Back to Journals » Drug Design, Development and Therapy » Volume 9
Ferrous sulfate, but not iron polymaltose complex, aggravates local and systemic inflammation and oxidative stress in dextran sodium sulfate-induced colitis in rats
Authors Toblli JE, Cao G, Angerosa M
Received 30 January 2015
Accepted for publication 13 March 2015
Published 7 May 2015 Volume 2015:9 Pages 2585—2597
DOI https://doi.org/10.2147/DDDT.S81863
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Jorge E Toblli, Gabriel Cao, Margarita Angerosa
Laboratory of Experimental Medicine, Hospital Alemán, School of Medicine, University of Buenos Aires, Buenos Aires, Argentina
Background and aims: Iron deficiency is common in inflammatory bowel disease, yet oral iron therapy may worsen the disease symptoms and increase systemic and local oxidative stress. The aim of this study was to compare the effects of oral ferrous sulfate and iron polymaltose complex on inflammatory and oxidative stress markers in colitic rats.
Methods: Animals were divided into four groups with ten animals each. Rats of three groups received dextran sodium sulfate to induce colitis and animals of two of these groups received 5 mg iron/kg of body weight a day, as ferrous sulfate or iron polymaltose complex, for 7 days. Gross colon anatomy, histology of colon and liver, stainings of L-ferritin, Prussian blue, hepcidin, tumor necrosis factor-α, and interleukin-6, as well serum levels of liver enzymes, inflammatory markers, and iron markers, were assessed.
Results: Body weight, gross anatomy, crypt injury and inflammation scores, inflammatory parameters in liver and colon, as well as serum and liver hepcidin levels were not significantly different between colitic animals without iron treatment and colitic animals treated with iron polymaltose complex. In contrast, ferrous sulfate treatment caused significant worsening of these parameters. As opposed to ferrous sulfate, iron polymaltose complex caused less or no additional oxidative stress in the colon and liver compared to colitic animals without iron treatment.
Conclusion: Iron polymaltose complex had negligible effects on colonic tissue erosion, local or systemic oxidative stress, and local or systemic inflammation, even at high therapeutic doses, and may thus represent a valuable oral treatment of iron deficiency in inflammatory bowel disease.
Keywords: preclinical, oral iron treatment, tolerability, colonic tissue erosion, inflammatory bowel disease
Introduction
Patients with inflammatory bowel disease (IBD) commonly develop iron deficiency and, if left untreated, they can develop iron deficiency anemia (IDA). In IBD, IDA is caused mainly by blood loss and/or impaired iron absorption from the gut,1 as well as by reduced iron availability for erythropoiesis.2 Under inflammatory conditions, iron sequestration is induced by an increased expression of hepcidin, which binds to and leads to degradation of the iron-export protein ferroportin and thus inhibits the release of iron from the enterocytes and from the macrophages of the reticuloendothelial system. As a consequence, the absorption and availability of iron is decreased.3 Oral iron, usually in the form of ferrous sulfate (FS) or ferrous fumarate (FF), is recommended as firstline therapy for IDA in IBD in some guidelines,4 although intravenous iron therapy is recommended as a preferred treatment option in other guidelines5 and in a number of recent publications.6–8
It has been extensively reported that overdoses of ferrous salts lead to systemic toxic effects9,10 as well as direct corrosive effects, which can result in mucosal necrosis and ulceration.11,12 However, therapeutic doses of ferrous salts have been shown to cause severe gastric and esophageal injury.13–15 In IBD patients, FS has been shown to worsen the inflammatory state in the colon16 and has been suggested to trigger ulcerative colitis.17 Similarly, FF has been shown to worsen the symptoms in patients affected by Crohn’s disease.18,19 Upon administration of a standard therapeutic dose of FS (60–100 mg Fe), rapid, uncontrolled absorption of iron causes high levels of serum iron and transferrin saturation (TSAT) and, thus, may lead to significant amounts of non–transferrin-bound iron (NTBI).20–23 In particular in chronic iron-overload disorders, NTBI has been shown to cause organ toxicity, as it is taken up unselectively by highly vascular tissues such as liver, heart, and endocrine system, where it may increase the intracellular labile iron pool and induce oxidative stress.24,25 Increased levels of oxidative stress markers (eg, thiobarbituric-acid-reactive substances [TBARS] and 8-hydroxy-2-deoxyguanosine) have been reported after therapeutic doses of FS combined with vitamin C in healthy volunteers.26,27 Similarly, increased markers of lipid peroxidation were observed in patients with iron deficiency after FS treatment.28,29 Thus, FS may exacerbate the oxidative stress that usually occurs under inflammatory conditions. Yet, IDA itself has also been shown to cause oxidative stress, due to (among other aspects) a shortage in iron-based antioxidant enzymes.25
Iron polymaltose complex (IPC) is a complex of a polynuclear iron(III)-oxyhydroxide core with polymaltose ligands that liberates ferric ions in a controlled way and thus assures a very low toxicity and good tolerability.30 For IPC, no cases of acute poisoning have been reported even when given accidentally at very high doses,30,31 and no cases of local injury of the mucosa have been linked to IPC treatment. Because iron from IPC is taken up through the enterocytes in a controlled manner, administration of a therapeutic dose of IPC does not lead to a fast increase in serum iron and TSAT levels and, thus, essentially no NTBI is observed.20,22,23 Gastrointestinal irritations may occur, but consistently lower rates are observed with IPC than with FS treatment.32 For instance, a clinical study in IBD patients comparing FS and IPC treatments over a course of 14 days showed an increase in lipid peroxidation markers with FS but not with IPC.33 Given that IDA in IBD patients tends to recur,34,35 and patients often undergo repeated regimens of iron therapy, possible long-term effects of the treatment should be considered. Thus, based on its better tolerability, IPC may represent a suitable treatment option.
Nonclinical studies in healthy rodents provided further insights into the toxicity, histological effects, quantitative results on local reactions, and oxidative stress caused by FS and IPC treatments.36,37 Moreover, the understanding of underlying principles in human IBD progressed with the use of rodent models with dextran sulfate sodium (DSS)-induced colitis.38,39 Previous studies investigated the effects of FF and iron pentacarbonyl treatments in rats with DSS-induced colitis,40,41 but, up until now, no direct comparison between FS and IPC was available in such a model. In the present study, we assessed the effects of iron therapy with FS and IPC in rats with DSS-induced colitis and compared their status with that of healthy animals as well as that of animals with colitis but without iron treatment. In particular, we assessed the potential of FS and IPC to further exacerbate the inflammation and to increase the oxidative stress markers in rats with DSS-induced colitis.
Materials and methods
Animals and treatments
All animal experiments were approved by the Hospital Aléman Ethics Committee and the Teaching and Research Committee, and were conducted according to the US National Institutes of Health Guide for the Care and Use of Laboratory Animals.42 All experiments were conducted with Sprague Dawley rats, aged 6 weeks old and weighing 200–250 g, at the Laboratory of Experimental Medicine, Hospital Aléman, Buenos Aires, Argentina. The rats were housed in a temperature-controlled room at 22°C±2°C and were fed standard rat chow (Cooperación, Buenos Aires, Argentina; iron content: 65 parts per million) ad libitum throughout the study. Forty rats were acclimatized for 7 days at the research laboratory and randomized into four groups (n=10) with equal proportions of male:female animals in each. Acute colitis was induced in all animals in groups 1–3 by addition of 30 g/L DSS (D6924; Sigma-Aldrich Co., St Louis, MO, USA; average molecular weight 9,000–20,000 Da) to the drinking water for 7 days. Group 4 had free access to tap water (control group). In parallel, groups 2 and 3 were administered 5 mg iron/kg of body weight as FS (ferrous sulfate heptahydrate ALT No 44982, batch BCBC3657, DSS + FS; Sigma-Aldrich Co.) or IPC (Maltofer® lot 92110011, DSS + IPC; Vifor Pharma, Zurich, Switzerland), respectively, by gavage for 7 days. A dose of 5 mg iron/kg of body weight corresponds to 300 mg iron for a 60 kg human adult, ie, this dosage is in the upper range of a recommended dose for oral iron therapy (100-300 mg iron per day). Group 4 received saline (control group) and group 1 did not receive any treatment in addition to DSS (DSS, acute colitis reference group). On day 8, blood samples were collected after a 14 hour fast, and the animals were sacrificed. Liver and bowel were perfused with ice cold saline solution and removed for further analysis.
