Back to Journals » Nanotechnology, Science and Applications » Volume 8
Factors determining the stability, size distribution, and cellular accumulation of small, monodisperse chitosan nanoparticles as candidate vectors for anticancer drug delivery: application to the passive encapsulation of [14C]-doxorubicin
Authors Masarudin MJ ![]() , Cutts S, Evison B, Phillips D, Pigram P
, Cutts S, Evison B, Phillips D, Pigram P
Received 4 July 2015
Accepted for publication 7 October 2015
Published 11 December 2015 Volume 2015:8 Pages 67—80
DOI https://doi.org/10.2147/NSA.S91785
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Israel Rubinstein
Mas Jaffri Masarudin,1 Suzanne M Cutts,2 Benny J Evison,3 Don R Phillips,2 Paul J Pigram4
1Department of Cell and Molecular Biology, Faculty of Biotechnology and Biomolecular Sciences, Universiti Putra Malaysia, Serdang, Malaysia; 2Department of Biochemistry, La Trobe University, Melbourne, Victoria, Australia; 3Department of Chemical Biology and Therapeutics, St Jude Children's Hospital, Memphis, TN, USA; 4Department of Physics, La Trobe University, Melbourne, Victoria, Australia
Abstract: Development of parameters for the fabrication of nanosized vectors is pivotal for its successful administration in therapeutic applications. In this study, homogeneously distributed chitosan nanoparticles (CNPs) with diameters as small as 62 nm and a polydispersity index (PDI) of 0.15 were synthesized and purified using a simple, robust method that was highly reproducible. Nanoparticles were synthesized using modified ionic gelation of the chitosan polymer with sodium tripolyphosphate. Using this method, larger aggregates were mechanically isolated from single particles in the nanoparticle population by selective efficient centrifugation. The presence of disaggregated monodisperse nanoparticles was confirmed using atomic force microscopy. Factors such as anions, pH, and concentration were found to affect the size and stability of nanoparticles directly. The smallest nanoparticle population was ~62 nm in hydrodynamic size, with a low PDI of 0.15, indicating high particle homogeneity. CNPs were highly stable and retained their monodisperse morphology in serum-supplemented media in cell culture conditions for up to 72 hours, before slowly degrading over 6 days. Cell viability assays demonstrated that cells remained viable following a 72-hour exposure to 1 mg/mL CNPs, suggesting that the nanoparticles are well tolerated and highly suited for biomedical applications. Cellular uptake studies using fluorescein isothiocyanate-labeled CNPs showed that cancer cells readily accumulate the nanoparticles 30 minutes posttreatment and that nanoparticles persisted within cells for up to 24 hours posttreatment. As a proof of principle for use in anticancer therapeutic applications, a [14C]-radiolabeled form of the anticancer agent doxorubicin was efficiently encapsulated within the CNP, confirming the feasibility of using this system as a drug delivery vector.
Keywords: nanobiotechnology, drug delivery, chitosan, chitosan nanoparticles, doxorubicin, FITC
Corrigendum for this paper has been
Introduction
Drug delivery is an issue of critical importance in the quest for more effective and more localized treatments and in the reduction of side effects associated with systemic modes of delivery. The fabrication of new types of drug delivery vehicles offers the prospect of increasing the potency and targeting of both currently approved drugs and emerging therapeutic materials. Nanoparticle drug delivery vehicles (diameters typically of the order of 10–200 nm) possess multiple potential advantages, including higher drug delivery efficiency,1 increased drug efficacy and cytotoxicity,2 as well as facilitation of penetration of various biological barriers such as the mucosal membrane and tumor vasculature.3 Moreover, the application of drug-loaded nanoparticles has been reported to overcome the drug-resistance characteristic of cancer cells, by facilitating entry into cells through endosomal uptake routes and surface binding, as opposed to the normal route of administration of the drug alone.4
In recent years, reports on the synthesis of chitosan-based nanoparticles have increased, especially in the field of medical science, reflecting the advantages of this system in biological applications. Chitosan is a carbohydrate polymer derived from the deacetylation of chitin and is known to be biodegradable and to possess unique immunological5,6 and antibacterial characteristics.7 Apart from being virtually nontoxic to cells, chitosan chains can undergo a range of chemical modifications, enabling the tailoring of properties to specific applications and may be tracked in cells through simple conjugation with fluorescent labels.8 Typical modifications include tailored functionalization of the chitosan chains, most commonly using amino groups (–NH2).
The synthesis of chitosan nanoparticles (CNPs) can occur through many routes, but the ionic gelation reaction of chitosan with anionic and polar molecules has been more thoroughly explored, mainly with glutaraldehyde,1,9 DNA,10 the chelator agent ethylenediaminetetraacetic acid,11 and sodium tripolyphosphate (TPP)12 as potential cross-linkers with chitosan. Calvo et al12 were among the first to describe an ionic gelation route for the synthesis of CNPs using TPP. While their methods produced nanoparticles in the size range of 300–1,000 nm, it was concluded that nanoparticle formation was feasible only for specific concentrations of both chitosan and its cross-linker. Because CNPs can be synthesized as delivery vectors or easily modified to confer features such as ligand targeting and chemical labeling, the initial size should preferably be very small in order to maximize loading efficiency.13 Encapsulation or further customization of the nanoparticles via synthesis usually leads to an expansion in size. Therefore, the ability to synthesize ultra-small CNPs (<100 nm) as delivery vectors is critical for drug delivery applications.
Contemporary CNP synthesis methodologies in the literature have limitations, in particular, relating to nanoparticle size control, which hinder applications as potential drug delivery vehicles. Conventional nanoparticle synthesis often leads to the formation of large particles or aggregates of smaller particles due to the mucoadhesive nature of chitosan.14 Currently, there exists only limited insight into what governs the size, formation, and distribution of CNPs obtained via ionic gelation with TPP. Factors such as pH, the fraction of free primary amino groups, solute concentration, and the inclusion of purification steps such as sonication and centrifugation all play a role in determining the physical characteristics and stability of nanoparticle formation. In this study, we describe a comprehensive optimization of these parameters for robust and repeatable CNP synthesis.
