Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 7
Finally sofosbuvir: an oral anti-HCV drug with wide performance capability
Received 10 May 2014
Accepted for publication 27 June 2014
Published 8 December 2014 Volume 2014:7 Pages 387—398
DOI https://doi.org/10.2147/PGPM.S52629
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Zeid Kayali,1 Warren N Schmidt2,3
1Division of Gastroenterology and Hepatology, University of Southern California, Los Angeles, CA, USA; 2Department of Internal Medicine and Research Service, Veterans Affairs Medical Center, Iowa City, IA, USA; 3Roy G and Lucille A Carver College of Medicine, University of Iowa, Iowa City, IA, USA
Abstract: Chronic Hepatitis C virus (HCV) infection is the leading cause of advanced liver disease worldwide. The virus successfully evades host immune detection and for many years has hampered efforts to find a safe, uncomplicated, and reliable oral antiviral therapy. Initially, interferon and ribavirin therapy was the treatment standard of care, but it offered limited performance across the wide spectrum of HCV disease and was fraught with excessive and often limiting side effects. Sofosbuvir (SOF) is a potent first-in-class nucleoside inhibitor that has recently been approved for treatment of HCV. The drug has low toxicity, a high resistance barrier, and minimal drug interactions with other HCV direct-acting antiviral agents such as protease inhibitors or anti-NS5A agents. SOF is safe and can be used across different viral genotypes, disease stages, and special patient groups, such as those coinfected with human immunodeficiency virus. When used in combination with ribavirin or another direct-acting antiviral agent, SOF has revolutionized the HCV treatment spectrum and set the stage for nearly universal HCV antiviral therapy. More so than any other anti-HCV drug developed to date, SOF offers the widest applicability for all infected patients, and new regimens will be tailored to maximize performance.
Keywords: Hepatitis C virus, sofosbuvir, polymerase inhibitors, interferon-free treatment
The clinical problem
Chronic Hepatitis C virus (HCV) infection is a serious major global health problem. Worldwide, over 170 million patients are estimated to be infected1 and suffer from chronic hepatitis, high rates of cirrhosis, end-stage liver disease, and increased risk for hepatocellular carcinoma.2 A smaller but significant number of infected patients also show extrahepatic complications such as mixed cryoglobulinemia, glomerulonephritis, arthritis, and some varieties of B-cell lymphoma.3 Following exposure to HCV, only a minority of patients clear the acute infection, whereas 80% persist with life-long chronic viremia if not successfully treated.4 The natural history of liver disease due to HCV is highly variable and profoundly influenced by patient cofactors such as type II diabetes, nontemperate alcohol consumption, fatty liver disease, and human immunodeficiency virus (HIV) coinfection.5
Since discovery of the virus, α-interferon (IFN) by injection has been the backbone drug for antiviral protocols, which have achieved only limited success. Regardless of the IFN preparation, the drug has been plagued with excessive toxicity, therapeutic limitations, and restricted patient availability. Recently, sofosbuvir (SOF), an oral antiviral agent with low toxicity and wide performance capability, has been approved by the US Food and Drug Administration (FDA) and European Union for treatment of chronic HCV infection. Because of the drug’s versatility, low side effect profile, and antiviral efficacy, it clearly is emerging as a first-in-class “breakthrough” drug that will replace IFN and become the new foundation for HCV therapy.
Description of the virus and structure
HCV is a small, 9,500-nucleotide, plus-stranded ribonucleic acid (RNA) virus that replicates in the cytoplasm with a single open-reading frame (Figure 1). The genome contains three major structural proteins and at least six nonstructural (NS) proteins that provide the protease, helicase, and RNA-dependent RNA polymerase (RdRp) activities of the virus.6,7 The plus-stranded viral RNA is first translated into a large polyprotein that is then cleaved by host and viral proteases into structural and enzymatic proteins, respectively.8,9 The small, error-prone RdRp is a major factor accounting for the high mutability and rapid acquisition of viral resistance to designer drugs that target viral sites such as the NS3/4A protease.
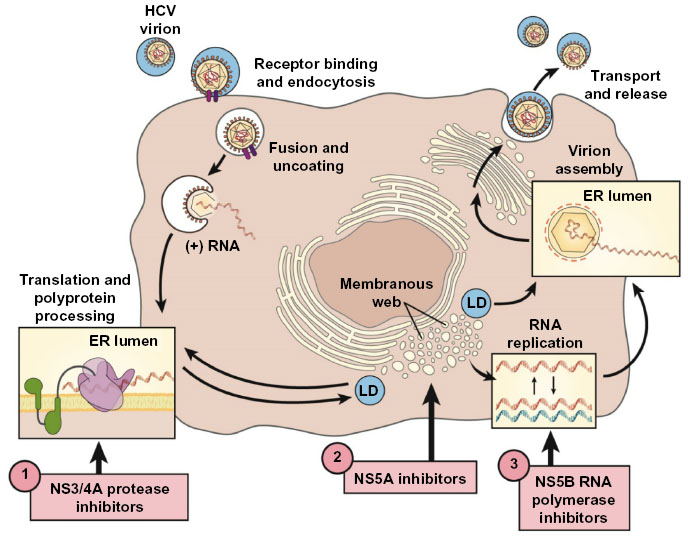 | Figure 1 Therapeutic targets of the HCV replication cycle. |
Early sequencing studies in patient populations established at least six major genotypes with clear differences in global geographic distribution and racial preferences.10 Although different genotypes show minimal overall differences in disease progression, they do exhibit marked differences in susceptibility to antiviral therapy, a fact that individualizes specific treatment paradigms depending on patient genotype. Cloning and sequencing of patient isolates also established that HCV circulates as a swarm of viruses, with small changes in gene sequences termed quasispecies, which arise naturally because of host innate antiviral pressures and the high replicative capacity of the error-prone RdRp.11
Development of effective antiviral therapies for HCV was initially challenging due to the failure of early attempts to pass the virus in cell culture or small animal models.8 Finally, the introduction of replicon systems and permissive cell types in 1999 greatly facilitated in vitro replication assays and boosted drug development.12–14 Replicon constructs enabled high-throughput in vitro testing for a wide variety of prototypical direct-acting antiviral agents (DAAs) that established the proof of concept that specific compounds could target viral proteins such as the NS3/4A protease and serve as antiviral drugs in vivo.15
Early antiviral therapy
Type I α-IFN was the first effective antiviral agent approved for use against chronic HCV infection in the early 1990s, although response rates were generally <20%.16 Sustained virological response (SVR) became the benchmark for successful therapy and is still defined as HCV becoming undetectable in blood during treatment and remaining so 24 weeks after the end of treatment. Recent data indicate that undetectable HCV RNA at 12 weeks after treatment is also highly reliable to predict SVR.17
Numerous clinical trials during the IFN era established guidelines for drug performance and treatment duration, which helped guide management of patients with different host and viral variables. These principles have continued to generate strategies for new DAA drug regimens, patient management, and expectations for patient responses. In spite of the fact that type I IFNs are effective natural antiviral agents, they are accompanied by rather severe side effects such as cytopenias, constitutional signs, and changes in mental status that limit their use in different patient populations.
