Back to Journals » International Journal of Women's Health » Volume 17
From Constitution to Disease: MicroRNA Signatures for the Early Prediction and Targeted Prevention of Polycystic Ovary Syndrome
Authors Shi N ![]() , Ji S, Zhang Y
, Ji S, Zhang Y ![]() , Qu H, Hou J, Jiang L, Liu HP
, Qu H, Hou J, Jiang L, Liu HP ![]()
Received 6 September 2025
Accepted for publication 9 December 2025
Published 12 December 2025 Volume 2025:17 Pages 5389—5398
DOI https://doi.org/10.2147/IJWH.S565574
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Matteo Frigerio
Na Shi, Shuhua Ji, Yan Zhang, Huiling Qu, Jinwei Hou, Longhuan Jiang, Hai-ping Liu
Department of Reproductive Medicine, the 960th Hospital of the PLA Joint Logistics Support Force, Jinan, People’s Republic of China
Correspondence: Hai-ping Liu, Email [email protected]
Background: The association between PCOS and the TCM concept of phlegm-damp constitution is well-documented, but their connection via miRNA remains unclear. This study sought to identify key miRNAs implicated in the transition from phlegm-damp constitution to PCOS and to elucidate their associated regulatory networks.
Methods: We conducted miRNA sequencing on granulosa cells from three groups: healthy controls with a neutral constitution (NP), healthy individuals with a phlegm-damp constitution (NB), and PCOS patients (PC). Principal component analysis (PCA) and differential expression analysis were utilized to pinpoint candidate miRNAs, which were subsequently validated using quantitative real-time PCR (qRT-PCR) in larger cohorts. Bioinformatics, including target prediction, protein-protein interaction (PPI) network construction, and pathway analysis, were employed to delineate the regulatory networks involved.
Results: PCA revealed distinct clustering patterns, with NB and PC groups exhibiting closer proximity compared to the NP group. Differential expression analysis identified 24 dysregulated miRNAs in the PC group (20 upregulated, 4 downregulated) and 14 in the NB group (11 upregulated, 3 downregulated), with both groups showing shared enrichment in metabolic, PI3K-Akt, and Ras pathways. Validation confirmed a progressive downregulation of hsa-miR-24-2-5p and hsa-miR-33a-5p across the NP, the NB, and PC groups (P< 0.05), whereas hsa-miR-374b-5p exhibited differential expression exclusively between NB and PC groups. Network analysis identified five hub genes (EGFR, MYC, KRAS, CREB1, SMAD4) and nine risk pathways, including MAPK and Wnt signaling pathways. The constructed miRNA-hub gene-pathway network comprised five miRNA-hub gene pairs and thirteen functional modules.
Conclusion: These findings elucidate shared miRNA regulatory mechanisms between phlegm-damp constitution and PCOS, highlighting hsa-miR-24-2-5p and hsa-miR-33a-5p as pivotal mediators in this pathological transition. This provides molecular evidence for the TCM “constitution-disease correlation” theory and identifies potential targets for the prevention and treatment of PCOS.
Keywords: polycystic ovary syndrome, phlegm-damp constitution, miRNA profiling, traditional Chinese medicine, constitution-disease correlation
Background
Polycystic ovary syndrome (PCOS), is one of the most prevalent endocrine and metabolic disorders affecting women of reproductive age. It is clinically characterized by hyperandrogenism, ovulatory dysfunction, and polycystic ovarian morphology, frequently accompanied by metabolic abnormalities such as insulin resistance and obesity.1 The “constitution-disease correlation” theory in Traditional Chinese Medicine (TCM) suggests that an individual’s constitutional predisposition serves as an intrinsic basis for disease development, with specific constitution types exhibiting distinct susceptibilities to particular diseases.2 This theory closely aligns with contemporary epigenetic concepts of “gene-environment interactions”,3 suggesting that TCM constitutions may influence disease pathogenesis through epigenetic regulatory mechanisms. Notably, the phlegm-damp constitution, due to its unique metabolic characteristic, has been identified as a significant risk factor for the onset of PCOS.
In TCM theory, phlegm-damp constitution is one of TCM’s nine basic constitution types, with its core characteristics being the dysregulation of fluid metabolism and the stagnation of Qi activity. This constitution is thought to arise from a deficiency in “Spleen” function, leading to the impaired transportation and transformation of body fluids, which then accumulate as “Phlegm” and “Dampness”. For a person with this constitution, the body’s internal milieu resembles a damp, viscous, and impure environment, which obstructs the flow of Qi and Blood and slows down metabolic processes. This pathophysiological profile has significant overlap with PCOS, a condition also fundamentally characterized by metabolic disturbances and chronic low-grade inflammation. This convergence suggests a shared molecular underpinning. MicroRNAs (miRNAs), as pivotal epigenetic regulators of metabolism, inflammation, and cell proliferation, are therefore ideal candidates for the molecular link that connects a phlegm-damp constitution to the pathogenesis of PCOS.4,5
Indeed, increasing evidence highlights distinctive miRNA expression profiles in the granulosa cells, follicular fluid, and peripheral blood of PCOS patients, suggesting that these differentially expressed miRNAs contribute to the pathogenesis of PCOS by impacting follicular development and steroidogenesis.6,7 Given the shared pathological features between phlegm-damp constitution and PCOS, it is highly plausible that a common set of miRNA-mediated regulatory networks is involved in both conditions.8 Nonetheless, comprehensive systematic comparisons of miRNA expression patterns and their functional associations between a phlegm-damp constitution and PCOS remain insufficiently explored.