This report describes a systematic approach to producing small, stable, and homogeneously dispersed CNPs using a simple, highly reproducible synthesis route. We aimed to rationalize the various factors that influence the formation of CNPs and then utilize this knowledge to increase the synthesis efficiency. The persistence and long-term stability of CNPs in vitro was also investigated by dispersing the nanoparticles in a cell culture environment for up to 6 days and observing changes in particle morphology using atomic force microscopy (AFM). Visualization of CNP uptake into cells was studied by fluorescein isothiocyanate (FITC) labeling, to demonstrate that the nanoparticle system could potentially enhance cellular uptake. The feasibility of using the synthesized CNPs as vectors in drug delivery systems was demonstrated in principle by the encapsulation of radiolabeled [14C]-doxorubicin.
Materials and methods
Materials
Chitosan (low molecular weight), TPP, sodium dodecyl sulfate (SDS), 2,4,6-trinitrobenzene sulfonic acid (TNBS or picrylsulfonic acid) solution (5% w/v), FITC, and phenazine methosulfate (PMS) were purchased in powder form from Sigma-Aldrich (St Louis, MO, USA) and were used without further purification. Hydrochloric acid, glacial acetic acid, and sodium hydroxide were purchased from Merck Chemicals (Darmstadt, Germany). MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) was obtained from Promega Corporation (Fitchburg, WI, USA). [14-14C]-Doxorubicin (55 mCi/mmol) was acquired from GE Healthcare Biosciences (Little Chalfont, UK) and was resuspended as a 2 mM stock solution in Milli-Q water and stored at −20°C. Bio-Spin 6 columns, used to purify the samples, were purchased from Bio-Rad Laboratories Inc. (Hercules, CA, USA). Silicon cantilever tips were used for AFM analysis and were obtained from Nanosensors (Neuchâtel, Switzerland). Unless stated otherwise, all solutions were prepared using distilled and deionized water.
Synthesis of CNPs
CNPs were prepared by ionic gelation reactions with TPP. The concentrations of both chitosan and TPP used for synthesis are shown in Table 1. To prepare the chitosan solution (CS), 50 mg of chitosan powder was dispersed in 50 mL deionized distilled water (1 mg/mL). Under vigorous stirring, 1% acetic acid was added dropwise until the powder was completely dissolved. The resulting CS was subsequently diluted to various predetermined concentrations (Table 1) before being adjusted to pH 5 using 0.5 M NaOH. TPP was dissolved separately in deionized distilled water to a concentration of 1 mg/mL and adjusted to pH 2 with 0.1 M HCl. CNPs were formed by mixing 600 μL of CS with increasing amounts of TPP solution (20–250 μL) and incubation for 5 minutes at room temperature. The nanoparticles were then collected by centrifugation at 16,000× g for 90 minutes and the supernatant was discarded. The pellet was resuspended in 1 mL of deionized distilled water and 0.1 M acetic acid (50:1) and dispersed in solution by vortexing. The suspension was subjected to further centrifugation at 16,000× g for 45 minutes. The top 500 μL of the supernatant (containing the monodisperse CNPs) was collected and used for further experimentation. Alternatively, the CNP-containing supernatant was freeze-dried to remove any excess acetic acid. The lyophilized CNP powder was then weighed before subsequent analysis.
 | Table 1 The parameters used for nanoparticle synthesis |
Synthesis of FITC-labeled CNPs
Fluorescently labeled chitosan solution (FITC-CS) was prepared using methods previously reported.8 Briefly, 1 mg/mL chitosan was prepared as described in the “Synthesis of CNPs” section and diluted to a working concentration of 0.5 mg/mL. The FITC solution was prepared by dissolving 0.00125 g of FITC powder in 5 mL of dimethyl sulfoxide (DMSO) (0.25 mg/mL) and stirred in dark at 25°C, before being diluted to a final concentration of 0.05 mg/mL in DMSO. The conjugation of FITC to CS was performed by the addition of 200 μL FITC (0.05 mg/mL) to 600 μL chitosan (1.0 mg/mL). The mixture was then homogeneously mixed using a pipette, prior to the addition of TPP to form FITC-CNP. The synthesis of FITC-CNP was carried out using ionic gelation methods as described in the “Synthesis of CNPs” section. A volume of 200 μL of TPP (0.7 mg/mL at pH 2.0) was added to 800 μL of FITC-CS and homogeneously mixed to ensure a uniform mixture is obtained. Finally, the resulting FITC-CNP solution was purified by centrifugation using the steps described earlier in the “Synthesis of CNPs” section.
Analysis of CNP size distribution by dynamic light scattering and laser Doppler velocimetry
The size distribution of the synthesized CNPs was evaluated by dynamic light scattering (DLS) and laser Doppler velocimetry using a Malvern Zetasizer Nano ZS instrument (Malvern Instruments, Malvern, UK). Samples were measured in disposable polystyrene zeta cuvettes at 25°C, with each measurement preceded by a 30-second equilibration time. Readings were replicated six times per analysis run, and each sample run was replicated independently six times to ensure consistency in data acquisition.
Analysis of CNP morphology and estimation of size by AFM
The morphology, size, and surface topology of the synthesized CNPs were analyzed using AFM on an Asylum MFP-3D AFM microscope (Asylum Research, Goleta, CA, USA). Lyophilized CNP pellets were initially resuspended in deionized distilled water, drop-coated onto microscope slides, and left to dry in covered Petri dishes for 12 hours at room temperature. The slides were then mounted onto the instrument for imaging. Imaging was performed using a silicon AFM tip at a scan rate of 1.5 Hz using the noncontact mode. All AFM images were processed and displayed using the Argyle Light application (Asylum Research).
