Back to Archived Journals » Research and Reports in Forensic Medical Science » Volume 4
Forensic DNA profiling: state of the art
Authors Linacre A, Templeton JEL
Received 12 May 2014
Accepted for publication 13 June 2014
Published 28 August 2014 Volume 2014:4 Pages 25—36
DOI https://doi.org/10.2147/RRFMS.S60955
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Adrian Linacre, Jennifer EL Templeton
School of Biological Sciences, Flinders University, Adelaide, SA, Australia
Abstract: DNA profiling is now a routine test, yet it has only been 26 years since its first use. During these intervening years, the science of DNA profiling has undergone many changes, and it will continue to do so in the future. DNA profiling in the laboratory starts with DNA extractions and is followed by quantification of the DNA, amplification of up to 23 human specific short tandem repeat loci, and separation on capillary columns. The final stage is interpretation of the data leading to reporting the DNA evidence to a court. Each of these steps has seen improvements leading to higher sensitivity from minute trace samples. Genetic markers on the X and Y chromosomes are now used for both criminal and civil investigations, along with mitochondrial DNA. Mitochondrial DNA has been used in forensic science since the 1990s because of its high copy number and particular maternal inheritance. These sex chromosome markers and mitochondrial DNA were used effectively to determine the identity or ancestry of individuals, and now other genetic markers can be used to determine certain phenotypic traits to a high degree of accuracy. Genetic testing adapted from medical and pharmaceutical sciences, such as next-generation DNA sequencing, will soon be applied to mainstream forensic science, opening new avenues in criminal investigations. This review aims to cover these key events and advancements in the field from both a historical view and current practice.
Keywords: forensic science, DNA profiling, short tandem repeats, mitochondrial DNA, phenotypic testing, next-generation sequencing
Opening comments
DNA profiling has revolutionized the process of forensic human identification. It is of note that this is a relatively recent addition to the forensic science tool kit and that it is constantly undergoing developments. The aim of this review is to take the reader through the processes of DNA typing, starting with DNA isolation from biological material through novel approaches and phenotypic testing. The review is written for those in forensic medicine; therefore, certain knowledge is assumed, but equally, aspects that are specific to forensic science will be emphasized and placed into the right context. For those who wish more information than can be provided within the scope of this review, there are books ranging from introductory texts1 to those that are more comprehensive.2,3
Introduction
The first use of DNA fingerprinting was to resolve an immigration dispute in 1985.4 A process of DNA fingerprinting had been published the year earlier in Nature, coauthored by Alec Jeffreys. This led to a local police force engaging Jeffreys (now Professor Sir Alec Jeffreys) to assist with an unsolved sexual assault and murder of two young girls in Leicestershire, England. DNA fingerprinting led originally to linking the two murders to the same perpetrator, then to the exoneration of a young man, and finally to the conviction of Colin Pitchfork for the crimes in 1988.5
The early test performed by Alec Jeffreys to identify an individual through DNA typing is very different from modern-day DNA testing; the only commonality is the word “DNA.” What had been noticed was that sections of DNA are hypervariable between people but conserved throughout the cells of an individual. DNA fingerprinting originally examined variation at many loci across the entire genome leading to multiple bands on a gel (an example of which can be seen in Figure 1), with the appearance of a barcode, and as everyone’s barcode appeared to be different, the term “DNA fingerprinting” was coined. Since 1994, all DNA profiling (as it is now termed) has centered on microsatellite DNA (termed short tandem repeats, or STRs, in this review), and in particular, on repeat units of four bases (see chapter 5 from Butler2 and chapter 6 from Goodwin et al1). The STR loci used in standard DNA profiling are all inherited in a Mendelian manner; are not genetically linked, and therefore are inherited independently; and crucially, are thought to have no known association with a disease state.
 | Figure 1 Steps involved in DNA profiling. Profiling starts with isolation of DNA by a process called DNA extraction, followed by quantification of the DNA in the sample, then amplification of short tandem repeat loci, separation of the short tandem repeat products, and finally, interpretation of the genotype data. |
The advent and application of polymerase chain reaction (PCR)6 to forensic genetics was timely and advantageous. PCR is a method of amplification used to produce millions of copies of a specific region of DNA. Tiny blood spots or single hairs could generate DNA profiles, thus increasing the sensitivity of the test to subnanogram levels of DNA. Allied to this were increasing powers of discrimination: the probability that another person shares the same DNA profile by chance. DNA profiling has been heralded as a game changer because of these high powers of discrimination (resulting from an increased number of loci being examined between individuals) and has been seen to override other forensic evidence for which the powers of association are many orders of magnitude less. Validation studies and interlaboratory proficiency testing to demonstrate as comprehensively as possible the robustness and reproducibility of the tests were undertaken because of the significance of DNA evidence in any criminal case. DNA frequency databases were generated for all populations to ensure an accurate (and frequently conservative) estimate of the frequency of the DNA profile. The chances that the DNA profile came from a close genetic relative were incorporated into statistical formulae, as were mechanisms to adjust the frequencies stated for small subpopulations. The current situation is that because of the extensive validation studies and challenges in courts in many jurisdictions, DNA is now considered a gold standard7 in forensic science, with the science underpinning the forensic application and processes of evaluation of the evidence being the standard to which other areas of forensic science should aspire.
The forensic DNA community has striven to achieve standardization to ensure the same core set of DNA loci are examined between jurisdictions. This will ensure there is commonality in the data obtained, allowing national and international sharing. This harmonization manifests itself in a number of ways, including, although far from exclusively, the basic steps in forensic DNA processing. These are illustrated in Figure 1. The process starts with DNA extraction and is followed by quantification of the DNA isolated and then amplification of the STR loci, separation of the STR alleles by capillary electrophoresis (CE), and finally, interpretation of the data.