Hematology and blood chemistry
Hemoglobin (Hb) levels were determined by Sysmex XT-1800i (Roche Diagnostic GmbH, Mannheim, Germany). Serum iron and the liver enzymes (aspartate aminotransferase [AST], alanine aminotransferase [ALT], and alkaline phosphatase [ALP]) were assessed by colorimetric and ultraviolet methods, respectively, with an Auto-analyzer Modular P800 with the corresponding reagents (Roche Diagnostic GmbH). Total iron binding capacity (TIBC) was determined with a colorimetric assay (Randox Laboratories Limited, Crumlin, Northern Ireland, UK). TSAT was calculated as follows: TSAT (%) = [serum iron concentration (μg/L)/TIBC (μg/L)] ×100. Serum hepcidin was assessed by a commercially available enzyme-linked immunosorbent assay kit for hepcidin (Catalog No E91979; Uscn Life Science Inc., Wuhan, People’s Republic of China).
Gross anatomy of the colon
The lesion index of the colon was determined according to the following scoring: 0= absence of lesion; 1= superficial, one to five hemorrhagic points; 2= superficial, six to ten hemorrhagic points; 3= submucosal hemorrhagic lesions with small erosions; and 4= severe hemorrhagic lesions and some invasive lesions. A measure of the extent of involvement in each score was expressed as follows: grade 1=1%–25% of the surface area examined; grade 2=26%–50%; grade 3=51%–75%; and grade 4=76%–100% of the surface area examined. The final score (cumulative score) was calculated as the product of the lesion index by the extent of involvement.
Light microscopy
Portions of colon and liver were fixed in phosphate-buffered 10% formaldehyde (pH 7.2) and embedded in paraffin. Three-micron sections were stained with hematoxylin–eosin. Prussian blue staining of the liver was performed according to Perls’ description,43,44 and were expressed as the percentage of positive immunostaining per area. All observations were performed with a Nikon E400 light microscope (Nikon Instruments, Melville, NY, USA).
Morphometric analysis
Histological sections from each animal were studied using an Image-Pro Plus version 4 image analyzer for Windows (Media Cybernetics, Inc., Rockville, MD, USA). Morphological analyses were performed at a magnification of ×100 or ×400 depending on the tissue evaluated, and the data were averaged. Crypt and inflammatory scores of the colon were determined by a pathologist blinded to the experimental group. The crypt injury was scored according to a validated scoring system:45 grade 0= intact crypt; grade 1= loss of bottom third of the crypt; grade 2= loss of bottom two-thirds of the crypt; grade 3= loss of entire crypt with the surface epithelium remaining intact; and grade 4= loss of the entire crypt and surface epithelium (erosion). The severity of the inflammation of the colon was scored according to reference:46 grade 0= normal; grade 1= focal inflammatory cell infiltration, including polymorphonuclear leukocytes; grade 2= inflammatory cells infiltration, gland dropout; and grade 3= crypt abscess. As for assessment of the gross anatomy of the colon, a measure of the extent of involvement of the examined surface area in each score was expressed as follows: grade 1=1%–25% of the surface area examined; grade 2=26%–50%; grade 3=51%–75%; and grade 4=76%–100% of the surface area examined. The final score is the product of either the inflammation or injury grade and the extent of involvement, ie, the inflammation cumulative score and the crypt injury cumulative score.
Immunohistochemical analysis
Tissue ferritin was determined with antiferritin light-chain goat polyclonal immunoglobulin G antibody (sc-14420; Santa Cruz Biotechnology Inc., Dallas, TX, USA) at 1:100 dilution, tissue hepcidin in liver and colon was determined with a rabbit polyclonal anti–hepcidin-25 antibody (ab30760; Abcam, Cambridge, MA, USA) at 1:100 dilution, tumor necrosis factor-α (TNF-α) was determined with a rat TNF-α antibody (AF-510-NA; R&D Systems, Inc., Minneapolis, MN, USA) at 1:50 dilution, and interleukin-6 (IL-6) was determined with an anti–IL-6 antibody (sc1265; Santa Cruz Biotechnology Inc.) at 1:100 dilution. Detection was carried out by a modified avidin–biotin–peroxidase complex technique (VECTASTAIN Universal Elite ABC kit; Vector Laboratories, Ltd., Peterborough, UK) as described previously.47 The ferritin immunostainings of the colon were evaluated by a semiquantitative score according to the following schedule: 0= absence of staining; 1= mild positive staining per villus; 2= moderate positive staining per villus; 3= intense positive staining per villus; and 4= very intense positive staining per villus. Ferritin immunostainings of the liver and hepcidin, IL-6, and TNF-α immunostainings of liver and colon were expressed as the percentage of positive immunostaining per area.
Western blotting and densitometric analysis
Tissue samples from colon and liver were solubilized in 1% triton detergent and analyzed by Western blotting as described previously.48 For assessing nitrosative stress, polyvinyl difluoride membranes with transferred proteins were probed with antinitrotyrosine antibody at 1:2,000 dilution (AB5411; EMD Millipore, Billerica, MA, USA). Inflammatory response was evaluated by probing for anti–TNF-α (AF-510-NA; R&D Systems, Inc.) at 1:1,000 dilution and IL-6 with an anti–IL-6 antibody (sc1265, Santa Cruz Biotechnology Inc.) at 1:1,000 dilution. Tissue hepcidin was probed with a rabbit polyclonal anti–hepcidin-25 antibody (ab30760; Abcam) at 1:1,000 dilution. After washing, membranes were probed with a horseradish peroxidase-conjugated goat antirabbit secondary antibody at dilution 1:20,000 (sc-2004; Santa Cruz Biotechnology Inc.). Specific bands were detected by enhanced chemiluminescence (ECL; Amersham Biosciences Corp., Piscataway, NJ, USA). Autoradiographs were scanned and band intensities were quantified by digital densitometry using Gel-Pro Analyzer 4.0 software (Media Cybernetics, Inc.). The data were recorded as mean optical density intensity. Equal protein loading of samples was confirmed by stripping polyvinyl difluoride membranes with a commercial re-blot solution (Chemicon International Inc., Billerica, MA, USA) and then re-probing with anti–β-actin primary antibody (A5316; Sigma-Aldrich Co.) at 1:5,000 dilution; and horseradish peroxidase-conjugated goat antimouse secondary antibody (sc-2005; Santa Cruz Biotechnology Inc.) at 1:20,000 dilution followed by chemiluminescent detection and densitometric quantification as described earlier.
Oxidative stress parameter evaluation in colon mucosa and liver
A fraction of the whole liver and colon mucosa were homogenized (1:3, w/v) in ice-cold 0.25 mol/L sucrose solution. Reduced glutathione (GSH) and glutathione disulfide (GSSG) levels were determined in the 10,000× g supernatant following methods as previously described.49–51 TBARS and the activity of catalase, copper–zinc superoxide dismutase (CuZn-SOD), and glutathione peroxidase (GPx) were measured as described earlier52 in different homogenates or their supernatants.