Determination of the fraction of free –NH2 groups in CNPs by the TNBS assay
The TNBS assay was performed to determine the fraction of free primary amino groups in the CNPs and to, subsequently, verify their role in the formation of the nanoparticles. TNBS reacts preferentially with primary amino groups to form a chromophore readily measured by colorimetric means at 335 nm. The assay used was a modification of methods previously described.15 Briefly, 150 μL of each CNP sample was mixed with an equal volume of 0.05% TNBS and then incubated at 37°C for 3 hours. Following incubation, 150 μL of 10% SDS and 125 μL 1 M HCl were added to terminate the reaction. A portion (200 μL) of the mixture was transferred to a 96-well plate and the absorbance was measured at 335 nm. The fraction of free amino groups in each CNP preparation was determined using the following equation:
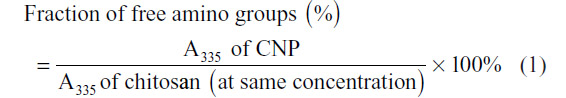
Cell culture and evaluation of CNP cytotoxicity in vitro using the MTS assay
Human kidney cancer cells (786-O) were purchased from the American Type Culture Collection (Manassas, VA, USA) and were maintained in RPMI medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal calf serum (Trade Scientific, Sigma-Aldrich, St Louis, MO, USA) in a humidified incubator at 37°C and 5% CO2. An MTS assay was performed to evaluate any toxicity of the CNP preparations toward the cells.
Approximately 100 μL of cells was seeded into 96-well plates at a density of 105 cells per well and allowed to grow for 24 hours prior to treatment. CNP samples were diluted serially from the highest concentration of 1 mg/mL. A volume of 50 μL of each concentration was pipetted into each well in quadruplicate. Treatment proceeded for 72 hours under cell growth conditions. Prior to the assay, MTS and PMS powders were dissolved separately in phosphate-buffered saline to a concentration of 1.6 mg/mL. Both solutions were sterile-filtered and mixed at a ratio of 6:1 in a Falcon tube. The solution was used immediately for the assay and 50 μL was pipetted into each well of the plate, before incubating for a further 3 hours at 37°C. The MTS absorbance was then measured on a microplate reader at 490 nm.
Time-dependent particle stability studies of synthesized CNPs in cell culture media
CNP samples were prepared as previously described in the “Synthesis of CNPs” section and were incubated at 37°C in RPMI medium (Thermo Fisher Scientific) for different time periods up to 72 hours. Following incubation, the medium was drop-coated onto microscope slides, dried, and analyzed using AFM through the methods noted in the “Analysis of CNP morphology and estimation of size by AFM” section.
Physical encapsulation of [14C]-doxorubicin within synthesized CNP
Radiolabeled [14C]-doxorubicin was used as a model drug for encapsulation to evaluate the potential of the CNP as a vehicle for drug delivery. Briefly, 1 mM [14C]-doxorubicin stock was prepared in deionized distilled water. The drug was added to 200 μL TPP (0.35 mg/mL, pH 2) to a final concentration of 3.0 μM and the mixture was stirred for 15 minutes at room temperature. CNP formation was then initiated by addition of 600 μL CS (0.25 mg/mL, pH 5). The CNP preparation was then purified through a Bio-Spin 6 column (Bio-Rad Laboratories Inc., Hercules, CA, USA) to remove unreacted CS and TPP molecules, as well as free [14C]-doxorubicin from solution. A sample of the resulting CNP-[14C]-doxorubicin was added to 1 mL of ReadySafe Scintillation Cocktail fluid (Beckman Coulter, Fullerton, CA, USA) and then analyzed using a Wallac 1410 Scintillation Counter (Pharmacia, Vantaa, Finland) to determine the amount of encapsulated [14C]-doxorubicin. The efficiency of encapsulation was calculated as follows:

Results and discussion
CNPs were successfully formed through ionic gelation of chitosan, with TPP acting as the cross-linking moiety. By modifying the methods previously described in the literature,12,16,17 we were able to synthesize homogeneously dispersed nanoparticles <100 nm in size, consistently with low polydispersity index (PDI) values, via a simple and easily reproducible synthesis route. However, it was interesting to note that although similar reports have been described for CNP synthesis, incongruence in particle size data can be observed. Therefore, it was concluded that while parameters for nanoparticle formation remain predominantly unchanged, particle size differed in most studies. This indicated that particle size was not determined by the concentration of chitosan or TPP used but is instead determined by various other factors. In order to determine the effects of chitosan and TPP concentrations on CNP size and distribution, three different parameter sets (termed CNP-F1, CNP-F2, and CNP-F3) were adapted for nanoparticle formation (Table 1). The PDI was used as an indicator for nanoparticle stability and uniformity of formation. The PDI value reflects the nanoparticle size distribution; samples with a wider range of particle sizes have higher PDI values, while samples consisting of evenly sized particles have lower PDI values. In our studies, a lower value indicates more monodisperse CNPs and greater particle stability, while higher values show lower particle stability and the presence of aggregated CNPs.
Formation of CNPs
CNP samples were formed through the cross-linking of chitosan chains by TPP via amino group ionic interactions. Figure 1 (inset) summarizes the fraction of free primary amino groups remaining in the CNPs, together with their respective size distributions and PDI values for all three chitosan:TPP ratios. These results showed that the fraction of free primary amino groups decreased with TPP, indicating an increase in the cross-linking interactions between the cationic amino groups of chitosan and the anionic TPP to form nanoparticles. DLS data showed that the formation of nanoparticles under 100 nm required a minimum of 50 μL of TPP, producing CNPs of sizes 96±1.4 nm, 83±1.0 nm, and 82±3.9 nm with PDI values of 0.35, 0.29, and 0.36, respectively (Figure 1A–C). The PDI further decreased with subsequent additions of TPP and resulted in the formation of more uniform and homogeneously distributed nanoparticles. At 200 μL TPP addition, nanoparticles with the smallest size and lowest PDI were formed for all three parameter sets (73.3±1.5 nm for CNP-F1, 61.76±0.13 nm for CNP-F2, and 62.2±0.9 nm for CNP-F3), while the PDI was 0.12, 0.15, and 0.15, respectively. Above 200 μL of TPP addition, the particle size and PDI increased significantly. At 250 μL TPP addition, PDI values increased to 0.63 in CNP-F1, 0.79 in CNP-F2, and 0.64 in CNP-F3, while particle size increased to 356±9 nm, 292±9 nm, and 267±13 nm in the CNP-F1, CNP-F2, and CNP-F3 formulations, respectively. On the basis of these observations, the optimal TPP volume (volume of TPP needed for synthesis of smallest, stable, and lowest-PDI-valued CNPs) for CNP synthesis was 200 μL (to 600 μL CS), giving a CS:TPP volume ratio of 3:1 for efficient CNP synthesis.
 | Figure 1 Influence of TPP volume on CNP size and polydispersity index. |
The striking decrease in particle size and PDI with TPP volume was consistent with the increased availability of TPP molecules to interact with the free amino groups of chitosan. As the nanoparticle forms, additional incorporation of the anion is suggested to further augment cross-linking between chitosan chains within the nanoparticle, thus explaining the decrease in CNP size with increasing TPP. This increase in internal cross-linking causes the chitosan chains to become more tightly bound within the particle, therefore condensing the particle further, leading to a gradual decrease in size. Because cross-linking also reduces the availability of free primary amino groups on chitosan, self-aggregation between different nanoparticles is prevented. This is consistent with the nanoparticles being more homogeneously distributed in size, along with lower PDI values. Such an interaction has been previously modeled in polymeric micelles,18,19 explaining the dynamics between the chitosan polymer and its cross-linker in our system.