Typically, the processes shown in Figure 1 take a minimum of 10 hours, using current standard technology. The aim of much research is to make this process quicker without compromising the quality of the data obtained. Each of these steps is described here.
DNA extraction
The ultimate aim of any DNA extraction process is for the final solution to contain DNA in sufficient purity and quantity to allow subsequent processing by PCR. The extraction process chosen may be modified depending on the biological tissue being analyzed. Blood contains DNA only within the white blood cells. Other cellular components need to be separated and purified from the DNA, such as proteins, lipids, and carbohydrates. Semen has the added complication that the DNA is encased within the spermatozoa head; this head (the acrosome) is made from a strong protein that is resistant to many naturally occurring enzymes. In contrast, the DNA from buccal and skin cells is more easily obtained, as these epithelial cells are easily lysed, exposing the DNA to the extraction buffer.
There are additional considerations for forensic science processing. For example, a forensic laboratory may process tens of thousands of samples every year. A manual procedure in which individual laboratory staff members pipette small volumes from one tube to another will be highly costly in staff time. Further, every time fluid is passed from one tube to another, it leaves open the potential for contamination or for adding fluid to a wrong or incorrectly labeled tube. The possibility of using automated extraction processes is therefore beneficial in terms of costs and ensuring that the wrong DNA profile is not attributed to an individual because of a sample mix-up.
The principal steps in any extraction process are to lyse the cellular membrane, using a solution typically containing sodium dodecyl sulfate; break open the nuclear envelope, using a solution containing a proteinase (typically proteinase K); separate and purify the DNA molecule (ethanol), making use of its negative charge; and elute the DNA into a fresh solution (low-salt buffer or water) relatively free from inhibitors and of sufficient purity to allow enzymatic amplification of the DNA.
The original method of DNA isolation was an organic extraction process8 in which, initially, a buffer containing sodium dodecyl sulfate, a chelating agent (ethylene diamine tetraacetic acid), and an enzyme to digest protein (proteinase K) is used to expose the native DNA molecule. The proteins are removed by addition of an equal volume of the solvent phenol (or a 1:1 mix of phenol and chloroform). Organic extractions are only used in current forensic practice for niche applications, as they are laborious, involve multiple tube changes in this process, and cannot be automated, and because phenol is a highly toxic chemical both to the user and the environment.
Chelex® (Bio-Rad Laboratories, Hercules, CA, USA) was introduced to forensic practice in the 1990s9 and quickly became part of a popular method. The process uses a suspension of Chelex® that binds any positive ion (such as Fe2+, Mg2+, and Ca2+), leaving the DNA behind. There are other biological materials that are negatively charged that will coextract with the DNA; hence, this method leaves the DNA dissolved finally in a fluid that is far from free of potential inhibitors. The use of Chelex® is not easy to automate for high-throughput laboratories and has been superseded by silica-based extraction methods, although Chelex® is still used for the isolation of DNA from buccal swabs, in which the DNA is in a less complex state compared with in blood or semen.
Although Chelex® uses a bead that binds positive ions, silica-based extraction processes make use of the negative charge carried by the DNA molecule. Silica-based extraction methods have become widely available as part of a commercial kit, making them more costly than either phenol or Chelex®, but they have the advantage of being open to automation. The silica-based material10 is embedded in a layer at the base of a spin column, through which fluid containing DNA can pass. The DNA binds to the silica membrane, but all the molecules that do not possess a strong negative charge pass through the membrane and are discarded. The DNA can ultimately be released from its bound state by altering the pH of the silica membrane, allowing DNA to be eluted as a relatively pure solution.
Magnetic bead technology11 acts in a similar way to silica-based methods, in that DNA binds to a biotin complex that is attracted to a streptavidin bound to a magnet. Other constituents of the cell that do not bind to this complex are flushed away or removed. Once this is complete, the DNA can be released from its binding and eluted into a relatively small volume of fluid. See Table 1 for a comparison of these methods.
 | Table 1 Four currently used DNA extraction methods |
DNA quantification
The amount of DNA isolated by the extraction step needs to be quantified to ensure the optimal amount of DNA template is added to a PCR. Typically, the amount of template required for PCR is 1 ng human DNA. Depending on the sample type, the total DNA isolated may include bacterial, fungal, or other nonhuman DNA.
The earliest method of DNA quantification involved the measurement of the absorbance of light at 260 nm, using an optical density spectrophotometer.8 Although this can be relatively accurate, it requires a large quantity of DNA, as the method is not sensitive, and hence most of the sample may be used. Further, this method detects all DNA and is not human-specific.
For a while in the 1990s, slot blots were used, in which DNA of known concentration was immobilized on a nylon membrane at specific locations, to act as standards. The DNA samples to be tested were immobilized near the standards. A short sequence of DNA found in humans only (actually an alpha satellite repeat sequence on chromosome 17) was used as a probe to bind to the immobilized DNA, and as the probe could be detected by chemiluminescence, the amount of human DNA present could be determined. This method was more sensitive than using an optical density spectrophotometer to measure the absorbance of light, but it took many hours to complete.
The current standard technology used in forensic science laboratories is real-time PCR.1,2 At each cycle in the PCR, there should be a doubling of each PCR product. As these PCR products are double-stranded molecules, any dye that binds to double-stranded DNA can be used to monitor the reaction as it occurs. If the PCR primers bind to human-specific DNA sequences, then the presence of only human DNA will be detected; this is a major benefit compared with other methods of DNA quantification. Equally, the amount of template DNA present at the start of the reaction can be determined by comparison with known standards. Using this real-time approach, investigators can monitor a reaction, and if suboptimal nuclear DNA is detected in a sample, they may choose to focus on targeting other markers in the genome that are more ideal for degraded DNA. Real-time PCR has a further advantage, in that the presence of inhibitors can be noted if the reaction fails. The only disadvantage is the cost of the equipment required, as it is significantly higher than the other methods.