Statistical methods
Values were expressed as mean ± standard deviation. All statistical analyses were performed using absolute values and were processed through GraphPad Prism, version 5.01 for Windows (GraphPad Software, Inc., La Jolla, CA, USA). Normality of the samples was determined by the Kolmogorov–Smirnov test. For parameters with a Gaussian distribution, the groups were compared by analysis of variance (ANOVA); for parameters such as histological data with non-Gaussian distribution, comparisons were performed by the Kruskal–Wallis test (nonparametric ANOVA) and Dunn’s multiple comparison test. A potential relationship between serum or tissue hepcidin with the TNF-α and IL-6 values was assessed by Spearman nonparametric correlation. A value of P<0.05 was considered significant.
Results
All animals that received DSS developed severe, acute colitis, which became obvious by the development of bloody diarrhea between day 5 and day 6. At the end of the experiment (day 8, presurgery), variable degrees of hemorrhagic proctitis were observed.
Body weight and food consumption
Body weight and food intake in the groups that received DSS decreased over the course of the experiment as expected and were lower than in the control group, which showed a net gain in body weight (Table 1). The animals in the DSS and DSS + IPC groups had comparable changes in body weight and food intake, whereas those in the DSS + FS group had a significantly lower food intake and lost more weight than the animals in the other groups. Water consumption was higher in the control group than in the DSS-receiving groups. The water consumption and thus the DSS intake did not differ significantly between the groups that received DSS (Table 1).
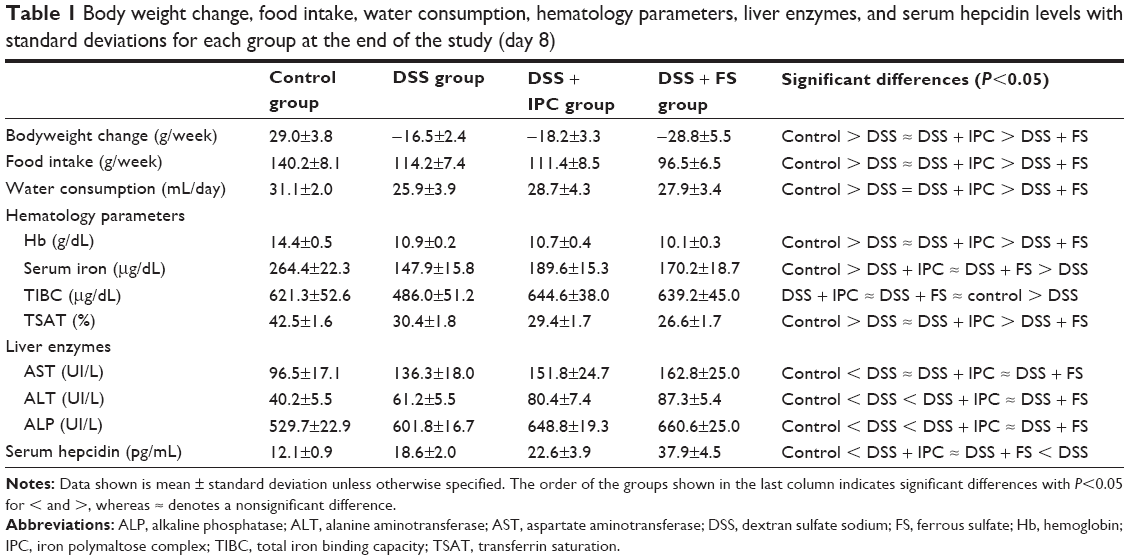 | Table 1 Body weight change, food intake, water consumption, hematology parameters, liver enzymes, and serum hepcidin levels with standard deviations for each group at the end of the study (day 8) |
Hematology parameters
Hemoglobin, as well as the markers of iron status, serum iron, and TSAT, were significantly higher in the control group than in all other groups. Both types of oral iron supplementation failed to maintain an adequate iron status in this model (Table 1). Rats treated with FS had significantly lower Hb values than those treated with IPC, and TSAT showed a corresponding significant difference. TIBC was maintained in the DSS + IPC and the DSS + FS groups at the same level as in the control group, whereas TIBC in the DSS group was significantly lower than in the other groups.
Pathology and immunohistochemistry findings in the colon
For all groups with acute colitis (DSS, DSS + IPC, DSS + FS), gross anatomy of the colon showed numerous areas with loss of mucosal folds and a variable degree of mucosal edema and congestion, along with submucosal hemorrhage and focal ulceration (Figure 1). The gross anatomy cumulative score of the colon indicated substantially more serious lesions in the DSS + FS group versus the DSS + IPC and DSS groups. The gross anatomy cumulative scores of the DSS + IPC and the DSS groups were not significantly different (significant differences between groups with P<0.05 in the following order: control < DSS ≈ DSS + IPC < DSS + FS). The gross anatomy lesion index and the extent of involvement showed the same relative order but with a nonsignificant difference of the extent of involvement between the two iron-supplemented groups (data not shown). Taken together, these data indicate that FS induced more severe macroscopic effects (based on the severity of the reaction or lesion index), and to a nonsignificant degree also more-widespread lesions, as shown by the extent of involvement. The injury was mainly located in the distal colon with crypt distortion and inflammatory cell infiltration, as indicated by the arrows in Figure 1. On the microscopic scale, the crypt injury cumulative score (Figure 1) as well as the crypt injury and extent of involvement scores (data not shown) showed a similar trend, with the DSS and DSS + IPC groups being significantly more inflamed than the control group but less so than the DSS + FS group (significant differences between groups with P<0.05: control < DSS ≈ DSS + IPC < DSS + FS). Also, the inflammation cumulative score of the colon yielded similar significant differences between groups (with P<0.05: control < DSS ≈ DSS + IPC < DSS + FS) (Figure 1).
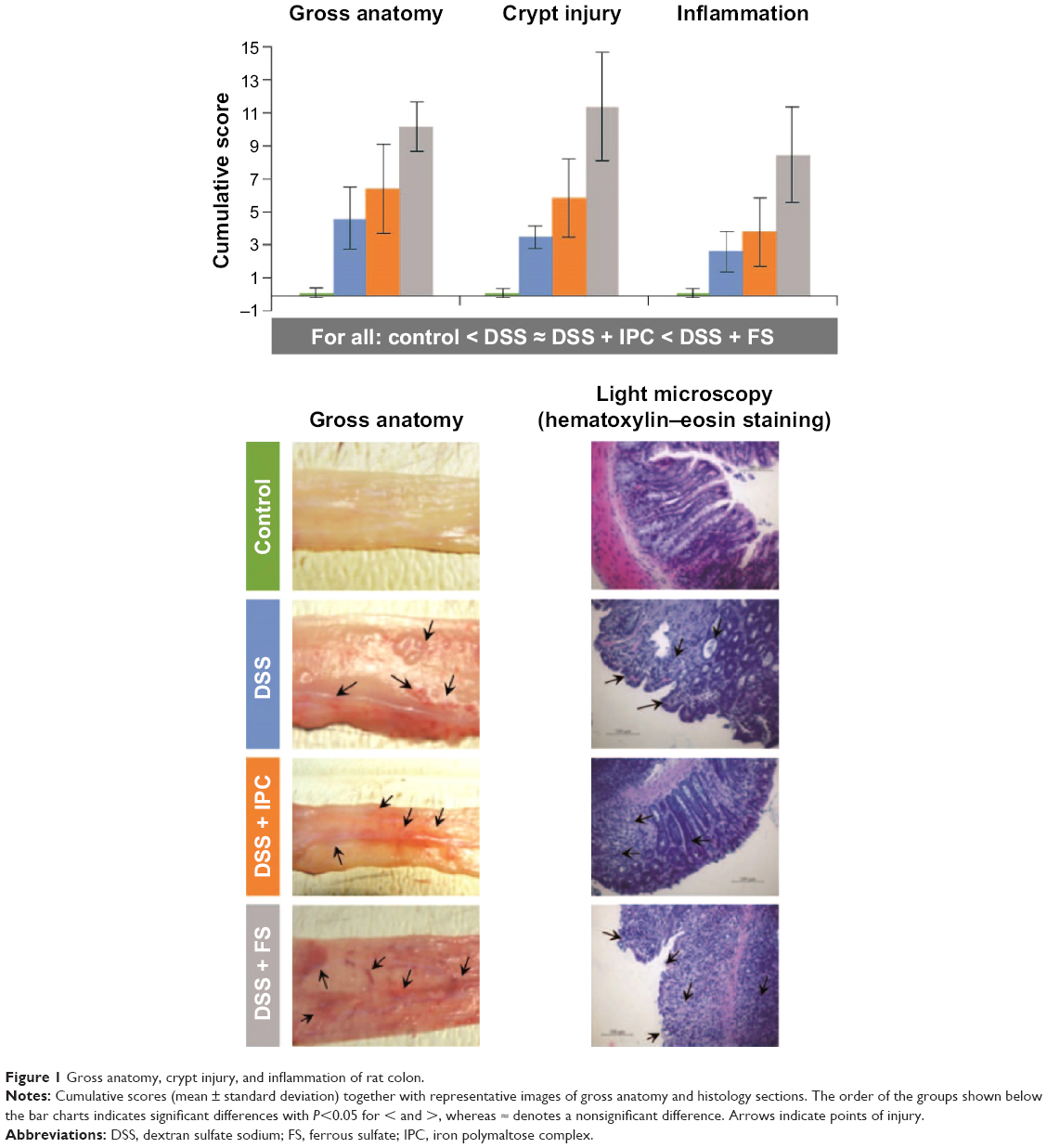 | Figure 1 Gross anatomy, crypt injury, and inflammation of rat colon. |
The expression of L-ferritin (as L-ferritin staining) in the colon villi was lowest in the DSS group, whereas it was not significantly different between the iron-receiving groups and the control group (significant differences between groups with P<0.05: control ≈ DSS + FS ≈ DSS + IPC; DSS < control ≈ DSS + FS) (Figure 2).