The pH of chitosan used also favored the formation of smaller-sized nanoparticles. Chitosan chains are more constricted at pH 5 compared to solutions with more acidic pH, as a result of the greater number of hydrogen bond interactions within its structure due to a lower degree of amine protonation.20 This compaction of chains allows for formation of much denser particles when cross-linked with TPP, as opposed to a more linear chitosan chain. However, the addition of TPP also decreases the pH of the CNP suspension further, causing the protonation of more amine groups (Figure 2). At higher levels of TPP (>200 μL), protonation may disrupt the ionic linkages between chitosan and TPP in the CNP, therefore causing the nanoparticles to aggregate.
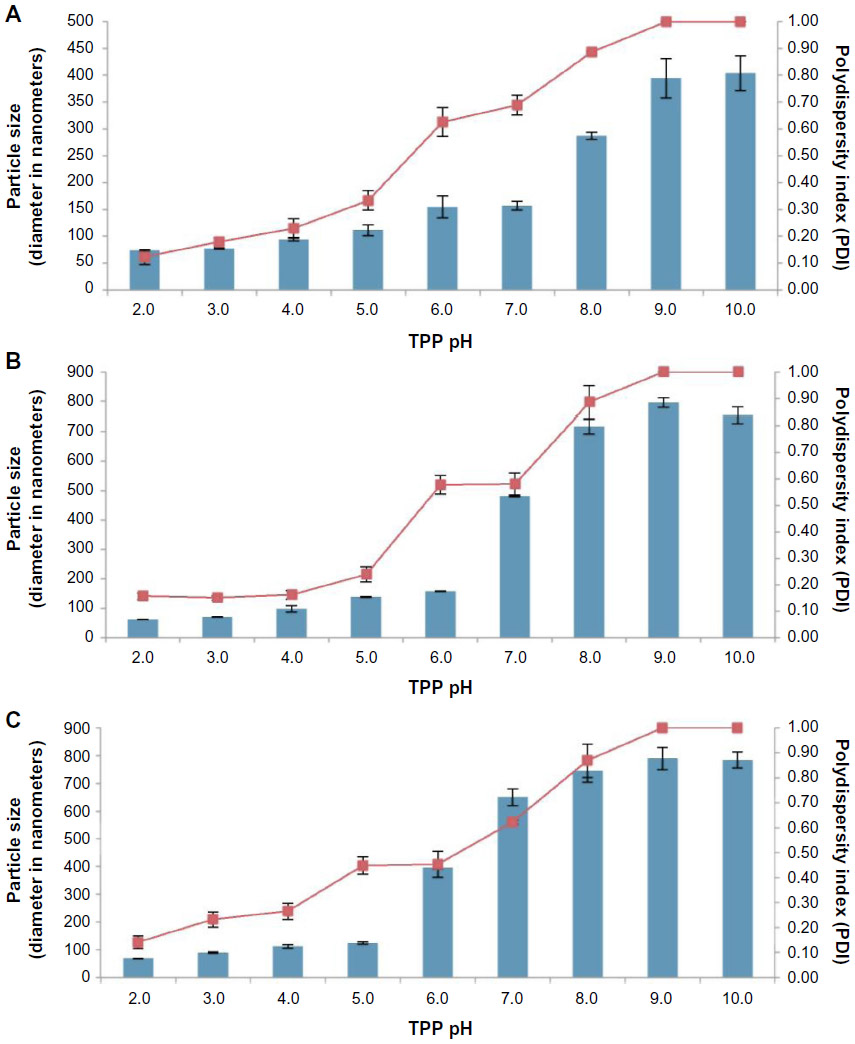 | Figure 2 Influence of TPP pH on CNP size and polydispersity index. |
In this study, we noted the simple yet pivotal role of applying various centrifugation steps in the synthesis route of CNP. Performing centrifugation steps at fixed intervals during nanoparticle synthesis was vital for the isolation of smaller and more homogeneously dispersed CNPs from the preformed particle aggregates. Due to Brownian motion, particles in the CNP colloidal solution sediment and collide with each other at different rates, according to size.21 During synthesis, the resulting CNP solution comprises both single and larger aggregated CNP particles. By considering the different sizes of the CNP, separation of smaller single, uniform nanoparticles from the larger, aggregated particles was accomplished by applying fixed centrifugation steps at various intervals of the synthesis route. When centrifugation was omitted from the synthesis route, we found that the CNP size significantly increased. The PDI and distribution peaks obtained from DLS were also noticeably higher without centrifugation, indicating the presence of CNP particles with a larger distribution in size, due to the presence of particle aggregates in the suspension. Such nanoparticle aggregation was also observed in samples visualized using AFM (data not shown), in which CNP preparations supplemented with centrifugation steps appeared as uniform, single nanoparticles compared to aggregated larger particles in CNP preparations that had not been subjected to centrifugation.
Influence of pH and concentration on CNP formation, stability, and particle size distribution
There have been numerous reports on the synthesis of chitosan and chitosan-modified nanoparticles in recent literature.22–24 In this study, we systematically explored the factors that influence CNP formation and stability. Although it has been postulated that nanoparticle size and formation is a direct consequence of the chitosan concentration used,25 there is no consistency in size distribution among these studies. We therefore investigated the role of TPP as a cross-linker and also TPP reactivity as a critical factor in the production of uniform CNP populations.
