Back to Archived Journals » Research and Reports in Biochemistry » Volume 4
Fragment-based drug discovery and protein–protein interactions
Authors Turnbull A, Boyd S, Walse B
Received 7 April 2014
Accepted for publication 20 May 2014
Published 18 September 2014 Volume 2014:4 Pages 13—26
DOI https://doi.org/10.2147/RRBC.S28428
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Andrew P Turnbull,1 Susan M Boyd,2 Björn Walse3
1CRT Discovery Laboratories, Department of Biological Sciences, Birkbeck, University of London, London, UK; 2IOTA Pharmaceuticals Ltd, Cambridge, UK; 3SARomics Biostructures AB, Lund, Sweden
Abstract: Protein–protein interactions (PPIs) are involved in many biological processes, with an estimated 400,000 PPIs within the human proteome. There is significant interest in exploiting the relatively unexplored potential of these interactions in drug discovery, driven by the need to find new therapeutic targets. Compared with classical drug discovery against targets with well-defined binding sites, developing small-molecule inhibitors against PPIs where the contact surfaces are frequently more extensive and comparatively flat, with most of the binding energy localized in “hot spots”, has proven far more challenging. However, despite the difficulties associated with targeting PPIs, important progress has been made in recent years with fragment-based drug discovery playing a pivotal role in improving their tractability. Computational and empirical approaches can be used to identify hot-spot regions and assess the druggability and ligandability of new targets, whilst fragment screening campaigns can detect low-affinity fragments that either directly or indirectly perturb the PPI. Once fragment hits have been identified and confirmed using biochemical and biophysical approaches, three-dimensional structural data derived from nuclear magnetic resonance or X-ray crystallography can be used to drive medicinal chemistry efforts towards the development of more potent inhibitors. A small-scale comparison presented in this review of “standard” fragments with those targeting PPIs has revealed that the latter tend to be larger, be more lipophilic, and contain more polar (acid/base) functionality, whereas three-dimensional descriptor data indicate that there is little difference in their three-dimensional character. These physiochemical properties can potentially be exploited in the rational design of PPI-specific fragment libraries and correlate well with optimized PPI inhibitors, which tend to have properties outside currently accepted guidelines for drug-likeness. Several examples of small-molecule PPI inhibitors derived from fragment-based drug discovery now exist and are described in this review, including navitoclax, a novel Bcl-2 family inhibitor which has entered Phase II clinical trials in patients with small-cell lung cancer and chronic lymphocytic leukemia.
Keywords: hot spot, druggability, ligandability
Introduction
Protein–protein interactions (PPIs) play major roles in many biological processes including signal transduction and the regulation of cellular function.1 As a result, PPIs represent attractive targets for therapeutic intervention in a range of diseases. However, the goal of developing potent and selective small-molecule modulators of PPIs is extremely challenging since PPI inhibitors tend to have properties that distinguish them from drugs acting against more classical targets such as kinases. PPI inhibitors can be subdivided into two main classes: orthosteric inhibitors, which directly interfere with the interface; and allosteric inhibitors, which bind away from the interface and cause or prevent conformational changes that perturb complex formation or engage transient pockets that are not present in the uncomplexed structures (Figure 1).2 Conventional tools and methodologies used in drug discovery have largely proven unsuccessful in identifying small-molecule inhibitors of PPIs. However, important progress has been made in recent years,3 with fragment-based drug discovery (FBDD) playing a pivotal role. Several examples of small-molecule PPI inhibitors now exist4 including the Bcl-xL inhibitor, ABT-263 (navitoclax),5 which is currently in Phase II clinical trials in patients with small-cell lung cancer and chronic lymphocytic leukemia. This review will focus on the application of FBDD to the development of potent and selective inhibitors of PPIs.
 | Figure 1 Orthosteric inhibition of a protein–protein interaction versus allosteric inhibition. |
Structural characteristics of PPIs
To date, studies on known, marketed drugs have demonstrated that only a very small number of biochemical targets have so far been exploited for their clinical application.6 Indeed, on average, only two new target classes are identified each year, representing an extremely small portion of the protein-encoding genes in the human genome. It has been estimated that there are around 400,000 PPIs within the human proteome, making this an attractive pool of largely untapped novel target matter yet to be explored.7
Of course, targeting PPIs is not necessarily straightforward. Typically, interaction sites of PPIs are larger than conventional small-molecule binding sites, often ranging in size from 800–3000 Å2 compared with a typical small-molecule binding site of 300–1000 Å2, with an average of 22 buried amino acid residues per binding partner.8 PPI interfaces may be flatter and less well-defined than standard binding sites, and the very nature of PPIs requires that often the interacting surfaces exhibit conformational plasticity in order to be able to interact with appropriate protein partners when needed. Indeed, some small molecules appear to induce conformational changes on an interacting surface, leading to the formation of a binding groove or transient pocket which was previously not observed.9 Many PPIs are driven by hydrophobicity, leading to additional challenges in identification of bioavailable small molecules directed towards disruption of such interactions, and many interfaces are comprised of non-contiguous regions of interaction, hindering mimetic design. Each residue at a PPI often contributes only a small amount to the binding energy, with typically only 5% of interface residues contributing more than 2 kcal/mol to the binding energy.10 These key interacting residues are often termed “hot spots” (Figure 2A) and frequently cluster at the center of interfaces surrounded by more polar residues that act as an energetically favorable solvent-excluding “O”-ring.11,12
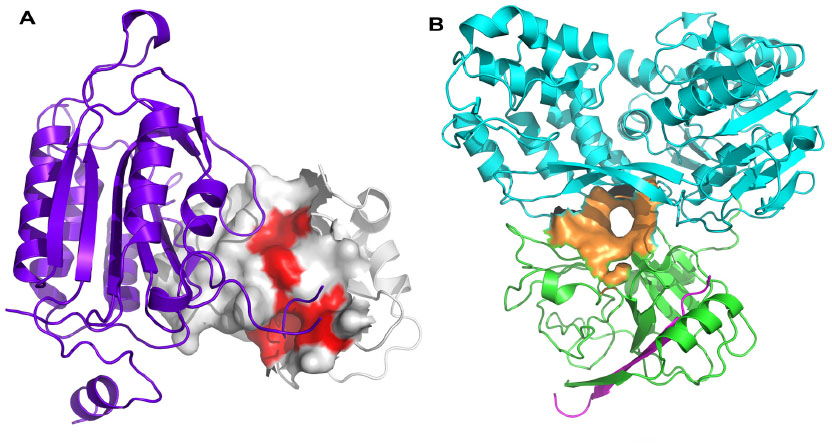 | Figure 2 Protein–protein interaction hot spots and allosteric sites. |
The “hot spot” concept
Hot spots tend to be observed on both interacting surfaces in a PPI and appear complementary to one another, forming hydrogen bonds, salt bridges, and hydrophobic interactions with the interacting partner protein. They are usually of a size which can be targeted using a small molecule (~600 Å2) and are also known to be adaptable. In many cases, a protein can interact with multiple partner proteins. Often the same hot spot is involved across the range of interactions, although it may alter its characteristics through side-chain rotameric forms, at backbone regions through movement of inter-domain hinge regions, and on interacting loops. Hot spots can be identified empirically using alanine scanning mutagenesis,13 whereby individual amino acids are systematically mutated to alanine, and the energetic contribution of the original side chain can be determined,14 or alternatively in silico alanine scanning approaches can be applied. It has been observed that hot-spot residues appear to be more structurally conserved and mutate more slowly than non-hot-spot residues of a protein.