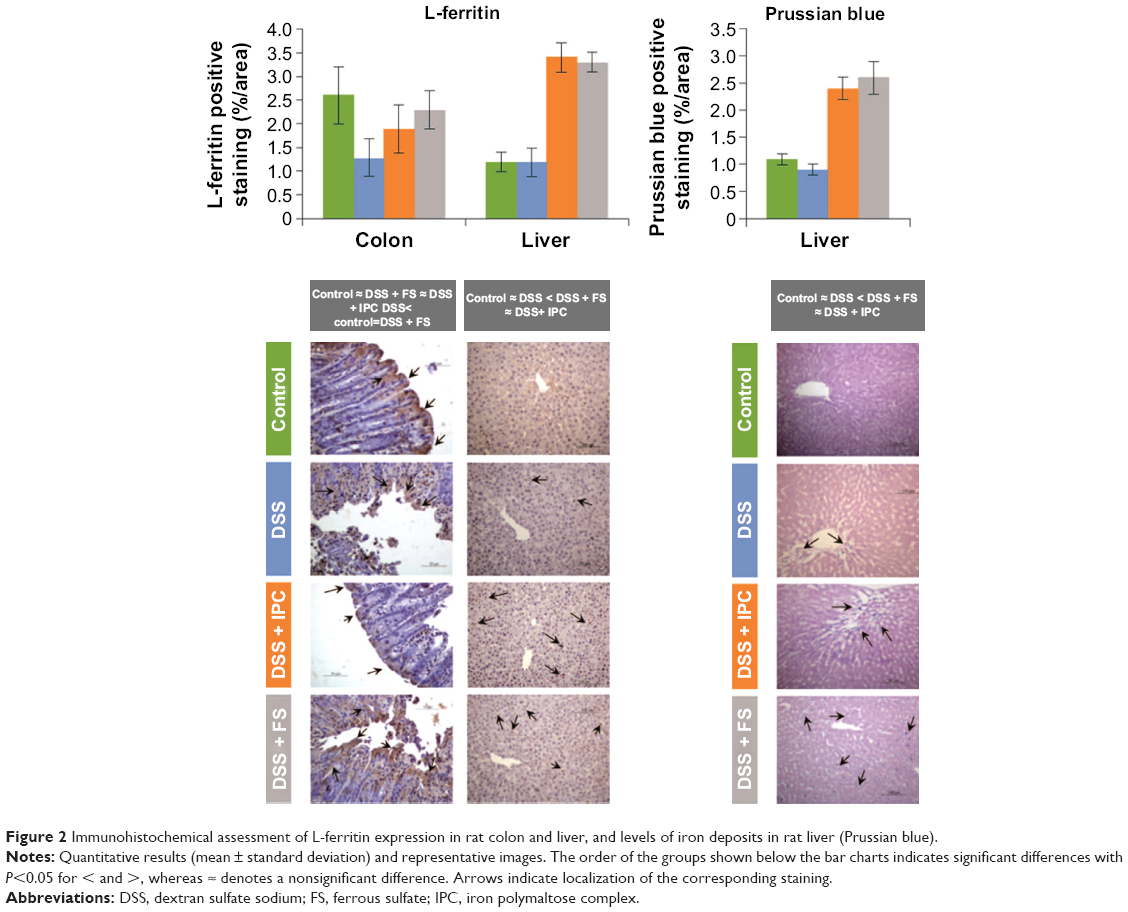 | Figure 2 Immunohistochemical assessment of L-ferritin expression in rat colon and liver, and levels of iron deposits in rat liver (Prussian blue). |
The comparative levels of oxidative stress parameters in the colon followed the same pattern, with increasing oxidative stress in the different groups in the order control < DSS ≤ DSS + IPC < DSS + FS (significant differences are given in Figure 3). Colon TBARS levels, a marker of lipid peroxidation, were significantly higher in the DSS versus the control group, and oral iron treatments caused an additional significant increase in TBARS level, with FS causing a significantly more pronounced increase than IPC (Figure 3). Similar relative behaviors were found for CuZn-SOD and GPx, although the difference between the DSS + IPC and DSS groups was not significant for GPx levels (Figure 3). GSH levels significantly decreased with increasing oxidative stress; this inverse behavior was further reflected in the GSH:GSSG ratio between groups in the order control > DSS > DSS + IPC > DSS + FS (Figure 3). In contrast to the other oxidative stress markers, the catalase activity was affected differently by acute colitis and oral iron supplementation. Whereas the DSS group showed significantly lower catalase activity than the control group, oral iron treatment caused a significant increase in catalase activity versus the control group. This increase was significantly higher for FS than for IPC (Figure 3).