Amplification of STR loci by PCR
The development in the enzyme that amplifies the STRs that is relevant to DNA typing is a “hot start” activity.12,13 Although the optimum temperature for Taq DNA polymerase is between 60°C and 72°C, it will work at room temperature, although relatively slowly. This can lead to poor amplification, and potentially amplification of the wrong fragments if there is a time delay between preparing the PCR and adding it to the thermal cycler. Hot start enzymes require heating to 95°C, during which time the pH of the buffer alters such that the enzyme becomes active. The end result is that there is more specific binding in the initial steps of the PCR, leading to more of the targeted PCR product.
STR loci
There has been an evolution in the number of STR loci amplified in one reaction. All the STRs used are noncoding, but most are close to a gene sequence or are within introns. The first STR loci were identified within introns and named after the gene with which the intron was associated. For instance, the STR locus called vWA is a repeat found in intron 40 of the von Willebrand Factor gene (named after the physician who noted a blood clotting disorder based on a mutation in this gene), which is on the short arm (p) of chromosome 12. TPOX is an STR within intron 10 of the thyroid peroxidase gene on the short arm of chromosome 2. A more simple nomenclature was adopted as more STR loci were incorporated into DNA profiling; for example, STR locus D3S1358, where D stands for DNA; 3 designates the chromosome; S indicates there is a single copy, rather than multiple copies; and the number indicates that this was the 1,358th section of chromosome 3 to be described.
Originally, there were four loci in 1994,14,15 and then six loci 1 year later.16–19 These reactions were assembled by the UK Forensic Science Service. Commercial companies saw there was a market and started to make their own STR multiplex kits; the main companies were Applied Biosystems (now part of Thermo Fisher Scientific, Waltham, MA, USA), and the Promega Corporation (Fitchburg, WI, USA). By 1997, there were kits that amplified nine STR loci plus a sex test, and then in 2000, a 16-STR locus test was introduced.20 At this time, the technology has improved to the stage at which 23 STR loci plus the sex test are amplified in a single reaction.
Another advance in STR technology is that the reaction buffer has seen improvements, such as an increased tolerance to inhibitors. Minor inhibitors of the reaction, which may have previously resulted in no amplification product, have less effect, leading to more complete DNA profiles. This has also increased the sensitivity of the PCR, with the optimal starting template being 500 pg DNA rather than 1 ng; 500 pg is approximately 84 human cells.
The primers used to amplify the STR loci have, in many instances, been designed to sequence closer to the start of the repeat unit. The aim of this was partly to ensure that these larger STRs would still amplify from highly degraded DNA, as previous empirical data had shown that larger STRs are more likely to be lost from the amplification as the amount of degradation of the DNA increases.
With Promega and Applied Biosystems offering different kits, and with occasional amplification of different STR loci, there had been little standardization of the STR data between countries. The adoption of the European Standard Set in 2009 led to 12 core STR loci being adopted by all European countries. This has opened up the exchange of DNA data. To accommodate the situation in the United States and lead to further standardization, the number of shared loci rose to 18.2 The new STR kits include all these 18 loci plus additional markers, allowing freer exchange of DNA data, such as allele frequencies, internationally.
Separation and detection of STR alleles
Polyacrylamide gel electrophoresis was the original method for separating STR alleles. In this method, DNA can be added to a gel matrix, and in the presence of an electric field, the negatively charged DNA will be separated according to size (to distinguish the STR alleles). Shorter molecules migrate faster through the matrix, and the separation of DNA allows for sizing of fragments against a known standard (ie, DNA ladder). CE was a major breakthrough, as the separating polymer is much safer to use. The only disadvantage was that the original CE machines separated only one or up to four samples at one time, unlike a gel with many lanes. Applied Biosystems produced a single-capillary machine (310 Genetic Analyzer) but quickly developed a 16-capillary machine (3130xlGenetic Analyzer), in which samples are separated on all 16 capillaries simultaneously. The runs take approximately 30 minutes but can be run overnight in batches, allowing 128 samples to be run in less than 5 hours.
Detection of the STR fragments is based on laser-induced fluorescence, as a dye can be attached to the 5′ end of one of the primers used in the PCR. Originally, there were four dyes available to allow three STR alleles to overlap in size, with the fourth dye set aside for an in-lane size standard. Previous technology meant that samples that overlapped in mass had to be separated in different lanes; hence, three dyes was a major advance, allowing up to ten STRs to all fit in a size range between 100 and 400 base pairs (this size window allowed nonincorporated primers to be separated from true products and also degraded DNA would still generate alleles at most of the ten loci). More recent technological advances have allowed a fifth dye to be used (and a sixth dye will be released at the end of 2014), such that up to 23 loci can be compressed into this small size range.
Criminal justice databases
The ability to generate STR profiles quickly and with a high power of discrimination led the UK government to consider the opportunity to retain the DNA profiles of any person found guilty of an offence punishable by imprisonment. This was in 1995, just as the six-loci test was introduced. The power of discrimination of this six-loci test was close to the population of the United Kingdom at the time, and hence the chance of a coincidental match should have been extremely unlikely. At the time of writing, because of changes in legislation affecting the retention and addition of DNA data, the number of people on the UK database (NDNAD) is more than 6 million. At this time, in the United Kingdom, approximately one in 20 members of the population are on the NDNAD. As approximately 80% of those on the NDNAD are men, and given that there are 30 million men in the United Kingdom, this means that approximately 12% of the entire male UK population is on the NDNAD. The most recent figures indicate there is a 58% chance that a DNA profile from a crime scene will match a DNA profile on the NDNAD (for more information, see chapter 10 of Goodwin et al1 and chapter 8 of Butler2).