In terms of amino acid composition, hot spots have been observed to be comparatively enriched with tryptophan, tyrosine, arginine, and isoleucine compared with other surface regions of proteins, whilst leucine, serine, threonine, and valine are found less frequently.15 This is most likely attributable to the ability of the more predominant residues to form a variety of comparatively strong interactions with protein-binding partners. Databases are available of experimentally identified hot spots, such as ASEdb,16 and also of computationally flagged regions likely to act as hot spots, such as HotRegion11 (http://prism.ccbb.ku.edu.tr/hotregion) and FTMap17 (http://ftmap.bu.edu). It has been estimated that only 9.5% of interfacial residues of PPIs are located in hot-spot regions.18 Fragment screening – both experimental and in silico – can be an appropriate approach to help identify hot-spot regions on protein surfaces.
Allosteric sites
Whilst many PPIs are targeted using a direct approach, where one protein partner is essentially mimicked by either a small molecule or a peptide-like construct, allosteric sites can sometimes provide an alternative strategy for targeting the PPI, particularly for cell-surface receptors. A compound which interacts at an allosteric site can effect conformational changes in the protein, which modulates its activity at the PPI-coupling site (Figure 1). The allosteric approach can offer some advantages over the direct approach. Because an allosteric site is saturatable, overdosing with a modulator is less likely. Allosteric modulators can be directed towards specific tissues by utilizing the physiological properties of the ligand and have a greater chance of presenting a favorable selectivity profile towards related receptors. Furthermore, allosteric sites are often more “typical” small-molecule binding sites than are the normal PPI interfaces.19 Identification of allosteric sites is often challenging. Sometimes sites are identified with the availability of a crystal structure, using phage display, tethering, or high-throughput or fragment screening combined with in silico docking and molecular dynamics (MD) approaches. There are now a number of successful examples of the targeting of allosteric sites using FBDD approaches, including hepatitis C virus (HCV) NS3 (Figure 2B) (see Case studies section).20
Computational approaches
Computational approaches can be applied to assist FBDD of PPI inhibitors from several angles. Initial studies may be employed to aid identification of suitable sites to target through small-molecule approaches, whilst automated docking and scoring approaches can be used to help predict or rationalize fragment binding and fragment elaboration. Such studies can also be useful to assist druggability/ligandability assessment of new targets. It has been noted that small molecules can sometimes mimic portions of a protein partner in a PPI, and that this can assist in the design of new inhibitors.21,22 Fragments also have the potential to act as mimics of small parts of a protein partner, and these approaches could similarly be applied to aid the FBDD process for PPIs.
Site identification
Many methods are available to identify clefts on proteins (eg, LigSite,23 Putative Active Sites with Spheres [PASS],24 and Fpocket25), but these rely on a reasonably accurate static conformation of the active protein surface, which is not always available for PPIs. Due to the inherent plasticity of many PPIs, often the simple “snapshot” of the interface provided by an X-ray crystallographic study will not give an accurate picture of how the protein surfaces may change on binding alternative protein partners or when the ligand-induced fit is affected by a small-molecule binding.
MD simulations can be used to predict various energetically favorable conformational states of the protein surfaces, for unbound surfaces or with binding partners. Studies of bound and unbound pairs of protein complexes for which three-dimensional structural information is available have suggested that in around half of the complexes, the unbound state at the interacting surface is perturbed to an observed bound state during MD simulation.26
Whilst MD can be useful in exploring local conformational variation in proteins, normal-mode analysis (NMA) is a more appropriate approach for prediction of bulk protein movement.27 NMA represents each amino acid as a bead, with proximal beads in the structure being computationally represented as springs. Solving a matrix version of Newton’s second law of motion allows derivation of the protein’s normal modes (and their associated frequencies), which describe the motion of the protein. The elNemo webserver (http://www.igs.cnrs-mrs.fr/elnemo/) will compute the low frequency normal modes of an input protein, which describe its overall large-scale movement. NMA does not, however, handle transitions between local minima, which could be relevant for PPIs. In such cases, hybrid MD/NMA approaches can be applied.28
Evolutionary trace analysis combines both sequence and structural data to locate biologically active sites on proteins.29 This bioinformatics approach uses sequence alignment data to identify conserved and variable residues across protein families, which are then mapped onto structural data.
Hot-spot detection
As described previously, identification of hot-spot regions can be pivotal to the success of PPI approaches and has been employed as a strategy in the design of PPI inhibitors.30 Computational prediction can take the form of support vector machine–based approaches,31 MD,32 in silico alanine scanning using molecular mechanics/Poisson Boltzmann surface area (MM/PBSA)33 or MD methods,34 and atom-based component analysis calculations to predict the individual contributions of amino acid residues to binding energy.35 Regression approaches using physicochemical properties including descriptors of protein flexibility have also been reported.36 DrugScorePPI (http://cpclab.uni-duesseldorf.de/dsppi) is a knowledge-based scoring function for in silico alanine scanning for hot-spot prediction in PPIs,37 and the database HotRegion (http://prism.ccbb.ku.edu.tr/hotregion) collects computationally predicted hot spots and highlights interface residues that are functionally and structurally important.11 The FTMap server (http://ftmap.bu.edu) identifies druggable hot spots of proteins using Fourier domain correlation techniques.17 Furthermore, HSPred31 (http://bioinf.cs.ucl.ac.uk/hspred) and Dr PIAS38 (Druggable Protein–protein Interaction Assessment System; http://www.drpias.net) represent systems for assessing the druggability of PPIs. In addition, 2P2Idb (http://2p2idb.cnrs-mrs.fr) is a hand-curated database dedicated to the structure of PPI complexes with known small-molecule inhibitors.39
Docking
Docking approaches can be applied against PPI targets, but the challenges of protein flexibility and dealing with solvent effects are usually more significant than for other structurally validated protein targets.
The comparatively flat surfaces of a PPI have fewer implicit steric constraints than do more standard binding-site cavities, and docking success will require more rigorous treatment of electrostatic and solvation effects. Some success has been reported with incorporating implicit solvation models into some docking algorithms,40 and more recent developments have included performing a multistep dock whereby initial docking results are re-scored using MM/PBSA approaches to increase the efficiency of the approach.41
Protein flexibility can currently only be handled in a limited manner, by using multiple input conformations of the protein (ensemble docking) or by allowing some limited movement of amino acid side chains during docking. MD can be useful in helping to identify a variety of protein conformations ahead of docking.
Pharmacophore approaches
In a similar manner to docking approaches, three-dimensional pharmacophore methods have also been used to assist the FBDD process, and of course, these approaches can be similarly used against PPI targets.42 A pharmacophore can be derived from the key binding interactions of known ligand or fragment-bound structures to help select a set of fragments for screening. An example (non-PPI) is the case of PARP-1, where a pharmacophore was derived from available X-ray data and was used to query a virtual database of fragments. The 8,000 hits from this exercise were then docked, leading to a subset of only 14 fragments being selected for biological testing, seven of which were active.43
Physicochemical properties
Whilst standard fragment libraries are now typically designed to the well-known “rule-of-3” physicochemical property criteria,44 little work has been reported to date on whether design of fragment sets to target PPIs should be modified to take account of the differing properties of these interfaces compared with typical enzyme-binding sites. However, a number of vendors offer focused libraries based on the properties of known PPI inhibitors, including Asinex Ltd. (Moscow, Russia) (11,000 compounds), Otava Ltd. (Vaughan, Canada) (IPPI Tree™: 1,300 compounds; iPPI Analogs™: 1,000 compounds), and Life Chemicals Inc. (Niagara-on-the-Lake, Canada) (“rule-of-4”: 4,300 compounds). In addition, Asinex Ltd. provides a specific PPI fragment library, which deviates from the “rule-of-3” criteria commonly used to prepare standard fragment libraries, comprising 1,200 compounds (mean molecular weight (MWT) =356 Da, mean cLogP =2.9, mean rotatable bond count =5.3, mean topological polar surface area (TPSA) =62 Å2, mean normalized principal moments of inertia ratios45 (NPR1 =0.26 and NPR2 =0.88)). Generally, these fragments include multiple hydrophobic cores, and both acidic and basic moieties have been added to increase solubility (acidic moieties containing COOH =55%, basic moieties containing basic N =24%).
