Back to Journals » Therapeutics and Clinical Risk Management » Volume 13
Four novel ARSA gene mutations with pathogenic impacts on metachromatic leukodystrophy: a bioinformatics approach to predict pathogenic mutations
Authors Dehghan Manshadi M, Kamalidehghan B, Aryani O, Khalili E, Dadgar S, Tondar M, Ahmadipour F, Yong Meng G, Houshmand M
Received 17 August 2016
Accepted for publication 18 January 2017
Published 16 June 2017 Volume 2017:13 Pages 725—731
DOI https://doi.org/10.2147/TCRM.S119967
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Masoumeh Dehghan Manshadi,1 Behnam Kamalidehghan,2,3 Omid Aryani,1 Elham Khalili,1 Sepideh Dadgar,1 Mahdi Tondar,4 Fatemeh Ahmadipour,5 Goh Yong Meng,6 Massoud Houshmand1,3
1Department of Medical Genetics, Special Medical Center, Tehran, Iran; 2Medical Genetics Department, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 3Department of Medical Genetics, National Institute for Genetic Engineering and Biotechnology, Tehran, Iran; 4Department of Biochemistry and Molecular & Cellular Biology, School of Medicine, Georgetown University, Washington, DC, USA; 5Department of Pharmacy, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia; 6Department of Veterinary Preclinical Sciences, Faculty of Veterinary Medicine, Universiti Putra Malaysia, Selangor, Malaysia
Abstract: Metachromatic leukodystrophy (MLD) disorder is a rare lysosomal storage disorder that leads to severe neurological symptoms and an early death. MLD occurs due to the deficiency of enzyme arylsulfatase A (ARSA) in leukocytes, and patients with MLD excrete sulfatide in their urine. In this study, the ARSA gene in 12 non-consanguineous MLD patients and 40 healthy individuals was examined using polymerase chain reaction sequencing. Furthermore, the structural and functional effects of new mutations on ARSA were analyzed using SIFT (sorting intolerant from tolerant), I-Mutant 2, and PolyPhen bioinformatics software. Here, 4 new pathogenic homozygous mutations c.585G>T, c.661T>A, c.849C>G, and c.911A>G were detected. The consequence of this study has extended the genotypic spectrum of MLD patients, paving way to a more effective method for carrier detection and genetic counseling.
Keywords: psychomotor regression, demyelinating, gait abnormality and impairment, metachromatic leukodystrophy (MLD), behavioral disturbances
Introduction
Metachromatic leukodystrophy (MLD) is a rare lysosomal storage disorder, caused by deficiency of the enzyme arylsulfatase A (ARSA, E.C. 3.1.6.1) with a frequency of approximately 1 per 40,000 worldwide.1 ARSA catalyzes initiative step of the metabolic pathway, sphingolipid 3′-o-sulfogalactosylceramide, known as sulfatide. Sulfatide is especially abundant in the myelin sheath of the nervous system.2 Mutations in the ARSA gene (Figure 1) (GenBank accession number NP_000478) could lead to a deficiency in ARSA activity, leading to accumulation of sulfatide, especially in the nervous system.3,4 This phenomenon causes a progressive demyelination that leads to different neurological symptoms including ataxia, an initially flaccid and later spastic paresis, optic atrophy, and dementia.5
 | Figure 1 The ARSA gene maps to chromosome 22q13 covers 3.2 kb of genomic DNA and includes 8 exons. |
To date, there is no effective treatment for MLD. However, bone marrow transplantation and stem cell therapy can be beneficial for patients with juvenile- and adult-onset forms in the early stages of the disease. In addition, gene and enzyme replacement therapies for the treatment of MLD have shown promising outcomes in mice models.6–11
ARSA deficiency is divided into 3 clinical subtypes: late-infantile (50%–60%), juvenile (20%–30%), and adult (15%–20%). The disorder course may range from 3 to 10 years or more in the late-infantile type and up to 20 years in the juvenile and adult types.9
Typical magnetic resonance imaging (MRI) alterations in MLD have been explained in the literature. T2-weighted MRI (T2W MRI) of the brain in MLD patients typically indicates butterfly-shaped confluent white matter hyperintensities with early involvement of the corpus callosum.12 In addition, there is elevated white matter involvement, including U-fibers and cerebellar white matter, as well as cerebral atrophy with progression of the MLD disease.13–16 In this study, the ARSA gene was examined in individuals who met the proposed clinical criteria for MLD in order to identify the pathogenic impact of the associated mutations in MLD.