The introduction of first-generation protease inhibitors (PIs) telaprevir and boceprevir, to be used in combination with pegylated IFN and ribavirin (RBV) (PR), provided the first demonstration that DAAs could increase SVR rates and shorten treatment duration. Either PI significantly boosted SVR rates in genotype 1 treatment-naïve (TN) or treatment-experienced (TE) patients when used with PR-based regimens.18–23 In spite of the success of first-generation PIs, there were major drawbacks. Both drugs have serious systemic side effects and a high daily pill burden, which contributed to suboptimal treatment adherence. Justified concerns for the tolerability of either PI led to yet newer patient barriers that further complicated an already rigorous PR regimen. Consequently, an unacceptable percentage of patients were barred from triple therapy, and many were simply unwilling to be treated, because of poor quality of life and anticipation for IFN-free protocols.
Because of the limitations of PR-based therapies, patient parameters were established to balance eligibility and potential benefits of treatment. A variety of host and viral cofactors were shown to significantly influence treatment outcome and needed to be taken into consideration when deciding to start a patient on IFN-based therapies.5 Poor prognostic factors included variables such as viral genotype 1 (especially 1a and 4 as compared with genotypes 1b and 2), high viral load ($800,000 IU/mL), lack of response or relapse to prior PR-based therapy, male sex, age >55 years, African American race, obese body mass index (BMI) (>30), IL28B carrying the T-allele,24 coinfection with HIV,25 and cirrhosis, especially with hepatic decompensation. These factors not only limited PR treatment decisions for both patient and provider but also tended to exclude “hard to treat” patient cohorts when testing new treatment paradigms.
The new DAA era
During the IFN era, it was obvious that future therapies would need to be less complicated, of shorter duration, and much less toxic to be applicable across the wide HCV disease spectrum. Theoretically, drug interference with any of the vital intracellular activities of the viral life cycle could be used to eradicate viral infection. DAAs of all the major classes were designed to directly inhibit viral enzymes and proteins. The NS proteins NS3/4A protease–helicase and NS5B RdRp and the NS5A protein all perform crucial activities for the viral life cycle and by far have been the favorite targets for development of new DAAs (Figure 1).26 A multitude of Phase II and III studies have pointed out advantages and disadvantages of the major classes of DAAs (Table 1).
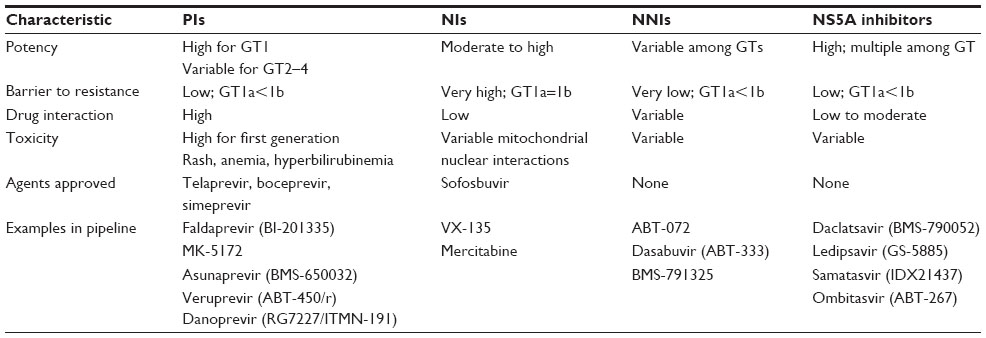 | Table 1 Major direct-acting antiviral drugs |
Nucleotide polymerase inhibitors
Nucleotide polymerase inhibitors (NIs) such as SOF represent some of the earliest DAAs designed for HCV therapy. Structurally, NIs are usually analogs of natural nucleosides and, mechanistically, they competitively inhibit attachment of natural nucleotides to the growing RNA chain, thus causing abrupt chain termination. Because the catalytic site of the RdRp is highly conserved, resistance mutations are poorly tolerated, and all NIs show broad pangenotypic antiviral activity with only limited half maximal inhibitory concentration differences between genotypes.27 Moreover, the most common resistance mutation (serine 282 to threonine [S282T]) has reduced viral fitness in tissue culture studies28,29 and in vivo does not appear to persist in treatment nonresponders when NI is discontinued. In chimpanzees30 as well as in patients, S282T is rarely observed and is not thought to be responsible for viral relapse.31,32
On the other hand, development of clinically useful drugs of this class has been problematic. Candidate NIs have shown a high attrition rate, and of 15 drugs that entered clinical development to date, only SOF has been approved for use. Most of the failed drugs were halted because of clinical toxicities and safety concerns but a few just performed poorly. The first two NI prototypes, NM283 and balapiravir (R1626), were stopped in Phase II development due to toxicity and lack of efficacy.33 Another drug, IDX184, has been placed on hold because of potential cardiac toxicity, although the drug was not given to patients. Testing of another NI, VX-135, at high potential monotherapy dosages was recently halted because of high liver enzyme abnormalities in patients. However, lower dosages of VX-135 are still being tested in Phase II studies in combination with either simeprevir (SMV) or daclatasvir (DCV). Another important NI currently in Phase II and III trials at this time is mericitabine (RG7128). This drug has moderate antiviral potency, a low side effect profile,34 and no in vivo resistance shown to date.35 In fact, the A Randomized, Placebo-controlled, Dose-ranging Study to Evaluate the Safety, Tolerability and Antiviral Activity of Combination Treatment With an HCV Polymerase Inhibitor (RO5024048) and an HCV Protease Inhibitor (RO5190591) in Genotype 1 Chronic Hepatitis C Patients (INFORM) trial used this drug in combination with the protease inhibitor danoprevir and provided the first proof of concept that oral antivirals could be safely coadministered to patients and suppress viral replication without IFN.36
Non-nucleoside polymerase inhibitors
Non-nucleoside polymerase inhibitors (NNIs) affect viral polymerase performance through allosteric binding and noncompetitive inhibition, in contrast to NIs, which competitively inhibit the enzyme catalytic site. Because NNIs affect only polymerase performance, their inhibition is relatively well tolerated by the virus, resulting in a low drug resistance barrier as compared with NIs, and viral breakthrough has frequently been observed in monotherapy studies.37,38 In spite of these shortcomings, Phase II and III trials of NNIs in combination with other DAAs are extremely promising.38 NNIs are likely effective in combination with PIs because there is no cross-resistance with common PI mutations.39 Multiple drugs are now in development (Table 1), and all will likely be used in multioral drug regimens to overcome their lower barrier to resistance.36
Protease inhibitors
The first-generation PIs boceprevir and telaprevir were characterized by narrow genotype 1 sensitivity, low barrier to viral resistance mutations, and relatively high side effect profile as compared with later second-generation PIs (such as SMV) and DAAs of other classes.37,40 Early-phase trials of prototypical antiproteases showed that resistance developed early when used as monotherapy.41 Consequently, all PIs must be used in combination with drugs of a different class, preferably one with a high resistance barrier, such as SOF.