The recently published “rule-of-4” would suggest that PPI inhibitors are typically larger and more lipophilic than inhibitors of more standard binding sites,46 and the key differences between PPI inhibitor space and non-PPI inhibitor space have been well documented in the literature,47 which is consistent with the observation that PPIs themselves are often more extensive and flatter than the well-defined, concave binding sites of most tractable protein targets. But does this paradigm extend to the fragment level, when fragments are used as the starting point for leads for a PPI target? It has been thought that increased three-dimensionality should be an advantage for PPI fragment libraries48 since druggable PPIs will usually either contain hot-spot regions which are nicely concave, or contain regions which involve interactions between helices, and both such situations could benefit from shape complexity in any fragment libraries used as an initial screen. To explore this assumption, a dataset of 100 fragments orthosterically active against PPI targets (a subset of data provided by LR Vidler and N Brown, Institute of Cancer Research, London, UK; unpublished data, August 2013) was compared with a set of 100 fragments which proved active against non-PPI targets (selected from the described larger set of 145 fragments by choosing inhibitors of active sites as being known not to act through a PPI mechanism)49 to examine how the physicochemical and shape properties of these two fragment sets may differ. For both the PPI and the non-PPI (“standard”) fragment datasets, a series of descriptors was calculated using the MOE modeling suite. Descriptors computed included MWT, logP, TPSA, and rotatable bond count, as well as descriptors of three-dimensional character including NPRs and molecular globularity. Functionality characteristics were also explored, including the percentage of acid-containing fragments, the percentage of base-containing fragments, and the proportion of hydrophobic atoms per molecule – defined as (hydrophobic atom count)/(heavy atom count). Ligand efficiency (LE) and lipophilic LE (LLE) were also examined.
From the data collected (Figure 3), it can be seen that the PPI fragments appear to be a little larger and more lipophilic than the standard fragments, but the three-dimensional descriptor data indicate that there is little difference in the three-dimensional character of the two datasets. LE was observed to be very similar for the PPI and standard sets, and given the increased MWT of the PPI fragments, this would suggest that the PPI fragments often bind their target with higher affinity than their standard counterparts, and this may be due to PPI fragments seeking out hot spots on the PPIs. Interestingly, a study of the functionality of the two fragment sets indicated that the PPI fragments had around double the number of acid- and base-containing fragments than the standard sets, and that the PPI fragments also contained a higher proportion of hydrophobic atoms than the standard set.
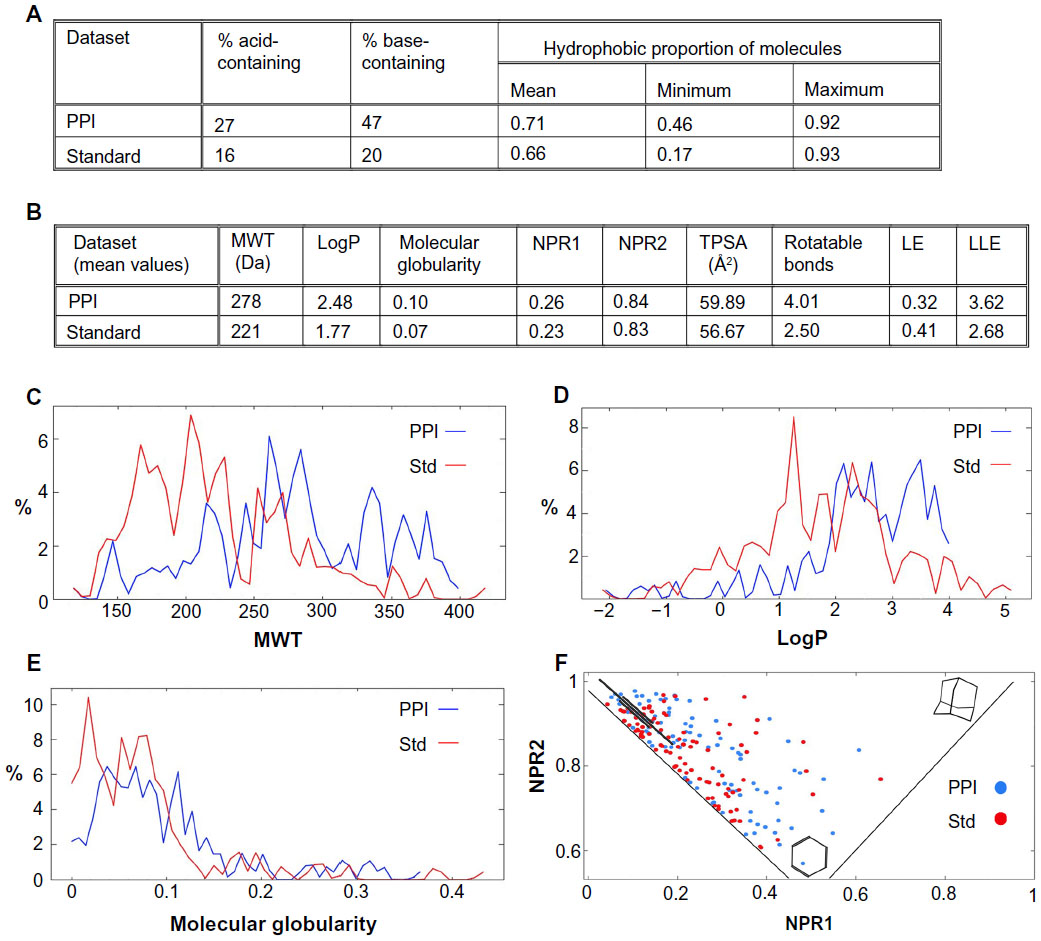 | Figure 3 Summary of selected properties and characteristics of fragment hits against PPI targets compared with fragment hits against typical enzyme targets (Std). |
The high frequency of acid and base moieties increases the solubility of fragments with a high proportion of hydrophobic atoms and additionally contributes to an increase in affinity. It has been shown that the most efficient protein−ligand complexes contain ligands that have one or more charge−charge interactions.50 Furthermore, desolvation of charged molecules is a barrier to binding, which can decrease LE for charged fragments that bind to deep pockets with bound water molecules. Therefore, the nature of PPI surfaces with only weakly bound waters on the surface could be more suitable for binding charged fragments.
So, whilst the data explored for this small study is quite limited, the output may nevertheless suggest that for the design of PPI fragment libraries, it could be useful to select slightly larger, more lipophilic fragments than for non-PPI fragment sets, and those which contain more polar (acid/base) functionality. Other groups have indicated that they are currently working on similar studies which encompass larger datasets than those publicly available at present. We await their findings with interest.