In 1998, in the United States, a similar database was established that now contains more than nine million STR profiles, making it the largest such database numerically (the one in the United Kingdom contains the most DNA profiles as a percentage of the population). Indeed, almost all countries with a forensic DNA capability have established such a database.
Criminal DNA databases have been a driving force for the standardization of STR loci across countries.
Lineage markers
All the STR loci discussed so far are on one of the 44 somatic chromosomes; hence, there is a copy inherited from the biological mother and one from the biological father. There are STR loci on the X and Y chromosomes, as well as polymorphic DNA sequences within the mitochondrial DNA. All three have found niche applications in forensic science for criminal casework, identification of victims in mass disasters, or paternity/immigration disputes. These same markers have been used for evolutionary purposes to reconstruct the movement of man since leaving Africa some 90,000 years ago.21,22 Table 2 illustrates the mode of inheritance of these loci.
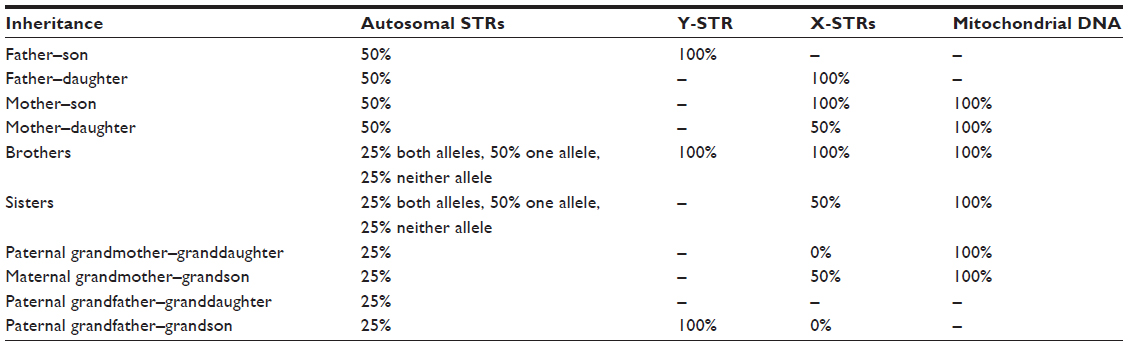 | Table 2 Inheritance and genetic sharing of autosomal STR markers, Y-chromosome STR markers, X-chromosome markers, and mitochondrial DNA |
Y-STR typing
The Y chromosome should be inherited as a complete chromosome from father to son. All male siblings should therefore also share the same Y chromosome. Unlike the 22 pairs of autosomal chromosomes, the Y chromosome does not recombine with any partner (only the tips make any contact with the X chromosome), and hence all the loci remain in the same order and linked from one generation to the next, to the next, to the next, and so on. The genetically linked DNA loci on the Y chromosome are termed a haplotype.
The characterization of highly variable STR markers on the Y chromosome led to commercially available kits that currently amplify 23 such male-specific loci.23,24 In forensic science, Y-STRs are particularly used in sexual assaults. Although there are methods to separate spermatozoa from epithelial cells, male epithelial cells will be detected, along with any female ones, leading to mixed DNA profiles. Frequently, in cases of alleged sexual assaults, samples are taken from fingernails. If taken from a female suspect, these samples will contain DNA from the epithelial cells of the female donor as well as epithelial cells from the male assailant. Any resulting DNA profile will be a mixture of the two and will be possibly difficult to interpret, but using Y-STR, markers of a single male DNA profile will be produced. The mixture of two or more men in the same sample from an alleged sexual assault, such as consensual intercourse followed a few hours later by an alleged rape, can also be problematic, using autosomal markers. This can be less of a problem using Y-STR loci if the Y-STR type from the consensual partner is known and can be removed from the mixed DNA profile.
One novel application of Y-STR loci is that some loci have been found to possess high levels of mutations, such that even close genetic relatives with a recent common paternal relative may be differentiated.25 Brothers should have the same Y-STR type, as is the case for most STRs on the Y chromosome, but some of these rapidly mutating loci may be different, allowing even brothers to be differentiated by Y-STR typing.23,26
X-STR typing
STR loci on the X chromosome are very new and have a limited but valuable niche. They are most useful in paternity testing or identification of victims of a mass disaster where the parents of offspring are unavailable. A woman will pass on her intact X chromosome to her son, who will pass the same X chromosome on to his daughter. Hence a grandchild will have the same X chromosome as a paternal grandmother. Such a linkage has been used to settle cases of incest,27 as well as help identify previously unknown individuals in cases that could be resolved only because of the particular inheritance of the X chromosome. Fourteen X-chromosome STRs have been characterized for forensic use28–30 by the Armed Forces DNA Laboratory in the United States with the potential for identification of the deceased to complement both Y-STR typing and mitochondrial markers.