HCV NS5A inhibitors
NS5A protein plays an important role in HCV replication, assembly, and release.6,42 Due to the conserved structural feature of domain 1 of NS5A, currently developed NS5A inhibitors are highly effective against all HCV genotypes, but they have a low genetic barrier to resistance, which results in rapid selection of resistant mutants (Table 1).43,44 Nevertheless, because of their pangenotypic activity, NS5A inhibitors show considerable promise for use in combination with drugs of a different DAA class, most likely an NI with broad genotypic coverage. In fact, the combination of ledipasvir (LDV) and SOF is currently awaiting approval by the FDA, and other NS5A and SOF combinations are in advanced testing.
Sofosbuvir: pharmacology, mode of action, and pharmacokinetics
SOF, originally called GS-7977 and now marketed with the trade name Sovaldi® (Gilead Corporation, Foster City, CA, USA), is a second-generation NI. The drug was approved by the FDA in December 2013 for treatment of HCV genotypes 2 and 3 in combination with RBV, and for genotypes 1 and 4 in combination with PR, based on data from nearly 3,000 patients studied in Phase II and III trials.45,46 SOF provides the first effective IFN-free regimen for genotype 2 and 3 patients and also offers a shorter treatment course (12 weeks) for patients with genotypes 1 or 4 when given in combination with PR. Most importantly, SOF alone has low toxicity and is highly compatible with antiviral drugs of other DAA classes, demonstrating that it is an attractive foundation drug for a variety of effective combinations and treatment regimens.
Structurally, SOF is a uridine nucleotide prodrug (Figure 2) with rapid intestinal absorption and is easily taken up by hepatocytes from the circulation. Intracellularly, the side chains on the phosphate are removed and the 2'-deoxy-2'-fluoro-2'-C-methyluridine monophosphate GS-606965 is converted into the uridine triphosphate form GS-461203 (Figure 2), which is now able to compete with endogenous uridine triphosphate for incorporation into the growing viral RNA chain (Figure 2).47 After binding of the nucleotide to the RNA chain, further addition of nucleotides is not possible and chain elongation is terminated. Fortunately, the uridine triphosphate form has limited, if any, binding affinity for host RNA or deoxyribonucleic acid polymerases.48
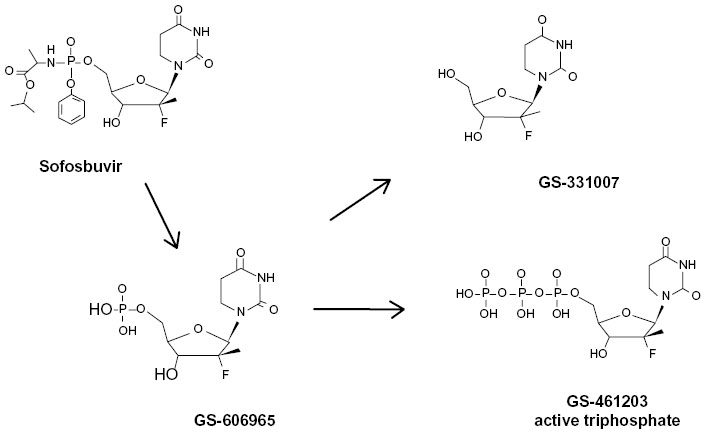 | Figure 2 Activation of sofosbuvir. |
The triphosphate metabolite is cleared by the kidney and there is no need to adjust SOF dosage in patients with glomerular filtration rate >30 mL/min/1.7 m2. The primary circulating metabolite is the GS-331007. Intracellular metabolism does not use P450 3A/4 (Cytochrome P450 [CYP] 3A/4), and no serious drug–drug interactions have been identified to date. The triphosphate metabolite has a very long intracellular half-life of >17.8 hours (from canine data), which permits the drug to be taken once daily.
Efficacy, safety, and tolerability of sofosbuvir
Oral sofosbuvir monotherapy with or without ribavirin: Phase II and III studies
Phase II studies of oral SOF with or without RBV (ELECTRON) provided novel and exciting early IFN-free treatment data (Table 2). Although the significance of ELECTRON was limited due to small sample sizes, it provided important proof of concept that a potent oral NI such as SOF, in combination with RBV, can produce SVR without the need for a type I IFN. Initially, SOF and RBV were given for 12 weeks to 35 genotype 1 patients of whom 25 were TN and ten TE.31 Two additional groups of ten genotype 2 and 3 patients were treated with SOF only or SOF + RBV. SVR rates of the genotype 1 TN and TE who received SOF + RBV were 84% and 10%, respectively. The high SVR in the TN genotype 1 group was surprising, considering that 88% of patients were subtype 1a and over half had unfavorable IL28B T-allele (CT or TT). In general, the latter finding has not been supported by data from larger fully randomized genotype 1 patient groups treated for 12 weeks.51 Moreover, in the group of ten genotype 1 TE patients who also received SOF and RBV, only one achieved SVR. Interestingly, treatment failure was entirely due to viral relapse, as all patients cleared virus while on therapy.31 In contrast, genotype 2 and 3 data were much more encouraging, as six of ten TN genotype 2/3 patients achieved SVR after 12 weeks of SOF even without RBV (Table 2).