In this study, we adopted an innovative methodology that integrates high-throughput miRNA sequencing with bioinformatics analyses to systematically compare healthy individuals with a neutral constitution, those with a phlegm-damp constitution, and patients with PCOS. By identifying differentially expressed miRNAs and characterizing key hub genes and signaling pathways, we aimed to elucidate the shared regulatory networks that drive the transition from a phlegm-damp constitution to PCOS. Our findings not only provide molecular-level validation for TCM theories of “constitution-disease correlation” and “constitutional regulation”, but also reveal novel potential targets for early prediction and personalized intervention in PCOS.
Materials and Methods
Study Participants and Sample Collection
The study cohort comprised women of reproductive age (20–40 years) undergoing IVF/ICSI treatment at the Reproductive Medicine Department of the 960th Hospital of the PLA Joint Logistics Support Force. Participants were categorized into three distinct groups: PCOS patients (PC group, n=3), healthy controls with a neutral constitution (NP group, n=3), and healthy controls with a phlegm-damp constitution (NB group, n=3). The healthy control groups (NP and NB) were diagnosed with male-factor infertility, had regular menstrual cycles (21–35 days), and exhibited no clinical or biochemical signs of PCOS or other endocrine disorders. Baseline demographic and clinical characteristics, including age, body mass index (BMI), and duration of infertility, were collected for all participants.
The diagnosis of PCOS was established according to the Rotterdam criteria. The classification of TCM constitution was independently conducted by two senior TCM practitioners utilizing standardized diagnostic criteria.
The diagnosis of PCOS was established according to the Rotterdam criteria.9 The classification of TCM constitution was independently conducted by two senior TCM practitioners according to the “Constitution Classification and Determination Table of TCM” (2009).10
Exclusion criteria were applied to all participants to minimize confounding factors. These included: (1) Failure to meet the established diagnostic criteria for PCOS or the standardized criteria for TCM constitution classification; (2) Age outside the specified inclusion range; (3) A history of steroid hormone therapy within the three months preceding the study; (4) Presence of other concomitant organic diseases; (5) Endocrine disorders other than PCOS; (6) Infertility attributable to known chromosomal abnormalities or other genetic factors; (7) Severe concomitant diseases, including cardiovascular, cerebrovascular, hematopoietic, hepatic, or renal diseases, or severe psychiatric disorders; (8) Incomplete clinical data or records that were insufficient for evaluation.
Granulosa Cell Isolation and RNA Extraction
Granulosa cells were isolated from follicular fluid using a density gradient centrifugation method, as previously described.11 Initial centrifugation was performed at 2000 rpm for 10 minutes using a centrifuge (Eppendorf, Germany), followed by washing with phosphate-buffered saline (PBS) and subsequent gradient centrifugation with lymphocyte separation medium to isolate the granulosa cell layer. Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA), and quality control measures were implemented to ensure RNA integrity (RNA Integrity Number, RIN >7.0) and purity (absorbance ratio at 260/280 nm: 1.8–2.0).
miRNA Sequencing and Bioinformatics Analysis
For the analysis of miRNA sequencing, small RNA libraries were constructed utilizing the TruSeq Kit (Illumina, San Diego, CA, USA) in accordance with the manufacturer’s guidelines. This was followed by 50 bp single-end sequencing on the Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) to produce raw sequencing data.
The sequencing data underwent a thorough bioinformatics analysis, commencing with the filtering of raw data to eliminate adapter sequences and low-quality reads, followed by size selection to retain miRNA fragments ranging from 18 to 26 nt. miRNA identification was executed using the ACGT101-miR software (version 4.2). Target gene prediction was then conducted using both the TargetScan and miRanda algorithms. The predicted target genes from both algorithms were intersected to generate a high-confidence list. Differential expression analysis was subsequently performed, applying thresholds of |log2 fold change| > 1 and p < 0.05. Target gene prediction was then conducted using both the TargetScan (version 5.0) and miRanda (version 3.3a) algorithms. Finally, functional enrichment analysis was performed using the DAVID database (version 2023q1) to identify significantly enriched biological pathways and processes.
qRT-PCR Validation
Three candidate miRNAs, specifically hsa-miR-374b-5p, hsa-miR-24-2-5p, and hsa-miR-33a-5p, were selected for quantitative validation by qRT-PCR in an expanded cohort (n=10 per group). Reverse transcription was performed using the MiRNA 1st Strand cDNA Synthesis Kit (Vazyme, Nanjing, China). Subsequently, qPCR was carried out using the SuperReal PreMix Plus (SYBR Green) (Vazyme, Nanjing, China) on a QuantStudio 5 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). U6 snRNA was utilized as the endogenous reference gene. Relative expression levels were calculated using the 2^-ΔΔCt method, with each sample measured in triplicate to ensure technical reproducibility.