Mitochondrial DNA
In reality, many biological samples submitted for forensic analysis are not of pristine quality, and some, by their nature, have very little nuclear DNA; for example, degraded bone fragments, teeth, and hair shafts. Highly compromised samples may contain damaged, degraded, and/or low-quantity DNA and fail to be characterized by normal autosomal STR DNA markers. In such cases, when nuclear DNA is considered low template (<100 pg), using the quantification methods described earlier, the analysis of mitochondrial DNA (mtDNA) can prove to be a valuable tool. mtDNA exists in a much higher copy number in the cell compared with nuclear DNA and is more likely to produce a result.31,32
Mitochondrial DNA is inherited down the maternal line and is particularly useful in cases of missing persons when maternal relatives separated by several generations can act as reference samples. It has also been used to identify the skeletal remains of war casualties33,34 and in the identification of the remains of Tzar Nicholas II of Russia.35,36 The Federal Bureau of Investigation crime laboratory in 1990 began research into the credibility of mtDNA to be used as evidence in casework, and in 1995, it published a validation study.37 Later, in September 1996, the first case of mtDNA evidence was used in a United States courtroom to prosecute a 27-year-old Tennessee murderer, Paul Ware. The defence claimed that the accused had been framed for the rape and murder of a 4-year-old girl, but mtDNA evidence had already linked the suspect to two hairs recovered from the victim. This mtDNA sequence had never been seen before and proved to be vital evidence in the case.
Forensic laboratories specializing in mtDNA typing generally focus on sequencing a small portion of mtDNA that is highly variable (changes occur by mutations), known as hypervariable regions I and II (HVSI/II) or the control region. The mtDNA control region random match probability (the chance that another unrelated individual will share the same sequence) is roughly one in 120 individuals, although it has still proven to be useful evidence in certain forensic cases.38 HVSI/II sequencing is, however, limited by the resolving power of this short sequence. Individuals with a similar maternal lineage will share the same haplotype and fail to be distinguished from each other. To maximize the information content from mtDNA, and to gain further resolution, the whole mitochondrial genome can now be sequenced with relative ease because of the advent of next-generation sequencing (NGS). Whole-mitochondrial-genome sequencing provides a high level of maternal lineage discrimination, and recent studies have shown that more than 70% of mtDNA variation lies outside HVSI/II for certain DNA sequence types (called haplogroups).39 Other studies highlight the advantage of sequencing the entire mitochondrial genome for human identification by detecting single nucleotide polymorphisms (SNPs) in the coding region.40–42
To succeed in sequencing whole mitochondrial genomes using traditional CE detection methods, a large number of amplicons are needed, which requires a large quantity of extracted DNA; this work is laborious, time-consuming, and prone to sequencing errors, as shown previously.43,44 To eliminate these problems, ancient DNA laboratories are now turning to a hybridization enrichment approach for sequencing whole mitochondrial genomes and have developed the ability to sequence samples that are thousands of years old.45,46 For hybridization enrichment, probe DNA of a known haplotype can be used to capture and enrich target DNA of interest. The unwanted microbial contaminant DNA is then washed away, using stringency washes of increased temperature and decreased salt concentration. This allows only sequences of perfect homology (in the 20–70 bp range) to be captured and sequenced using NGS platforms.39,47
This approach has the potential to be adopted into mainstream forensic practice to assist with large-scale identification efforts in cases of mass disasters where the DNA is severely damaged, fragmented, and in low copy number. However, first a worldwide effort is required to produce large-scale mtDNA databases48 compatible with such platforms.
To improve current use of mitochondrial DNA in forensic science, the quality of mtDNA sequencing and its corresponding database has been improved by the implementation of a quality assurance check of the data called the EMPOP project (http://empop.org).49 EMPOP stands for European Mitochondrial DNA population database. Software inherent in the database screens sequencing data prone to errors, resulting in high-quality output data. Certain forensic journals now stipulate that mtDNA population studies need to be quality control checked by EMPOP first, to improve the overall quality of results before publication.
Ancestrally informative genetic markers
Genetic markers such as the Y chromosome (passed down the paternal line only), mitochondrial genome (maternally inherited only), and X chromosome (inherited from a father to his son or daughter or from a mother to her daughter only) can all be used to predict a person’s ancestral lineage. In the Western developed world, up until the advent of canals in the mid-1700s and then the railways in the mid- to late 1800s, few people could travel far, and hence populations in towns and villages were made of people who shared recent common ancestors. More recently, mass transport has become available, but still many people remain close to the town or city where the rest of their family lives. The net result is that some DNA types are much more common in some local populations than in others, as these people share recent common ancestors. Sampling populations around the world has led to the observation of clustering of certain DNA types. There can be differences, however, between mitochondrial DNA types and Y-STR frequencies, as in many cultures, males and females move differently. An example is that males often inherit land from their fathers, and thus remain in the same place, but they may marry a woman from a nearby village, who will then move. Conversely, men in the historical past may have been parts of armies or colonizers, who then fathered children in the new local population. By looking at a wide range of mitochondrial, Y-chromosome, and nuclear markers from current populations, it is possible to predict from where a person most likely originated.
Variation in the genetic make-up of populations was first noticed in World War I, when the frequencies of the ABO blood group system were recorded and it was noted that soldiers from Western Europe had different frequencies than those from Eastern Europe (in fact, ABO B type is relatively common in Eastern Asia but becomes rarer moving westward to Europe, whereas the frequency of ABO A increases).50 Leaping forward to current technology, with which many genetic markers can be examined, a more comprehensive picture of the genetic ancestry of a person can be developed.
This type of technology is particularly informative if a DNA profile from a body fluid stain (such as blood or semen) matches no known suspect or no one on any DNA database. A number of forensic cases highlight this use, including the identification of a serial rapist in London in the United Kingdom. DNA samples from multiple rape cases were found to match each other, indicating a single rapist, and by looking at ancestrally informative DNA markers, it was proposed that the rapist most likely came from West Indian heritage, leading the police to ultimately find the suspect.51
Phenotypic markers
All the genetic markers discussed so far have no known phenotypic affect. The STR markers chosen originally were selected specifically such that a person’s DNA profile would be a digital code that is meaningless unless compared with another digital code. However, as knowledge of the human genome has increased, so has the elucidation of certain genes that affect our appearance. At this time, these genes are limited to those for eye color and hair color, but other traits encoded genetically may soon be developed.