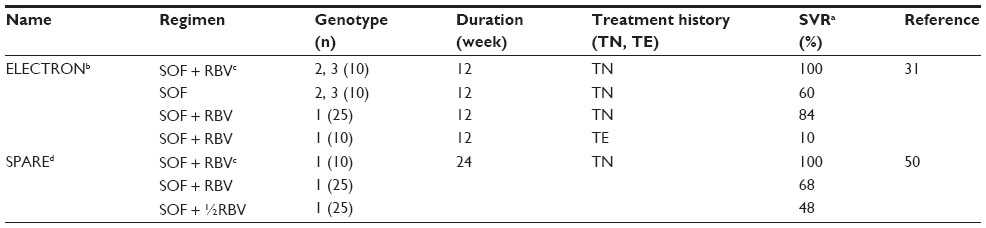 | Table 2 Sofosbuvir (SOF) Phase II trials |
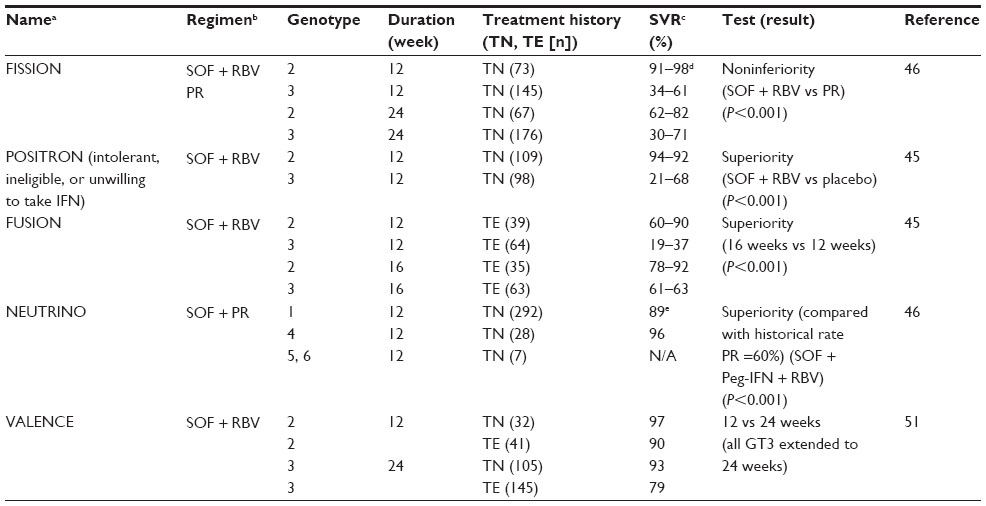
Collectively, ELECTRON findings suggested important treatment parameters that would eventually be verified by Phase III studies. First, the data showed that an additional antiviral, in this case RBV, enhances SVR, similar to the effect seen much earlier with IFN and RBV.49 Second, SOF in combination with RBV was suggested to elicit superior rates of SVR in genotype 2 and 3 patients as compared with conventional PR therapy. Finally, ELECTRON also showed that retreating TE patients with an IFN-free regimen can still result in a high viral relapse rate, in spite of the fact that most patients cleared virus on oral therapy. This implied that longer treatment durations would likely be more efficacious in these patients, a concept that was finally documented with Phase III data (Table 3).45
 | Table 3 Sofosbuvir (SOF) ± ribavirin (RBV) Phase III trials |
Assessment of the adverse events (AEs) encountered in initial Phase I as well as in Phase II testing of SOF (Table 2) showed that the drug is nontoxic and widely tolerable. There were few serious AEs, and reported side effects were mostly minor constitutional signs that did not lead to high noncompliance or discontinuation of drug (1%). For these reasons, SOF could be tested over the most diverse and hard to treat populations of patients ever used in treatment trials of HCV-infected patients. A significant proportion of patients from the SOF studies would have been excluded from participation in IFN-based trials. Consequently, the safety and reliability of the drug in HCV special patient populations have been reassuring.
The National Institutes of Health SPARE trial purposely tested hard to treat TN patients with 24 weeks of oral SOF in combination with full or half dosage RBV in 60 subjects.50 Overall, patients were 70% viral subtype 1a, 62% male, 83% African American, 80% non-CC IL28 genotype, 48% BMI >30, 23% with advanced stage F3 or F4 liver disease, and >60% had high viral loads. Of ten patients with early fibrosis scores and minimal liver disease who received SOF and full dosage RBV, SVR was 100%. In contrast, of 24 patients with higher fibrosis scores, only 67% achieved SVR with SOF and full dosage RBV and only 48% achieved SVR when given SOF and half dosage RBV. As all patients in the SPARE trial had undetectable viral levels at the end of drug therapy, differences in SVR outcome were due to relapse. These findings suggest that important variables for relapse, such as fibrosis score and use of RBV, are also important for SOF protocols, similar to earlier data seen for PR therapy. Nevertheless, differences in relapse across patient groups can be nearly eliminated with SOF in combination with another DAA, as discussed in the following section (also see Table 4).
 | Table 4 Sofosbuvir (SOF) in combination ± ribavirin (RBV) Phase II–III trials |
Five Phase III trials studied genotype 1, 2, and 3-infected patients when treated with SOF in combination with RBV or PR (FISSION, POSITRON, FUSION, NEUTRINO, and VALENCE, Table 3).45,46,51 Although the majority of Phase III studies were performed on TN patients, no trial specifically looked at TE genotype 1 patients, only two studies looked at TE genotype 2 and 3 (FUSION and VALENCE), and the latter included relatively small patient numbers. On the other hand, it is important to point out that a wide diversity of individuals were studied here for the first time, many of whom were intolerant, unwilling, or ineligible to receive IFN and would not have been included in conventional PR trials. Furthermore, FISSION, POSITRON, FUSION, and NEUTRINO contained a generous mix of cirrhotic patients: 20%, 16%, 34%, and 17%, respectively. All in all, these findings were crucial for the FDA decision to fully approve SOF.