Materials and methods
Patient collection and ethical statement
In this study, 12 Iranian non-consanguineous MLD patients, with a mean age of 3.5 years, were diagnosed between January 2009 and November 2012. Clinical characteristics of MLD patients are summarized in Table 1. Blood samples from 12 MLD patients and 40 healthy individuals were obtained from the Special Medical Center (SMC), Tehran, Iran. Written informed consent for genetic study and molecular analysis and consent to publish results was obtained from the healthy controls, patients, and parents on behalf of their children. The clinical ethics committee of SMC specifically approved this study (Approval No ML-41-1224) in December 2012. The exclusion criteria for healthy individuals were any history of familial and sporadic cancers, hereditary and non-hereditary metabolic disorders, and nuclear and mitochondrial DNA-associated disorders.
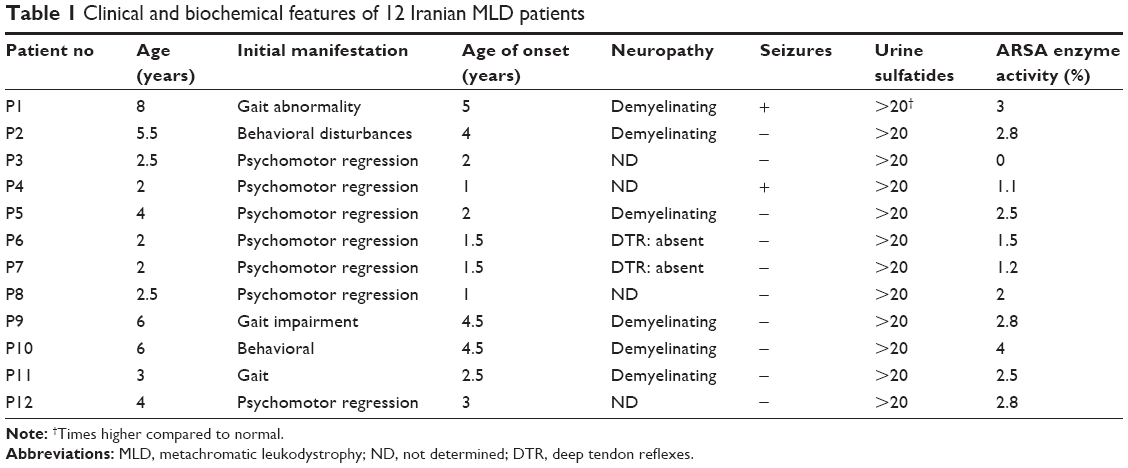 | Table 1 Clinical and biochemical features of 12 Iranian MLD patients |
Enzymatic ARSA activity assay
ARSA activity was determined in leukocytes using p-nitrocatechol sulfate as described previously by Molzer et al.17
Neuroimaging (MRI) analysis
T2-weighted spin-echo sequences of the brain were carried out using a Siemens Magnetom Avanto 1.5 Tesla MRI (Munich, Germany). Images were evaluated in a blind fashion by a neuroradiologist.
DNA extraction and polymerase chain reaction (PCR)
The genomic DNA was extracted from the blood samples of MLD patients by the QIAamp DNA Micro Kit (Qiagen #56304). PCR primers18 for amplification of exons 1–8 of the ARSA gene are as indicated in Table 2. Briefly, PCR was carried out in final volumes of 25 μL, containing 100–200 ng of total genomic DNA, 10 pmol of forward and reverse primers, 2.5 mM of MgCl2, 200 mM of each deoxyribonucleoside triphosphates (dNTP), and 1 U of super Taq DNA polymerase (Roche Diagnostics, Mannheim, Germany). The PCR mixture was cycled for 35 times at 95°C for 1 minute; annealing temperature was based on temperature (TM) (°C) of forward and reverse primers (Table 2). The PCR products were examined on 2% agarose gel electrophoresis (Figure 2) in 0.5× Tris-borate-EDTA (TBE) buffer at 110 V for 50 minutes, and then stained with 0.002 mg/mL EtBr solution and visualized using ultraviolet light.