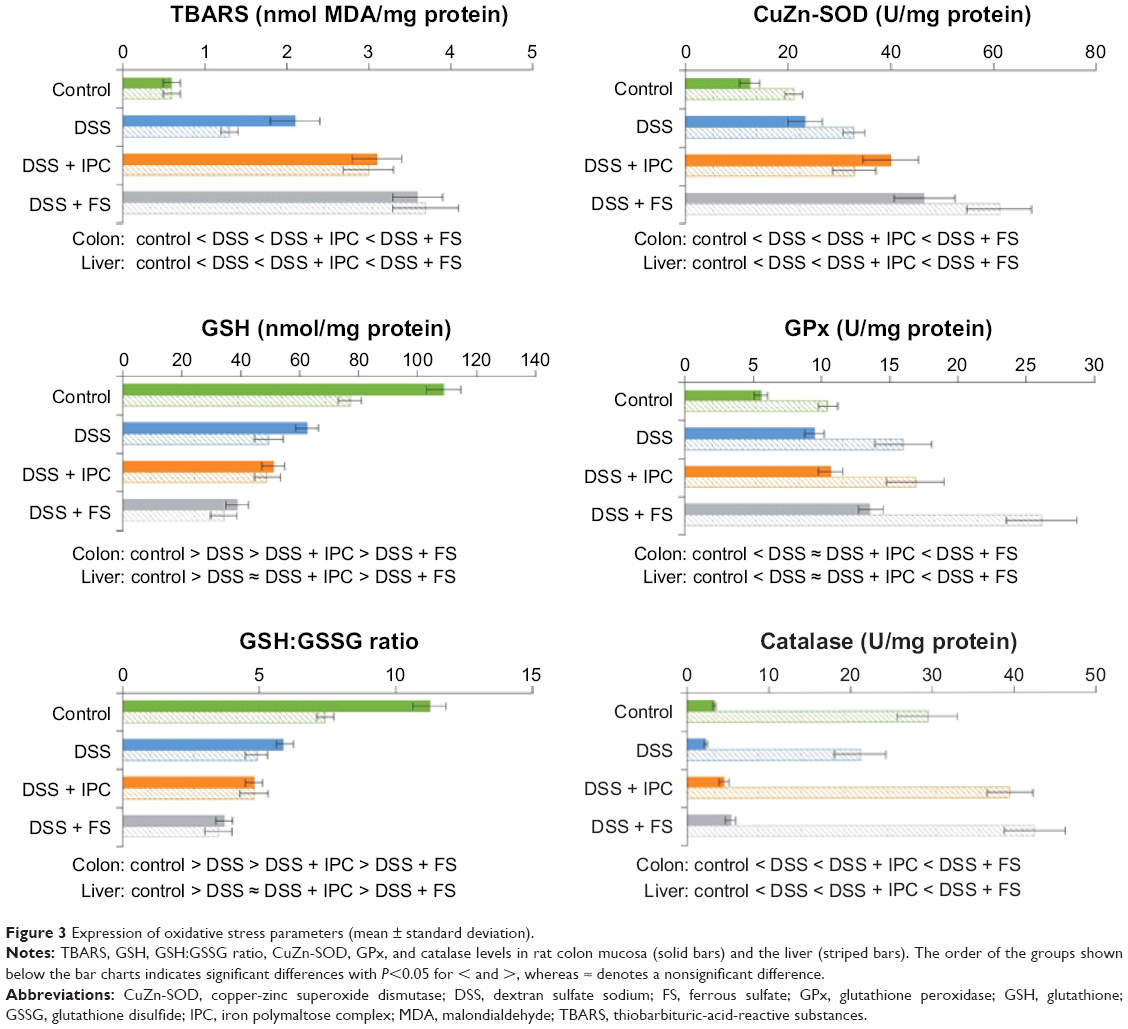 | Figure 3 Expression of oxidative stress parameters (mean ± standard deviation). |
Pathology and immunohistochemistry findings in the liver, and liver enzymes
Liver morphology did not reveal any major differences between the groups (data not shown). L-ferritin expression and iron deposits (as Prussian blue staining) were significantly higher in the two groups that had received oral iron compounds than in the control and DSS groups, with no difference between the two iron treatments (significant differences between groups with P<0.05: control ≈ DSS < DSS + FS ≈ DSS + IPC) (Figure 2).
The three tested liver enzymes, ALT, AST, and ALP, were significantly higher in the groups that received DSS versus the control group. For ALP and ALT, the DSS + IPC and DSS + FS groups also showed significantly higher values than the DSS group (Table 1).
Trends for oxidative stress parameters in the liver were similar to those observed in the colon mucosa, and oxidative stress increased in the order control < DSS ≤ DSS + IPC < DSS + FS (significant differences given in Figure 3). As also described for the colon, the catalase activity in the liver was affected differently by acute colitis and oral iron supplementation, ie, the catalase activity in the control group was significantly higher than that in the DSS group, and iron supplementation with FS or IPC resulted in a significant increase of catalase activity not only versus the DSS group but also versus the control group.
Inflammatory markers and hepcidin
All groups with acute colitis presented a significant increase in TNF-α and IL-6 expression in the colon and in the liver, compared with the control group (Figures 4A and B). This increase was significantly exacerbated in the DSS + FS group, while the DSS + IPC group was not significantly different from the DSS group, resulting in significant differences between the groups in the following order: control < DSS ≈ DSS + IPC < DSS + FS.
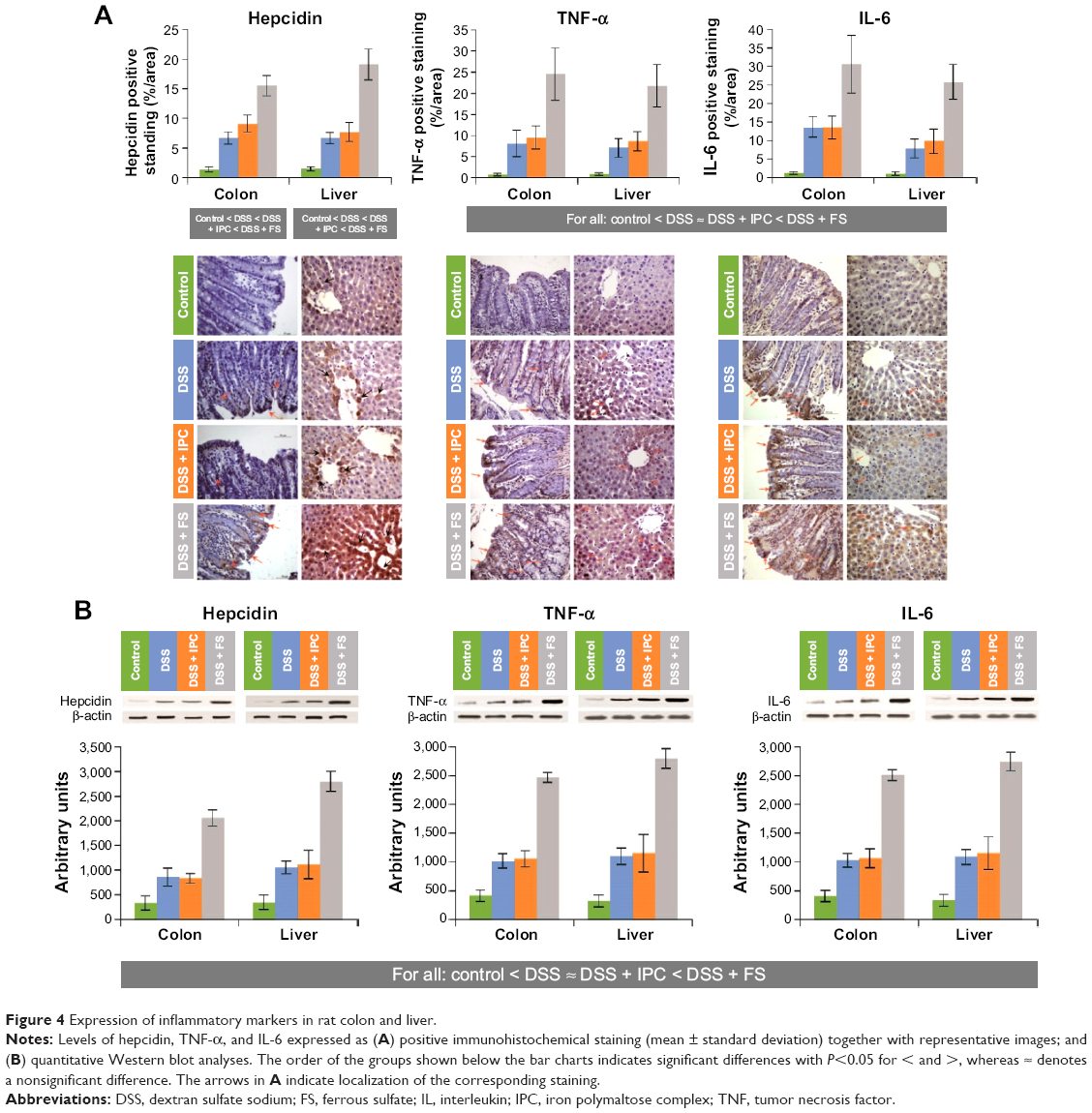 | Figure 4 Expression of inflammatory markers in rat colon and liver. |
Hepcidin immunostaining and Western blot analysis from liver samples (Figures 4A and B) and hepcidin levels in the serum (Table 1) behaved similarly, with the same order of significantly increasing hepcidin levels between the groups, ie, control < DSS ≈ DSS + IPC < DSS + FS.