PPI screening
A significant challenge to targeting PPIs using FBDD is the ability to reliably detect low affinity (typically high μM to low mM) fragment binders.51 High-throughput screening techniques are required to first detect fragment binders in a primary screening phase followed by a hit validation phase to confirm and characterize the interactions (Table 1). Primary screening techniques include protein- and ligand-observed nuclear magnetic resonance (NMR), X-ray crystallography, surface plasmon resonance, thermal shift assay (TSA), and biochemical assays. Notably, two-dimensional heteronuclear single quantum coherence (HSQC) NMR and X-ray crystallography, employed as frontline approaches at Abbott Laboratories (Abbott Park, IL, USA) and Astex Pharmaceuticals (Cambridge, UK), respectively, deliver detailed information about fragment-binding modes at the earliest possible stage in the fragment-screening campaign. Biochemical assays offer the advantage of being the highest throughput primary screening techniques and are capable of directly detecting fragments that perturb PPIs through proximity assays such as fluorescence resonance energy transfer (FRET) and AlphaScreen® (PerkinElmer Inc., Waltham, MA, USA), or assays reporting the size of species in solution, such as fluorescence polarization (FP) assays. In addition, other biophysical methods including capillary electrophoresis,52 target immobilized NMR screening,53 and microscale thermophoresis (NanoTemper Technologies GmbH, München, Germany)54 have also been used to successfully prosecute PPI targets. Primary screens, with the exception of 2D-HSQC NMR and X-ray crystallography, are prone to false positives. Hence, it is essential that hits identified in the primary screen or using a structure-based virtual screening approach are subsequently cross-validated using orthogonal screening techniques. Several screening methods are capable of being run as competition assays to identify fragment hits that perturb the PPI. Once fragment hits have been confirmed, it is usual to determine structural information relating to their binding modes using NMR or X-ray crystallography, which is subsequently used to drive medicinal chemistry efforts towards the development of more potent and selective inhibitors.
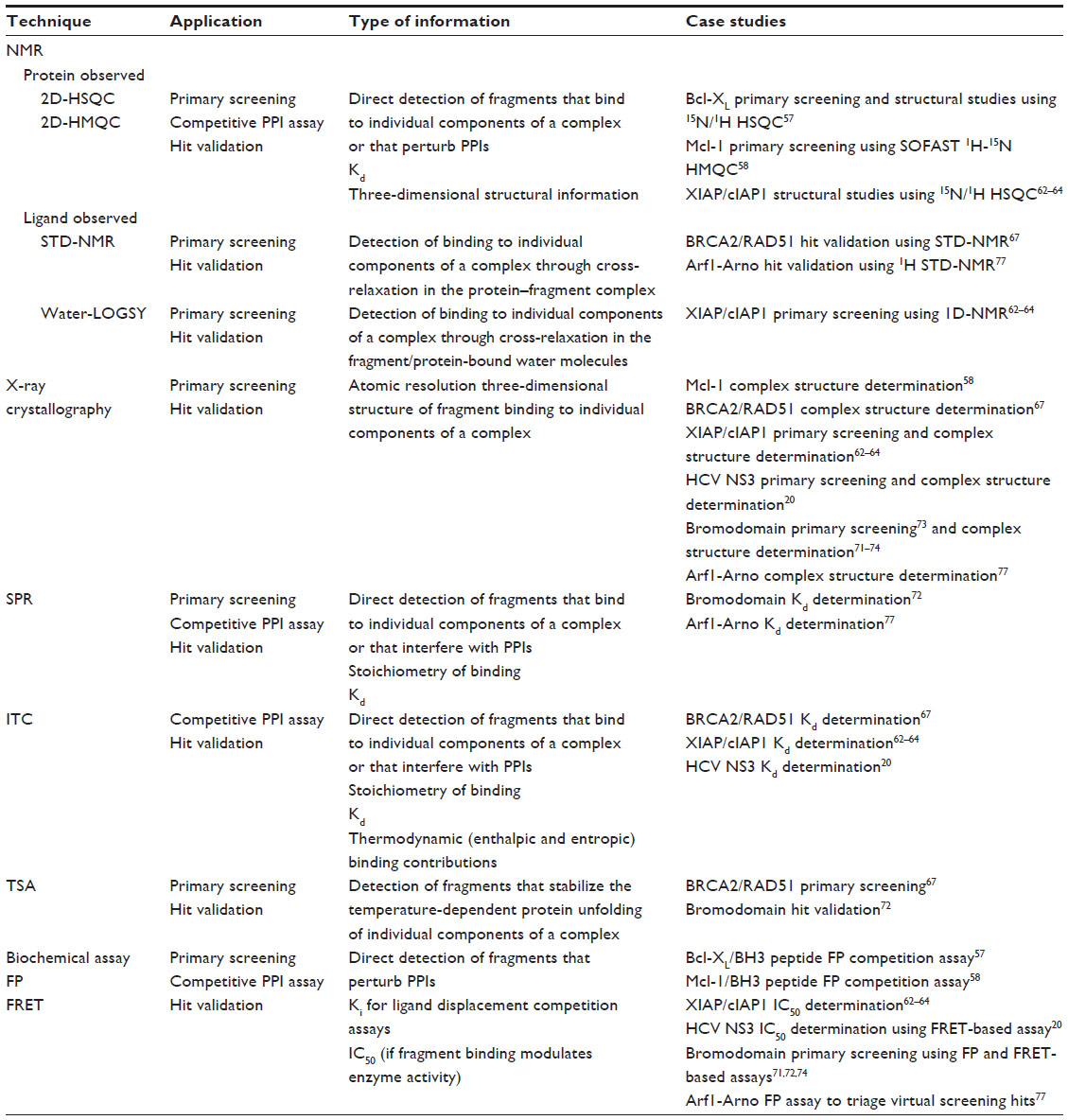 | Table 1 Major screening and validation techniques used to detect PPI fragment hits |
Case studies
FBDD techniques have been applied successfully to develop small-molecule inhibitors targeting a number of PPIs, with representative examples described in this section. This is not an exhaustive list, and other notable examples, such as the use of X-ray techniques with FBDD in the discovery of HIV-1/LEDGF inhibitors55 are not described in detail in the interests of brevity.
Bcl-xL
The anti-apoptotic protein Bcl-xL is a member of the B-cell lymphoma 2 (Bcl-2) family and represents an attractive target for the development of anticancer agents. Bcl-xL has a deep hydrophobic groove on its surface, which acts as the binding site for a key α-helix on other Bcl-2 proteins such as Bak. Abbott Laboratories carried out a fragment screening campaign using the structure-activity relationship (SAR) by NMR approach56 to identify potent inhibitors that bind to the hydrophobic groove of Bcl-xL and disrupt this PPI (Table 2).57 Initially, a screen of 10,000 fragments using 15N-HSQC NMR identified fluoro-biaryl acid (Table 2, fragment 1) as a fragment hit (binding affinity [Kd] =300±30 μM). NMR structural analysis showed that this fragment binds at the center of the hydrophobic groove, with a proximal second-site hot spot available. A second-site NMR screen of 3,500 fragments identified a naphthyl analogue (Table 2, fragment 2) (Kd =4,300±1600 μM) that bound simultaneously to Bcl-xL in the presence of fragment 1. The NMR structure of the ternary complex of Bcl-xL with fragments 1 and 2 was used to guide the development of an acylsulfonamide linker to merge the two fragments into a single lead molecule (Bcl-xL FP assay inhibition constant [Ki] =36±2 nM). Subsequent intense synthetic chemistry efforts led to the development of the highly potent (Bcl-xL FP assay Ki <5 nM) inhibitor ABT-263 (navitoclax) (Table 2, compound 3), which has entered Phase II clinical trials in patients with small-cell lung cancer and B-cell malignancies.5 Navitoclax is larger and more liphophilic than non-PPI inhibitors, violating three “rule-of-5” criteria (MWT =975 Da; cLogP =12; number of hydrogen bond acceptors =12).
