Back to Journals » International Journal of Nanomedicine » Volume 13
Fabrication and evaluation of a γ-PGA-based self-assembly transferrin receptor-targeting anticancer drug carrier
Authors Zhang L ![]() , Zhu X, Wu S, Chen Y, Tan S, Liu Y, Jiang W
, Zhu X, Wu S, Chen Y, Tan S, Liu Y, Jiang W ![]() , Huang J
, Huang J
Received 23 July 2018
Accepted for publication 11 October 2018
Published 22 November 2018 Volume 2018:13 Pages 7873—7889
DOI https://doi.org/10.2147/IJN.S181121
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Li Zhang, Xiaoyu Zhu, Shijia Wu, Yazhou Chen, Shiming Tan, Yingjie Liu, Wenzheng Jiang, Jing Huang
School of Life Sciences, East China Normal University, Shanghai 200241, China
Background: cis-Dichlorodiamineplatinum (CDDP) was one of the most common used drugs in clinic for cancer treatment. However, CDDP caused a variety of side effects. The abundant carboxyl groups on the surface of poly glutamic acid (PGA) could be modified with various kinds of targeted ligands. PGA delivery system loaded CDDP for cancer therapies possesses potential to overcome the side effects.
Materials and methods: In this study, we constructed a safe and efficient anticancer drug delivery system PGA–Asp–maleimide–cisplatin–peptide complex (PAMCP), which was loaded with CDDP and conjugated with the transferrin receptor (TFR)-targeting peptide through a maleimide functional linker. The size of PAMCP was identified by transmission electron microscopy (TEM) and dynamic light scattering (DLS). Fluorescence microscopy and flow cytometry methods were used to detect the cell targeting ability in vitro. The MTT assay was used to detect targeted toxicity in vitro. The in vivo acute toxicity was tested in Kun Ming (KM) mice. The tumor suppression activity and drug distribution was analyzed in nude mice bearing with HeLa tumor cells.
Results: The nano-size was 110±28 nm detected with TEM and 89±18 nm detected with DLS, respectively. Fluorescence microscopy and flow cytometry methods indicated that PAMCP possessed excellent cell targeting ability in vitro. The MTT assay suggested that PAMCP was excellent for targeted toxicity. The acute in vivo toxicity study revealed that the body mass index and serum index in the PAMCP-treated group were superior to those in the CDDP-treated group (P<0.001), and no obvious differences were detected on comparing with the PBS- or PGA–Asp–maleimide–P8 (PAMP)-treated groups. PAMCP reduced the toxicity of CDDP, suppressed tumor cell growth, and achieved efficient anti-tumor effects in vivo. After PAMCP treatment, the toxicity of CDDP was reduced and tumor growth was more remarkably inhibited compared with the free CDDP treatment group (P<0.01). Much stronger (5–10 folds) fluorescence intensity in tumor tissue was detected compared with the irrelevant-peptide group for drug distribution analysis detected with the frozen section approach.
Conclusion: Our data highlighted that PAMCP reduced the side effects of CDDP and exhibited stronger anti-tumor effects. Therefore, PAMCP presented the potential to be a safe and effective anticancer pharmaceutical formulation for future clinical applications.
Keywords: cisplatin, tumor suppression, side effects, targeted drug delivery system
Introduction
Currently, chemotherapy is one of the most commonly used anticancer treatments in clinics. However, the anticancer effect is accompanied by severe side effects on normal tissues. The anticancer drug cisplatin (CDDP) has been widely used in the clinic for many years for the treatment of various cancer, such as metastatic testicular tumors, metastatic ovarian tumors, advanced bladder cancer, and so on.1 However, the shortcomings of CDDP, such as bone marrow growth inhibition,2 severe renal toxicity, nephrotoxicity, and lack of tissue-targeting specificity limit its clinical usage.3 CDDP-loaded drug delivery systems for cancer therapy possess potential clinical research value and are promising novel candidates for clinical antitumor therapy.