Figure 2 shows the influence of pH of TPP on the formation and stability of CNPs based on their PDI and size data obtained from DLS. These results show that formation of stable and homogeneously dispersed CNPs depended significantly on the pH of TPP used for synthesis. CNP synthesis using TPP at pH 2 produced the smallest particle size (73.3 nm, 62.2 nm, and 67.9 nm for parameter sets CNP-F1, CNP-F2, and CNP-F3, respectively). TPP at pH 10 produced the largest nanoparticles for CNP-F1 (403.7 nm), while CNP synthesized using TPP at pH 9 was the largest for CNP-F2 (796.0 nm) and CNP-F3 (789.0 nm). Collectively, particle size and PDI indexes were found to increase with both pH and the concentration of chitosan and TPP used, but they were more prominently influenced by the pH.
It is postulated that the pH of TPP affects the electronegative potential of the molecule in its reaction with free amine groups in chitosan. At lower pH values, TPP becomes less reactive for chemical interactions with chitosan because it is buffered by more positive ions in solution (H3O+ and H+). TPP therefore reacts with fewer amino groups of chitosan (NH3+), leading to the formation of smaller-sized nanoparticles that are more monodisperse. However, TPP at more basic pH is more reactive in solution due to lower positive ion buffering, which increases its affinity for interaction not only with the free amine groups of chitosan but also with the free amine groups of already formed CNPs (Figure 3). Size and PDI data obtained from DLS showed that TPP was most reactive at ≥ pH 7 for all three CNP formulations. Cross-linking between the nanoparticles by TPP causes agglomeration and hence a higher PDI value as the size distribution increases due to the presence of both the nanoparticles and their aggregates in solution.
 | Figure 3 The influence of pH on TPP reactivity. |
Morphology and appearance of CNPs
Figure 4 shows the AFM images of nanoparticles with a spherical morphology <100 nm in size. The size distribution of CNPs obtained from AFM was slightly smaller than the equivalent size data obtained from DLS analysis; DLS measures the hydrodynamic diameter of particles, whereas AFM sizes arise from direct tip–particle interactions. Analysis of the AFM data indicated size ranges of 68–75 nm for CNP-F1, 48–61 nm for CNP-F2, and 45–65 nm for CNP-F3 (Figure 4A–C, respectively) at a CS:TPP volume ratio of 3:1.
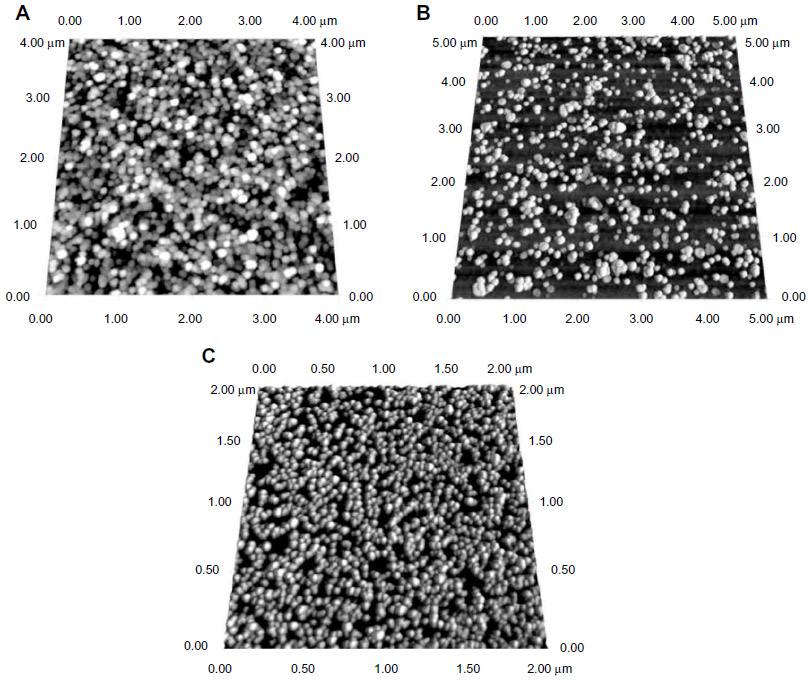 | Figure 4 Surface morphology of synthesized (A) CNP-F1, (B) CNP-F2+ and (C) CNP-F3 analyzed using AFM. |
While some aggregates were evident in the AFM images (Figure 4), this appears to be a consequence of the sample-drying procedure, arising from the decrease in solvent volume surrounding the nanoparticles. The nanoparticles synthesized at parameter sets CNP-F1, CNP-F2, and CNP-F3 were consistently similar in shape and were distributed as discrete spherical nanoparticles. Particle size was largest in CNP-F1 and smallest for CNP-F3, and was influenced by the concentration of both the chitosan chain and its cross-linker.
A homogeneous distribution of nanoparticles was apparent in samples purified with the extra centrifugation step during synthesis, while nanoparticle aggregation was apparent in CNP samples not subjected to centrifugation. CNPs purified through centrifugation showed smaller sizes with a much lower PDI, and these were observed with AFM as homogeneously distributed nanoparticles. In contrast, CNP samples prepared without centrifugation were observed as clearly larger, aggregated nanoparticles. For aggregated nanoparticles, the PDI values were typically >0.5 (data not shown).
Stability of synthesized CNPs in cell culture media
Both in vitro and in vivo nanoparticle stability is a key factor for the efficiency of delivery vectors. The nanoparticles need to remain stable in the extra- and intracellular environments long enough to be able to release their cargo into the cell or target tissue and to provide sufficient protection for the drug cargo on its journey to the target. In order to determine nanoparticle structure stability over time, the synthesized CNPs were incubated in RPMI media supplemented with serum for 72 hours under standard cell growth conditions. Following incubation, surface morphology analysis was performed using AFM, as shown in Figure 5.
 | Figures 5 The persistence and stability of CNPs in vitro. |
These results showed that the synthesized nanoparticles maintained their spherical structure in the serum-supplemented RPMI media for up to 72 hours. The size distribution of the nanoparticles (determined via AFM imaging) showed an increase after incubation. The nanoparticles continued to swell for up to 96 hours of incubation (data not shown), which was most likely due to the adsorption of water or other surrounding molecules through external surface pores.7 At 48–72 hours incubation, nanoparticle size had increased more than three-fold. The synthesized CNPs were found to be stable in the media for at least 5 days posttreatment with cells in vitro, a time frame that is anticipated to be sufficient for the delivery of biomolecules such as drugs and nucleic acids in cellular treatments. Following 6-days’ incubation, a fraction of the nanoparticles started to form hollow ring-like structures and considerable formation of aggregates was observed (Figure 5), indicating an initiation of nanoparticle degradation in the media. The presence of CNPs in media declined further at subsequent time points and was not detected 30 days posttreatment as the nanoparticles had been dissolved into the media (data not shown).