POSITRON established that an oral regimen of SOF + RBV for 12 weeks in TN and TE genotype 2 or 3 patients was superior to placebo. In FISSION, study of TN genotype 2 or 3 patients demonstrated that the all oral regimen was noninferior to historical PR therapy for 24 weeks, the standard of care. Remarkably, SOF + RBV elicited 92% SVR in genotype 2 patients but only 56% SVR in genotype 3 patients. Lower SVR rates for TE genotype 3 patients when given 12 or 16 weeks of all oral therapy were again documented in FUSION. Nevertheless, the poor response of genotype 3 patients could be remedied with even longer treatment times of 24 weeks, as shown in the VALENCE trial. TN and TE genotype 3 patients achieved SVR of 93% and 79% if treated with SOF + RBV for 24 weeks. NEUTRINO demonstrated that SOF can also be used to great effect when combined with PR in shortened (12 week) regimens to treat genotypes 1 and 4 (Table 3). However, widespread use of a PR + SOF combination appears unlikely, as the field is dramatically changing into completely IFN-free regimens with the anticipated approval of SOF in combination with other DAAs such as PIs and anti-NS5A drugs for genotype 1 patients (Table 4). Finally, here again, cirrhotic patients consistently had lower SVR12 rates as compared with noncirrhotics regardless of treatment history. This was more evident in genotype 3 compared with genotype 2 patients, with the exception of POSITRON, where SVR12 for cirrhotics was 94% versus 92% for noncirrhotics. SVR12 for cirrhotics in NEUTRINO was 80% versus 92% for noncirrhotics.
Sofosbuvir in combination with simeprevir, daclatasvir, or ledipasvir
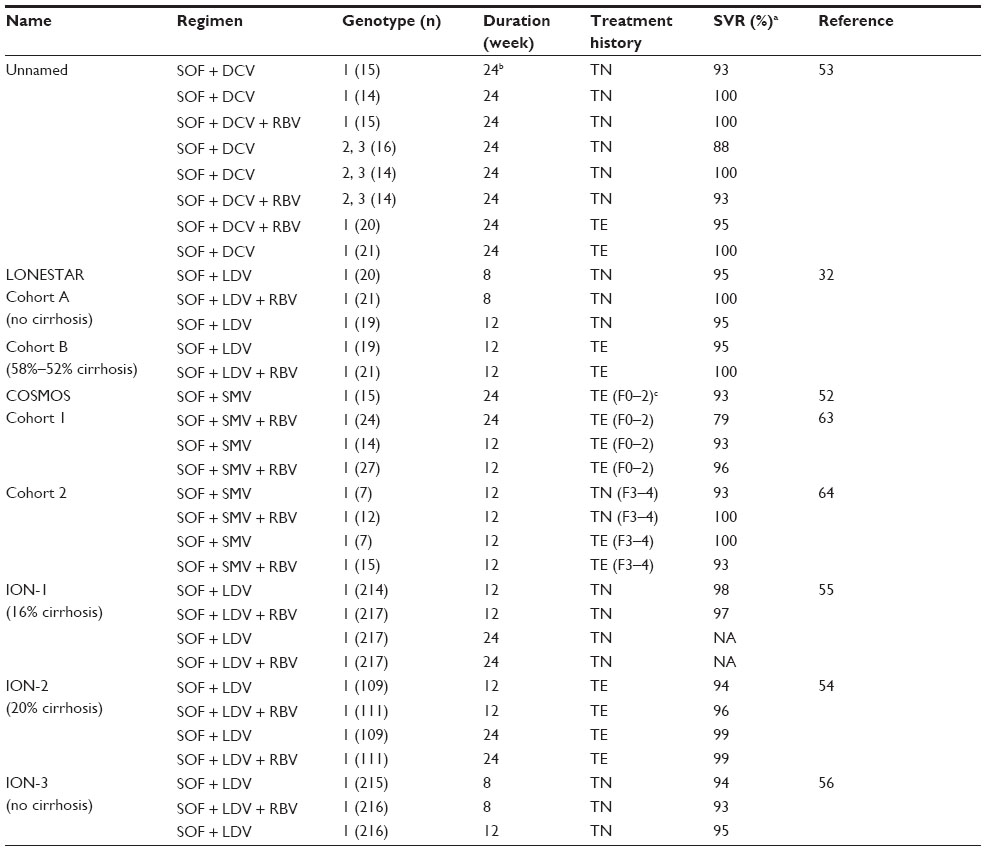
Phase II and III trials of SOF in combination with a DAA from another drug class have shown startling SVR rates at close to 100% for nearly all treated patients (Table 4). In these studies, SOF was combined with the PI SMV (COSMOS)52 or either of the anti-NS5A drugs DCV53 or LDV (LONESTAR32 and ION54–56). The two DAA drug combinations, regardless of whether RBV was included, performed impressively and led to SVR rates between 79% and 100%.
The recent approval of SMV, an effective second-generation PI, presented the perfect opportunity for a combination trial with SOF (Table 4). The COSMOS study included TN and TE cohorts with many patients having advanced fibrosis. There was no difference in SVR12 rates for advanced fibrosis patients (F3 and F4) when treated with SOF + SMV for 24 weeks with or without RBV (both 100%). There was also no difference in SVR12 rates for F3 and F4 patients treated for 12 or 24 weeks with SOF + SMV. TE and TN patients showed no difference in SVR12 when 12 and 24 weeks of SOF + SMV were compared. Finally, SVR12 in all groups was not affected by the presence of Q80K polymorphism and genotype 1 subtype.
The necessity for RBV to decrease relapse seen in the SOF + RBV studies (Tables 2 and 3) was obviated in the SOF + DAA studies of Table 4, regardless of whether the codrug was the PI SMV or either of the anti-NS5A drugs. Where tested (COSMOS and ION), 12 weeks of therapy was noninferior to 24 weeks to achieve SVR. Furthermore, it now appears that SOF in combination with LDV for 8 weeks may be just as effective as 12 weeks in genotype 1 patients without cirrhosis, as established by LONESTAR and ION-3 data (Table 4). Of the four major two-DAA combinations shown in Table 4, only SOF + DCV was tested against genotypes 2 and 3. However, Phase II data (ELECTRON 2) recently shown at European Association for Study of Liver Disease (EASLD) in 2014 demonstrated that 100% (26/26) of TN genotype 3 patients achieved SVR12 after 12 weeks of LDV + SOF + RBV.57 However, the LDV + SOF combination was not as effective if RBV was excluded in genotype 3 patients. Only 16 of 25 TN genotype 3 patients achieved SVR12 after 12 weeks of LDV/SOF without RBV.
