Back to Journals » Vascular Health and Risk Management » Volume 17
Familial Hypercholesterolemia: A Narrative Review on Diagnosis and Management Strategies for Children and Adolescents
Authors Tada H, Takamura M, Kawashiri M
Received 13 December 2020
Accepted for publication 28 January 2021
Published 17 February 2021 Volume 2021:17 Pages 59—67
DOI https://doi.org/10.2147/VHRM.S266249
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Pietro Scicchitano
Hayato Tada, Masayuki Takamura, Masa-aki Kawashiri
Department of Cardiology, Kanazawa University Graduate School of Medicine, Kanazawa, Japan
Correspondence: Hayato Tada
Department of Cardiology, Kanazawa University Graduate School of Medicine, 13-1 Takara-Machi, Kanazawa, 920-8641, Japan
Tel +81-76-265-2000 (2251)
Fax +81-76-234-4251
Email [email protected]
Abstract: Familial hypercholesterolemia (FH) is a relatively common inherited disorder caused by deleterious mutation(s) in the low-density lipoprotein (LDL) receptor or its associated genes. Given its nature as a heritable disease, any useful screening scheme, including universal, and cascade screening, allows for the early identification of patients with FH. Another important aspect to note is that early diagnosis associated with appropriate treatment can promote better prognosis. However, most clinical diagnostic criteria for adults have adopted clinical elements, such as physical xanthomas and family history, both of which are usually obscure and/or difficult to obtain in children and adolescents. Moreover, LDL cholesterol levels fluctuating considerably during adolescence, hindering the timely diagnosis of FH. In addition, recent advancements in human genetics have revealed several types of FH, including conventional monogenic FH, polygenic FH caused by common single nucleotide variations (SNV) accumulation associated with elevated LDL cholesterol, and oligogenic FH with multiple deleterious genetic variations leading to substantially elevated LDL cholesterol. The aforementioned findings collectively suggest the need for amassing information related to genetics and imaging, in addition to classical clinical elements, for the accurate diagnosis of FH in this era of personalized medicine. The current narrative review summarizes the current status of the clinical and genetic diagnosis of FH in children and adolescents, as well as provide useful management strategies for FH in children and adolescents based on currently available clinical evidence.
Keywords: familial hypercholesterolemia, cardiovascular disease, statin, children, adolescents
Introduction
Low-density lipoprotein (LDL) cholesterol has been identified as the causative factor for atherosclerotic cardiovascular disease (ASCVD) based on a variety of evidence obtained from epidemiology,1 human pathology,2 human genetics,3 and clinical trials.4–7 Familial hypercholesterolemia (FH), an inherited hyper-LDL cholesterolemia, has often been associated with tendon and cutaneous xanthomas and premature ASCVD.8 This disorder has been regarded as a Mendelian autosomal dominant disease caused by rare genetic mutation(s) in the LDL receptor or its associated genes. Theoretically, this disease can be diagnosed at the early stages of life, even as early as pregnancy. Currently, two major methods have been proposed for the screening of FH worldwide.9 The first approach involves cascade screening where a diagnosis of FH in new cases, typically younger relatives, is triggered by the diagnosis of the index cases. The second approach involves universal screening where LDL cholesterol measurements are conducted universally at a certain age, after which detailed assessments, including genetic analyses, are subsequently performed to confirm their diagnoses. However, diagnosing FH in children and adolescents is often quite difficult given that physical xanthomas and family histories are usually obscure and/or difficult to obtain among such young patients.
Apart from the difficulties in diagnosing FH among children and adolescents, debates regarding when, how, and who to treat at this early stage of life have remained ongoing. In this regard, recent advancements in human genetics have revealed that personalized medicine can also be applicable to FH, where patients with deleterious genetic mutations and/or signs of premature atherosclerosis development should be treated earlier and more aggressively.10 On the other hand, children and adolescents with FH who had started early treatment exhibited excellent prognosis even under mild treatment,11 suggesting the importance of earlier treatment in the management of FH.
The current review outlines the current status of clinical and genetic diagnosis of FH in children and adolescents while also providing useful management strategies for FH in children and adolescents based on currently available clinical evidence.