Network Analysis
The PPI network was developed utilizing the STRING database, applying a confidence score threshold greater than 0.4. Subsequent identification of hub genes was conducted through topological analysis using the cytoHubba plugin within Cytoscape software (version 3.7.2). This methodical approach facilitated the identification of pivotal regulatory genes within the interaction network.
In the investigation of the miRNA-hub gene-risk pathway network, an integrated analysis was conducted by amalgamating differentially expressed miRNAs with their predicted target genes. Key pathways were discerned through KEGG enrichment analysis, and the resultant network relationships were visualized utilizing Cytoscape version 3.7.2. This approach facilitated the elucidation of the intricate regulatory interactions among miRNAs, hub genes, and biological pathways.
Statistical Analysis
Statistical analyses were performed utilizing SPSS 25.0 and GraphPad Prism 8.0.2. Continuous variables are presented as mean ± standard deviation (SD). The Shapiro–Wilk test was applied to evaluate the normality of the data. Based on the normality assessment, either parametric tests (Student’s t-test or ANOVA) or non-parametric tests (Mann–Whitney U or Kruskal–Wallis tests) were employed for group comparisons. A significance level of p < 0.05 was established a priori.
Results
The baseline demographic and clinical characteristics of the participants are summarized in Table 1. There were no statistically significant differences in age, body mass index (BMI), or duration of infertility among the three groups, ensuring the comparability of the cohorts.
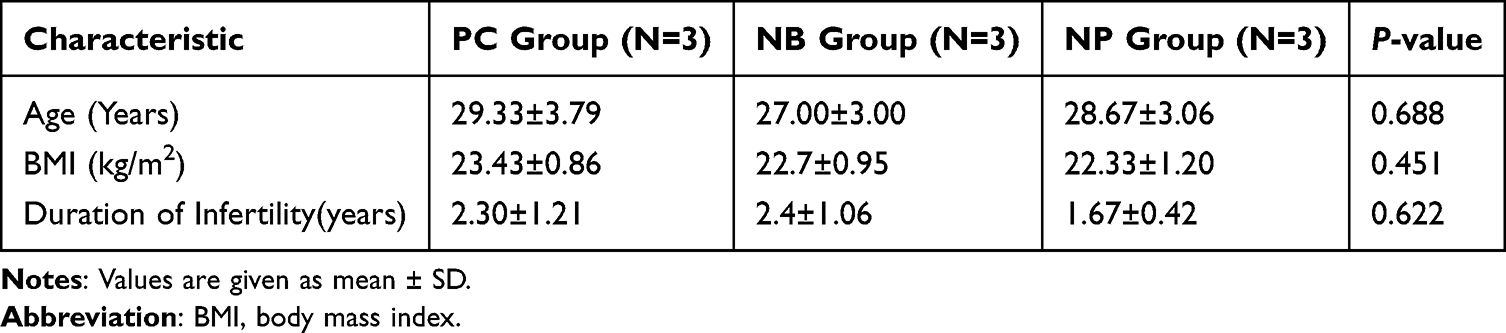 |
Table 1 Baseline Characteristics (with Percentages Rounded for Clarity) |
RNA Quality Assessment and Sequencing Data Analysis
All samples adhered to the predefined quality criteria for RNA sequencing. The extracted total RNA exhibited the following parameters: a concentration range of 0.12–0.60 μg/μL, 260/280 ratios between 2.06 and 2.19, and RNA integrity numbers (RIN) spanning from 5.9 to 9.8. The sequencing data obtained were of high quality, with total reads ranging from 8.13 to 20.81 million per sample, Q20 scores exceeding 98.6%, and Q30 scores surpassing 95.8%.
Principal Component Analysis (PCA)
PCA demonstrated distinct clustering patterns, as illustrated in Figure 1. The NB group exhibited closer proximity to the PC group, whereas the NP group constituted a separate cluster. The NB group occupied an intermediate position between the NP and PC groups, indicating.
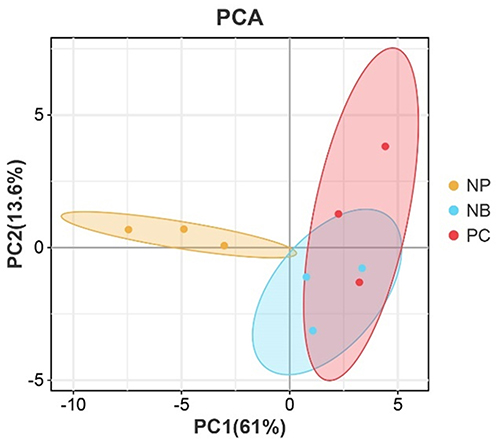 |
Figure 1 PCA plot of miRNA expression patterns across NP (yellow), NB (green), and PC (red) groups. |
Differential miRNA Expression Analysis
A comparative analysis identified significant differential miRNA expression (|log2FC| > 1, p < 0.05) between the groups. The PC group demonstrated 24 differentially expressed miRNAs in comparison to the NP group, consisting of 20 upregulated and 4 downregulated miRNAs. Likewise, the NB group exhibited 14 differentially expressed miRNAs relative to the NP group, with 11 upregulated and 3 downregulated miRNAs (refer to Tables 2 and 3).