Spearman nonparametric correlation yielded significant correlations between serum hepcidin and the hepcidin expression in the liver in all groups (data not shown). A positive correlation was also found between hepcidin expression in the liver and both inflammatory markers (TNF-α and IL-6, colon and liver) in all groups. Although serum and liver hepcidin correlated significantly with each other, liver hepcidin expression correlated more strongly with the inflammatory markers than serum hepcidin.
Discussion
A previous study with healthy, nonanemic rats treated with FS or IPC during 4 weeks or 4 months36 demonstrated important acute as well as early and late gastrointestinal tract and liver toxicity effects induced by FS but not by IPC. Among others, FS caused mucosal erosions, increased liver enzymes, significantly altered oxidative stress markers, and had a more than tenfold lower median lethal dose value than did IPC. In another study, the effects of FS, IPC, and FF treatments on anemic, pregnant rats were investigated.37 It was demonstrated that, although all treatments corrected anemia, FS and FF, but not IPC, increased oxidative stress markers and elicited hepatic damage in pregnant rats and fetuses. Moreover, it was shown that IPC, but not FS or FF, significantly reduced the levels of the oxidative stress and inflammatory markers, which were elevated by the anemic status of the animals before treatment.37
The effect of oral iron supplementation in DSS-induced colitis models has been investigated previously for FF, pentacarbonyl iron, and ethylenediaminetetraacetic acid iron(III). It was reported that: 1) FF caused an exacerbation of the intestinal inflammation but did not affect the level of plasma malondialdehyde, plasma antioxidant vitamins, or plasma aminothiols;41 2) pentacarbonyl iron not only worsened the disease activity but also increased oxidative stress;40,53 and 3) ethylenediaminetetraacetic acid iron(III) increased disease activity, oxidative and nitrosative stress in the colon, as well as the risk of colorectal carcinoma in a long-term experiment (255 days).54
The present study was carried out to assess the potential of FS and IPC to further exacerbate the inflammation and oxidative stress induced by colitis in a rat model. DSS-induced acute colitis in rodents typically induces enhanced expression of inflammatory markers including IL-6 and TNF-α, reduced food intake, weight loss, and bloody diarrhea.38,55 As expected, all these effects were observed in the present study. The sharp decrease of Hb, TSAT, and serum iron levels after the relatively short period of 8 days may be mainly attributed to intestinal blood loss, although iron sequestration due to the severe inflammatory response may also have contributed to this condition. Because of the short treatment time, neither FS nor IPC efficiently corrected the Hb level, and they also failed to keep the serum markers of iron status, TSAT and serum iron, at an adequate level. Yet, serum iron and TSAT were significantly higher in the DSS + iron groups versus the DSS group, indicating that iron absorption from IPC and FS took place to some extent in spite of the highly inflammatory conditions.
The effects of DSS on the colon were clearly visible in the gross anatomy and histology of the colon crypts and have been described in earlier studies.38,40,41,53 Treatment with FS but not IPC caused substantially more severe lesions and more serious inflammation than observed in the untreated DSS group (Figure 1). This observation was mirrored in a markedly stronger local inflammatory response in the colons of the DSS + FS group than in those of the DSS + IPC and DSS groups, as indicated by the staining of the inflammatory markers TNF-α and IL-6 (Figure 4). Also, hepcidin staining in the colon was more intense for the DSS + FS group than for the DSS + IPC group. The role of colon hepcidin is still unclear,56 but these data support a correlation between the colon hepcidin levels and the degree of inflammation, which was higher with FS than with IPC treatment. Yet, it remains to be established whether hepcidin expression represents a defensive or a pathological response to intestinal inflammation.56
The less-intense ferritin staining of the colon villi of the animals in the DSS group as compared to those of the control group suggests that substantial amounts of ferritin were lost into the intestinal lumen as a consequence of the damage to the colon mucosa, and/or that iron was exported into the blood and not stored in the enterocytes due to the severe IDA (Figure 2). In contrast, previous studies in similar models showed colon iron deposits by Prussian blue stainings in the colitis groups but not in control animals.54,56 Yet, the different methodologies to assess tissue iron levels and the severity of the disease in our study may well explain these discrepancies. The villus tips of the iron-receiving groups, however, showed a ferritin staining similar to that of the control group. It is likely that the relatively high iron dose (5 mg iron/kg of body weight) allowed for iron absorption and storage within the enterocytes at the villus tips in spite of the damaged colon mucosa.
In the liver, a strong inflammatory response was also observed. After DSS treatment, the levels of the markers TNF-α and IL-6 as well as hepcidin were strongly enhanced, and a further exacerbation was induced by FS but not by IPC treatment (Figure 4). Some degree of hepatic damage in all the DSS-receiving groups was indicated by the elevated liver enzymes, ALT, AST, and ALP (Table 1). We consider that the most likely reason for the increase in liver enzymes observed with IPC and FS treatment was due to the inflammatory status present in the underlying DSS-induced animal colitis model; the small increase in liver enzymes induced by IPC and FS was not significantly different from that induced by DSS only. Consequently, from the liver enzyme results in the treated groups, it is not possible to conclude that iron treatment by itself exacerbates liver injury. The increased liver ferritin and Prussian blue stainings are characteristic of iron sequestration under inflammatory conditions, which leads to increased iron storage in the liver. In the DSS group, the ferritin and Prussian blue liver stainings were normal despite the inflammation, an observation that can probably be explained by the relatively severe IDA in this group.
As mentioned above, local hepcidin expression in the colon may cause local iron sequestration and thus increase local ferritin levels, but it does not necessarily correlate with increased serum or liver hepcidin levels.56 In our study, however, the severity of the colitis most likely also triggered the systemic response of hepcidin expression in the liver and thus led to increased serum hepcidin levels. As expected, the levels of serum and liver hepcidin correlated with each other and with the levels of inflammatory markers in liver and colon in the DSS-receiving groups. The higher serum hepcidin levels found in the DSS + FS group versus the untreated DSS group could arise from the more-severe inflammation as well as from the less-severe IDA present in the DSS + FS group. However, the observations that the iron parameters in the DSS + IPC group were similar to those in the DSS + FS group, but that the levels of hepcidin and inflammatory markers were lower after IPC treatment, rather support the interpretation that the higher hepcidin levels in the DSS + FS group are a consequence of the more severe inflammatory state. Yet, the role of serum hepcidin in the IBD setting is not clear.57–62
Recent studies support the importance of IL-6 signaling in the development of IBD; however how this pathway causes intestinal inflammation is not fully understood.63 Circulating and intestinal levels of IL-6 as well as levels of the soluble IL-6 receptor (sIL-6R) are increased in patients with IBD and there is growing evidence that increased formation of IL-6/sIL-6R complexes interacting with membrane-bound gp130 on T cells via trans-signaling leads to the development and perpetuation of IBD.63
The local effects of FS have been ascribed to the formation of reactive oxygen species in the inflamed tissue, leading to increased inflammatory response and even carcinogenesis,16 whereas increased serum levels of iron promote the generation of NTBI, which could provoke oxidative stress in the cells of a number of different compartments such as the heart and liver.20,22,23,25 In comparison to FS, treatment with IPC in addition to DSS caused less (TBARS, GSH:GSSG colon) or no (GSH:GSSG liver, CuZn-SOD liver, GPx) additional oxidative stress. The physicochemical properties of IPC and its mechanism of iron uptake are different from those of ferrous salts, such as FS. IPC is a polynuclear iron(III)-oxyhydroxide complex stabilized by a polymaltose shell. This complex is stable throughout the whole gastrointestinal tract, a property that allows for a controlled release, and thus uptake, of iron. In contrast, soluble ferrous salts release large amounts of iron(II) ions that can be taken up by paracellular diffusion, which can lead to increased levels of NTBI and therefore increased levels of oxidative stress. The different behavior of the catalase concentrations in liver and colon, which was lowest in the DSS group and only slightly higher in the iron-receiving groups than in the control group, could be explained by the severe IDA in the DSS group, which may hinder the synthesis of this iron-containing enzyme. These observations agree well with the effect of FS and IPC treatment in pregnant, anemic rats, where FS but not IPC elicited oxidative stress in the pregnant rats, fetuses, and placentas.37
Conclusion
Our study shows that, in contrast to FS, IPC treatment during DSS-induced colitis in rats does not significantly exacerbate colonic tissue erosion, local or systemic oxidative stress, or local or systemic inflammation, even at high therapeutic doses. This result confirms the favorable properties of IPC, which does not release large amounts of ionic iron30,64 in the gastrointestinal tract and thus does not provoke local reactions in the colon. Moreover, iron from IPC is taken up in a controlled way, a feature which prevents NTBI formation.23,37,64 Up until now, IPC is the only oral iron supplement that has been shown to have no major detrimental effect on disease activity, oxidative stress, and inflammatory markers in rodent models of colitis and may thus represent a valuable oral treatment for IDA in IBD patients. The favorable properties of IPC are especially relevant with regard to the long duration of a course of oral iron substitution and the fact that IDA tends to be a reemergent condition in IBD patients.35
Acknowledgments
Scientific writing support was provided by Dr Susann Neiser of Vifor (International) Ltd., and support for graphics and illustrations was given by Tracey Nichols of Tracey Nichols Graphic Design Ltd. Financial support for this work was received from Vifor Pharma Ltd.