Familial Hypercholesterolemia and Its Importance
FH is characterized by the clinical triad of primary hyper-LDL cholesterolemia, tendon xanthomas, and premature ASCVD.12 The first documentation of FH dates back as far as 1873,13 during which it had been described as xanthomatous disease. By the 1930s, FH had started to be considered as an inherited disease,14 with Prof. Brown and Goldstein later discovering genetic abnormalities in the LDL receptor as the principal cause of this condition by the 1970s.15 Subsequently, other genes, including apolipoprotein B (APOB)16 and proprotein convertase subtilisin/kexin type 9 (PCSK9)17 genes, have also been identified to cause this disease. Since its initial documentation, FH has long been described to have a prevalence of 1 in 500 individuals among the general population. In 2011, our group found that FH had a prevalence of 1 in 208 based on genetic epidemiology of homozygous FH in the Hokuriku district of Japan.18 Following our report, similar estimates have been obtained in the United States and Europe.19,20 Currently, FH is considered to have a prevalence of 1 in ~300 individuals among the general population.21 By such estimates, only one monogenic mutation causes the critical phenotype. Monogenic FH is considered the “standard” form of FH, wherein the mutation status of affected genes is associated with increased likelihood of developing ASCVD, independent of LDL cholesterol values.22 This disease appears to account for at least a portion of patients with ASCVD (estimated to be ~1 in 31 individuals), especially those with premature ASCVD (estimated to be ~1 in 15 individuals). No other single disorder can be responsible for such proportions of ASCVD, which has been identified as the leading cause of mortality worldwide.23 Moreover, timely diagnosis and treatment of children and adolescents with FH have been shown to promote a favorable prognosis.11 Accordingly, children and adolescents with require better awareness and more attempts at diagnosing FH compared to adults.
Screening for Familial Hypercholesterolemia: Universal Screening, Opportunistic Screening, Cascade Screening, and Reverse Cascade Screening
As stated previously, timely diagnosis and treatment has been shown to prevent ASCVD events in patients with FH. As such, identifying patients with FH at a younger age is of particular importance given that this leads to prompt treatment initiation and prevention of premature ASCVD. However, diagnosing FH in younger individuals is somewhat difficult considering that they typically do not exhibit increased Achilles tendon thickness, which has been used as one of the major diagnostic criteria for adult FH worldwide. One proposed screening method for FH is universal screening at an age when FH can be effectively identified.24,25 On the other hand, opportunistic screening, utilizing every opportunity to screen patients for FH, is also effective to find FH.26–28 For example, measurement of LDL cholesterol is common practice, and we can find patients with FH when LDL cholesterol level is over a threshold irrespective of the primary aim of its measurements. Another effective screening method for FH is cascade screening, which has been recommended by many organizations around the world. Indeed, countries where dedicated cascade screening programs have been implemented have identified a notably higher number of patients with FHs. For instance, the Netherlands and Norway have diagnosed 71% and 43% of FH cases, respectively.29 In addition, we had demonstrated that cascade screening is significantly associated with better prognoses among patients with FH30 One of the major factors contributing to our results is the notion that an earlier diagnosis promotes better outcomes, which is especially true for patients with FH. As such, although numerous studies have shown the efficacy of LDL-lowering therapies among patients with FH, the magnitude of the benefits obtained from such therapies appear to vary according to the timing of therapy commencement, with far greater benefits having been observed among children than among adults in secondary prevention settings.11,30,31 Accordingly, we firmly believe that earlier diagnosis either via cascade or universal screening and timely LDL-lowering therapies could be beneficial for patients with FH. Alternative way of screening for FH is reverse cascade screening where the index case is a child, and then parents are diagnosed as FH.32,33 It is usually associated with universal screening. However, studying the parents first has a high diagnostic yield.34
Clinical Diagnosis of Familial Hypercholesterolemia in Children and Adolescents
Several different types of clinical diagnostic criteria have been established for FH globally, including the Dutch Lipid Clinical Network (DLCN),35 Make Early Diagnosis to Prevent Early Deaths (MEDPED) diagnostic criteria,36 Japan Atherosclerosis Society (JAS) FH diagnostic criteria,37 and Simon Broome diagnostic criteria for FH.35 Each of the aforementioned diagnostic criteria for pediatric FH has specific cutoffs for LDL cholesterol (Boxes 1 and 2; Tables 1 and 2) given the considerable variability in its levels within this group, especially among adolescents.38 Moreover, children and adolescents with FH barely exhibit physical xanthomas, which is one of the major clinical diagnostic criteria for adults. Nonetheless, care should be exercised when using lower LDL cholesterol thresholds for screening young patients with FH, with family history being much more important in pediatric than in adult cases. In this regard, clinical diagnostic criteria for pediatric FH by JAS appears to be quite useful. Because it is quite simple to use (there are only 2 elements), and it really put weight on their family history (of parents). In order to diagnose them as FH, (reverse) cascade screening for FH will be conducted, and then at least 2, or perhaps even more patients with FH can be identified.