Hair color
The natural color of our hair is based on the DNA sequence of the melanocortin-1 receptor (MCIR). A number of single base changes can determine whether a person has red, black, or blonde hair. These single base changes (an example of a SNP) can be detected in a simple test.52
Eye color
The color of the iris is affected by SNPs within a range of genes. These SNPs are combined into a test called IrisPlex, which can be performed on any body fluid or tissue and lead to a prediction of the donor’s eye color. The SNPs for eye color and hair color have been combined into one single SNP-based test called Hirisplex.53,54 This test includes 24 SNPs from eleven different genes. In a multilaboratory trial of this assay, hair color had an accuracy of 69.5% correct calls for blonde, 78.5% for brown, 87.5% for black, and 80% for red hair.53 In 92.5% of cases, the genetic eye color prediction test agreed with the eye color phenotype of the individual.53 As more SNPs are found that affect either eye color or hair color, they can be added to increase the accuracy of the SNP test.
Accreditation: ISO17025
One of the greatest changes to have occurred very recently in the area of forensic science is not a new test per se but, rather, the introduction of accreditation to all operational laboratories. The ISO standard to which analytical science laboratories are assessed is ISO17025 (see http://www.iso.org/iso/home.html for more details). This standard sets in place standard operating procedures (often abbreviated to SOPs) that must be adhered to at all times, as well as competency tests. Examples of competency tests are proficiency tests where known samples are provided by a testing agency to whom the correct answers should be returned. These types of tests demonstrate confidence in the method and the ability of the staff.
Although miscarriages of justice can never be eradicated completely, as any test that involves a human is open to experimental error, such events are very rare in laboratories that operate to ISO 17025 standards. DNA profiling has been undoubtedly a spur toward accreditation, as suddenly there is a test that provides overwhelming powers of discrimination overshadowing other areas of traditional forensic science. If the DNA typing was in error, then a wrong verdict may be provided by the court, regardless of other scientific or circumstantial evidence. Not surprisingly, DNA has undergone extensive validation and verification, as recognized by the US National Academy of Sciences.7
NGS for forensic human identification
The use of DNA technology for the purpose of identification continues to promote interest in more powerful and novel methods of analysis. The latest tool in the field of genetic research and human identification testing is the application of NGS. Advances in genomic technology enable more and more information to be extracted from samples containing limited, degraded, and poor-quality DNA. Although CE detection methods work by distinguishing STR alleles according to the size of the amplicons, NGS can distinguish between alleles that are similar in length by providing information on the sequence content of the DNA (by typing Intra-STR SNPs) in addition to the length of the allele. NGS has the ability to target a larger number of PCR amplicons in a single assay by performing simultaneous analysis of different polymorphism types (eg, identity, ancestry, and phenotypic SNPs). The ability to barcode (individualize) samples55 enables multiple samples to be processed in a single run, increasing the multiplexing and high-throughput capacity,56 another major advantage to forensic labs. Small-scale bench top NGS machines57,58 are already proving to be useful in certain forensic applications, as they are affordable, fast, easy-to-use and provide a large amount of data (10 megabases to several gigabases) in a relatively short time.
The workflow for NGS involves three fundamental steps: library preparation, sequencing, and data interpretation. The first stage in processing biological samples requires the amplification of forensically informative loci and the creation of an amplicon library (collection of DNA fragments) of SNPs, STRs, and/or mtDNA to immortalize the extract. Library preparation involves ligating (adding on) adaptors (small sequences of synthetic DNA) to the ends of the fragments or amplicons of DNA once they are the correct sequencing length (200–400 bp). Before sequencing, library DNA is then amplified (to generate many copies), using primers (synthetic DNA of a known sequence) complementary to adaptor sequences. This library DNA can then serve as a template for the subsequent sequencing reaction. The postenriched library DNA is then sequenced to determine the order of nucleotide bases in the DNA sequence and the length of the fragments; current platforms include Illumina MiSeq/HiSeq, Life Technologies Ion Torrent PGM, and Roche GS Junior/454.58,59 These platforms are capable of revealing the DNA sequence of thousands of individual molecules generated from library DNA. Signals received by the detection instrument are then translated into sequence, and the raw data can be analyzed using bioinformatic pipelines. Both sequence depth and coverage provided by NGS exceeds that generated by traditional CE detection methods by many orders of magnitude and, as discussed previously, is currently the best method of sequencing DNA that is highly degraded (<100 base pairs in length).46
NGS sequencing is the driving force behind ancient DNA research, in which the DNA may have been subjected to extreme environmental conditions for so long it appears damaged, degraded, and in low amounts compared with the unwanted microbial content. Ancient mitochondrial genomes are now being routinely sequenced using NGS,60–62 and it is currently being used to advance metagenomic analysis of environmental samples, another important aspect in forensic science.
The potential does exist for NGS to drive forensic DNA research as the price of sequencing continues to drop. However, refinement and rigorous testing are necessary before it can be implemented in a forensic DNA testing laboratory. Robust validation processes are required before any new procedure can be adopted in an accredited forensic laboratory, and this ultimately slows down the whole process. In addition, selecting the right sequencing platform that meets the demands of sample size, coverage, cost, and accuracy is also a fundamental requirement for forensic DNA applications. In the meantime, research in this exciting new field will continue to advance.