Across special difficult to treat populations such as cirrhotics, in patients with IL-28b T-alleles and viral genotypes (1a and 3 where tested), the SOF combinations elicited a high SVR without significant differences between groups. Ironically, important predictors for success with PR-based therapies and, to a lesser degree, SOF + RBV (Tables 2 and 3) were not significant in the data of the SOF + DAA studies listed in Table 4. A possible exception occurred in the TE patients of ION-2 where a small but significant difference was noted in the SVR of cirrhotics (82%–86%) versus noncirrhotics (95%–100%) treated for 12 weeks.54 This difference was not observed for the same groups treated for 24 weeks, suggesting that cirrhotics may benefit from longer treatment times, at least if given SOF + LDV. Finally, even important “on treatment” PR response predictors such as early clearance of virus by 4 weeks of therapy were not found to be statistically significant in the SOF and DAA combination trials. As it was well demonstrated that even SOF monotherapy usually leads to undetectable virus at end of therapy, the major increase in SVR was due to a reduction of patient relapse in susceptible groups. How the extra drug, either PI or anti-NS5A, so effectively influences patient relapse is not clear at this time and should be evaluated further. Finally, study of earlier treatment intervals (≤8 weeks in all patient groups) may actually reveal even shorter but adequate treatment periods for some individuals.
The study of SOF in combination with either RBV or another DAA in other traditionally hard to treat patient groups such as liver transplant recipients or those coinfected with HIV has also been impressive. Although these investigations are not as far along, recent findings indicate that overall rates of SVR are very similar for HCV coinfected patients as compared with monoinfected genotype 1, 2, and 3 patients.58 Furthermore, early studies have looked at the effects of SOF in patient groups both pre- and postliver transplant. Historically, these patients are notoriously hard to treat with PR-based therapies. In the pretransplant setting, 61 patients of different genotypes received up to 48 weeks of SOF +RBV right up to transplant time. Nearly all patients cleared virus, and recurrent HCV was effectively prevented in 64% of transplanted individuals.59 In the post-transplant setting of recurrent HCV graft infection, 77% of 40 patients with genotypes 1 (83%), 3 (15%), or 4 (3%) treated with SOF + RBV for 24 weeks achieved at least SVR4.60
In all the SOF + DAA trials noted to date, the drug combinations were well tolerated. The most common AEs included constitutional signs such as intermittent fatigue, headache, nausea, and insomnia. By far the highest AEs were seen in patients who also received RBV, and, as might be expected, those patients had high incidences of hemolytic anemia. Serious AEs were few and included single events, without a trend to be connected with SOF.
SOF has relatively few drug–drug interactions.61,62 The GS-331007 metabolite (Figure 2) is cleared renally, and neither the prodrug nor metabolites are a substrate or inducer of CYP 450 pathways. However, the parent drug SOF is a substrate of intestinal drug transport P-glycoprotein and breast cancer-resistant protein, although the renal cleared metabolite (GS-331007) is not. Consequently, use of SOF with potent inducers of P-glycoprotein such as rifampin or St John’s Wort would likely decrease plasma levels of the parent drug and is not recommended. Furthermore, coadministration of SOF with anticonvulsants such as phenytoin, carbamazepine, phenobarbital, and oxcarbazepine; antimycobacterials such as rifabutin and rifapentine; and antiretrovirals such as tipranavir/ritonavir are also not recommended for similar reasons.
Overall, SOF appears to be an attractive choice to serve as a backbone for future DAA combinations. Recently, the FDA granted priority review to Gilead’s New Drug Application for a once-daily fixed-dose combination of SOF (400 mg) and LDV (90 mg) for the treatment of chronic hepatitis C genotype 1 infection in adults. As of this writing, the FDA has set a target action date under the Prescription Drug User Fee Act of October 10, 2014 for approval decision.
Resistance to sofosbuvir
SOF has been studied in several thousand patients to date, and effective in vivo drug resistance leading to viral escape and relapse in response to therapy appears to be, at best, a rare event. In the registration Phase III studies of Table 3, no genotypic or phenotypic resistance was detected. In Phase II trials, S282T mutation was detected in only one relapse patient, and this occurred after SOF monotherapy. The patient was then treated successfully with SOF + RBV.31 One patient in the LONESTAR study had S282T mutation at relapse.32 However, of the multitude of patients seen in the other trials of SOF ± DCV, SMV, or LDV, no S282T was detected.53–56 In contrast, viral mutations in the respective SOF codrugs occurred much more commonly in as much as 20% of patients. However, none was suspected to be responsible for relapse, and the vast majority of patients reached SVR. Consequently, in contrast to the problems noted with first-generation PIs, SOF resistance does not appear to affect patient outcome, especially when SOF is used in combination with another DAA.
Overall, SOF has eliminated most of the treatment barriers for patients that were present in the IFN era. Nevertheless, making treatment decisions among patients still requires thoughtful consideration, as host factors such as cirrhosis and viral factors such as genotype still have significant influences on treatment times and, in some cases, outcome. An important principle is that monotherapy with SOF is not recommended.
Patient care and quality of life
The introduction of a safe drug with the potential to cure a serious infection that affects millions of people worldwide is indeed a rare event in medicine history. For health care providers who have worked in the HCV treatment arena since the virus’s discovery over 24 years ago, this is truly momentous. Remarkably, the proof of concept has now been well demonstrated. SOF is a nontoxic but very effective antiviral agent that is heralding a new era of all oral IFN-free therapy for the management of most, if not all, chronic HCV infection. There is no doubt that the complicated and highly toxic PR-based protocols for HCV therapy will be largely replaced with a SOF-based or equivalent combination of oral drugs that will benefit the vast majority of HCV patients. SOF is the first in kind of a wide array of oral drugs that offer multiple advantages: eg, single daily dosage, reduced treatment durations, and few, if any, dosing restrictions recognized to date. In combination, SOF elicits high SVR rates across multiple patient cohorts, including hard to treat patients, with little evidence for viral resistance to cause treatment failure. It is ironic that the toxicity and only modest efficacy of PR protocols restricted access and the hope for cure among the majority of HCV-positive individuals, yet a simple and far more tolerable oral drug is now available to greatly expand the HCV treatment spectrum.
SOF can be used in many patients who were previously IFN ineligible, such as those with severe depression, decompensated cirrhosis, and marginal platelet counts. Currently, the FDA-approved regimens allow many of these patients to receive SOF + RBV for 24 weeks with noninferior efficacy as compared with IFN regimens. Patients with anemia or hemoglobin disorders who cannot receive RBV are now eligible to be treated with RBV-free regimens such as SOF +SMV, which are available now although off label, as well as SOF +LDV, which is anticipated in the near future. Although specific Phase III or IV trials treating IFN-ineligible patients are yet forthcoming, it is likely that multiple DAA regimens will become available and tailored for use in these populations.