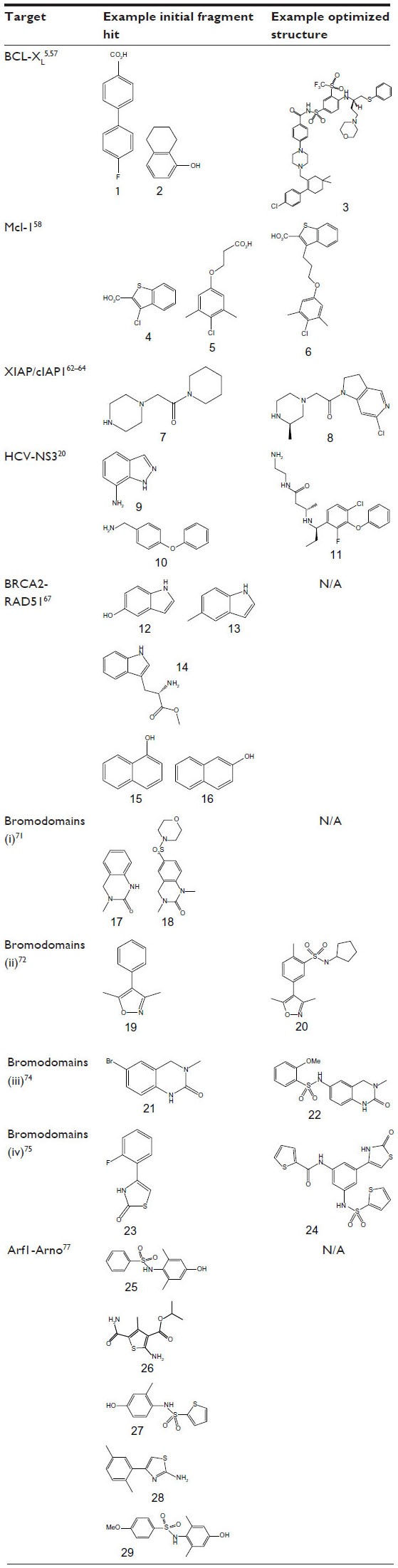 | Table 2 Initial fragment hits and current leads for protein–protein interaction case studies |
Mcl-1
Overexpression of another member of the Bcl-2 family, Mcl-1, is associated with resistance to ABT-263 (navitoclax) and a number of other anticancer therapies. Mcl-1 exerts its activity through PPIs involving a large binding surface, and direct inhibition of BH3-containing peptides from binding to Mcl-1 represents an effective point of intervention for the development of anticancer therapeutics.
Recently, a combination of fragment-based methods and structure-based design was applied to target Mcl-1.58 A curated library comprising in excess of 13,800 fragments was screened using the SAR by NMR approach56 in pools of 12 compounds. Deconvolution of these mixtures yielded 132 hits, with more than a quarter demonstrating an LE greater than 0.25. Ki values were determined by measuring the disruption of the interaction between Mcl-1- and BH3-containing peptides using a competition FP assay. Based on affinity and chemical tractability, two fragment classes (eg, FP assay Ki =131 μM, LE =0.33 [Table 2, fragment 4] and FP assay Ki =60 μM, LE =0.26 [Table 2, fragment 5]) were selected and pursued further. Interestingly, both classes comprised carboxylic acids attached to a 6,5-fused heterocyclic system or tethered through a linker to a hydrophobic aromatic system. NMR studies were applied to elucidate the binding mode of these fragments using NOE (Nuclear Overhauser Effect) distance information combined with docking. These studies showed that the fragments bound to non-overlapping but similar regions on Mcl-1, and attempts to merge them were initiated. A small library of compounds with various linker lengths was put together (particularly four-atom linked compounds) and led to a significant increase in potency. Further SAR elaboration of the linked fragments resulted in a dramatic increase in potency, yielding nanomolar lead molecules with increased selectivity over Bcl-XL and Bcl-2 (eg, FP assay Ki =0.32 μM; LE =0.25; Table 2, compound 6). Subsequent structural analysis of selected compounds using X-ray crystallography explained the SAR and has suggested new routes for the development of clinically useful Mcl-1-selective inhibitors.
XIAP/cIAP1
Inhibitors of apoptosis (IAP) protein-family members are frequently overexpressed in cancer and promote tumor cell survival and proliferation, making them attractive targets for cancer therapy.59,60 IAP proteins possess one or more baculoviral IAP repeat (BIR) domains – a PPI domain that is essential for the anti-apoptopic activity of most IAPs. BIR domains typically contain a surface peptide groove capable of specifically interacting with the N-terminus of a number of important apoptopic proteins such as caspases (Figure 2A), which contributes to oncogenesis and resistance to therapy. This interaction can be inhibited by the “AVPI” tetrapeptide binding motif on the N-terminus of Smac (second mitochondrial activator of caspases), an endogenous inhibitor of IAPs.61 The interaction between Smac and the BIR domain of IAPs provides an ideal site for the design of small-molecule inhibitors. Several peptidomimetic IAP antagonists are currently in clinical trials as new anticancer agents including birinapant (TetraLogic Pharmaceuticals, Malvern, PA, USA), which has entered Phase II clinical trials for both solid tumors and hematological malignancies. Recently, Astex Pharmaceuticals used FBDD to identify novel non-peptidomimetic IAP antagonists against XIAP and cIAP1.62–64 The Astex fragment library (~1,100 compounds) and a targeted set (~100 compounds) were screened against the XIAP-BIR3 domain using X-ray crystallography and 1D-NMR. Hits were further evaluated using X-ray crystallography and two-dimensional NMR to determine their precise binding modes, and with isothermal titration calorimetry (ITC) and an FP assay to determine their binding affinities. Initial fragment hits were found to bind very weakly to cIAP1-BIR3 and XIAP-BIR3 with mM potencies (XIAP-BIR3 half maximal inhibitory concentration [IC50] >5 mM cIAP1-BIR3 IC50 >5 mM) (Table 2, fragment 7). Subsequently, the X-ray structures of the fragment hits were used to guide fragment optimization, leading to more potent structures (eg, Table 2, compound 8), which were developed to a series of dual XIAP and cIAP1 antagonists with nM potency (XIAP-BIR3 IC50 <40 nM; cIAP1-BIR3 IC50 <10 nM; structures undisclosed).
HCV NS3
The HCV NS3 protein is a bifunctional enzyme comprising an N-terminal serine protease domain and C-terminal helicase domain, with both domains capable of interacting and affecting each other’s activities. NS3 has been the subject of intensive research for the development of HCV treatments, with two inhibitors targeting the N-terminal protease domain – telaprevir (Vertex Pharmaceuticals Inc., Boston, MA, USA) and boceprevir (Merck and Co., Inc., Whitehouse Station, NJ, USA) – gaining FDA approval in 2011. Recently, Astex Pharmaceuticals carried out a screening campaign against full length NS3 to identify drug leads with a different chemical and biological profile compared with the protease inhibitors.20 An X-ray fragment screen was performed using crystals of full length NS3 in complex with the peptide cofactor NS4a.65 The high sensitivity of X-ray crystallography facilitated the detection of very weak fragment hits with acceptable LE. Of the 176 fragments screened, 16 bound to a novel site located at the interface between the two domains, inhibiting the protease activity via an allosteric mechanism (Figure 2B). Subsequently, structure-based optimization was used to elaborate the initial low affinity fragment hits, including fragment 9 in Table 2 (NS3–NS4a FRET-based protease assay IC50 >5 mM; LE <0.3) and fragment 10 in Table 2 (IC50 ~500 μM; LE =0.3) into a potent chemical lead compound (compound 11 in Table 2) (IC50 <0.01 μM; LE ~0.39) that stabilizes an auto-inhibited conformation by binding at the allosteric site. The discovery of this allosteric site on NS3 may facilitate the development of a novel class of potent HCV antiviral agents.