 | Table 2 PCR primers for amplification of ARSA gene in MLD patients |
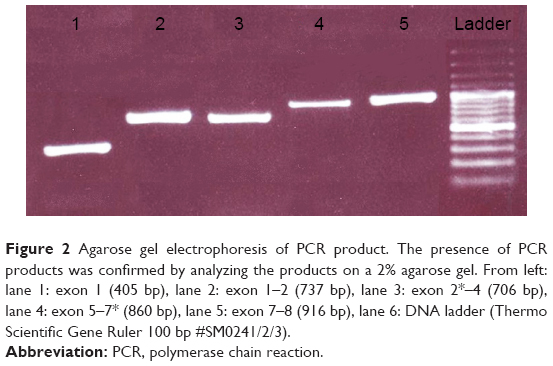 | Figure 2 Agarose gel electrophoresis of PCR product. The presence of PCR products was confirmed by analyzing the products on a 2% agarose gel. From left: lane 1: exon 1 (405 bp), lane 2: exon 1–2 (737 bp), lane 3: exon 2*–4 (706 bp), lane 4: exon 5–7* (860 bp), lane 5: exon 7–8 (916 bp), lane 6: DNA ladder (Thermo Scientific Gene Ruler 100 bp #SM0241/2/3). |
DNA sequencing and bioinformatics analysis
The PCR products were sequenced by forward or reverse primers on an ABI 3700 sequencer (Kosar Company, Tehran, Iran) and the results were compared using Finch TV program and were then analyzed on the NCBI website (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Moreover, the target sequence of patient was compared with normal reference sequence and ARSA gene mutations in exons and the splicing sites of the introns were identified. The functional and structural impacts of identified novel mutations in ARSA gene were assessed using in silico prediction algorithms including SIFT,19 PolyPhen-2,20 and I-Mutant 2.0 (http://folding.biofold.org/i-mutant/i-mutant2.0.html).
Statistical analysis
The chi-square test was used with Statistical Package for the Social Sciences (version 13) to examine the association between patient and healthy control samples, whereas P-value <0.05 was considered statistically significant.
Results
At T2W MRI, MLD patients demonstrated symmetric confluent areas of high signal intensity in the periventricular white matter (Figure 3: arrows in A–C) with sparing of the subcortical U-fibers (Figure 3: arrows in A). No enhancement is evident in computed tomography or MRI. The tigroid (Figure 3: arrows in D) and leopard skin (Figure 3: arrows in E and F) patterns