There is little discussion of the mechanisms of CNP degradation in the current literature. We speculate that nanoparticle degradation in this study was caused by changes in the physiological environment,19 such as pH and temperature, over time. Nanoparticle deterioration is thought to occur predominantly through a reversed cross-linking process of the NH2–TPP linkage, due to changes in pH of the media. Changes in media pH over time alter the ionization of –NH3+ groups of chitosan (pKa =6.3) and at higher pH, the electrostatic affinity between the amine group and TPP decreases.26 This reduced affinity then lowers their cross-linking interaction. This causes large pores and gaps to form at the nanoparticle surface, which in turn allow the ingress of water molecules and hence an increase in particle size over time. Collisions associated with Brownian motion promote the possibility of aggregation of unstable CNPs over time. Degradation may also be caused by nanoparticle leaching via the hydrolysis of chitosan chains throughout prolonged incubation times. Such a phenomenon has been described in previous literature and is more prominent in nanoparticles incubated at higher temperatures.20,27
Intracellular FITC–CNP accumulation
In order to determine whether the small, monodispersed CNPs conferred enhanced particle accumulation in treated cells, the nanoparticles were fluorescently labeled with FITC for visualization of cellular entry. Cells were treated with FITC–CNP and viewed at three specific time intervals: 30 minutes, 6 hours, and 24 hours, to observe any time-dependent cellular uptake. Figure 6 shows that intracellular nanoparticle uptake occurred as early as 30 minutes post-cell treatment. FITC-labeled CNP (FITC-CNP) was visualized as spherical green dots under ultraviolet excitation, while similar fluorescence was not observed in control cells without treatment (Figure 6D). At the 30-minutes’ treatment interval, FITC–CNP appears to reside in close proximity to the cell, outlining the cellular membrane, with a few particles appearing inside cells (Figure 6A). However, significant accumulation of intracellular FITC–CNP was observed at 6 hours posttreatment, surrounding the nucleus or persisting in the cytoplasm. Interestingly, a stronger fluorescence and cell smearing was observed as shown in Figure 6B. While this observation was unique, the increase in FITC intensity between early and late particle uptake was expected, from previously related reports.28 The intensity of fluorescence and cell smearing significantly increased 24 hours posttransfection (Figure 6C), and the persistence of spherical green dots suggesting the presence of FITC–CNP became less apparent. This transition from spherical dots to smears was thought to be contributed by the cleavage of FITC from CNP, thus releasing the fluorophore into intracellular space. Because FITC–CNP efficiently resides in cells, the FITC smearing was only observed inside the cells and not in the extracellular matrix. Therefore, the results could lead toward an elucidation of cell uptake routes that involve a time-dependent, dynamic physiological change such as an endocytosis-based pathway. Intracellular smearing due to FITC cleavage from FITC–CNP would also reinforce our suggested nanoparticle degradation pathway previously discussed in the “Stability of synthesized CNPs in cell culture media” section.
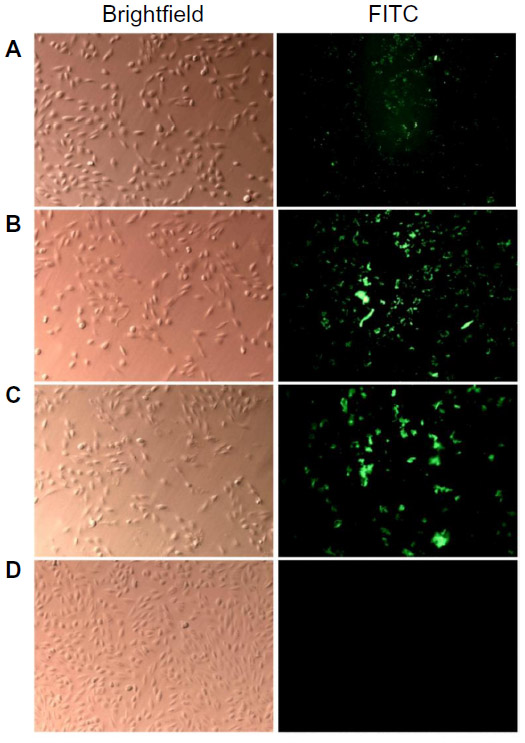 | Figure 6 Studies on uptake of FITC-labeled CNPs into 786-0 human kidney cancer cells. |
Cell cytotoxicity analysis of CNPs
The MTS assay was performed on human kidney cancer 786-O cells treated with CNP preparations to measure any inherent cytotoxicity of the nanoparticles in vitro. Cells were treated with different concentrations of the nanoparticles for 72 hours and were assessed for cell viability as shown in Figure 7. On the basis of the cell viability results obtained, all three CNP formulations were found to be nontoxic to the 786-O cells. At the maximum treatment dosage of 1.0 mg/mL, cell viability was 78%, 85%, and 89% for CNP formulations F1, F2, and F3, respectively. Cell viability appeared to reach a plateau >1.0 mg/mL; treatment beyond this concentration did not reduce cell viability (data not shown).