Conclusion
As more is understood about the functioning of the human genome, then inevitably, aspects of this new knowledge will be adapted for use in forensic science. New technologies such as NGS and phenotypic typing are bound to open potential new avenues of forensic genetic typing. Over the last 2 decades, we have seen DNA typing progress from the examination of only a few genetic loci in a matter of days to sequencing the entire human genome in only a single day. A well-informed debate will be needed to determine how much knowledge law enforcement agencies should be allowed to acquire and who should have access. An individual’s genome has always been seen as a very personal thing, yet both medical and forensic genetic testing can tell much about the individual. Although many would accept that there is very good reason for the police to determine information about an unknown suspect’s potential ancestral background and some aspects of their complexion, many might find the potential to determine genetic dispositions to certain disorders as unacceptable. These new technologies need to be used wisely and for the reasons that they are intended. Despite these comments of concern, undoubtedly forensic DNA profiling has led to both exonerations of the innocent and implication of the guilty on a scale never seen before in the criminal justice system. It would be wrong not to embrace new technologies as they become available in the cause of justice.
Disclosure
The authors report no conflicts of interest in this work.
References
Goodwin W, Linacre A, Hadi S. An Introduction to Forensic Genetics. Chichester, West Sussex, UK: Wiley-Blackwell; 2011. | |
Butler J. Advanced Topics in Forensic DNA Typing: Methodology. Waltham, MA: Academic Press; 2012. | |
Liu RH. Forensic DNA Applications. Boca Raton, FL: CRC Press; 2014. | |
Jeffreys AJ, Brookfield JFY, Semeonoff R. Positive identification of an immigration test-case using human DNA fingerprints. Nature. 1985;317(6040):818–819. | |
Seton C. Life for sex killer who sent decoy to take genetic test. The Times. January 23, 1988. | |
Mullis KB, Faloona FA. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 1987;155:335–350. | |
Academies NRCotN. Strengthening Forensic Science in the United States: A Path Forward. Washington, DC: National Academies Press; 2009. | |
Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. | |
Walsh PS, Metzger DA, Higuchi R. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques. 1991;10(4):506–513. | |
Castella V, Dimo-Simonin N, Brandt-Casadevall C, Mangin P. Forensic evaluation of the QIAshredder/QIAamp DNA extraction procedure. Forensic Sci Int. 2006;156(1):70–73. | |
Witt S, Neumann J, Zierdt H, Gébel G, Röscheisen C. Establishing a novel automated magnetic bead-based method for the extraction of DNA from a variety of forensic samples. Forensic Sci Int Genet. 2012;6(5):539–547. | |
Birch DE. Simplified hot start PCR. Nature. 1996;381(6581):445–446. | |
Moretti T, Koons B, Budowle B. Enhancement of PCR amplification yield and specificity using AmpliTaq Gold DNA polymerase. Biotechniques. 1998;25(4):716–722. | |
Kimpton C, Fisher D, Watson S, et al. Evaluation of an automated DNA profiling system employing multiplex amplification of four tetrameric STR loci. Int J Legal Med. 1994;106(6):302–311. | |
Lygo JE, Johnson PE, Holdaway DJ, et al. The validation of short tandem repeat (STR) loci for use in forensic casework. Int J Legal Med. 1994;107(2):77–89. | |
Gill P, Sparkes R, Kimpton C. Development of guidelines to designate alleles using an STR multiplex system. Forensic Sci Int. 1997;89(3):185–197. | |
Kimpton CP, Oldroyd NJ, Watson SK, et al. Validation of highly discriminating multiplex short tandem repeat amplification systems for individual identification. Electrophoresis. 1996;17(8):1283–1293. | |
Sparkes R, Kimpton C, Gilbard S, et al. The validation of a 7-locus multiplex STR test for use in forensic casework. (II), Artefacts, casework studies and success rates. Int J Legal Med. 1996; 109(4):195–204. | |
Sparkes R, Kimpton C, Watson S, et al. The validation of a 7-locus multiplex STR test for use in forensic casework. (I). Mixtures, ageing, degradation and species studies. Int J Legal Med. 1996;109(4):186–194. | |
Krenke BE, Tereba A, Anderson SJ, et al. Validation of a 16-locus fluorescent multiplex system. J Forensic Sci. 2002;47(4):773–785. | |
Templeton A. Out of Africa again and again. Nature. 2002;416(6876):45–51. | |
Ingman M, Kaessmann H, Pääbo S, Gyllensten U. Mitochondrial genome variation and the origin of modern humans. Nature. 2000;408(6813):708–713. | |
Thompson JM, Ewing MM, Frank WE, et al. Developmental validation of the PowerPlex® Y23 System: a single multiplex Y-STR analysis system for casework and database samples. Forensic Sci Int Genet. 2013;7(2):240–250. | |
Turrina S, Ferrian M, Caratti S, De Leo D. Evaluation of genetic parameters of 22 autosomal STR loci (PowerPlex® Fusion System) in a population sample from Northern Italy. Int J Legal Med. 2014;128(2):281–283. | |
Ballantyne KN, Keerl V, Wollstein A, et al. A new future of forensic Y-chromosome analysis: rapidly mutating Y-STRs for differentiating male relatives and paternal lineages. Forensic Sci Int Genet. 2012;6(2):208–218. | |
Bosch E, Calafell F, Santos FR, et al. Variation in short tandem repeats is deeply structured by genetic background on the human Y chromosome. Am J Hum Genet. 1999;65(6):1623–1638. | |