of demyelination, suggesting sparing of the perivascular white matter, are identified in the periventricular white matter and centrum semiovale. The corpus callosum, corticospinal, and internal capsule tracts are also frequently involved. The cerebellar white matter may appear hyperintense at T2W MRI. In the later step of MLD, corticosubcortical atrophy is often ascertained, particularly after involvement of the subcortical white matter.
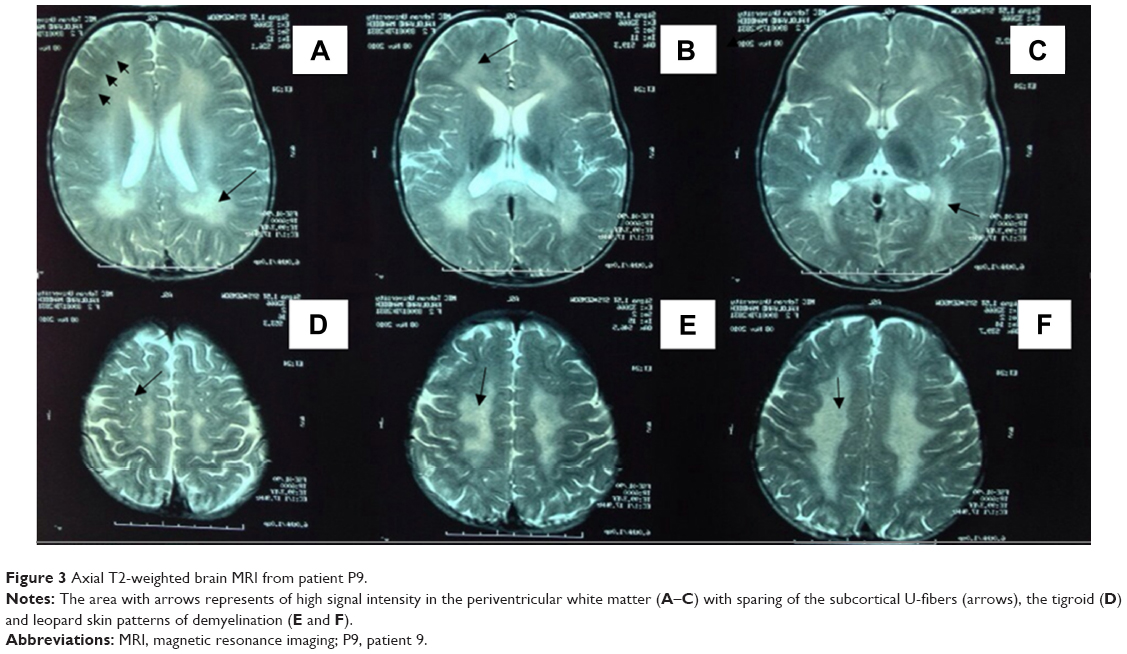 | Figure 3 Axial T2-weighted brain MRI from patient P9. |
Eight exons of the ARSA gene were examined in 12 patients with MLD symptoms. The c.100G>A homozygous mutation in exon 1, c.661T>A homozygous mutation in exon 3, c.739G>A homozygous mutation in exon 4, c.827C>T homozygous mutation in exon 4, c.911A>G homozygous mutation in exon 5, c.931G>A homozygous mutation in exon 5, c.849C>G homozygous mutation in exon 4, A>G 96 relative to termination codon homozygous mutation, W195C homozygous mutation in exon 3, and c.978+1G>A homozygous mutation in intron 5 were detected as presented in Table 3. The 4 new mutations c.585G>T, c.661T>A, c.849C>G, and c.911A>G were significantly (P<0.05) identified in 4 patients (Table 4 and Figure 4A–D). The possible structural and functional effects of identified new mutations in ARSA were examined using the bioinformatics SIFT, PolyPhen, and I-Mutant 2.0 software. Here, SIFT outcomes showed that W195C, F221I, D283E, and K340R mutations were determined as deleterious with scores of −0.734, −5.852, −3.908, and −2.931, respectively. I-Mutant analysis, based on the free energy change value (sign of DDG), demonstrated that p.W195C, p.F221I, p.D283E, and p.K340R mutations decreased protein stability. According to the PolyPhen score, the c.585G>T, c.661T>A, c.849C>G, and c.911A>G mutations were determined as probably damaging the protein structure and function with scores of 0.994, 1.000, 1.000 and 1.000, respectively (Table 4).