Disclosure
Vifor (International) Ltd. supported this study with a research grant but did not contribute to the collection, analysis, or interpretation of data. Professor Jorge E Toblli has received research grants and consultancy fees from Vifor Pharma Ltd. The authors report no other conflicts of interest in this work.
References
Bergamaschi G, Di Sabatino A, Albertini R, et al. Prevalence and pathogenesis of anemia in inflammatory bowel disease. Influence of anti-tumor necrosis factor-alpha treatment. Haematologica. 2010;95(2):199–205. | ||
Gomollón F, Gisbert JP. Current management of iron deficiency anemia in inflammatory bowel diseases: a practical guide. Drugs. 2013;73(16):1761–1770. | ||
Goodnough LT, Nemeth E, Ganz T. Detection, evaluation, and management of iron-restricted erythropoiesis. Blood. 2010;116(23):4754–4761. | ||
Mowat C, Cole A, Windsor A, et al; IBD Section of the British Society of Gastroenterology. Guidelines for the management of inflammatory bowel disease in adults. Gut. 2011;60(5):571–607. | ||
Gasche C, Berstad A, Befrits R, et al. Guidelines on the diagnosis and management of iron deficiency and anemia in inflammatory bowel diseases. Inflamm Bowel Dis. 2007;13(12):1545–1553. | ||
Evstatiev R, Gasche C. Diagnosis and management of anemia in IBD. Inflamm Bowel Dis Monit. 2011;11(4):152–159. | ||
Stein J, Hartmann F, Dignass AU. Diagnosis and management of iron deficiency anemia in patients with IBD. Nat Rev Gastroenterol Hepatol. 2010;7(11):599–610. | ||
Stein J, Dignass AU. Management of iron deficiency anemia in inflammatory bowel disease – a practical approach. Ann Gastroenterol. 2013;26(2):104–113. | ||
Bronstein AC, Spyker DA, Cantilena LR, Green JL, Rumack BH, Giffin SL. 2008 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 26th Annual Report. Clin Toxicol (Phila). 2009;47(10):911–1084. | ||
Manoguerra AS, Erdman AR, Booze LL, et al. Iron ingestion: an evidence-based consensus guideline for out-of-hospital management. Clin Toxicol (Phila). 2005;43(6):553–570. | ||
Tenenbein M, Littman C, Stimpson RE. Gastrointestinal pathology in adult iron overdose. J Toxicol Clin Toxicol. 1990;28(3):311–320. | ||
Pestaner JP, Ishak KG, Mullick FG, Centeno JA. Ferrous sulfate toxicity: a review of autopsy findings. Biol Trace Elem Res. 1999;69(3):191–198. | ||
Areia M, Gradiz R, Souto P, et al. Iron-induced esophageal ulceration. Endoscopy. 2007;39 Suppl 1:E326. | ||
Patel JR, Sahota O, Kaye PV. Fatal esophageal perforation caused by oral iron. Scand J Gastroenterol. 2010;45(5):635–636. | ||
Zhang X, Ouyang J, Wieczorek R, DeSoto F. Iron medication-induced gastric mucosal injury. Pathol Res Pract. 2009;205(8):579–581. | ||
Kulnigg S, Gasche C. Systematic review: managing anaemia in Crohn’s disease. Aliment Pharmacol Ther. 2006;24(11–12):1507–1523. | ||
Kawai M, Sumimoto S, Kasajima Y, Hamamoto T. A case of ulcerative colitis induced by oral ferrous sulfate. Acta Paediatr Jpn. 1992;34(4):476–478. | ||
Erichsen K, Hausken T, Ulvik RJ, Svardal A, Berstad A, Berge RK. Ferrous fumarate deteriorated plasma antioxidant status in patients with Crohn disease. Scand J Gastroenterol. 2003;38(5):543–548. | ||
Erichsen K, Ulvik RJ, Nysaeter G, et al. Oral ferrous fumarate or intravenous iron sucrose for patients with inflammatory bowel disease. Scand J Gastroenterol. 2005;40(9):1058–1065. | ||
Dresow B, Petersen D, Fischer R, Nielsen P. Non-transferrin-bound iron in plasma following administration of oral iron drugs. Biometals. 2008;21(3):273–276. | ||
Hutchinson C, Al-Ashgar W, Liu DY, Hider RC, Powell JJ, Geissler CA. Oral ferrous sulphate leads to a marked increase in pro-oxidant nontransferrin-bound iron. Eur J Clin Invest. 2004;34(11):782–784. | ||
Schümann K, Kroll S, Romero-Abal ME, et al. Impact of oral iron challenges on circulating non-transferrin-bound iron in healthy Guatemalan males. Ann Nutr Metab. 2012;60(2):98–107. | ||
Schümann K, Solomons NW, Romero-Abal ME, Orozco M, Weiss G, Marx J. Oral administration of ferrous sulfate, but not of iron polymaltose or sodium iron ethylenediaminetetraacetic acid (NaFeEDTA), results in a substantial increase of non-transferrin-bound iron in healthy iron-adequate men. Food Nutr Bull. 2012;33(2):128–136. | ||
Brissot P, Ropert M, Le Lan C, Loréal O. Non-transferrin bound iron: a key role in iron overload and iron toxicity. Biochim Biophys Acta. 2012;1820(3):403–410. | ||
Koskenkorva-Frank TS, Weiss G, Koppenol WH, Burckhardt S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic Biol Med. 2013;65:1174–1194. | ||
Lachili B, Hininger I, Faure H, et al. Increased lipid peroxidation in pregnant women after iron and vitamin C supplementation. Biol Trace Elem Res. 2001;83(2):103–110. | ||
Schümann K, Kroll S, Weiss G, et al. Monitoring of hematological, inflammatory and oxidative reactions to acute oral iron exposure in human volunteers: preliminary screening for selection of potentially-responsive biomarkers. Toxicology. 2005;212(1):10–23. | ||
King SM, Donangelo CM, Knutson MD, et al. Daily supplementation with iron increases lipid peroxidation in young women with low iron stores. Exp Biol Med (Maywood). 2008;233(6):701–707. | ||
Tuomainen TP, Nyyssönen K, Porkkala-Sarataho E, et al. Oral supplementation with ferrous sulfate but not with non-ionic iron polymaltose complex increases the susceptibility of plasma lipoproteins to oxidation. Nutr Res. 1999;19(8):1121–1132. | ||
Geisser P. Safety and efficacy of iron(III)-hydroxide polymaltose complex/a review of over 25 years experience. Arzneimittelforschung. 2007;57(6A):439–452. | ||
Geisser P. Iron therapy, oxidative stress and immunology. In: Chandra RK, editor. Nutrition and Immunology in the 21st Century. Guragon, India: TSAR Health; 2004:54–65. | ||
Toblli JE, Brignoli R. Iron(III)-hydroxide polymaltose complex in iron deficiency anemia/review and meta-analysis. Arzneimittelforschung. 2007;57(6A):431–438. | ||