Schmidtke J, Kühnau W, Wand D, Edelmann J, Szibor R, Krawczak M. Prenatal exclusion without involving the putative fathers of an incestuous father-daughter parenthood. Prenat Diagn. 2004;24(8):662–664. | |
Diegoli TM, Coble MD. Development and characterization of two mini-X chromosomal short tandem repeat multiplexes. Forensic Sci Int Genet. 2011;5(5):415–421. | |
Diegoli TM, Linacre A, Coble MD. Population genetic data for 15 X chromosomal short tandem repeat markers in three US populations. Forensic Sci Int Genet. 2014;8(1):64–67. | |
Schanfield MS, Tiesma D, Diegoli T, Coble M, Crawford M. Preliminary report on the anthropology of 15 X STR loci. Am J Phys Anthropol. 2013;150:242–242. | |
Cavelier L, Jazin E, Jalonen P, Gyllensten U. MtDNA substitution rate and segregation of heteroplasmy in coding and noncoding regions. Hum Genet. 2000;107(1):45–50. | |
Garrido N, Griparic L, Jokitalo E, Wartiovaara J, van der Bliek AM, Spelbrink JN. Composition and dynamics of human mitochondrial nucleoids. Mol Biol Cell. 2003;14(4):1583–1596. | |
Holland MM, Fisher DL, Mitchell LG, et al. Mitochondrial DNA sequence analysis of human skeletal remains: identification of remains from the Vietnam War. J Forensic Sci. 1993;38(3):542–553. | |
Zupanič Pajnic I, Gornjak Pogorelc B, Balažic J. Molecular genetic identification of skeletal remains from the Second World War Konfin I mass grave in Slovenia. Int J Legal Med. 2010;124(4):307–317. | |
Gill P, Ivanov PL, Kimpton C, et al. Identification of the remains of the Romanov family by DNA analysis. Nat Genet. 1994;6(2):130–135. | |
Coble MD, Loreille OM, Wadhams MJ, et al. Mystery solved: the identification of the two missing Romanov children using DNA analysis. PLoS ONE. 2009;4(3):e4838. | |
Wilson MR, Holland MM, Stoneking M, DiZinno JA, Budowle B. Guidelines for the use of mitochondrial DNA sequencing in forensic science. Crime Lab Digest. 1993;20(4):68–77. | |
Parson W, Bandelt HJ. Extended guidelines for mtDNA typing of population data in forensic science. Forensic Sci Int Genet. 2007;1(1):13–19. | |
Brotherton P, Haak W, Templeton J, et al; Genographic Consortium. Neolithic mitochondrial haplogroup H genomes and the genetic origins of Europeans. Nat Commun. 2013;4:1764. | |
Just RS, Leney MD, Barritt SM, et al. The use of mitochondrial DNA single nucleotide polymorphisms to assist in the resolution of three challenging forensic cases. J Forensic Sci. 2009;54(4):887–891. | |
Just RS, Loreille OM, Molto JE, et al. Titanic’s unknown child: the critical role of the mitochondrial DNA coding region in a re-identification effort. Forensic Sci Int Genet. 2011;5(3):231–235. | |
Melton T, Clifford S, Kayser M, Nasidze I, Batzer M, Stoneking M. Diversity and heterogeneity in mitochondrial DNA of North American populations. J Forensic Sci. 2001;46(1):46–52. | |
Bandelt HJ, Salas A, Lutz-Bonengel S. Artificial recombination in forensic mtDNA population databases. Int J Legal Med. 2004;118(5):267–273. | |
Forster P. To err is human. Ann Hum Genet. 2003;67(Pt 1):2–4. | |
Briggs AW, Good JM, Green RE, et al. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science. 2009;325(5938):318–321. | |
Maricic T, Whitten M, Pääbo S. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS ONE. 2010;5(11):e14004. | |
Templeton JE, Brotherton PM, Llamas B, et al. DNA capture and next-generation sequencing can recover whole mitochondrial genomes from highly degraded samples for human identification. Investig Genet. 2013;4(1):26. | |
Irwin JA, Parson W, Coble MD, Just RS. mtGenome reference population databases and the future of forensic mtDNA analysis. Forensic Sci Int Genet. 2011;5(3):222–225. | |
Parson W, Dür A. EMPOP – a forensic mtDNA database. Forensic Sci Int Genet. 2007;1(2):88–92. | |
Sykes B. Blood of the Isles. London, UK: Bantam Press; 2006. | |
DNA profiling used to track rapist. Independent. March 24, 2011. Available from: http://www.independent.co.uk/news/uk/crime/dna-profiling-used-to-track-rapist-2251753.html. Accessed August 8, 2014. | |
Liu F, Struchalin MV, Duijn K, et al. Detecting low frequent loss-of-function alleles in genome wide association studies with red hair color as example. PLoS ONE. 2011;6(11):e28145. | |
Walsh S, Liu F, Ballantyne KN, van Oven M, Lao O, Kayser M. IrisPlex: a sensitive DNA tool for accurate prediction of blue and brown eye colour in the absence of ancestry information. Forensic Sci Int Genet. 2011;5(3):170–180. | |
Kayser M, de Knijff P. Improving human forensics through advances in genetics, genomics and molecular biology. Nat Rev Genet. 2011;12(3):179–192. | |
Meyer M, Stenzel U, Myles S, Prüfer K, Hofreiter M. Targeted high-throughput sequencing of tagged nucleic acid samples. Nucleic Acids Res. 2007;35(15):e97. | |
Gnirke A, Melnikov A, Maguire J, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27(2):182–189. | |
Rothberg JM, Hinz W, Rearick TM, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011; 475(7356):348–352. | |
Loman NJ, Misra RV, Dallman TJ, et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012; 30(5):434–439. | |
Merriman B; Ion Torrent R&D Team, Rothberg JM. Progress in ion torrent semiconductor chip based sequencing. Electrophoresis. 2012;33(23):3397–3417. | |
Green RE, Krause J, Briggs AW, et al. A draft sequence of the Neandertal genome. Science. 2010;328(5979):710–722. | |
Krause J, Fu Q, Good JM, et al. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature. 2010;464(7290):894–897. | |
Fu Q, Rudan P, Pääbo S, Krause J. Complete mitochondrial genomes reveal neolithic expansion into Europe. PLoS ONE. 2012;7(3):e32473. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.