 |
Box 1 Diagnosis of Familial Hypercholesterolemia in Children and Adolescents (EAS) |
 |
Box 2 Pediatric Familial Hypercholesterolemia Diagnostic Criteria (JAS) |
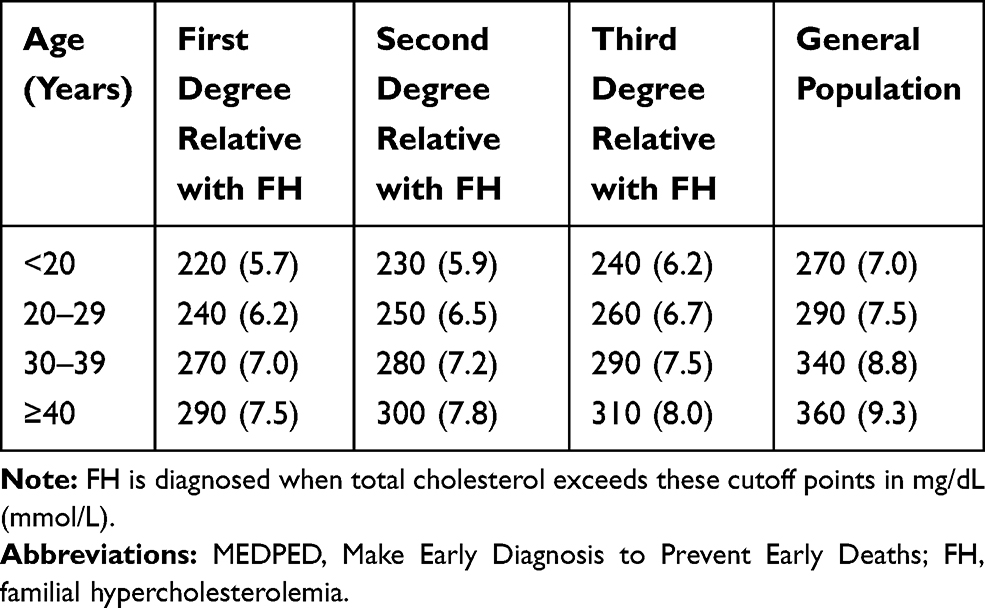 |
Table 1 Diagnosis of Familial Hypercholesterolemia (MEDPED) |
 |
Table 2 Simon Broome Diagnostic Criteria for FH |
Genetic Diagnosis of Familial Hypercholesterolemia in Children and Adolescents
To establish a diagnosis of FH in children and adolescents, genetic testing may be quite useful, although ethical aspects should be carefully considered.39 However, we also need to be careful for what is FH. A few years ago, an useful classification has been proposed regarding the classification of FH. According to this, FH can be classified into heterozygous FH (caused by a deleterious mutation in FH-gene), homozygous FH (caused by double deleterious mutations in FH-gene), polygenic FH (caused by LDL-associated common genetic variations), and polygenic FH plus hypertriglyceridemia (caused by LDL-, and TG-associated common genetic variations).40 In terms of genetic diagnosis, it is still quite difficult to diagnose polygenic state of FH. Accordingly, genetic diagnosis of FH is usually referring to genetic testing for rare genetic variations of FH-genes. In addition, it is also important to think differently between heterozygous FH and homozygous FH irrespective of ages. As stated above, the prevalence of “heterozygous” FH is 1 in ~300 among general population, which is a common disorder, and difficult to diagnose them as FH in children adolescents because of reasons stated previously. On the other hand, homozygous FH is a rare condition, the prevalence of which is estimated to 1 in 160,000 among general population. However, it is of note that state of homozygous FH is an emergent condition, where cardiovascular complications are observed in their adolescence.8 There are several special treatments for homozygous FH, including microsomal triglyceride transfer protein (MTTP) inhibitor, LDL apheresis, and liver transplantation.41–45 Genetic diagnosis for homozygous FH is very important not just because of their diagnosis, but can be useful for their phenotyping. It has been shown that PCSK9 inhibitor, which is quite useful for heterozygous FH, has minimal effect to reduce LDL cholesterol level among the patients with homozygous FH caused by null-type of mutations of LDLR.46 Other useful points for this matter include assessment of responsiveness to dietary intervention, although dietary interventions typically have minimal influence on LDL cholesterol levels among pediatric patients with FH.47 Moreover, an important differential diagnosis worth considering in pediatric FH includes sitosterolemia,48,49 a disease found to be a phenocopy of homozygous FH. Although patients with sitosterolemia usually exhibit physical xanthomas associated with elevated LDL cholesterol, sitosterolemia is a recessive disorder, with dietary interventions being quite useful for reducing LDL cholesterol levels.50 Sitosterolemia can be distinguished from FH based on the mentioned important clinical manifestations.