Erichsen K, Ulvik RJ, Grimstad T, Berstad A, Berge RK, Hausken T. Effects of ferrous sulphate and non-ionic iron-polymaltose complex on markers of oxidative tissue damage in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2005;22(9):831–838. | ||
Gasche C, Evstatiev R, Haas T, et al. [Diagnosis and treatment of iron deficiency and anaemia in inflammatory bowel diseases. Consensus of the Austrian IBD Working Party]. Z Gastroenterol. 2011;49(5):627–632. German. | ||
Kulnigg S, Teischinger L, Dejaco C, Waldhör T, Gasche C. Rapid recurrence of IBD-associated anemia and iron deficiency after intravenous iron sucrose and erythropoietin treatment. Am J Gastroenterol. 2009;104(6):1460–1467. | ||
Toblli JE, Cao G, Olivieri L, Angerosa M. Comparative study of gastrointestinal tract and liver toxicity of ferrous sulfate, iron amino chelate and iron polymaltose complex in normal rats. Pharmacology. 2008;82(2):127–137. | ||
Toblli JE, Cao G, Oliveri L, Angerosa M. Effects of iron polymaltose complex, ferrous fumarate and ferrous sulfate treatments in anemic pregnant rats, their fetuses and placentas. Inflamm Allergy Drug Targets. 2013;12(3):190–198. | ||
Chassaing B, Koren O, Carvalho FA, Ley RE, Gewirtz AT. AIEC pathobiont instigates chronic colitis in susceptible hosts by altering microbiota composition. Gut. 2014;63(7):1069–1080. | ||
Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98(3):694–702. | ||
Carrier J, Aghdassi E, Platt I, Cullen J, Allard JP. Effect of oral iron supplementation on oxidative stress and colonic inflammation in rats with induced colitis. Aliment Pharmacol Ther. 2001;15(12):1989–1999. | ||
Erichsen K, Milde AM, Arslan G, et al. Low-dose oral ferrous fumarate aggravated intestinal inflammation in rats with DSS-induced colitis. Inflamm Bowel Dis. 2005;11(8):744–748. | ||
Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Institute for Laboratory Animal Research. Division on Earth and Life Studies. National Research Council. Guide for the Care and use of Laboratory Animals. 8th ed. Washington, DC: The National Academies Press, 2011. | ||
Perls M. Nachweis von Eisenoxyd in gewissen Pigmenten. Virchows Arch Pathol Anat. 1867;39:42–48. German. | ||
Cook HC. Manual of Histological Demonstration Techniques. London, UK: The Butterworth Group; 1974. | ||
Murthy SN, Cooper HS, Shim H, Shah RS, Ibrahim SA, Sedergran DJ. Treatment of dextran sulfate sodium-induced murine colitis by intracolonic cyclosporin. Dig Dis Sci. 1993;38(9):1722–1734. | ||
Onderdonk AB, Bartlett JG. Bacteriological studies of experimental ulcerative colitis. Am J Clin Nutr. 1979;32(1):258–265. | ||
Toblli JE, Cao G, Angerosa M. Comparison of early gastrointestinal tract and liver toxicity of the originator iron polymaltose complex (IPC) and an IPC similar preparation in non-anemic rats. Int J Clin Pharmacol Ther. 2012;50(8):573–583. | ||
Giani JF, Gironacci MM, Muñoz MC, Peña C, Turyn D, Dominici FP. Angiotensin-(17) stimulates the phosphorylation of JAK2, IRS-1 and Akt in rat heart in vivo: role of the AT1 and Mas receptors. Am J Physiol Heart Circ Physiol. 2007;293(2):H1154–H1163. | ||
Akerboom TP, Sies H. Assay of glutathione, glutathione disulfide, and glutathione mixed disulfides in biological samples. Methods Enzymol. 1981;77:373–382. | ||
Owen JB, Butterfield DA. Measurement of oxidized/reduced glutathione ratio. Methods Mol Biol. 2010;648:269–277. | ||
Rossi R, Cardaioli E, Scaloni A, Amiconi G, Di Simplicio P. Thiol groups in proteins as endogenous reductants to determine glutathione-protein mixed disulphides in biological systems. Biochim Biophys Acta. 1995;1243(2):230–238. | ||
Toblli JE, Cao G, Olivieri L, Angerosa M. Comparison of the renal, cardiovascular and hepatic toxicity data of original intravenous iron compounds. Nephrol Dial Transplant. 2010;25(11):3631–3640. | ||
Carrier J, Aghdassi E, Cullen J, Allard JP. Iron supplementation increases disease activity and vitamin E ameliorates the effect in rats with dextran sulfate sodium-induced colitis. J Nutr. 2002;132(10):3146–3150. | ||
Seril DN, Liao J, Ho KL, Warsi A, Yang CS, Yang GY. Dietary iron supplementation enhances DSS-induced colitis and associated colorectal carcinoma development in mice. Dig Dis Sci. 2002;47(6):1266–1278. | ||
Dothel G, Vasina V, Barbara G, De Ponti F. Animal models of chemically induced intestinal inflammation: predictivity and ethical issues. Pharmacol Ther. 2013;139(1):71–86. | ||
Gotardo ÉM, Ribeiro Gde A, Clemente TR, et al. Hepcidin expression in colon during trinitrobenzene sulfonic acid-induced colitis in rats. World J Gastroenterol. 2014;20(15):4345–4352. | ||
Bergamaschi G, Di Sabatino A, Albertini R, et al. Serum hepcidin in inflammatory bowel diseases: biological and clinical significance. Inflamm Bowel Dis. 2013;19(10):2166–2172. | ||
Dudkowiak R, Neubauer K, Poniewierka E. Hepcidin and its role in inflammatory bowel disease. Adv Clin Exp Med. 2013;22(4):585–591. | ||
Mecklenburg I, Reznik D, Fasler-Kan E, Drewe J, Beglinger C, Hruz P; Swiss IBD Cohort Study Group. Serum hepcidin concentrations correlate with ferritin in patients with inflammatory bowel disease. J Crohns Colitis. 2014;8(11):1392–1397. | ||
Oustamanolakis P, Koutroubakis IE, Messaritakis I, Malliaraki N, Sfiridaki A, Kouroumalis EA. Serum hepcidin and prohepcidin concentrations in inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2011;23(3):262–268. | ||
Oustamanolakis P, Koutroubakis IE, Kouroumalis EA. Diagnosing anemia in inflammatory bowel disease: beyond the established markers. J Crohns Colitis. 2011;5(5):381–391. | ||
Sukumaran A, James J, Janardhan HP, et al. Expression of iron-related proteins in the duodenum is up-regulated in patients with chronic inflammatory disorders. Br J Nutr. 2014;111(6):1059–1068. | ||
Mitsuyama K, Sata M, Rose-John S. Interleukin-6 trans-signaling in inflammatory bowel disease. Cytokine Growth Factor Rev. 2006;17(6):451–461. | ||
Geisser P, Burckhardt S. The pharmacokinetics and pharmacodynamics of iron preparations. Pharmaceutics. 2011;3(1):12–33. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.