Over the past decades, tumor therapy has shifted from nontargeted chemotherapy to targeted therapies based on tumor-specific pathogenesis. Many research reports have focused on tumor-associated antigens that are overexpressed or absent in normal tissue.4–6 Based on the tumor microenvironment and tumor-specific antigens, monoclonal antibodies (mAbs)-based targeted molecular therapy has attracted more and more attention.7,8 Targeted therapy has become the focus in the field of tumor therapy because of its high efficacy and less number of side effects.9 The mAbs therapies include cellular growth factor receptors10 and cell surface antigens.10 However, mAbs have relatively larger molecular weights and the protein structure occupies more space, which make it hard for the mAb to penetrate into solid tumor tissues to achieve its therapeutic effects.11,12 Many researchers have screened tumor antigen-specific-targeting peptides using phage display technology.13 The screened peptides have smaller molecular weights, which enables penetration into the solid tumor tissues and maintenance of a relatively high affinity.14 Moreover, the short peptides can be easily obtained from chemical synthesis approaches, which could reduce the cost of treatment.
Transferrin receptor (TFR), as one of the tumor-specific antigens, is overexpressed in tumor tissues than in normal cells.15–18 In vivo phage display methods have effectively discovered that peptides could target TFR and significantly enhance the targeting ability for tumor therapy.19–21 A peptide of sequence HAIYPRH was obtained by the phage display method from human TFR and it harbored high affinity.22,23 CDDP has poor solubility. However, as previously reported, an aspartylated (Asp) poly-γ-glutamic acid (PGA) drug carrier system has good hydrophilic properties and could implement high CDDP loading efficiency.24,25 The existing carboxyl groups on the surface could be modified with targeting ligands to implement active targeting.26–29 CDDP-loaded PGA-based drug delivery systems conjugated with TFR-targeted peptides for cancer therapy possess important preclinical research value.