Development of Atherosclerotic Cardiovascular Diseases Among Children and Adolescents with Familial Hypercholesterolemia
FH has been considered one of the major causes of premature ASCVD, with carotid ultrasound being one of the most popular and non-invasive methods for assessing atherosclerosis among pediatric patients with FH. Carotid intima-media thickness (IMT) is often used as a surrogate marker for systemic atherosclerosis among not only the general population but also pediatric patients with FH.51,52 Moreover, coronary and/or aortic calcium scores have been used to assess early subclinical atherosclerosis,53 apart from actual plaque accumulation in the coronary artery.54 Furthermore, arterial stiffness assessed through brachial-ankle pulse wave velocity had been found to be significantly associated with the presence of ASCVD in patients with FH.55 According to accumulated evidence obtained thus far, the development of ASCVD among patients with FH appears to start during adolescence. These findings have motivated us to consider initiating LDL cholesterol-lowering treatments at an earlier stage of life.
Management of Familial Hypercholesterolemia in Children and Adolescents
Lifestyle interventions should be the fundamental strategy for managing FH in children and adolescents at any age. Statins can be introduced according to guidelines or recommendations. For instance, pitavastatin can be used for Japanese pediatric patients with FH (age ≥10 years) whose LDL cholesterol levels remain ≥180 mg/dL under lifestyle interventions, with the optimal target being set at <140 mg/dL, especially among those with diabetes or a family history of premature ASCVD (Figure 1).37 There are many studies showing the efficacy and safety regarding the use of statins for children and adolescents, and a meta-analysis and a systemic review are suggesting that it is true.56,57 Adherence should be closely monitored among those with poor response to statins before increasing the dose. Adolescent girls should be counseled to suspend statin therapy when contemplating pregnancy. Other medications, such as ezetimibe and resin, can be considered when needed. Notably, the European Atherosclerosis Society had proposed a similar strategy in Europe where “high-risk” pediatric patients with FH aged 8–10 years are recommended to start statins to reduce LDL cholesterol (Figure 2).10 Moreover, the National Lipid Association expert panel on FH had recommended similar management approaches (Box 3).58 In addition, resin, and ezetimibe are also shown to effectively reduce LDL cholesterol among the pediatric FH patients.59,60 More recently, it has been shown that evolocumab reduced the LDL cholesterol level and other lipid variables among them.61 Notably, all of the mentioned recommendations have acknowledged the need for actively attempting to diagnose/identify FH in children and adolescents and considering lowering LDL cholesterol levels through lifestyle intervention and statins.62
 |
Figure 1 Strategies for the management of pediatric familial hypercholesterolemia (FH) (JAS). Green arrows indicate “Yes”; blue arrows indicate “No.” The essential message is that the pediatric patients with FH aged 10 or greater who have low-density lipoprotein cholesterol levels ≥180 mg/dL under appropriate lifestyle intervention may be treated using statins. Reproduced from Harada-Shiba M, Ohta T, Ohtake A, et al. Joint Working Group by Japan Pediatric Society and Japan Atherosclerosis Society for Making Guidance of Pediatric Familial Hypercholesterolemia. Guidance for Pediatric Familial Hypercholesterolemia 2017. J Atheroscler Thromb. 2018;25(6):539–553.37 |
 |
Figure 2 Strategies for the diagnosis and management of familial hypercholesterolemia (FH) in children and adolescents (EAS). Premature coronary heart disease is defined as a coronary event before age 55 and 60 years in men and women, respectively. Definite FH is defined as genetic confirmation of at least one FH-causing genetic mutation. Close relative is defined as 1st or 2nd degree relatives. Highly probable FH is based on clinical presentation (ie, phenotypic FH): either an elevated low-density lipoprotein cholesterol (LDL-C) level ≥5 mmol/L in a child after dietary intervention or a LDL-C level ≥4 mmol/L in a child with a family history of premature coronary heart disease in close relatives and/or high baseline cholesterol in one parent. Cascade screening from an index case with a FH-causing mutation may identify a child with elevated LDL-C levels ≥3.5 mmol/L. Reproduced with permission from Wiegman A, Gidding SS, Watts GF, et al. European atherosclerosis society consensus panel. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J. 2015;36(36):2425–2437.10 |
On the other hand, there are other strategies, including MTTP inhibitor (lomitapide), APOB inhibitor (mipomersen), LDL apheresis, and liver transplantation for the cases with homozygous FH. Another potential medical therapy is Angiopoietin-like 3 (ANGPTL3) inhibitor, whose efficacy and safety for adult patients with homozygous FH has been shown.63
 |
Box 3 Summary Recommendations from the National Lipid Association Expert Panel on Treatments for Pediatric Familial Hypercholesterolemia |
Motivation for Genetic Analyses of Familial Hypercholesterolemia and Personalized Medicine for Familial Hypercholesterolemia in Current Clinical Settings
Regardless of its definition, genetic analyses for patients with FH should have clear indications beyond clinical diagnosis. As such, we herein highlight the several advantages of genetic background analysis in FH. First, quite a few patients with hypercholesterolemia have obscure or marginal clinical diagnoses of FH. Moreover, their family history is sometimes quite challenging to obtain. Genetic analyses can definitively identify patients with FH, especially those with traditional monogenic FH. Second, genetic analysis allows us to determine whether patients are heterozygous or homozygous. Several special medical therapies, such as MTTP inhibitors and LDL apheresis, have typically only been used for homozygous FH in many parts of the world. Moreover, homozygous FH is one of the designated intractable diseases where all medical costs can be covered by the Japanese government, thereby increasing the importance of diagnosis. Third, a portion of patients with FH may have been misdiagnosed as such and actually have another diagnosis, such as sitosterolemia. In such cases, ezetimibe instead of statins is recommended. Fourth, the genetic status of patients with FH has been found to be associated with increased risk for ASCVD. Accordingly, determining the genetic status can lead to better risk stratification. Fifth, genetic status determination can lead to better cascade screening and consequently better prognoses. Panel sequencing covering FH genes appears to be the current gold standard for determining the genetic status.64 However, determining the pathogenicity of the identified genetic variations has remained challenging for us.