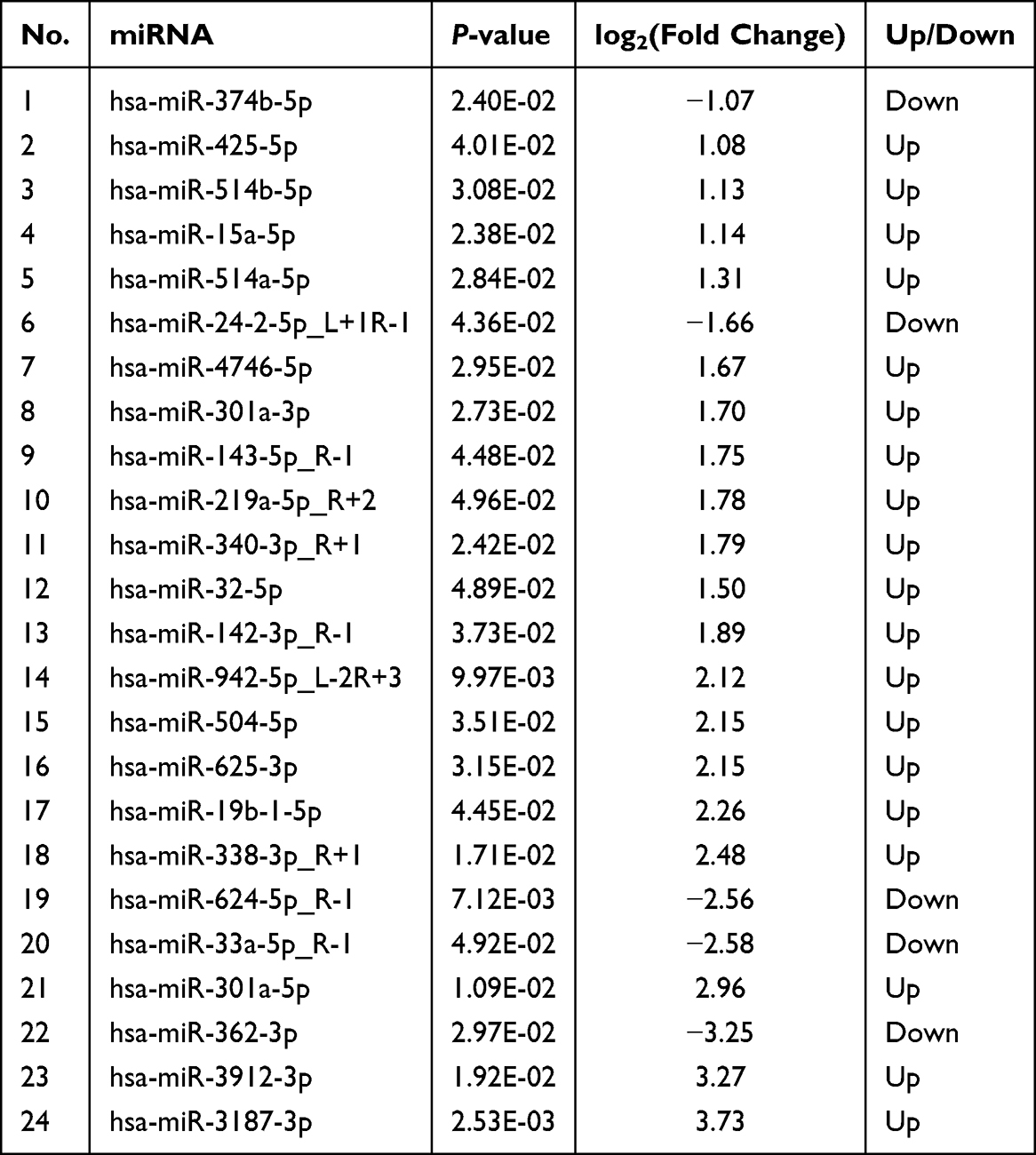 |
Table 2 Differentially Expressed miRNAs in PC Group Versus the NP Group |
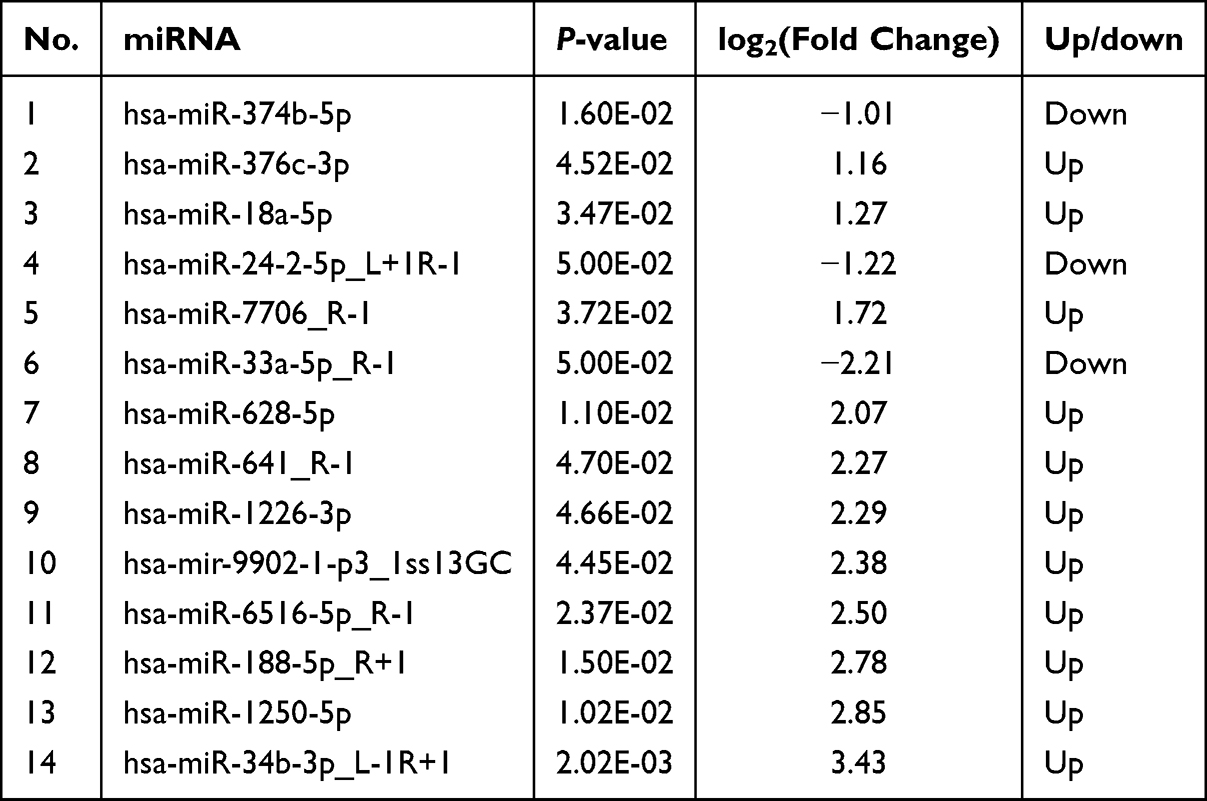 |
Table 3 Differentially Expressed miRNAs in NB Group Versus the NP Group |
Identification and Validation of miRNAs Associated with the Transition From Phlegm-Damp Constitution to PCOS
A comparative analysis of differentially expressed miRNAs between NB vs NP and PC vs NP groups identified three miRNAs — hsa-miR-374b-5p, hsa-miR-24-2-5p, and hsa-miR-33a-5p — that were consistently downregulated in granulosa cells from both phlegm-damp constitution subjects and PCOS patients (Figure 2).
 |
Figure 2 (A) Venn diagram showing overlapping differentially expressed miRNAs between comparison groups; (B) Heatmap analysis of differentially expressed miRNAs. Red indicates high expression; blue indicates low expression. |
Validation in expanded cohorts (n=10 per group) demonstrated significant gradient decreases in the expression of hsa-miR-24-2-5p and hsa-miR-33a-5p across the NP, NB, PC groups (P<0.05, Figure 3). In contrast, hsa-miR-374b-5p exhibited differential expression specifically between the NB and PC groups (P<0.05), with no significant variation observed between the NP and NB groups.
 |
Figure 3 Validation of candidate miRNA expression by qRT-PCR. Data are mean ± SD. Significance: *P < 0.05, **P < 0.01, ***P < 0.001, ns: not significant. |
Functional Analysis of miRNA Target Genes in Phlegm-Damp Constitution to PCOS Transition
Through the application of rigorous bioinformatics criteria (TargetScan score ≥ 50 and miRanda energy < −15), we identified 729 and 779 potential target genes for miR-24-2-5p and miR-33a-5p, respectively. After eliminating duplicates, this resulted in a total of 1,433 unique target genes.
The functional enrichment analysis of the predicted target genes indicated their predominant involvement in biological processes such as signal transduction, RNA polymerase II-mediated transcriptional regulation, DNA-templated transcription, and ion transport. These genes were primarily localized to membrane systems, as well as cytoplasmic and nuclear compartments. The molecular function analysis revealed significant enrichment in protein, metal ion, and nucleic acid binding, along with transferase activity (Figure 4A). Additionally, KEGG pathway analysis identified 34 significantly enriched signaling pathways, with the MAPK, calcium, and cGMP-PKG signaling cascades being particularly notable (Figure 4B). A total of 127 target genes were found to collectively represent core network components within the top 10 risk pathways (Figure 5A).