In this research, we used maleimide-fixed Asp-γ-PGA complex to load the bulk of CDDP by a self-assembly strategy. Maleimide conjugates with different kinds of ligands containing the thiol group through a thio-Michael reaction to form a thioether bond.30 The CDDP-loaded nanosized drug carrier was further conjugated with the TFR-targeted peptide with cysteine through the click reaction (Scheme 1). The tumor cell targeting and antitumor effects of the nanodrug system were evaluated in vitro and in vivo.
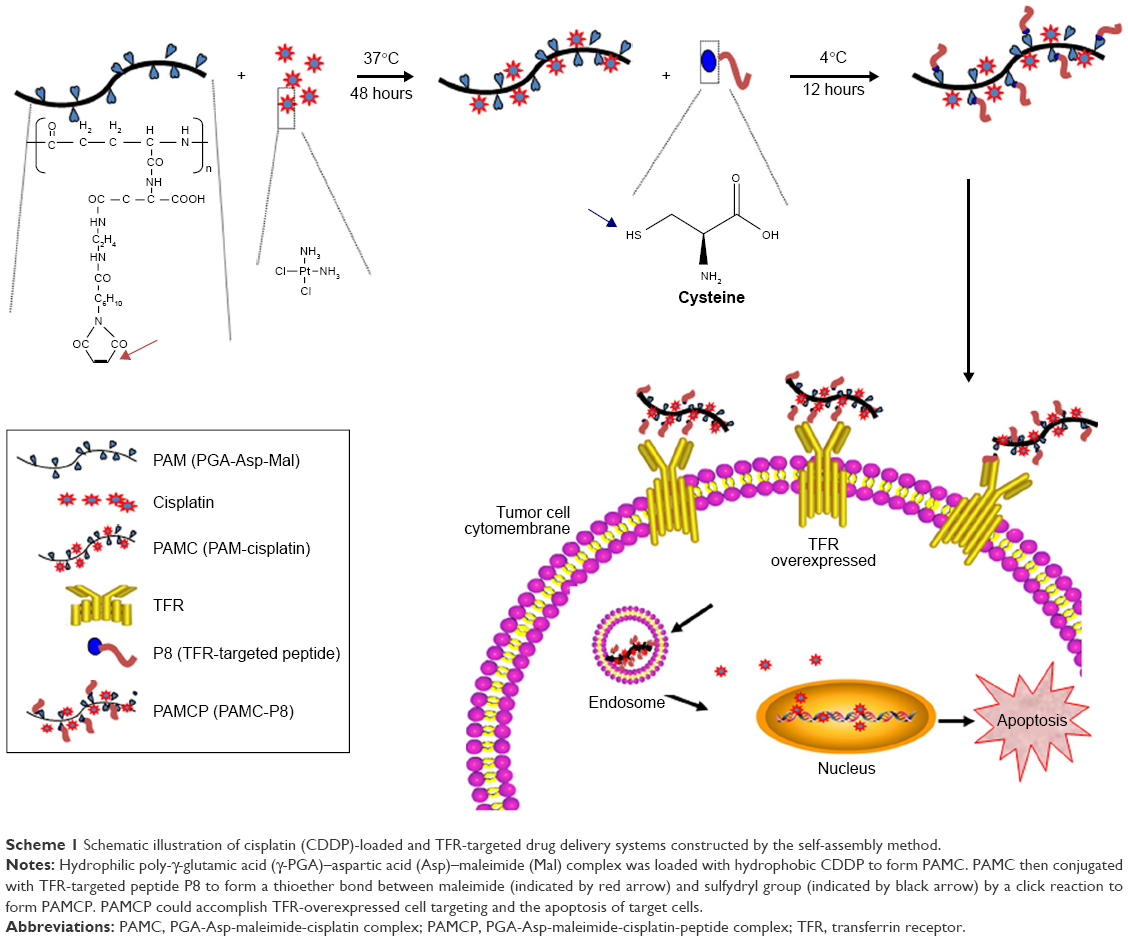 | Scheme 1 Schematic illustration of cisplatin (CDDP)-loaded and TFR-targeted drug delivery systems constructed by the self-assembly method. |
Materials and methods
Materials
PGA was purchased from Zijin Harbor Biological Company (Haining, China); H-Asp(OtBu)-OtBu.HCl(Asp) was bought from GL Biochem Ltd (Shanghai, China). Anti-TFR antibody was purchased from Abcam (Cambridge, MA, USA). Enhanced chemiluminescence (ECL) kit was purchased from Beyotime (Shanghai, China). Endothelial cell culture medium (ECM) was from ScienCell (Santiago Canyon, CA, USA) TUNEL kit (used for detecting apoptosis) was bought from Keygentec Co, Ltd (Nanjing, China). MTT was from Amerosco (Solon, OH, USA). Water-soluble amidation catalyst 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI) was purchased from Medpep (Shanghai, China). N-(maleimidohexanoyl)-ethylenediamine (Mal) was bought from Heowns (Tianjin, China). Protein content detection kit was from Thermo Fisher Scientific (Waltham, MA, USA), fluorescein isothiocyanate (FITC)-labeled TFR-targeting peptide P8 (sequence: CHAIYPRH) and FITC-labeled peptide P8X (sequence: CPIAHRHY, used as the control) were synthesized by Qiangyao Ltd (Shanghai, China). Trypsin, penicillin-and-streptomycin (P/S), FBS, and CDDP were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Human cervical cancer cell line (HeLa) and human umbilical vein endothelial cells (HUVECs) were purchased from the Cell Bank of Shanghai Academy of Chinese Academy of Sciences.
Cell culture
HeLa cells were cultured in DMEM cell culture medium with 10% FBS and 1% P/S. HUVECs were cultured in ECM with 10% FBS and 1% P/S. All cells were incubated in a cell incubator with 5% CO2 at 37°C.