Prime Time for Redefining Familial Hypercholesterolemia
We currently face an important dilemma regarding the definition of FH. If we adhere to clinical manifestations, such as tendon xanthomas, we believe that it is too late considering that earlier interventions based on earlier diagnosis have already been proposed. We believe that at least two different types of diagnostic criteria can be established. The first criteria, which would aim to diagnose “definite” FH, can be rather strict and have high diagnostic specificity, whereas the second one, which would aim to diagnose “potential” FH, can have high diagnostic sensitivity. Nonetheless, a diagnosis of FH needs to be ultimately established and adequately treated as early as possible before pediatric patients grow into adults.
Future Perspectives
Data science and personalized medicine are two major keywords describing medical innovations in the coming 10 years. In the management of FH, genetic analyses involving genes associated with not only LDL cholesterol itself but also ASCVD will become standard. Moreover, target, timing, and LDL cholesterol-lowering therapies will become quite individualized based on genotype, lifestyle, environmental factors, and belief systems. For the earlier identification of patients with FH, nation-wide mass screening, similar to that currently conducted for several other inherited metabolic diseases, such as newborn screening, phenylketonuria, and homocystinuria, will become standard. Furthermore, family history will be automatically assessed to accumulate a huge dataset over the years, which will help us minimize the risk of overlooking children and adolescents with FH (and other inherited diseases).
Conclusions
Given that FH is an inherited disease, early diagnosis and intervention can lead to excellent prognosis. Cascade and universal screening appear to be practical strategies for the early identification of patients with FH; however, we need to consider that certain clinical approaches can promote better identification of children and adolescents with FH. Clinical practices and genetic analyses will certainly help improve not only diagnostic accuracy but also risk stratification for personalized medicine.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kannel WB, Castelli WP, Gordon T, McNamara PM. Serum cholesterol, lipoproteins, and the risk of coronary heart disease. The Framingham study. Ann Intern Med. 1971;74(1):1–12. doi:10.7326/0003-4819-74-1-1
2. Friedman M. Pathogenesis of the spontaneous atherosclerotic plaque. A study on the A/Friedman M: pathogenesis of the spontaneous atherosclerotic plaque. A study on the cholesterol-fed rabbit. Arch Pathol. 1963;76:318–329.
3. Tada H, Usui S, Sakata K, Takamura M, Kawashiri MA. Low-density lipoprotein cholesterol level cannot be too low: considerations from clinical trials, human genetics, and biology. J Atheroscler Thromb. 2020;27(6):489–498. doi:10.5551/jat.RV17040
4. Baigent C, Blackwell L, Emberson J, Cholesterol Treatment Trialists’ (CTT) Collaboration, et al.. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681.
5. Cannon CP, Blazing MA, Giugliano RP, IMPROVE-IT Investigators, et al.. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372(25):2387–2397. doi:10.1056/NEJMoa1410489
6. Sabatine MS, Giugliano RP, Keech AC, et al. FOURIER steering committee and investigators. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713–1722. doi:10.1056/NEJMoa1615664
7. Schwartz GG, Steg PG, Szarek M, ODYSSEY OUTCOMES Committees and Investigators, et al.. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379(22):2097–2107. doi:10.1056/NEJMoa1801174
8. Mabuchi H. Half a century tales of familial hypercholesterolemia (FH) in Japan. J Atheroscler Thromb. 2017;24(3):189–207. doi:10.5551/jat.RV16008
9. Gidding SS, Champagne MA, de Ferranti SD, et al. American heart association atherosclerosis, hypertension, and obesity in young committee of council on cardiovascular disease in young, council on cardiovascular and stroke nursing, council on functional genomics and translational biology, and council on lifestyle and cardiometabolic health. The agenda for familial hypercholesterolemia: a scientific statement from the American heart association. Circulation. 2015;132(22):2167–2192. doi:10.1161/CIR.0000000000000297
10. Wiegman A, Gidding SS, Watts GF, et al. European atherosclerosis society consensus panel. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J. 2015;36(36):2425–2437. doi:10.1093/eurheartj/ehv157
11. Luirink IK, Wiegman A, Kusters DM, et al. 20-Year follow-up of statins in children with familial hypercholesterolemia. N Engl J Med. 2019;381(16):1547–1556. doi:10.1056/NEJMoa1816454
12. Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001:2863e913.