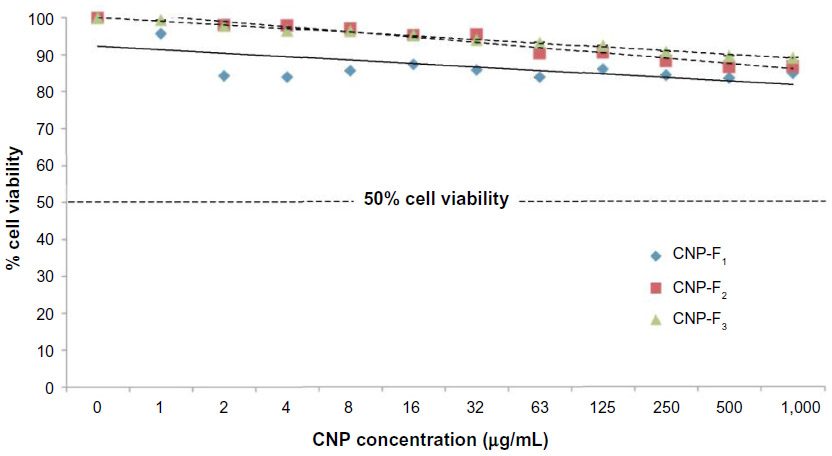 | Figure 7 Cell viability testing of 786-O cells by the MTS assay. |
The slight decrease of cell viability at higher concentrations of CNP was most probably caused by material leaching from the nanoparticles. While CNP stability studies showed that nanoparticles retain their structure throughout the assay period, it is worth noting that during this time frame, changes in cell culture environments, such as pH, may lead to slow degradation of the particles.
Physical encapsulation of [14C]-doxorubicin within synthesized CNPs
[14C]-Doxorubicin was physically encapsulated using the formulation CNP-F2 to demonstrate the potential of CNP as a drug delivery vehicle. The use of [14C]-doxorubicin allows a more accurate quantification of the amount of drug encapsulated in nanoparticles, compared to less-sensitive fluorometric detection methods. In this study, 3.0 μM of the drug was initially complexed with either CS or TPP before CNP formation. Ideally, during cross-linking between CS and TPP, the drug becomes physically entrapped within the CNP. The amount of [14C]-doxorubicin detected by scintillation analysis before and after encapsulation is shown in Figure 8. Interestingly, our findings suggest that encapsulation of [14C]-doxorubicin only occurs when the drug is complexed with the anion TPP prior to CNP formation. Results indicate that 0.51 μM [14C]-doxorubicin was encapsulated in the synthesized CNP following purification, a 17% efficiency in encapsulation. No encapsulation was observed when the drug was complexed with CS before CNP formation or when added to the solution after CNP formation (Figure 8). Encapsulation of the drug also led to a substantial increase in CNP size, as shown in Figure 8. Particle size increased from 143.2±1.0 nm in CNP to 397.0±0.4 nm in CNP-[14C]-doxorubicin samples, and PDI increased from 0.223 to 0.433 in CNP-[14C]-doxorubicin samples as compared to CNP without the drug. When the particles were purified by centrifugation to remove aggregates and to preserve only single nanoparticles, size distribution exhibited a four-fold increase from 63.5±1.3 nm to 270.3±25.6 nm between purified CNP and CNP-[14C]-doxorubicin samples, respectively (Figure 9).
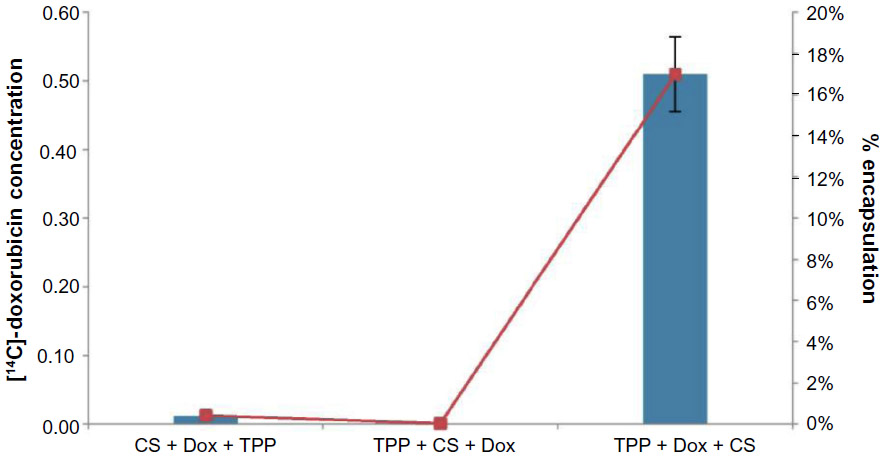 | Figure 8 Encapsulation of14C-Dox into CNP. |
 | Figure 9 Difference in nanoparticle size and PDI value before and after addition of 14C-Dox, represented by line (particle size) and bar (polydispersity index) graphs. |
Scintillation analysis has revealed that the physical entrapment of [14C]-doxorubicin into CNP relied significantly on the interaction and proximity of oppositely charged molecules present in solution during CNP formation. Doxorubicin, which is positively charged, becomes closer in proximity with the anionic molecule TPP when complexed in solution, due to the interactions between their charges. Thus, during CNP formation, the drug has a greater probability of passive encapsulation within the nanoparticle. However, when the drug is precomplexed with the cationic molecule chitosan, encapsulation is not possible due to repulsions between the two molecules that precede nanoparticle formation. Such charge interactions between doxorubicin and chitosan have been previously observed, and it has also been reported that the inclusion of a polyanion increased doxorubicin encapsulation efficiency into CNPs.17
Conclusion
We have successfully devised a highly reproducible, simple, and inexpensive route for the synthesis of monodisperse CNPs. Nanoparticles synthesized by all three formulations described herein were <100 nm in size, as confirmed by both DLS and AFM analysis. Parameters such as TPP concentration, pH, and chitosan concentration were found to significantly affect the formation of the nanoparticles. The nanoparticles were persistent in cell culture medium for up to 6 days posttreatment and diffused into media from 7 days posttreatment. Nanoparticles entered cells as early as 30 minutes and efficiently accumulated in cells within 24 hours posttransfection, without any designation of cell efflux. The synthesized CNPs were able to efficiently encapsulate [14C]-doxorubicin, and encapsulation was critically dependent on the sequence of components added during synthesis. Further formulations of these monodisperse nanoparticles, with respect to chemical modifications such as polyethylene glycol conjugation and the addition of tumor-targeted peptides, are anticipated to yield vectors suitable for delivery of drugs and small interfering RNAs to tumors in vivo.