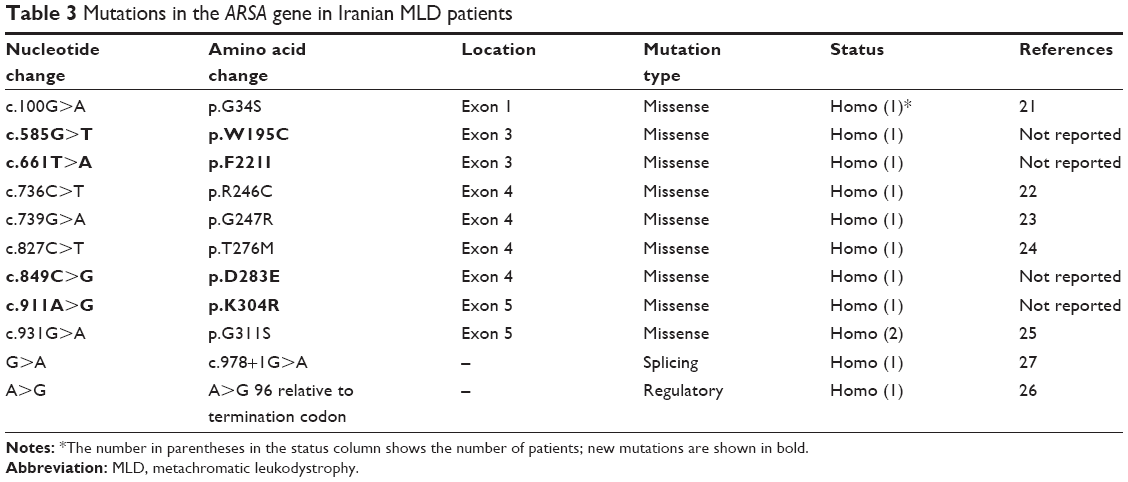 | Table 3 Mutations in the ARSA gene in Iranian MLD patients |
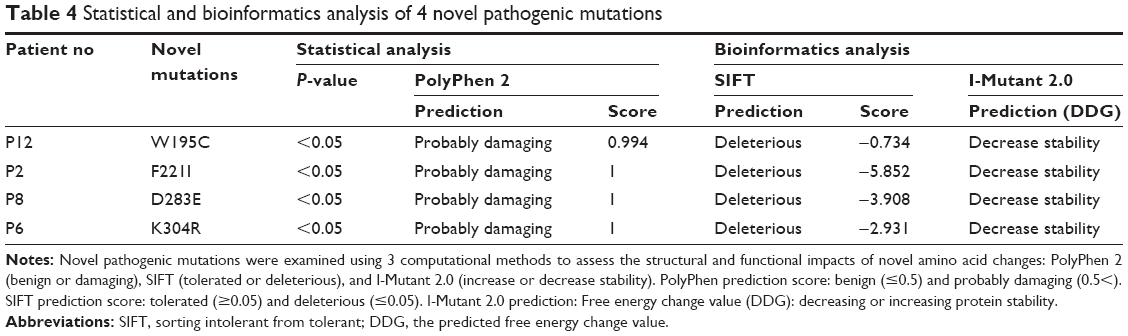 | Table 4 Statistical and bioinformatics analysis of 4 novel pathogenic mutations |
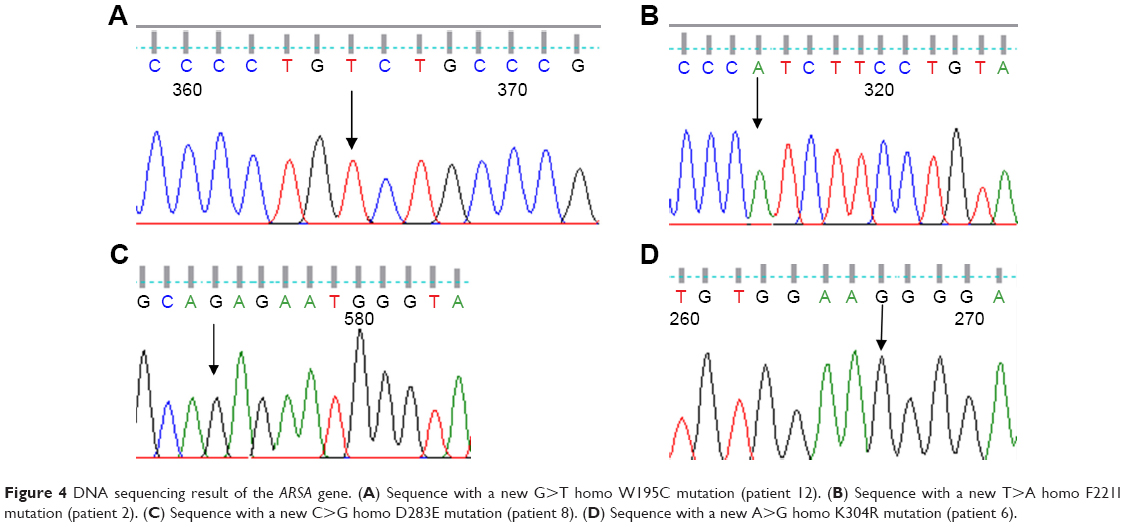 | Figure 4 DNA sequencing result of the ARSA gene. (A) Sequence with a new G>T homo W195C mutation (patient 12). (B) Sequence with a new T>A homo F221I mutation (patient 2). (C) Sequence with a new C>G homo D283E mutation (patient 8). (D) Sequence with a new A>G homo K304R mutation (patient 6). |
Discussion
In this study, 4 novel pathogenic mutations including c.585G>T, c.661T>A, c.849C>G, and c.911A>G in the ARSA gene were identified among 12 unrelated Iranian MLD patients. The previous reported mutations including the c.100G>A mutation in patient 1 (P1), c.736C>T mutation in P3, c.739G>A mutation in P4, c.827C>T mutation in P5, c.931G>A mutation in P7 and P9, A>G 96 relative to the termination codon in P15, and c.978+1G>A mutation in P11 that were reported by Gort et al,21 Gieselmann et al,22 Hasegawa et al,23 Harvey et al,24 Kreysing et al,25 Gieselmann et al,26 and Eng et al,27 respectively.
Hence, additional research is required to affirm the biological role of these pathogenic mutations and confirm the in silico bioinformatics findings that have shown possible effects on protein structure and/or function in the absence of the ARSA enzyme.
The ARSA gene has 8 exons, which are located on chromosome 22 (22q13.33).25 At present, more than 150 mutations were identified in the ARSA gene according to the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ARSA) (Figure 5).