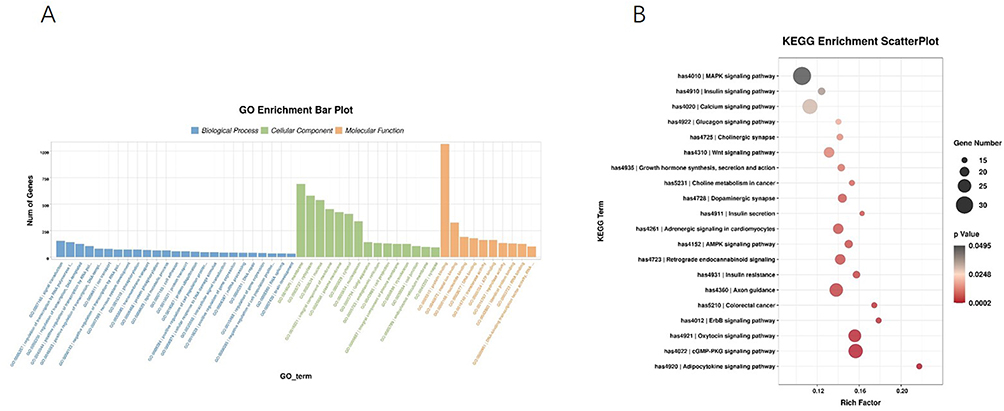 |
Figure 4 Functional enrichment analysis of miRNA target genes: (A) Significantly enriched GO terms of target genes; (B) Bubble chart of KEGG pathway enrichment analysis, where bubble size indicates gene count and color indicates P-value. |
 |
Figure 5 Functional characterization of core target genes: (A) Venn diagram of 127 target genes enriched in the top 10 risk pathways; (B) PPI network of predicted target genes. |
PPI Network Construction and Core Hub Gene Identification
A PPI network was constructed using 1,433 predicted target genes, resulting in a network comprising 1,411 nodes and 6,230 edges following the removal of unconnected nodes through analysis with the STRING database (confidence score >0.4; Figure 5B). To further elucidate key regulatory factors, the cytoHubba plugin in Cytoscape software was utilized, employing five distinct algorithms (MCC, Degree, DMNC, EPC, and MNC) to identify the top 15 candidate genes from each algorithmic ranking (Figure 6A). Through a multi-algorithm intersection analysis, six core hub genes were identified: EGFR, MYC, KRAS, CREB1, RPS20, and SMAD4. Notably, an intersection analysis between these core hub genes and the 127 target genes enriched in the top 10 high-risk pathways revealed that five hub genes (EGFR, MYC, KRAS, CREB1, and SMAD4) were co-enriched in critical signaling pathways.
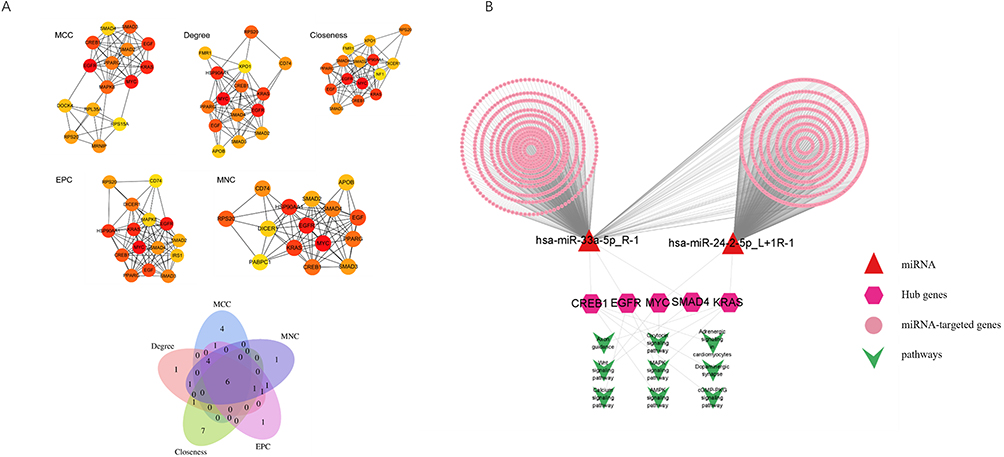 |
Figure 6 (A) Venn diagram of top 15 genes identified by five cytoHubba algorithms and their overlapping hub genes; (B) Integrated miRNA-hub gene-risk pathway interaction network. |
Hub Gene-Mediated miRNA-Signaling Pathway Regulatory Network
Based on the five identified hub genes (EGFR, MYC, KRAS, CREB1, and SMAD4), we developed an integrated miRNA-hub gene-risk pathway regulatory network (Figure 6B). Bioinformatics analysis indicated a significant enrichment of these hub genes within nine critical signaling pathways: MAPK, calcium ion, oxytocin, Wnt, axon guidance, cGMP-PKG, cardiomyocyte adrenergic, dopaminergic synapse, and AMPK signaling pathways. The regulatory network predominantly consisted of two principal types of interactions. Firstly, five miRNA-hub gene regulatory pairs were identified: miR-24-2-5p-MYC, miR-24-2-5p-KRAS, miR-33a-5p-CREB1, miR-33a-5p-EGFR, and miR-33a-5p-SMAD4. Secondly, thirteen miRNA-hub gene-pathway ternary regulatory modules were characterized, with the MAPK signaling pathway demonstrating the most extensive regulation through three key interactions: miR-24-2-5p-MYC, miR-24-2-5p-KRAS, and miR-33a-5p-EGFR.