BRCA2-RAD51
The tumor suppressor BRCA2 binds to the recombinase RAD51 via eight conserved BRC repeats. The BRC repeat–RAD51 interaction is essential for DNA repair and disruption of this interaction is likely to sensitize tumor cells to DNA damaging agents. The crystal structure of human RAD51 in complex with one of the BRC repeats, BRC4 (Protein Data Bank code =1N0W), revealed that BRC4 uses an evolutionary conserved “FXXA” motif to bind to RAD51.66 The hot spot for this interaction comprises small, well defined pockets for the phenylalanine and alanine side chains of this conserved motif.
A fragment-screening campaign using an array of biophysical approaches (TSA, ITC, and saturation transfer difference–NMR [STD–NMR]) and X-ray crystallography was run to target the interaction site of the BRC4 repeat of human BRCA2 on RAD51.67 Attempts to generate stable, unpartnered RAD51 with an exposed “FXXA” binding region were unsuccessful. Therefore, the RAD51 ortholog, Pyrococcus furiosus RadA, which could be produced in a suitable form for fragment screening was used as a surrogate system. Humanized RadA was screened against a set of 1,249 fragments using TSA, which led to the identification of two top hits (Table 2, fragments 12 and 13) sharing a common indole core. ITC experiments determined that both these fragments bind with a Kd of 2.1 mM and X-ray analysis revealed that they bind in the phenylalanine pocket of the “FXXA” motif. Subsequently, a small library of 42 compounds designed to investigate SAR around the indole scaffold was screened using competitive STD–NMR, followed by a second screen of a 120 fragment library selected by in silico screening and inspection of commercially available analogues. STD–NMR screening led to the identification of an additional six fragment hits, including L-methyl ester tryptophan (Table 2, fragment 14) with the lowest LE (0.28) and highest LLE (2.7), and two naphthols (Table 2, fragments 15 and 16), that bind in the hot spot region with improved ( μM) potency compared with the original indole hits. Synthetic elaboration of these fragments to a lead series may result in the development of more potent molecules targeting the BRCA2-RAD51 interaction.
Bromodomains
A PPI of substantial biological interest is the molecular recognition of epigenetic marks on covalently modified histone proteins. Epigenetic reading proteins containing one or more evolutionary conserved effector modules that carry out these processes and their association with chromatin leads to transcriptional activation. Bromodomain-containing proteins are one such class of epigenetic reading proteins that recognize acetylated lysine residues (AcK) on histone tails and comprise a left-handed four-helix bundle forming a conserved hydrophobic pocket that accommodates the AcK side chain.68 The AcK pocket can bind selective small-molecule inhibitors with low nanomolar affinity,69,70 and several efforts have been pursued to identify candidate inhibitors through FBDD.71–75 In these studies, the fragment hits interact with conserved water molecules and residues by mimicking the AcK head group, whilst lipophilic parts of the fragments engage with hydrophobic side chains.
Chung et al71 used proprietary information from several cocrystal structures of bromodomain-inhibitor complexes at GlaxoSmithKline plc (Stevenage, UK) and assembled a chemically diverse focused fragment library of AcK mimetics, which was screened using an FP ligand displacement assay. Of 1,376 compounds tested, 132 showed more than 30% displacement of the fluorogenic ligand in the FP assay. Direct target engagement towards the N-terminal bromodomain of BRD2 was confirmed using X-ray crystallography, and 40 different complexes were analyzed. An interesting sulfonamide analogue (Table 2, fragment 18) to fragment hit 17 (Table 2) was identified with detectable binding and improved potency, although not sufficient for it to act as a cellular probe of bromodomain function (Table 2, bromodomains [i]); IC50 =30–40 μM against BRD2, BRD3, and BRD4, corresponding to an LE =0.25–0.27). Subsequently, isoxazole containing fragment 19 (Table 2) was selected as an alternative starting point for hit elaboration (Table 2, bromodomains [ii]).72 Whilst fragment 19 was far from the most potent fragment detected (IC50 ≈200 μM), it was less directly related to AcK. Its binding mode was investigated using X-ray crystallography in order to optimize its potency further, and a three-dimensional pharmacophore model was constructed and used to search a database of commercially available compounds. Several isoxazole analogues with sulfonamide substituents on the phenyl ring meta to the isoxazole were identified with low micromolar potency. The sulfonamide moiety introduces a bend in the molecule which promotes favorable interactions in the binding pocket. These sulfonamide substituents were further optimized using structure-based design leading to compounds (such as 20 in Table 2) showing anti-inflammatory activity in cellular assays and thus demonstrating the tractability of the bromodomain target class to FBDD and structure-based lead optimization. In addition, Gehling et al73 also describe the identification of an isoxazole-containing fragment similar to 19 (amino isoxazole), which was elaborated into a potent and selective inhibitor using structure-based design methods.
A subsequent screening campaign to identify a chemical probe towards BRD4 used a 6-brominated equivalent fragment 21 (Table 2) of 3,4-dihydro-3methyl-2(1H)-quinazolinone (Table 2, fragment 17), first identified by Chung et al,71 as an efficient starting point for FBDD.74 Elucidation of the binding mode by X-ray crystallography directed the design towards replacing the bromine in fragment 21 with sulfonamide substitutions in order to achieve a kink necessary for productive interactions with the mouth of the pocket. From a small library with reversed sulfonamide moieties, compound 22 (Table 2) with a methoxy substituted aryl group was identified, which was found to have good credentials for acting as a chemical probe for the BET-family of bromodomains (Table 2, bromodomains [iii]).
A recent study identified fragments active against BRD4 comprising new chemotypes mimicking the AcK binding moiety.75 A diverse “rule-of-3” compliant fragment library was assembled by filtering the entire ZINC database76 of commercially available compounds ready for virtual screening and cherry picking of 500 fragments. This library was subsequently docked into BRD4, and 41 fragments were selected for cocrystallization trials. The X-ray structures for nine different complexes were determined: four are described in the paper and bind in a similar position to AcK despite having different chemical characteristics. One of the fragment hits (Table 2, fragment 23) with a 2-thiazolidinone core was further optimized using a structure-based approach, and a sulfonamide group was introduced to create a kink in the compound’s shape to introduce hydrophobic moieties towards the mouth of the binding pocket. Structural information indicated that one of the sulfonamide substituents extended in another direction and, by using this information, improved analogues were designed that both reach out towards the mouth and into other areas of the pocket. Thus, the potency was improved 100-fold from 24 μM for the initial fragment hit 23 to 0.23 μM for the lead compound 24 (Table 2, bromodomains [iv]).
Arf1-Arno
Arf1 is a G-protein belonging to the ARF (ADP-ribosylation factor) family. Arf1 nucleotide exchange is regulated through the interaction with the catalytic Sec7 domain of guanine nucleotide exchange factors such as Arno. A fragment-screening campaign was established to identify fragments that interfere with the Arf1-Arno Sec7 domain interaction and inhibit GDP to GTP nucleotide exchange.77 This represents a challenging PPI target, since the Arf1–Arno interface comprises several hot spots disseminated over a surface area close to 1500 Å2.