From the patient perspective, an optimal quality of life during therapy as well as improved mortality and reduced morbidity are now feasible and realistic. Although it appears that the present DAA combinations are starting to normalize, showing a “one size fits all” treatment program, it is likely that new drugs in the treatment pipeline will open the door for more personalized care, such as shorter treatment intervals and more focused therapeutic options depending on clinical necessity. This will have to be determined empirically with large patient access trials and/or Phase IV studies.
Conclusion
With the advent of oral regimens such as SOF that clearly obviate the use of IFN, we have entered the final, long-awaited chapter of HCV therapeutics. IFN-free therapy is a major milestone in liver disease and not only eliminates the IFN side effects barrier but expands patient treatment options, eligibility, comfort, convenience, and efficacy. SOF has clearly helped establish the important proof of concept that chronic HCV infection can be cured in most, if not all, affected individuals.
Acknowledgments
Supported by a Merit Review grant from the Veterans Affairs Medical Center, Iowa City, IA, USA (BX000159) and the Doriann Foundation for Hepatitis Research, University of Iowa, Iowa City, IA, USA (WNS).
Author contributions
Warren N Schmidt and Zeid Kayali both contributed to the study design, concepts, data management, manuscript draft, and revisions.
Disclosure
Zeid Kayali has received research grants and provided consultant services for Abbvie, Gilead, Merck, and Bristol Meyer Squibb. Warren N Schmidt has served as an advisor for Gilead and Merck.
References
Hope VD, Eramova I, Capurro D, et al. Prevalence and estimation of hepatitis B and C infections in the WHO European Region: a review of data focusing on the countries outside the European Union and the European Free Trade Association. Epidemiol Infect. 2014;142: 270–286. | |
Naggie S, Patel K, McHutchison J. Hepatitis C virus directly acting antivirals: current developments with NS3/4A HCV serine protease inhibitors. J Antimicrob Chemother. 2010;65:2063–2069. | |
Kayali Z, Labrecque DR, Schmidt WN. Treatment of hepatitis C cryoglobulinemia: mission and challenges. Curr Treat Options Gastroenterol. 2006;9:497–507. | |
Hoofnagle JH. Hepatitis C: the clinical spectrum of disease. Hepatology. 1997;26:S15–S20. | |
Rosen HR. Chronic hepatitis C infection. N Engl J Med. 2011;364: 2429–2438. | |
Lindenbach BD, Rice CM. Unravelling hepatitis C virus replication from genome to function. Nature. 2005;436:933–938. | |
Major ME, Feinstone SM. The molecular virology of hepatitis C. Hepatology. 1997;25:1527–1538. | |
Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol. 2000;81:1631–1648. | |
Lindenbach BD, Evans MJ, Syder AJ, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. | |
Simmonds P, Holmes EC, Cha TA, et al. Classification of hepatitis C virus into 6 major genotypes and a series of subtypes by phylogenetic analysis of the Ns-5 region. J Gen Virol. 1993;74:2391–2399. | |
Domingo E, Holland JJ. RNA virus mutations and fitness for survival. Annu Rev Microbiol. 1997;51:151–178. | |
Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. 2002;76:13001–13014. | |
Lohmann V, Korner F, Koch JO, et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285: 110–113. | |
Pietschmann T, Lohmann V, Rutter G, et al. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J Virol. 2001;75: 1252–1264. | |
Hinrichsen H, Benhamou Y, Wedemeyer H, et al. Short-term antiviral efficacy of BILN 2061, a hepatitis C virus serine protease inhibitor, in hepatitis C genotype 1 patients. Gastroenterology. 2004;127:1347–1355. | |
Hoofnagle JH, Mullen KD, Jones DB, et al. Treatment of chronic non-A, non-B hepatitis with recombinant human alpha interferon. A preliminary report. N Engl J Med. 1986;315:1575–1578. | |
Martinot-Peignoux M, Stern C, Maylin S, et al. Twelve weeks posttreatment follow-up is as relevant as 24 weeks to determine the sustained virologic response in patients with hepatitis C virus receiving pegylated interferon and ribavirin. Hepatology. 2010;51:1122–1126. | |
Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364:2405–2416. | |
Poordad F, McCone J Jr, Bacon BR, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011;364:1195–1206. | |
Sherman KE, Flamm SL, Afdhal NH, et al. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med. 2011;365:1014–1024. | |
McHutchison JG, Manns MP, Muir AJ, et al. Telaprevir for previously treated chronic HCV infection. N Engl J Med. 2010;362:1292–1303. | |
Muir AJ, Poordad FF, McHutchison JG, et al. Retreatment with telaprevir combination therapy in hepatitis C patients with well- characterized prior treatment response. Hepatology. 2011;54: 1538–1546. | |
Bacon BR, Gordon SC, Lawitz E, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med. 2011;364: 1207–1217. | |
Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology. 2010;139:120–178. | |
Koziel MJ, Peters MG. Viral hepatitis in HIV infection. N Engl J Med. 2007;356:1445–1454. | |
Soriano V, Peters MG, Zeuzem S. New therapies for hepatitis C virus infection. Clin Infect Dis. 2009;48:313–320. | |
Sarrazin C, Zeuzem S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology. 2010;138:447–462. | |
Migliaccio G, Tomassini JE, Carroll SS, et al. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J Biol Chem. 2003;278:49164–49170. | |
Lam AM, Espiritu C, Bansal S, et al. Genotype and subtype profiling of PSI-7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob Agents Chemother. 2012;56:3359–3368. | |
Carroll SS, Ludmerer S, Handt L, et al. Robust antiviral efficacy upon administration of a nucleoside analog to hepatitis C virus-infected chimpanzees. Antimicrob Agents Chemother. 2009;53:926–934. | |
Gane EJ, Stedman CA, Hyland RH, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med. 2013;368:34–44. | |
Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet. 2014;383:515–523. | |