13. Fagge CH. Xanthomatous disease of the skin. I. general xanthelasma of vitiligoides. Trans Pathol Soc. 1873;24:242–250.
14. Thannhauser SJ, Magendantz H. The different clinical groups of xanthomatous diseases: a clinical physiological study of 22 cases. Ann Intern Med. 1938;11(9):1162. doi:10.7326/0003-4819-11-9-1662
15. Brown MS, Goldstein JL. Familial hypercholesterolemia: defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3–hydroxy-3-methylglutaryl coenzyme A reductase activity. Proc Natl Acad Sci U S A. 1974;71(3):788–792. doi:10.1073/pnas.71.3.788
16. Innerarity TL, Weisgraber KH, Arnold KS, et al. Familial defective apolipoprotein B-100: low density lipoproteins with abnormal receptor binding. Proc Natl Acad Sci U S A. 1987;84(19):6919–6923. doi:10.1073/pnas.84.19.6919
17. Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154–156. doi:10.1038/ng1161
18. Mabuchi H, Nohara A, Noguchi T, et al. Molecular genetic epidemiology of homozygous familial hypercholesterolemia in the Hokuriku district of Japan. Atherosclerosis. 2011;214(2):404–407. doi:10.1016/j.atherosclerosis.2010.11.005
19. Benn M, Watts GF, Tybjærg-Hansen A, Nordestgaard BG. Mutations causative of familial hypercholesterolaemia: screening of 98 098 individuals from the Copenhagen general population study estimated a prevalence of 1 in 217. Eur Heart J. 2016;37(17):1384–1394. doi:10.1093/eurheartj/ehw028
20. de Ferranti SD, Rodday AM, Mendelson MM, et al. Prevalence of familial hypercholesterolemia in the 1999 to 2012 United States national health and nutrition examination surveys (NHANES). Circulation. 2016;133(11):1067–1072. doi:10.1161/CIRCULATIONAHA.115.018791
21. Beheshti SO, Madsen CM, Varbo A, Nordestgaard BG. Worldwide prevalence of familial hypercholesterolemia: meta-analyses of 11 million subjects. J Am Coll Cardiol. 2020;75(20):2553–2566. doi:10.1016/j.jacc.2020.03.057
22. Tada H, Kawashiri MA, Nohara A, Inazu A, Mabuchi H, Yamagishi M. Impact of clinical signs and genetic diagnosis of familial hypercholesterolaemia on the prevalence of coronary artery disease in patients with severe hypercholesterolaemia. Eur Heart J. 2017;38(20):1573–1579. doi:10.1093/eurheartj/ehx004
23. Virani SS, Alonso A, Benjamin EJ, et al. American heart association council on epidemiology and prevention statistics committee and stroke statistics subcommittee. Heart disease and stroke statistics-2020 update: a report from the American heart association. Circulation. 2020;141:e139–e596.