First, virtual screening of approximately 3,000 “rule-of-3” compliant fragments from the ChemBridge library led to the selection of 33 fragments that could potentially interact with the hot spots (identified via in silico alanine scanning) on the Arno Sec7 domain binding surface. Subsequently, an FP assay established that four of these fragments (Table 2, fragments 25, 26, 27 and 28) had very strong inhibitory effects on nucleotide exchange. The binding of three of these fragments (25, 26, and 28) to the Arno Sec7 domain was confirmed by surface plasmon resonance and STD–NMR. Finally, the X-ray structures for the complexes with fragment 25 (Kiapp =3.7 mM) and a more potent analogue (Table 2, fragment 29) (Kiapp =1.6 mM) were determined, which enabled the binding mode to be visualized. Despite their low affinity, these fragment hits represent useful starting points for the development of higher affinity inhibitors targeting the Arf1–Arno interaction.
Summary
PPIs represent attractive targets for therapeutic intervention in a range of diseases. However, the goal of developing potent and selective small-molecule modulators of PPIs is extremely challenging, since PPI inhibitors tend to have properties that distinguish them from drugs acting against “standard” enzyme targets such as kinases. Important progress has been made in recent years, with FBDD playing a pivotal role, driven in part by advancements in the throughput and sensitivity of fragment-screening technologies, and computational techniques to identify hot spots. As a result, low affinity fragments binding to hot-spot regions within the protein–protein interface can be detected and subsequently elaborated into potent PPI inhibitors via medicinal chemistry. A small-scale comparison of fragments binding to “standard” and PPI targets presented in this review has revealed that the latter tend to be larger, more lipophilic, and contain more polar (acid/base) functionality, whereas three-dimensional descriptor data indicate that there is little difference in their three-dimensional character. The findings from this study and a number of similar studies encompassing larger datasets currently underway can be used to guide the design of PPI-focused fragment libraries to increase the likelihood of success for future PPI-screening campaigns. Targeting PPIs represents one of the most exciting and challenging areas of FBDD, and several small-molecule PPI inhibitors are currently in development. Notably, navitoclax (ABT-263), which was derived using an FBDD approach, has entered Phase II clinical trials for the treatment of various lymphomas and leukemias.
Acknowledgments
The PPI fragment dataset used in the analysis of the differences between PPI and non-PPI fragments was kindly provided by Dr Lewis R Vidler and Dr Nathan Brown, Institute of Cancer Research, London, UK.
Disclosure
The authors report no conflicts of interest in this work.
References
Toogood PL. Inhibition of protein–protein association by small molecules: approaches and progress. J Med Chem. 2002;45: 1543–1558. | |
Eyrisch S, Helms V. Transient pockets on protein surfaces involved in protein–protein interaction. J Med Chem. 2007;50:3457–3464. | |
Nero TL, Morton CJ, Holien JK, Wielens J, Parker MW. Oncogenic protein interfaces: small molecules, big challenges. Nat Rev Cancer. 2014;14(4):248–262. | |
Arkin MR, Wells JA. Small-molecule inhibitors of protein–protein interactions: progressing towards the dream. Nature Rev Drug Discov. 2004;3:301–317. | |
Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. | |
Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5(12):993–996. | |
Ramani AK, Bunescu RC, Mooney RJ, Marcotte EM. Consolidating the set of known human protein–protein interactions in preparation for large-scale mapping of the human interactome. Genome Biol. 2005;6(5):R40. | |
Conte LL, Chothia C, Janin J. The atomic structure of protein–protein recognition sites. J Mol Biol. 1999;285:2177–2198. | |
Arkin MR, Randal M, DeLano WL, et al. Binding of small molecules to an adaptive protein–protein interface. PNAS. 2003;100(4):1603–1608. | |
Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. | |
Cukuroglu E, Gursoy A, Keskin O. HotRegion: a database of predicted hot spot clusters. Nucleic Acids Res. 2012;40:D829–D833. | |
Winter C, Henschel A, Tuukkanen A, Schroeder M. Protein interactions in 3D: from interface evolution to drug discovery. J Struct Biol. 2012;179(3):347–358. | |
Cunningham BC, Wells JA. Minimized proteins. Curr Opin Struct Biol. 1997;7(4):457–462. | |
Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267(5196):383–386. | |
Metz A, Ciglia E, Gohlke H. Modulating protein–protein interactions: from structural determinants of binding to druggability prediction to application. Curr Pharmaceutical Design. 2012;18:4630–4647. | |
Thorn KS, Bogan AA. ASEdb: a database of alanine mutations and their effects on the free energy of binding in protein interactions mutations and their effects on the free energy of binding in protein interactions. Bioinformatics. 2001;17:284–285. | |
Brenke R, Kozakov D, Chuang G-Y, et al. Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics. 2009;25(5):621–627. | |
Moreira IS, Fernandes PA, Ramos MJ. Hot spots–a review of the protein–protein interface determinant amino-acid residues. Proteins. 2007;68:803–812. | |
Fuentes G, Oyarzabal J, Rojas AM. Databases of protein–protein interactions and their use in drug discovery. Curr Opin Drug Discov Devel. 2009;12(3):358–366. | |
Saalau-Bethell SM, Woodhead AJ, Chessari G, et al. Discovery of an allosteric mechanism for the regulation of HCV NS3 protein function. Nature Chem Biol. 2012;8:920–925. | |
Fry DC. Drug-like inhibitors of protein–protein interactions: a structural examination of effective protein mimicry. Curr Protein Pept Sci. 2008;9:240–247. | |
Fry DC. Small-molecule inhibitors of protein–protein interactions: how to mimic a protein partner. Curr Pharm Des. 2012;18:4679–4684. | |
Hendlich M, Rippmann F, Barnickel G. LIGSITE: automatic and efficient detection of potential small molecule binding sites in proteins. J Mol Graph. 1997;15:359–363. | |
Brady GP, Stouten PF. Fast prediction and visualization of protein binding pockets with PASS. J Comput Aided Mol Des. 2000;14(4):383–401. | |
Le Guilloux V, Schmidtke P, Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics. 2009;10:168. | |
Smith GR, Sternberg MJ, Bates PA. The relationship between the flexibility of proteins and their conformational states on forming protein–protein complexes with an application to protein–protein docking. J Mol Biol. 2005;347(5):1077–1101. | |
Bray JK, Weiss DR, Levitt M. Optimized torsion-angle normal modes reproduce conformational changes more accurately than cartesian modes. Biophys J. 2011;101(12):2966–2969. | |
Zhang Z, Shi Y, Liu H. Molecular dynamics simulations of peptides and proteins with amplified collective motions. Biophys J. 2003;84(6):3583–3593. | |
Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257:342–358. | |
Guo W, Wisniewski JA, Ji H. Hot spot-based design of small-molecule inhibitors for protein–protein interactions. Bioorg Med Chem Lett. 2014;24:2546–2554. | |
