticoid receptor. Gut. 2004;53(1):78–84. PMID: 14684580; PMCID: PMC1773940. doi:10.1136/gut.53.1.78
76. Nguyen TT, Ung TT, Kim NH, Jung YD. Role of bile acids in colon carcinogenesis. World J Clin Cases. 2018;6(13):577–588. PMID: 30430113; PMCID: PMC6232560. doi:10.12998/wjcc.v6.i13.577
77. Ambros-Rudolph CM, Glatz M, Trauner M, Kerl H, Müllegger RR. The importance of serum bile acid level analysis and treatment with ursodeoxycholic acid in intrahepatic cholestasis of pregnancy: a case series from central Europe. Arch Dermatol. 2007;143(6):757–762. PMID: 17576942. doi:10.1001/archderm.143.6.757
78. Anwer MS, Meyer DJ. Bile acids in the diagnosis, pathology, and therapy of hepatobiliary diseases. Vet Clin North Am Small Anim Pract. 1995;25(2):503–517. PMID: 7785176. doi:10.1016/s0195-5616(95)50039-7
79. Schwarz HP, Paumgartner G, Preisig R. Diagnostische Bedeutung der Serumgallensäuren [Diagnostic significance of serum bile acids]. Schweiz Med Wochenschr. 1975;105(17):533–535. German. PMID: 1153981.
80. Liu N, Feng J, Lv Y, et al. Role of bile acids in the diagnosis and progression of liver cirrhosis: a prospective observational study. Exp Ther Med. 2019;18(5):4058–4066. PMID: 31611941; PMCID: PMC6781791. doi:10.3892/etm.2019.8011
81. Thomas CE, Luu HN, Wang R, et al. Association between pre-diagnostic serum bile acids and hepatocellular carcinoma: the Singapore Chinese health study. Cancers. 2021;13(11):2648. PMID: 34071196; PMCID: PMC8198655. doi:10.3390/cancers13112648
82. Caspary WF, Reimold WV. Klinische bedeutung des 14C-glykocholat-atemtests in der gastroenterologischen diagnostik bei erkrankungen mit gesteigerter dekonjugation von Gallensäuren [clinical significance of the 14C-glycocholate breath test in the diagnosis of gastro-enterological diseases (author’s transl)]. Dtsch Med Wochenschr. 1976. 101(10):353–360. German. PMID: 1248374. doi:10.1055/s-0028-1104088
83. Baumgartner U, Schölmerich J, Kremer B, et al. Early detection of graft dysfunction after orthotopic liver transplantation in man by serum and biliary bile acid analysis. Hepatogastroenterology. 1995;42(6):950–960. PMID: 8847051.
84. Paumgartner G, Beuers U. Mechanisms of action and therapeutic efficacy of ursodeoxycholic acid in cholestatic liver disease. Clin Liver Dis. 2004;8(1):67–81. PMID: 15062194. doi:10.1016/S1089-3261(03)00135-1
85. Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology. 2002;36(3):525–531. PMID: 12198643. doi:10.1053/jhep.2002.36088
86. Tint GS, Salen G, Colalillo A, et al. Ursodeoxycholic acid: a safe and effective agent for dissolving cholesterol gallstones. Ann Intern Med. 1982;97(3):351–356. PMID: 7051912. doi:10.7326/0003-4819-97-3-351
87. Novotny K, Hapshy V, Nguyen H, Parmar M. Obeticholic acid. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2021. PMID: 33620812.