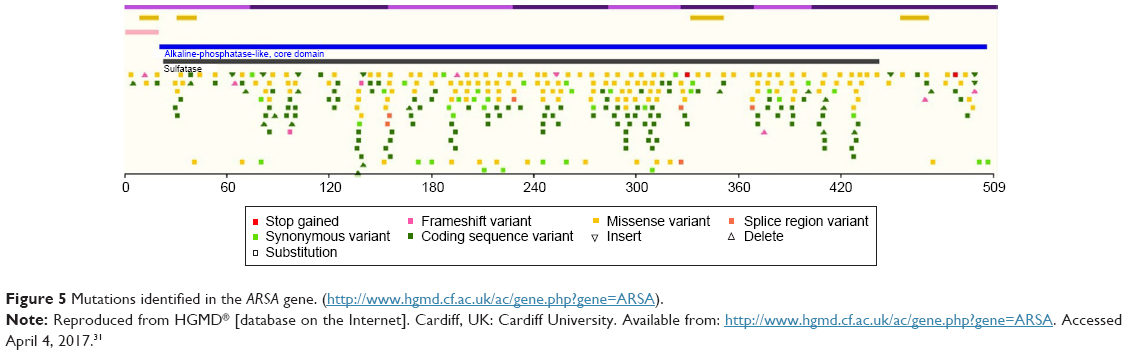 | Figure 5 Mutations identified in the ARSA gene. (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ARSA). |
MLD is a heterogeneous disease with 3 frequent defective alleles including a missense mutation that leads to a Ilel79Ser substitution, a splice donor-site mutation at the exon 2/intron 2 border, and a missense mutation that causes a Pro246Leu substitution, which account for 12%, 25% (among European patients), and 25% of all defective alleles, respectively.28,29 Other mutant alleles were reported in only a few or single patients.30
Conclusion
The result of this research has broadened the genotypic spectrum of Iranian patients with MLD, paving way to a more effective method for career detection, genetic diagnosis, and counseling of Iranian patients with MLD disorder.
Acknowledgments
The authors of this study would like to appreciate all Iranian patients with MLD disorder from the Medical Genetics Department of the Special Medical Center, Tehran, Iran, for blood donation. The authors would like to express their utmost gratitude to SMC for providing financial support (MLD-6102) to conduct this study. Additionally, the funders of this study had no role in study design, data collection and analysis, decision to publish, or manuscript preparation.
Author contributions
MH conceived and designed the experiments. OA, EK, and SD performed the experiments and contributed to reagents/materials/analysis tools. FA, GYM, and MT analyzed the data. BK wrote the manuscript, contributed to the discussion, and reviewed the manuscript. All the authors read and approved the final manuscript. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Gustavson KH, Hagberg B. The incidence and genetics of metachromatic leucodystrophy in northern Sweden. Acta Paediatr Scand. 1971;60(5):585–590. | ||
Gieselmann V, Franken S, Klein D, et al. Metachromatic leukodystrophy: consequences of sulphatide accumulation. Acta Paediatr Suppl. 2003;92(443):74–79; discussion 45. | ||
van Rappard DF, Boelens JJ, Wolf NI. Metachromatic leukodystrophy: disease spectrum and approaches for treatment. Best Pract Res Clin Endocrinol Metab. 2015;29(2):261–273. | ||
Gieselmann V, Krägeloh-Mann I. Metachromatic leukodystrophy – an update. Neuropediatrics. 2010;41(1):1–6. | ||
Costello DJ, Eichler AF, Eichler FS. Leukodystrophies: classification, diagnosis, and treatment. Neurologist. 2009;15(6):319–328. | ||
Batzios SP, Zafeiriou DI. Developing treatment options for metachromatic leukodystrophy. Mol Genet Metab. 2012;105(1):56–63. | ||
Solders M, Martin DA, Andersson C, et al. Hematopoietic SCT: a useful treatment for late metachromatic leukodystrophy. Bone Marrow Transplant. 2014;49(8):1046–1051. | ||
van Egmond ME, Pouwels PJ, Boelens JJ, et al. Improvement of white matter changes on neuroimaging modalities after stem cell transplant in metachromatic leukodystrophy. JAMA Neurol. 2013;70(6):779–782. | ||
Wang RY, Bodamer OA, Watson MS, Wilcox WR; ACMG Work Group on Diagnostic Confirmation of Lysosomal Storage Diseases. Lysosomal storage diseases: diagnostic confirmation and management of presymptomatic individuals. Genet Med. 2011;13(5):457–484. | ||
Biffi A, Naldini L. Novel candidate disease for gene therapy: metachromatic leukodystrophy. Expert Opin Biol Ther. 2007;7(8):1193–1205. | ||
Sevin C, Aubourg P, Cartier N. Enzyme, cell and gene-based therapies for metachromatic leukodystrophy. J Inherit Metab Dis. 2007;30(2):175–183. | ||