Nelson DR, Zeuzem S, Andreone P, et al. Balapiravir plus peginterferon alfa-2a (40KD)/ribavirin in a randomized trial of hepatitis C genotype 1 patients. Ann Hepatol. 2012;11:15–31. | |
Pockros P, Jensen D, Tsai N, et al. First SVR data with the nucleoside analogue polymerase inhibitor mericitabine (RG7128) combined with peginterferon/ribavirin in treatment-naive HCV G1/4 patients: interim analysis from the JUMP-C trial. J Hepatol. 2011;54:S538–S538. | |
Pawlotsky JM, Najera I, Jacobson I. Resistance to mericitabine, a nucleoside analogue inhibitor of HCV RNA-dependent RNA polymerase. Antivir Ther. 2012;17:411–423. | |
Gane EJ, Roberts SK, Stedman CAM, et al. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet. 2010;376:1467–1475. | |
Pawlotsky JM. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology. 2011;53: 1742–1751. | |
Schmidt WN, Nelson DR, Pawlotsky JM, et al. Direct acting antiviral agents and the path to interferon independence. Clin Gastroenterol Hepatol. 2014;12(5):728–737. | |
Susser S, Welsch C, Wang YL, et al. Characterization of resistance to the protease inhibitor boceprevir in hepatitis C virus-infected patients. Hepatology. 2009;50:1709–1718. | |
Bartenschlager R, Lohmann V, Penin F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat Rev Microbiol. 2013;11:482–496. | |
Kieffer TL, Sarrazin C, Miller JS, et al. Telaprevir and pegylated interferon-alpha–2a inhibit wild-type and resistant genotype 1 hepatitis C virus replication in patients. Hepatology. 2007;46:631–639. | |
Targett-Adams P, Graham EJ, Middleton J, et al. Small molecules targeting hepatitis C virus-encoded NS5A cause subcellular redistribution of their target: insights into compound modes of action. J Virol. 2011;85:6353–6368. | |
Gao M, Nettles RE, Belema M, et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465:96–108. | |
Wang CF, Huang HC, Valera L, et al. Hepatitis C virus RNA elimination and development of resistance in replicon cells treated with BMS-790052. Antimicrob Agents Chemother. 2012;56: 1350–1358. | |
Jacobson IM, Gordon SC, Kowdley KV, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med. 2013;368(20):1867–1877. | |
Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 2013;368:1878–1887. | |
Murakami E, Tolstykh T, Bao H, et al. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J Biol Chem. 2010;285: 34337–34347. | |
Lam AM, Murakami E, Espiritu C, et al. PSI–7851, a pronucleotide of beta-D–2'-deoxy–2'-fluoro–2'-C-methyluridine monophosphate, is a potent and pan-genotype inhibitor of hepatitis c virus replication. Antimicrob Agents Chemother. 2010;54:3187–3196. | |
Davis GL, Esteban-Mur R, Rustgi V, et al. Interferon alfa-2b alone or in combination with ribavirin for the treatment of relapse of chronic hepatitis C. International Hepatitis Interventional Therapy Group. N Engl J Med. 1998;339:1493–1499. | |
Osinusi A, Meissner EG, Lee YJ, et al. Sofosbuvir and ribavirin for hepatitis C genotype 1 in patients with unfavorable treatment characteristics a randomized clinical trial. JAMA. 2013;310: 804–811. | |
Zeuzem S, Dusheiko GM, Salupere R, et al. Sofosbuvir plus ribavirin for 12 or 24 weeks for patients with HCV genotype 2 or 3: the VALENCE trial. Hepatology. 2013;58:733a–734a. | |
Jacobson IM, Ghalib RH, Rodriguez-Torres M, et al. SVR results of a once-daily regimen of simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naive and prior null responder patients: the COSMOS study. Hepatology. 2013;58:1379a–1380a. | |
Sulkowski MS, Gardiner DF, Rodriguez-Torres M, et al. Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N Engl J Med. 2014;370:211–221. | |
Afdhal N, Reddy KR, Nelson DR, et al. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med. 2014;370:1483–1493. | |
Afdhal N, Zeuzem S, Kwo P, et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med. 2014;370: 1889–1898. | |
Kowdley KV, Gordon SC, Reddy KR, et al. Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med. 2014;370:1889–1898. | |
Gane EJ, Hyland RH, An D, et al. Sofosbuvir/Ledipasvir fixed dose combination is safe and effective in difficult-to-treat populations including genotype-3 patients, decompensated genotype-1 patients, and genotype-1 patients with prior Sofosbuvir treatment experience. J Hepatol. 2014;60:S3. | |
Naggie S, Sulkowski M, Lalezari J, et al. Sofosbuvir plus ribavirin for HCV genotype 1–3 infection in HIV coinfected patients (PHOTON-1). Conference on Retroviruses and Opportunistic Infections; March 3–6, 2014; Boston, MA, USA. Abstract 26. | |
Curry MP, Forns X, Chung RT, et al. Pretransplant sofosbuvir and ribavirin to prevent recurrence of HCV infection after liver transplantation. Hepatology. 2013;58:314a–315a. | |
Charlton MR, Gane EJ, Manns MP, et al. Sofosbuvir and ribavirin for the treatment of established recurrent hepatitis c infection after liver transplantation: preliminary results of a prospective, multicenter study. Hepatology. 2013;58:1378a–1378a. | |
Koff RS. Review article: the efficacy and safety of sofosbuvir, a novel, oral nucleotide NS5B polymerase inhibitor, in the treatment of chronic hepatitis C virus infection. Aliment Pharmacol Ther. 2014;39:478–487. | |
Karageorgopoulos DE, El-Sherif O, Bhagani S, et al. Drug interactions between antiretrovirals and new or emerging direct-acting antivirals in HIV/hepatitis C virus coinfection. Curr Opin Infect Dis. 2014;27:36–45. | |
Sulkowski M, Jacobson IM, Ghalib R, et al. Once-daily Simeprevir (TMC435) plus Sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with METAVIR FO-2: COSMOS Study subgroup analysis. J Hepatol. 2014;60:S4. | |
Lawitz E, Ghalib R, Rodriguez-Torres M, et al. Simeprevir plus sofosbuvir with/without ribavirin in HCV genotype-1 prior null-responder / treatment-naïve patients (COSMOS study): primary endpoint (SVR12) results in patients with METAVIR F3-4 (Cohort 2). J Hepatol. 2014;60:S524. | |
Sofosbuvir for Treatment of Chronic Hepatitis C Infection, Antiviral Drugs Advisory Committee Meeting Briefing Document. Gilead Sciences, Inc.; 2013. Available from: http://www.natap.org/2013/HCV/20131025-AVDAC-B1-02-Gilead.pdf. Accessed August 25, 2014. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.