88. Kjærgaard K, Frisch K, Sørensen M, et al. Obeticholic acid improves hepatic bile acid excretion in patients with primary biliary cholangitis. J Hepatol. 2021;74(1):58–65. PMID: 32717289. doi:10.1016/j.jhep.2020.07.028
89. Insull W Jr. Clinical utility of bile acid sequestrants in the treatment of dyslipidemia: a scientific review. South Med J. 2006;99(3):257–273. PMID: 16553100. doi:10.1097/01.smj.0000208120.73327.db
90. Lee KJ. Pharmacologic agents for chronic diarrhea. Intest Res. 2015;13(4):306–312. PMID: 26576135; PMCID: PMC4641856. doi:10.5217/ir.2015.13.4.306
91. Al-Dury S, Marschall HU. Ileal bile acid transporter inhibition for the treatment of chronic constipation, cholestatic pruritus, and NASH. Front Pharmacol. 2018;9:931. PMID: 30186169; PMCID: PMC6111463. doi:10.3389/fphar.2018.00931
92. Rao A, Kosters A, Mells JE, et al. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Sci Transl Med. 2016;8(357):357ra122. PMID: 27655848; PMCID: PMC5056562. doi:10.1126/scitranslmed.aaf4823
93. Chedid V, Vijayvargiya P, Camilleri M. Elobixibat for the treatment of constipation. Expert Rev Gastroenterol Hepatol. 2018;12(10):951–960. PMID: 30204504; PMCID: PMC6386599. doi:10.1080/17474124.2018.1522248
94. Halpern MD, Weitkamp JH, Mount Patrick SK, et al. Apical sodium-dependent bile acid transporter upregulation is associated with necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol. 2010;299(3):G623–31. PMID: 20616306; PMCID: PMC2950692. doi:10.1152/ajpgi.00242.2010
95. Saveleva EE, Tyutrina ES, Nakanishi T, Tamai I, Salmina AB. Ingibitory natriĭ-zavisimogo perenoschika zhelchnykh kislot (ASBT) kak perspektivnye lekarstvennye sredstva [The inhibitors of the apical sodium-dependent bile acid transporter (ASBT) as promising drugs]. Biomed Khim. 2020. 66(3):185–195. Russian. PMID: 32588824. doi:10.18097/PBMC20206603185
96. Agarwal S, Patil A, Aware U, et al. Discovery of a potent and orally efficacious TGR5 receptor agonist. ACS Med Chem Lett. 2015;7(1):51–55. PMID: 26819665; PMCID: PMC4716599. doi:10.1021/acsmedchemlett.5b00323
97. Hodge RJ, Nunez DJ. Therapeutic potential of Takeda-G-protein-receptor-5 (TGR5) agonists. Hope or hype? Diabetes Obes Metab. 2016;18(5):439–443. PMID: 26818602. doi:10.1111/dom.12636
98. Zhou M, Learned RM, Rossi SJ, DePaoli AM, Tian H, Ling L. Engineered FGF19 eliminates bile acid toxicity and lipotoxicity leading to resolution of steatohepatitis and fibrosis in mice. Hepatol Commun. 2017;1(10):1024–1042. PMID: 29404440; PMCID: PMC5721409. doi:10.1002/hep4.1108
99. Moon B, Kim MC, Park JS. Synthetic CDCA derivatives-induced apoptosis of stomach cancer cell line SNU-1 cells. Cancer Res Treat. 2004;36(2):132–139. PMID: 20396553; PMCID: PMC2855097. doi:10.4143/crt.2004.36.2.132
100. Xu M, Zhao Q, Shao D, Liu H, Qi J, Qin C. Chenodeoxycholic acid derivative HS-1200 inhibits hepatocarcinogenesis and improves liver function in diethylnitrosamine-exposed rats by downregulating MTH1. Biomed Res Int. 2017;2017:1465912. PMID: 28261604; PMCID: PMC5316462. doi:10.1155/2017/1465912
101. Im EO, Choi YH, Paik KJ, et al. Novel bile acid derivatives induce apoptosis via a p53-independent pathway in human breast carcinoma cells. Cancer Lett. 2001;163(1):83–93. PMID: 11163111. doi:10.1016/s0304-3835(00)00671-6
102. Šarenac T, Mikov M. Cervical cancer, different treatments and importance of bile acids as therapeutic agents in this disease. Front Pharmacol. 2019;10:484. PMID: 31214018; PMCID: PMC6558109. doi:10.3389/fphar.2019.00484
103. Choi YH, Im EO, Suh H, Jin Y, Yoo YH, Kim ND. Apoptosis and modulation of cell cycle control by synthetic derivatives of ursodeoxycholic acid and chenodeoxycholic acid in human prostate cancer cells. Cancer Lett. 2003;199(2):157–167. PMID: 12969788. doi:10.1016/s0304-3835(03)00351-3
104. Choi YH, Im EO, Suh H, et al. Apoptotic activity of novel bile acid derivatives in human leukemic T cells through the activation of caspases. Int J Oncol. 2001;18(5):979–984. PMID: 11295044. doi:10.3892/ijo.18.5.979
105. Pavlović N, Goločorbin-Kon S, Ðanić M, et al. Bile acids and their derivatives as potential modifiers of drug release and pharmacokinetic profiles. Front Pharmacol. 2018;9:1283. PMID: 30467479; PMCID: PMC6237018. doi:10.3389/fphar.2018.01283
106. Nurunnabi M, Khatun Z, Revuri V, et al. Design and strategies for bile acid mediated therapy and imaging. RSC Adv. 2016;6(78):73986–74002. doi:10.1039/C6RA10978K
107. Ziv E, Eldor A, Kleinman Y, Bar-On H, Kidron M. Bile salts facilitate the absorption of heparin from the intestine. Biochem Pharmacol. 1983;32(5):773–776. PMID: 6838625. doi:10.1016/0006-2952(83)90575-0
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.