Significantly, CREB1 was identified as a crucial regulatory node involved in the modulation of four essential metabolic pathways: cGMP-PKG signaling, cardiomyocyte adrenergic signaling, dopaminergic synapse, and AMPK signaling.
Discussion
This study elucidates the scientific underpinnings of the TCM theory of “constitution-disease correlation” in the pathogenesis of PCOS from an epigenetic standpoint, thereby offering essential theoretical support for the application of the “preventive treatment” principle in managing PCOS. As a multifaceted endocrine-metabolic disorder, PCOS exhibits a significant association with the phlegm-damp constitution, a finding that is in remarkable concordance with classical TCM doctrines. From the foundational concepts articulated in the Yellow Emperor’s Inner Canon (“When genuine qi exists internally, pathogens cannot invade”) to the clinical observations of Qing Dynasty physicians (“Obese individuals frequently suffer from wind-stroke syndromes”) and contemporary Professor Wang Qi’s “susceptibility-transformation” theory,12 this study provides the first epigenomic evidence linking the TCM concept of phlegm-damp constitution to the pathogenesis of PCOS, substantiating the “constitution-disease correlation” theory at a molecular level. Our key findings, including the identification of 14 and 24 differentially expressed miRNAs in the NB and PC groups respectively, and PCA results showing the NB group as an intermediate molecular state, suggest phlegm-damp constitution is a pre-disease state that predisposes individuals to PCOS.
We confirmed the progressive downregulation of hsa-miR-24-2-5p and hsa-miR-33a-5p across the NP→NB→PC continuum. Mechanistically, these miRNAs are well-positioned to drive this transition. hsa-miR-33a-5p, a known regulator of cholesterol metabolism via SREBP-2 and ABCA1, connects directly to the dyslipidemia common in both phlegm-damp constitution and PCOS.13,14 Therefore, the downregulation of hsa-miR-33a-5p that we observed in our data provides a direct mechanistic explanation for the metabolic disturbances that characterize the phlegm-damp constitution-to-PCOS transition. Meanwhile, hsa-miR-24-2-5p, a regulator of cell cycle and apoptosis, could disrupt granulosa cell dynamics, a central feature of PCOS pathology.15 Consequently, the reduced expression of hsa-miR-24-2-5p identified in our study offers a direct molecular link to the follicular developmental arrest and anovulation seen in PCOS patients. This is because, based on previous studies, hsa-miR-24-2-5p is known to function as a regulator of cell cycle and apoptosis, processes that are critical for normal follicular development. The coordinated dysregulation thus provides a compelling molecular link between constitutional predisposition and disease state.
Our functional enrichment analysis supports this, highlighting the coordinated dysregulation of key signaling pathways. Specifically, aberrant activation of MAPK and Wnt pathways can explain disruptions in follicular development and chronic inflammation,16,17 while dysregulation of AMPK and cGMP/PKG pathways points to underlying metabolic and energy imbalances.18,19 These findings align with TCM theories of phlegm-dampness and provide a novel molecular framework for PCOS development.
Despite these novel findings, our study has limitations. The small sample size of the discovery cohort and cross-sectional design mean our results are preliminary and require validation in larger, longitudinal studies to establish causality. Furthermore, our focus on granulosa cells may not capture the full systemic nature of the phlegm-damp constitution-PCOS transition.
Conclusion
This study offers a powerful mechanistic framework for the TCM “constitution-disease correlation” theory in PCOS. Our findings reveal a remarkable congruence in miRNA expression patterns between individuals with a phlegm-damp constitution and PCOS patients, particularly within neuro-endocrine-metabolic pathways, providing molecular validation for conceptualizing the phlegm-damp constitution as a “fertile soil” conducive to the development of PCOS. Furthermore, we identified a progressive decline in the expression of hsa-miR-24-2-5p, a regulator of the cell cycle, and hsa-miR-33a-5p, a modulator of cholesterol metabolism, across the continuum from a neutral constitution to a phlegm-damp constitution and finally to PCOS. This positions these miRNAs as both potential biomarkers and mechanistic mediators of the disease transition. Finally, our network analysis highlights aberrant activation of the MAPK and Wnt signaling pathways as critical molecular drivers in this process. While these preliminary findings require further validation in larger longitudinal cohorts, they identify new targets for early prediction and personalized prevention of PCOS. Future studies are warranted to confirm their predictive value in a clinical setting.