Cheon JE, Kim IO, Hwang YS, et al. Leukodystrophy in children: a pictorial review of MR imaging features. Radiographics. 2002;22(3):461–476. | ||
Van der Knaap MS, Pouwels PJW. Magnetic resonance spectroscopy: basic principles and application in white matter disorders. In: van der Knaap MS, Valk J, editors. Magnetic resonance of myelination and myelin disorders. Berlin:Springer; 2005: 859–880. | ||
Faerber EN, Melvin J, Smergel EM. MRI appearances of metachromatic leukodystrophy. Pediatr Radiol. 1999;29(9):669–672. | ||
Kim TS, Kim IO, Kim WS, et al. MR of childhood metachromatic leukodystrophy. AJNR Am J Neuroradiol. 1997;18(4):733–738. | ||
Eichler F, Grodd W, Grant E, et al. Metachromatic leukodystrophy: a scoring system for brain MR imaging observations. AJNR Am J Neuroradiol. 2009;30(10):1893–1897. | ||
Molzer B, Sundt-Heller R, Kainz-Korschinsky M, Zobel M. Elevated sulfatide excretion in heterozygotes of metachromatic leukodystrophy: dependence on reduction of arylsulfatase A activity. Am J Med Genet. 1992;44(4):523–526. | ||
Wang J, Zhang W, Pan H, et al. ARSA gene mutations in five Chinese metachromatic leukodystrophy patients. Pediatr Neurol. 2007;36(6):397–401. | ||
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. | ||
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. | ||
Gort L, Coll MJ, Chabás A. Identification of 12 novel mutations and two new polymorphisms in the arylsulfatase A gene: haplotype and genotype-phenotype correlation studies in Spanish metachromatic leukodystrophy patients. Hum Mutat. 1999;14(3):240–248. | ||
Gieselmann V, Zlotogora J, Harris A, Wenger DA, Morris CP. Molecular genetics of metachromatic leukodystrophy. Hum Mutat. 1994;4(4):233–242. | ||
Hasegawa Y, Kawame H, Eto Y. Mutations in the arylsulfatase A gene of Japanese patients with metachromatic leukodystrophy. DNA Cell Biol. 1993;12(6):493–498. | ||
Harvey JS, Nelson PV, Carey WF, Robertson EF, Morris CP. An arylsulfatase A (ARSA) missense mutation (T274M) causing late-infantile metachromatic leukodystrophy. Hum Mutat. 1993;2(4): 261–267. | ||
Kreysing J, von Figura K, Gieselmann V. Structure of the arylsulfatase A gene. Eur J Biochem. 1990;191(3):627–631. | ||
Gieselmann V, Polten A, Kreysing J, von Figura K. Arylsulfatase A pseudodeficiency: loss of a polyadenylation signal and N-glycosylation site. Proc Natl Acad Sci U S A. 1989;86(23):9436–9440. | ||
Eng B, Nakamura LN, O’Reilly N, et al. Identification of nine novel arylsulfatase a (ARSA) gene mutations in patients with metachromatic leukodystrophy (MLD). Hum Mutat. 2003;22(5):418–419. | ||
Polten A, Fluharty AL, Fluharty CB, Kappler J, von Figura K, Gieselmann V. Molecular basis of different forms of metachromatic leukodystrophy. N Engl J Med. 1991;324(1):18–22. | ||
Berger J, Löschl B, Bernheimer H, et al. Occurrence, distribution, and phenotype of arylsulfatase A mutations in patients with metachromatic leukodystrophy. Am J Med Genet. 1997;69(3):335–340. | ||
Schestag F, Yaghootfam A, Habetha M, et al. The functional consequences of mis-sense mutations affecting an intra-molecular salt bridge in arylsulphatase A. Biochem J. 2002;367(Pt 2):499–504. | ||
HGMD® [database on the Internet]. Cardiff, UK: Cardiff University. Available from: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ARSA. Accessed April 4, 2017. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.