Acknowledgments
MJM would like to thank the Malaysian Ministry of Higher Education and Universiti Putra Malaysia for kindly providing a scholarship and provisions for study leave. SMC acknowledges the Australian Research Council for a Future Fellowship. This work was performed in part at the Melbourne Centre for Nanofabrication in the Victorian Node of the Australian National Fabrication Facility.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Mitra S, Gaur U, Ghosh PC, Mitra AN. Tumour targeted delivery of encapsulated dextran-doxorubicin conjugate using chitosan nanoparticles as carrier. J Control Release. 2001;74:317–323. | |
Park J, Fong PM, Lu J, et al. PEGylated PLGA nanoparticles for the improved delivery of doxorubicin. Nanomedicine. 2009;5:410–418. | |
Krasnici S, Werner A, Eichhorn ME, et al. Effect of the surface charge of liposomes on their uptake by angiogenic tumor vessels. Int J Cancer. 2003;105:561–567. | |
Riganti C, Voena C, Kopecka J, et al. Liposome-encapsulated doxorubicin reverses drug resistance by inhibiting p-glycoprotein in human cancer cells. Mol Pharm. 2011;8:683–700. | |
Song S, Zhou F, Nordquist RE, Carubelli R, Liu H, Chen WR. Glycated chitosan as a new non-toxic immunological stimulant. Immunopharmacol Immunotoxicol. 2009;31:202–208. | |
Zaharoff DA, Rogers CJ, Hance KW, Schlom J, Greiner JW. Chitosan solution enhances both humoral and cell-mediated immune responses to subcutaneous vaccination. Vaccine. 2007;25:2085–2094. | |
Qi L, Xu Z, Jiang X, Hu C, Zou X. Preparation and antibacterial activity of chitosan nanoparticles. Carbohydr Res. 2004;339:2693–2700. | |
Jia X, Chen X, Xu Y, Han X, Xu Z. Tracing transport of chitosan nanoparticles and molecules in Caco-2 cells by fluorescent labeling. Carbohydr Polym. 2009;78:323–329. | |
Kulkarni VH, Kulkarni PV, Keshavayya J. Glutaraldehyde-crosslinked chitosan beads for controlled release of diclofenac sodium. J Appl Polym Sci. 2007;103:211–217. | |
Lee D, Lockey R, Mohapatra S. Folate receptor-mediated cancer cell specific gene delivery using folic acid-conjugated oligochitosans. J Nanosci Nanotechnol. 2006;6:2860–2866. | |
Loretz B, Bernkop-Schnurch A. In vitro evaluation of chitosan-EDTA conjugate polyplexes as a nanoparticulate gene delivery system. AAPS J. 2006;8:756–764. | |
Calvo P, Remunan-Lopez C, Vila-Jato JL, Alonso MJ. Novel hydrophilic chitosan-polyethylene oxide nanoparticles as protein carriers. J Appl Polym Sci. 1997;63:125–132. | |
Türk M, Hils P, Helfgen B, Schaber K, Martin HJ, Wahl MA. Micronization of pharmaceutical substances by the Rapid Expansion of Supercritical Solutions (RESS): a promising method to improve bioavailability of poorly soluble pharmaceutical agents. J Supercrit Fluids. 2002;22:75–84. | |
Sajomsang W, Gonil P, Ruktanonchai UR, et al. Self-aggregates formation and mucoadhesive property of water-soluble ß-cyclodextrin grafted with chitosan. Int J Biol Macromol. 2011;48:589–595. | |
Bubnis WA, Ill CMO. The determination of e-amino groups in soluble and poorly soluble proteinaceous materials by a spectrophotometric method using trinitrobenzenesulfonic acid. Anal Biochem. 1992;207:129–133. | |
Dudhani AR, Kosaraju SL. Bioadhesive chitosan nanoparticles: preparation and characterization. Carbohydr Polym. 2010;81:243–251. | |
Janes KA, Fresneau MP, Marazuela A, Fabra A, Alonsoa MJ. Chitosan nanoparticles as delivery systems for doxorubicin. J Control Release. 2001;73:255–267. | |
Chen H, Ruckenstein E. Formation and degradation of multicomponent multicore micelles: insights from dissipative particle dynamics simulation. Langmuir. 2013;29:5428–5434. | |
Chen H, Ruckenstein E. Formation of complex colloidal particles: morphologies and mechanisms. Soft Matter. 2012;8:8911–8916. | |
Tsai ML, Chen RH, Bai SW, Chen WY. The storage stability of chitosan/tripolyphosphate nanoparticles in a phosphate buffer. Carbohydr Polym. 2011;84:756–761. | |
Zhang X, Davis RH. The rate of collisions due to Brownian or gravitational motion of small drops. J Fluid Mech. 1991;230:479–504. | |
Alishahi A, Mirvaghefi A, Tehrani MR, Farahmand H, Koshio S, Dorkoosh FA. Chitosan nanoparticle to carry vitamin C through the gastrointestinal tract and induce the non-specific immunity system of rainbow trout (Oncorhynchus mykiss). Carbohydr Polym. 2011;86:142–146. | |
Anitha A, Rani VVD, Krishna R, et al. Synthesis, characterization, cytotoxicity and antibacterial studies of chitosan, O-carboxymethyl and N,O-carboxymethyl chitosan nanoparticles. Carbohydr Polym. 2009;78:672–677. | |
Wang JW, Chen CY, Kuo YM. Preparation and characterization of chitosan-coated hydroxyapatite nanoparticles as a promising non-viral vector for gene delivery. J Appl Polym Sci. 2011;121:3531–3540. | |
Katas H, Alpar HO. Development and characterisation of chitosan nanoparticles for siRNA delivery. J Control Release. 2006;115:216–225. | |
Shu XZ, Zhu KJ. A novel approach to prepare tripolyphosphate/chitosan complex beads for controlled release drug delivery. Int J Pharm. 2000;201:51–58. | |
Morris GA, Castile J, Smith A, Adams GG, Harding SE. The effect of prolonged storage at different temperatures on the particle size distribution of tripolyphosphate (TPP) – chitosan nanoparticles. Carbohydr Polym. 2011;84:1430–1434. | |
Zhang J, Chen XG, Peng WB, Liu CS. Uptake of oleoyl-chitosan nanoparticles by A549 cells. Nanomedicine. 2008;4:208–214. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.