Data Sharing Statement
The sequencing raw data from this study has been archived at the NCBI Sequence Read Archive [PRJNA: 1296788] (https://www.ncbi.nlm.nih.gov/sra/PRJNA1296788, release date 31 Jan. 2028).
Ethical Approval
This study was approved by the Research Ethics Committee of the 960th Hospital of the People’s Liberation Army (Approval No. 2025-015). The Study complies with the Declaration of Helsinki.
Informed Consent
Written informed consent was obtained from all individual participants included in the study. Participants were informed of the study’s purpose, procedures, potential risks, and benefits, and their right to withdraw at any time without consequence.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors affirm that this research was conducted without any commercial or financial affiliations that could be perceived as potential conflicts of interest.
References
1. Wang J, Gui R, Li Y, et al. SFRP4 contributes to insulin resistance-induced polycystic ovary syndrome by triggering ovarian granulosa cell hyperandrogenism and apoptosis through the nuclear β-catenin/IL-6 signaling axis. Biochim Biophys Acta Mol Cell Res. 2024;1871(7):119822. doi:10.1016/j.bbamcr.2024.119822
2. Wang Q. Traditional Chinese medicine constitution decoding life sciences by applying complex systematic and scientific thinking. J Beijing Univ Trad Chin Med. 2023;46(07):889–896.
3. Watanabe M. Twin study method: unlocking genetic and environmental interactions. Endocr J. 2025;72(10):1061–1068. doi:10.1507/endocrj.EJ25-0188
4. Amin MMJ, Trevelyan CJ, Turner NA. MicroRNA-214 in health and disease. Cells. 2021;10(12):3274. doi:10.3390/cells10123274
5. Zorlu U, Turgay B, Yılmazer-Zorlu SN, Halilzade Mİ, Halilzade İ, Yalçın HR. Investigation of the relationship between clinical features and the sonographic appearance of the ovaries with the delta neutrophil index in polycystic ovary syndrome. JBRA Assist Reprod. 2025;29(2):333–337. doi:10.5935/1518-0557.20250014
6. Nasser JS, Altahoo N, Almosawi S, Alhermi A, Butler AE. The role of MicroRNA, long non-coding RNA and circular RNA in the pathogenesis of polycystic ovary syndrome: a literature review. Int J Mol Sci. 2024;25(2):903. doi:10.3390/ijms25020903
7. Chen B, Xu P, Wang J, Zhang C. The role of MiRNA in polycystic ovary syndrome (PCOS). Gene. 2019;706:91–96. doi:10.1016/j.gene.2019.04.082
8. Huang YC, Zhou JQ, Guo SS, Wang ZY, Zhu XH, Ren SC. Objective molecular diagnostic model integrating serum metabolites and immunocytokines for Phlegm-Dampness constitution. Pharmacol Clin Chin Materia Medica. 2021;37(06):133–138.
9. Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod. 2004;19(1):41–47. doi:10.1093/humrep/deh098
10. China Association of Chinese Medicine. Classification and determination of constitution in traditional Chinese medicine. World J Integrat Trad Western Med. 2009;4(4):303–304.
11. Yang X, Wang Q, Wang Y, et al. LRH-1 high expression in the ovarian granulosa cells of PCOS patients. Endocrine. 2021;74(2):413–420. doi:10.1007/s12020-021-02774-2
12. Wang Q. A new perspective on constitution-disease relation from the perspective of pathogenesis. Tianjin J Trad Chin Med. 2019;36(01):7–12.
13. Marquart TJ, Allen RM, Ory DS, Baldán A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc Natl Acad Sci USA. 2010;107(27):12228–12232. doi:10.1073/pnas.1005191107
14. Huang K, Pitman M, Oladosu O, et al. The impact of MiR-33a-5p inhibition in pro-inflammatory endothelial cells. Diseases. 2023;11(3):88. doi:10.3390/diseases11030088
15. Mukherjee S, Shelar B, Krishna S. Versatile role of miR-24/24-1*/24-2* expression in cancer and other human diseases. Am J Transl Res. 2022;14(1):20–54.
16. Zhao H, Dinh TH, Wang Y, Yang Y. The roles of MAPK signaling pathway in ovarian folliculogenesis. J Ovarian Res. 2025;18(1):152. doi:10.1186/s13048-025-01737-9
17. Wu XQ, Wang YQ, Xu SM, et al. The WNT/β-catenin signaling pathway may be involved in granulosa cell apoptosis from patients with PCOS in North China. J Gynecol Obstet Hum Reprod. 2017;46(1):93–99. doi:10.1016/j.jgyn.2015.08.013
18. Liu M, Guo S, Li X, et al. Semaglutide alleviates ovary inflammation via the AMPK/SIRT1/NF‑κB signaling pathway in polycystic ovary syndrome mice. Drug Des Devel Ther. 2024;18:3925–3938. doi:10.2147/DDDT.S484531
19. Tian Y, Heng D, Xu K, et al. cGMP/PKG-I pathway-mediated GLUT1/4 regulation by NO in female rat granulosa cells. Endocrinology. 2018;159(2):1147–1158. doi:10.1210/en.2017-00863
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.