24. Lozano P, Henrikson NB, Dunn J, et al. Lipid screening in childhood and adolescence for detection of familial hypercholesterolemia: evidence report and systematic review for the US preventive services task force. JAMA. 2016;316(6):645–655. doi:10.1001/jama.2016.6176
25. Groselj U, Kovac J, Sustar U, et al. Universal screening for familial hypercholesterolemia in children: the Slovenian model and literature review. Atherosclerosis. 2018;277:383–391. doi:10.1016/j.atherosclerosis.2018.06.858
26. Bell DA, Hooper AJ, Bender R, et al. Opportunistic screening for familial hypercholesterolaemia via a community laboratory. Ann Clin Biochem. 2012;49(6):534–537. doi:10.1258/acb.2012.012002
27. Scicali R, Di Pino A, Platania R, et al. Detecting familial hypercholesterolemia by serum lipid profile screening in a hospital setting: clinical, genetic and atherosclerotic burden profile. Nutr Metab Cardiovasc Dis. 2018;28(1):35–43. doi:10.1016/j.numecd.2017.07.003
28. Mirzaee S, Choy KW, Doery JCG, Zaman S, Cameron JD, Nasis A. The tertiary hospital laboratory; a novel avenue of opportunistic screening of familial hypercholesterolemia. Int J Cardiol Heart Vasc. 2019;23:100354. doi:10.1016/j.ijcha.2019.100354
29. Besseling J, Sjouke B, Kastelein JJ. Screening and treatment of familial hypercholesterolemia - lessons from the past and opportunities for the future (based on the anitschkow lecture 2014). Atherosclerosis. 2015;241(2):597–606. doi:10.1016/j.atherosclerosis.2015.06.011
30. Tada H, Okada H, Nomura A, et al. Prognostic impact of cascade screening for familial hypercholesterolemia on cardiovascular events. J Clin Lipidol. 2021. doi:10.1016/j.jacl.2020.12.012
31. Rodenburg J, Vissers MN, Wiegman A, et al. Statin treatment in children with familial hypercholesterolemia: the younger, the better. Circulation. 2007;116(6):664–668. doi:10.1161/CIRCULATIONAHA.106.671016
32. Wu X, Pang J, Wang X, et al. Reverse cascade screening for familial hypercholesterolemia in high-risk Chinese families. Clin Cardiol. 2017;40(11):1169–1173. doi:10.1002/clc.22809
33. Vinson A, Guerra L, Hamilton L, Wilson DP. Reverse cascade screening for familial hypercholesterolemia. J Pediatr Nurs. 2019;44:50–55. doi:10.1016/j.pedn.2018.09.011
34. Ibarretxe D, Rodríguez-Borjabad C, Feliu A, Bilbao JÁ, Masana L, Plana N. Detecting familial hypercholesterolemia earlier in life by actively searching for affected children: the DECOPIN project. Atherosclerosis. 2018;278:210–216. doi:10.1016/j.atherosclerosis.2018.09.039
35. Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol. 2004;160(5):407–420. doi:10.1093/aje/kwh236
36. Williams RR, Hunt SC, Schumacher MC, et al. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol. 1993;72(2):171–176. doi:10.1016/0002-9149(93)90155-6
37. Harada-Shiba M, Ohta T, Ohtake A, et al. Joint Working Group by Japan Pediatric Society and Japan Atherosclerosis Society for Making Guidance of Pediatric Familial Hypercholesterolemia. Guidance for Pediatric Familial Hypercholesterolemia 2017. J Atheroscler Thromb. 2018 ;25(6):539–553.
38. Eissa MA, Mihalopoulos NL, Holubkov R, Dai S, Labarthe DR. Changes in fasting lipids during puberty. J Pediatr. 2016;170:199–205. doi:10.1016/j.jpeds.2015.11.018
39. Tada H, Nohara A, Kawashiri MA. Monogenic, polygenic, and oligogenic familial hypercholesterolemia. Curr Opin Lipidol. 2019;30(4):300–306. doi:10.1097/MOL.0000000000000609
40. Masana L, Ibarretxe D, Rodríguez-Borjabad C, et al. Expert group from the Spanish arteriosclerosis society. Toward a new clinical classification of patients with familial hypercholesterolemia: one perspective from Spain. Atherosclerosis. 2019;287:89–92. doi:10.1016/j.atherosclerosis.2019.06.905
41. France M. Homozygous familial hypercholesterolaemia: update on management. Paediatr Int Child Health. 2016;36(4):243–247. doi:10.1080/20469047.2016.1246640
42. Raal FJ, Hovingh GK, Catapano AL. Familial hypercholesterolemia treatments: guidelines and new therapies. Atherosclerosis. 2018;277:483–492. doi:10.1016/j.atherosclerosis.2018.06.859
43. Pottle A, Thompson G, Barbir M, et al. Lipoprotein apheresis efficacy, challenges and outcomes: a descriptive analysis from the UK lipoprotein apheresis registry, 1989–2017. Atherosclerosis. 2019;290:44–51. doi:10.1016/j.atherosclerosis.2019.09.006
44. Kawagishi N, Satoh K, Akamatsu Y, et al. Long-term outcome after living donor liver transplantation for two cases of homozygous familial hypercholesterolemia from a heterozygous donor. J Atheroscler Thromb. 2007;14(2):94–98. doi:10.5551/jat.14.94
45. Mlinaric M, Bratanic N, Dragos V, et al. Case report: liver transplantation in homozygous familial hypercholesterolemia (HoFH)-long-term follow-up of a patient and literature review. Front Pediatr. 2020;8:567895. doi:10.3389/fped.2020.567895
46. Santos RD, Stein EA, Hovingh GK, et al. Long-term evolocumab in patients with familial hypercholesterolemia. J Am Coll Cardiol. 2020;75(6):565–574. doi:10.1016/j.jacc.2019.12.020
47. Malhotra A, Shafiq N, Arora A, Singh M, Kumar R, Malhotra S. Dietary interventions (plant sterols, stanols, omega-3 fatty acids, soy protein and dietary fibers) for familial hypercholesterolaemia. Cochrane Database Syst Rev. 1975;2014;2014(3):CD001918. doi:10.1152/ajplegacy.1975.229.3.570
48. Tada H, Nohara A, Inazu A, Sakuma N, Mabuchi H, Kawashiri M-A. Sitosterolemia, hypercholesterolemia, and coronary artery disease. J Atheroscler Thromb. 2018;25(9):783–789. doi:10.5551/jat.RV17024
49. Tada H, Okada H, Nomura A, et al. Rare and deleterious mutations in ABCG5/ABCG8 genes contribute to mimicking and worsening of familial hypercholesterolemia phenotype. Circ J. 2019;83(9):1917–1924. doi:10.1253/circj.CJ-19-0317
50. Tada H, Kawashiri MA, Takata M, et al. Infantile cases of sitosterolaemia with novel mutations in the ABCG5 gene: extreme hypercholesterolaemia is exacerbated by breastfeeding. JIMD Rep. 2015;21:115–122.