Lise S, Buchan D, Pontil M, Jones DT. Predictions of hot spot residues at protein–protein interfaces using support vector machines. PLoS One. 2011;6(2):e16774. | |
DeLano WL. Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struct Biol. 2002;12(1):14–20. | |
Srinivasan J, Cheatham TE, Cieplak P, Kollman PA, Case DA. Continuum solvent studies of the stability of DNA, RNA and phosphoramidate-DNA helices. J Am Chem Soc. 1998;120:9401–9409. | |
Moreira IS, Fernandes PA, Ramos MJ. Computational alanine scanning mutagenesis – an improved methodological approach. J Comput Chem. 2007;28:644–654. | |
Gohlke H, Kiel C, Case DA. Insights into protein–protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J Mol Biol. 2003;330(4):891–913. | |
Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33(Web Server issue):W382–W388. | |
Krüger DM, Gohlke H. DrugScorePPI webserver: fast and accurate in silico alanine scanning for scoring protein–protein interactions. Nucleic Acids Res. 2010;38(Web Server issue):W480–W486. | |
Sugaya N, Kanai S, Furuya T. Dr PIAS2.0: an update of a database of druggable protein–protein interactions. Database (Oxford). 2012; 2012:bas034. | |
Basse MJ, Betzi S, Bourgeas R, et al. 2P2Idb: a structural database dedicated to orthosteric modulation of protein–protein interactions. Nucleic Acid Res. 2013;41:824–827. | |
Krumrine J, Raubacher F, Brooijmans N, Kuntz I. Principles and methods of docking and ligand design. Methods Biochem Anal. 2003;44: 443–476. | |
Case DA, Cheatham TE, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688. | |
Voet A, Zhang KYJ. Pharmacophore modelling as a virtual screening tool for the discovery of small molecule protein–protein interaction inhibitors. Curr Pharm Des. 2012;18:4586–4598. | |
Singh SS, Jarp S, Narasu L. Fragment based design, docking and biological evaluation to identify novel PARP1 inhibitors and their role in cancer. Int J Comput Med Chem. 2013;1:35–43. | |
Congreve M, Carr R, Murray C, Jhoti H. A “rule of three” for fragment-based lead discovery? Drug Discov Today. 2003;8(19):876–877. | |
Sauer WH, Schwarz MK. Molecular shape diversity of combinatorial libraries: a prerequisite for broad bioactivity. J Chem Inf Comput Sci. 2003;43(3):987–1003. | |
Morelli X, Bourgeas R, Roche P. Chemical and structural lessons from recent successes in protein–protein interaction inhibition (2P2I). Curr Opin Chem Biol. 2011;15:475–481. | |
Sperandio O, Reynès CH, Camproux A-C, Villoutreix BO. Rationalizing the chemical space of protein–protein interaction inhibitors. Drug Discov Today. 2010;15:220–229. | |
Bower J, Pannifer A. Using fragment-based technologies to target protein–protein interactions. Curr Pharma Design. 2012;18: 4685–4696. | |
Ferenczy GG, Keseru GM. How are fragments optimized? A retrospective analysis of 145 fragment optimizations. J Med Chem. 2013;56: 2478–2486. | |
Smith RD, Engdahl AL, Dunbar JB Jr, Carlson HA. Biophysical limits of protein–ligand binding. J Chem Inf Model. 2012;52(8):2098–2106. | |
Syafrizayanti, Betzen C, Hoheisel JD, Kastelic D. Methods for analyzing and quantifying protein–protein interaction. Expert Rev Proteomics. 2014;11(1):107–120. | |
Pierceall WE, Zhang L, Hughes DE. Affinity capillary electrophoresis analyses of protein–protein interactions in target-directed drug discovery. Methods Mol Biol. 2004;261:187–198. | |
Vanwetswinkel S, Heetebrij RJ, van Duynhoven J, et al. TINS, target immobilized NMR screening: an efficient and sensitive method for ligand discovery. Chem Biol. 2005;12(2):207–216. | |
Jerabek-Willemsen M, Wanner TAR, Roth HM, Duhr S, Baaske P, Breitsprecher D. MicroScale Thermophoresis: interaction analysis and beyond. J Mol Struct. Epub March 16, 2014. | |
Peat TS, Rhodes DI, Vandegraaff N, et al. Small molecule inhibitors of the LEDGF site of human immunodeficiency virus integrase identified by fragment screening and structure based design. PLoS One. 2012;7(7):e40147. | |
Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274: 1531–1534. | |
Petros AM, Dinges J, Augeri DJ, et al. NMR-derived structure of the inhibitor N-(4″-fluorobiphenyl-4-ylcarbonyl)-3-nitro-4-(2-phenylsulfanylethyl amino)benzenesulfonamide bound to the antiapoptotic protein Bcl-xL. J Med Chem. 2006;49:656–663. | |
Friberg A, Vigil D, Zhao B, et al. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J Med Chem. 2013;56(1):15–30. | |
Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nature Rev Drug Disc. 2012;11:109–124. | |
Darding M, Meier P. IAPs: guardians of RIPK. Cell Death Differ. 2012;19:58–66. | |
Shiozaki EN, Shi Y. Caspases, IAPs and Smac/DIABLO: mechanisms from structural biology. Trends Biochem Sci. 2004;29:486–494. | |
Chessari G, Buck I, Coyle J, et al. Novel small molecule antagonists of XIAP, cIAP1/2 generated by fragment-based drug discovery. Poster presented at the ESH International Conference on Mechanisms of Cell Death and Disease: Advances in Therapeutic Intervention and Drug Development; October 14–18, 2010; Cascais, Portugal. | |
Chessari G, Ahn M, Buck I, et al. AT-IAP, a dual cIAP1 and XIAP antagonist with oral antitumor activity in melanoma models. Poster presented at AACR Annual Meeting; April 6–10, 2013; Washington, DC. | |
Ahn M, Ward G, Chessari G, et al. Potent, dual cIAP1/XIAP antagonists induce apoptosis in a melanoma stem cell population. Poster presented at AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Conference; October 19–23, 2013; Boston, MA. | |
Yao N, Reichert P, Taremi SS, Prosise WW, Weber PC. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure. 1999;7:1353–1363. | |
Pellegrini L, Yu DS, Lo T, et al. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420: 287–293. | |
Scott DE, Ehebauer MT, Pukala T, et al. Using a fragment-based approach to target protein–protein interactions. ChemBioChem. 2013;14:332–342. | |
Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11(5):384–400. | |
Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. | |
Nicodeme E, Jeffrey KL, Schaefer U, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468(7327):1119–1123. | |
Chung CW, Dean AW, Woolven JM, Bamborough P. Fragment-based discovery of bromodomain inhibitors part 1: inhibitor binding modes and implications for lead discovery. J Med Chem. 2012;55(2):576–586. | |
Bamborough P, Diallo H, Goodacre JD, et al. Fragment-based discovery of bromodomain inhibitors part 2: optimization of phenylisoxazole sulfonamides. J Med Chem. 2012;55(2):587–596. | |
Gehling VS, Hewitt MC, Vaswani RG, et al. Discovery, design, and optimization of isoxazole azepine BET inhibitors. ACS Med Chem Lett. 2013;4:835–840. | |
Fish PV, Filippakopoulos P, Bish G, et al. Identification of a chemical probe for bromo and extra C-terminal bromodomain inhibition through optimization of a fragment-derived hit. J Med Chem. 2012;55(22):9831–9837. | |
Zhao L, Cao D, Chen T, et al. Fragment-based drug discovery of 2-thiazolidinones as inhibitors of the histone reader BRD4 bromodomain. J Med Chem. 2013;56(10):3833–3851. | |
Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model. 2012;52(7):1757–1768. | |
Rouhana J, Hoh F, Estaran S, et al. Fragment-based identification of a locus in the Sec7 domain of Arno for the design of protein–protein interaction inhibitors. J Med Chem. 2013;56:8497–8511. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.