Preparation of a self-assembly biopolymer PGA–Asp–maleimide–cisplatin–peptide complex (PAMCP)
The biopolymer PGA-Asp-Mal (PAM) was fabricated as previously described.31,32 Low-molecular-weight PGA was prepared by acid degradation (pH 3.0) in 98°C for 20 minutes. Low-molecular-weight PGA (4.25 g, 33 mmol carboxyl sites), Asp (8.5 g, 30 mmol), and EDCI (6.375 g, 33 mmol) were dissolved in ultrapure water (pH 6.7), and the reaction was maintained at room temperature for 24 hours. The mixture was purified by the dialysis method against ultrapure water with a dialysis bag (molecular weight cutoff [MWCO]: 10,000 Da) at pH 2.0 for 48 hours. The PGA-Asp complex (111 mg, 0.90 mmol carboxyl groups) was dissolved in 10 mL dimethyl sulfoxide (DMSO) for 1 hour at 37°C. EDCI (1.8 mmol) was added into 2 mL DMSO with PGA-Asp. Triethylamine (TEA, 30 μL) was added, and then Mal (0.3 mmol) was dropped into the reaction mixture. The reaction was mixed under magnetic stirring with nitrogen at room temperature for 24 hours. The product PAM was extracted using diethyl ether and freeze-dried in a vacuum. The complex PGA–Asp–Mal–CDDP (PAMC) was obtained by adding CDDP (30 mg) to the PAM (15 mg) aqueous solution (the final pH value was adjusted to 7.0) and shaken for 48 hours in the dark at 37°C, the free CDDP was removed by dialysis (MW, 10 kDa) against ultrapure water for 48 hours, then freeze-dried in a vacuum. Eventually, the P8 peptide (1 mg) was added into PAM aqueous solution (20 mg) and conjugated with PAM through the click reaction, and the reaction was conducted for 12 hours at 4°C to form the empty drug carrier PGA-Asp–maleimide-P8 (PAMP) without loading CDDP. P8 peptide (1 mg) was further added into PAMC aqueous solution (37 mg) to form the drug targeting system PAMCP. Unbound peptide was removed by dialysis, and then the final product was freeze-dried in a freeze dryer (−40°C, vacuum). CDDP packaged in PAMCP was detected with colorimetric o-phenylenediamine method.33 The drug-loading efficiency was calculated using the equation EE = M1/M2. M1 represents the weight of CDDP in PAMCP, and M2 represents the total weight of CDDP used in the equation.
Particle size analysis
A light scattering (Zetasizer Nano-ZS; Malvern Instruments, Malvern, UK) instrument was used to detect the PAMCP nanoparticle size and polydispersity index (PDI) in triplicates. Characterization of PAMCP shape was carried out with transmission electron microscopy (TEM; JEM-2100; JEOL, Tokyo, Japan).
The release behavior of CDDP from PAMCP
CDDP release behavior from PAMCP was investigated by dialysis.34 CDDP released from PAMCP was examined in phosphate buffer solution (PBS) (pH 7.4) or acetate-buffered saline (ABS, pH 5.5, 0.14 M NaCl) using a dialysis bag with MWCO of 8,000 Da. Furthermore, 5 mL of PAMCP or free CDDP solution was added in a dialysis bag immersed in 100 mL dialysis buffer and was laid on a shaker with a speed of 150 rpm at 37°C. At the given time, 200 μL of sample was acquired from the external dialysis fluid, and an equal amount of fresh dialysis fluid was reloaded. The quantity of CDDP released was detected with o-diaminobenzene method. The light absorption value was detected by a microplate reader (Epoch; BioTek, Winooski, VT, USA) at 702 nm.
Cell targeting assay
HeLa and HUVECs were cultured in a 24-well plate (5×104 cells per well). P8 (30 μg/mL), and PAMP (peptide concentration equaled 30 μg/mL, quantified by bicinchoninic acid [BCA] protein quantification kit) and P8X (peptide concentration equaled 30 μg/mL) were added into each well, respectively. The cells were incubated at 37°C for 30 minutes, and the cells were washed with PBS and stained with DAPI. The cells were observed under an inverted fluorescence microscope (DMI4000B; Leica Microsystems, Wetzlar, Germany).
The cell-binding ability of PAMP was further detected with flow cytometery (FACScan; Becton Dickinson, Franklin Lakes, NJ, USA). HeLa and HUVECs were digested with 0.25% trypsin and harvested with centrifugation (800× g, 5 minutes). The TFR low-expression HUVECs were incubated with P8 (50 μg/mL); then, HeLa cells were added in 2 mL PBS or incubated with P8 (50 μg/mL), P8X (50 μg/mL, negative control peptide group), PAMP (peptide concentration was equivalent to 50 μg/mL, quantified by BCA protein quantification kit) for 30 minutes at 37°C. PBS-treated group was used as control. Cells were washed three times with PBS. The mean fluorescence intensity was detected to evaluate the targeting effect. flowJo 7.6 software was used to analyze the cell-targeting ability.