51. Tada H, Kawashiri MA, Okada H, et al. Assessments of carotid artery plaque burden in patients with familial hypercholesterolemia. Am J Cardiol. 2017;120(11):1955–1960. doi:10.1016/j.amjcard.2017.08.012
52. Tada H, Nakagawa T, Okada H, et al. Clinical impact of carotid plaque score rather than carotid intima-media thickness on recurrence of atherosclerotic cardiovascular disease events. J Atheroscler Thromb. 2020;27(1):38–46. doi:10.5551/jat.49551
53. Okada H, Tada H, Hayashi K, et al. Aortic root calcification score as an independent factor for predicting major adverse cardiac events in familial hypercholesterolemia. J Atheroscler Thromb. 2018;25(7):634–642. doi:10.5551/jat.42705
54. Tada H, Kawashiri MA, Okada H, et al. Assessment of coronary atherosclerosis in patients with familial hypercholesterolemia by coronary computed tomography angiography. Am J Cardiol. 2015;115(6):724–729. doi:10.1016/j.amjcard.2014.12.034
55. Tada H, Kawashiri MA, Nohara A, Inazu A, Mabuchi H, Yamagishi M. Assessment of arterial stiffness in patients with familial hypercholesterolemia. J Clin Lipidol. 2018;12(2):397–402. doi:10.1016/j.jacl.2017.12.002
56. Dombalis S, Nash A. The effect of statins in children and adolescents with familial hypercholesterolemia: a systematic review. J Pediatr Health Care. 2020;
57. Anagnostis P, Vaitsi K, Kleitsioti P, et al. Efficacy and safety of statin use in children and adolescents with familial hypercholesterolaemia: a systematic review and meta-analysis of randomized-controlled trials. Endocrine. 2020;69(2):249–261. doi:10.1007/s12020-020-02302-8
58. Goldberg AC, Hopkins PN, Toth PP, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the national lipid association expert panel on familial hypercholesterolemia. J Clin Lipidol. 2011;5(3):133–140. doi:10.1016/j.jacl.2011.03.001
59. Glueck CJ, Mellies MJ, Dine M, Perry T, Laskarzewski P. Safety and efficacy of long-term diet and diet plus bile acid-binding resin cholesterol-lowering therapy in 73 children heterozygous for familial hypercholesterolemia. Pediatrics. 1986;78(2):338–348.
60. van der Graaf A, Cuffie-Jackson C, Vissers MN, et al. Efficacy and safety of coadministration of ezetimibe and simvastatin in adolescents with heterozygous familial hypercholesterolemia. J Am Coll Cardiol. 2008;52(17):1421–1429. doi:10.1016/j.jacc.2008.09.002
61. Santos RD, Ruzza A, Hovingh GK, HAUSER-RCT Investigators, et al.. Evolocumab in pediatric heterozygous familial hypercholesterolemia. N Engl J Med. 2020;383(14):1317–1327. doi:10.1056/NEJMoa2019910
62. Ramaswami U, Futema M, Bogsrud MP, Holven KB. Comparison of the characteristics at diagnosis and treatment of children with heterozygous familial hypercholesterolaemia (FH) from eight European countries. Atherosclerosis. 2020;292:178–187. doi:10.1016/j.atherosclerosis.2019.11.012
63. Raal FJ, Rosenson RS, Reeskamp LF, ELIPSE HoFH Investigators, et al.. Evinacumab for homozygous familial hypercholesterolemia. N Engl J Med. 2020;383(8):711–720. doi:10.1056/NEJMoa2004215
64. Tada H, Kawashiri MA, Nomura A, et al. Oligogenic familial hypercholesterolemia, LDL cholesterol, and coronary artery disease. J Clin Lipidol. 2018;12(6):1436–1444. doi:10.1016/j.jacl.2018.08.006
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.