In vitro cytotoxicity assay
HeLa cells were grown in 96-well plates at a density of 5×103 cells per well. The cytotoxicity was tested by general and specific cytotoxicity methods. In the general cytotoxicity assay, the culture medium was replaced with serial dilutions of PAMCP (CDDP concentration equal to 200, 100, 50, 25, 12.5, 6.25, 3.125 μg/mL), PAMC (CDDP concentration equal to 200, 100, 50, 25, 12.5, 6.25, 3.125 μg/mL), or CDDP (200, 100, 50, 25, 12.5, 6.25, 3.125 μg/mL) in fresh medium. The incubation lasted for another 24 hours or 48 hours. In the specific cytotoxicity method, after 10 minutes of incubation, the PAMCP-, PAMC-, and CDDP-containing culture medium was removed and incubated with fresh medium for another 24 hours or 48 hours. Then, MTT solution (5 mg/mL) was dropped into each well (20 μL per well) for another 4 hours. The supernatant was removed, 100 μL DMSO was added to each well, and the plate was vibrated for 5 minutes. The OD value of each well was measured at 570 nm with a spectrophotometer (Thermo Fisher Scientific).
In vivo toxicity
The in vivo toxicity was tested on Kun Ming (KM) mice from Shanghai Laboratory Animal Center (SLAC) model animal center. KM mice (n=40; 5–7 weeks old) were divided into 4 groups (n=10 per group) according to body weight. Two KM mice groups received CDDP (4 mg/kg) or PAMCP (CDDP equivalent dose of 4 mg/kg) by tail vein injection, respectively. The remaining groups received PAMP (equal to 200 μL PAMCP) or PBS (200 μL). Five injections for each group were given every other day. Mice survival rate examination lasted for 16 days. As an indicator of systemic toxicity, the body weight was measured every day. Finally, the mice were anesthetized and the sera were collected. White blood cells (WBCs) and red blood cells (RBCs) were counted using a hemocytometer for the hematological assessment.35 Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and creatinine (CRE) were tested using a routine blood biochemical analyzer (BM704; Hitachi Ltd., Tokyo, Japan) for renal function (BUN and CRE) and hepatic function (ALT and AST).
The kidney, spleen, and liver were harvested and fixed in 10% neutral formalin and then processed with dehydration, paraffin embedding, and section cutting. To detect the tissue apoptosis, an in situ apoptosis detection TUNEL kit was processed according to the manufacturer’s instruction. The animal experiments conducted in the study were approved by the Animal Ethics Committee of East China Normal University and all animal experiments followed the guidelines of East China Normal University Animal Care and Use Committee.
The tumor suppression activity
BALB/c nude mice (female, 6 weeks old) from SLAC were divided into 4 groups (n=8). To develop tumor-bearing mice, 1×107 HeLa cells were injected into the right femoral region of mice by hypodermic injection. CDDP or PAMCP (containing 4 mg CDDP/kg) was administered by caudal vein injection every 4 days for 5 injections and the experiment lasted for 25 days. The remaining groups were given PBS or PAMP solution through caudal vein injection. The following equation was used to measure the tumor volume:
|
|
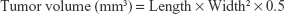
In vivo PAMCP distribution
The distributions of PAMCP in different organs were detected with fluorescence microscope after freezing the sections. PAMCP (peptide concentration 6.6 mg/kg, quantified by BCA protein quantification kit) and P8 (peptide concentration 6.6 mg/kg) were administrated into HeLa tumor-bearing nude mice (n=4 for each group) by tail vein injection. After 24 hours, mice were anesthetized, which was followed by heart perfusion. The tumor, spleen, kidney, brain, lung, heart, and liver tissues were harvested for frozen sectioning. Then, selected tissues were dyed with DAPI and then mounted in 80% glycerol. Different tissue sections were observed under a fluorescence microscope (DMLB; Leica). The fluorescence intensities of different organs were analyzed by Carestream MI SE molecular imaging software.
Data analysis
The data analysis of different groups was conducted with one-way ANOVA with GraphPad Prism 5 software. A value of P<0.05 was determined to be a significant statistical difference. All data are presented as mean ± SD.
Results
Characterization of PAMCP
The mean particle diameter of PAMCP was 89±18 nm, which is presented in Figure 1A, and the nanosize was in the ideal ranges36,37 reported for the EPR effect in cancer treatment. The PAMCP showed a relatively wide monomodal distribution, and the PDI value of PAMCP was 0.401, which suggested that the PAMCP had a relatively wide dispersion. The mean particle diameter of PAMCP detected by TEM was 110±28 nm (Figure 1B). The drug-loading efficiency was 37%±6%. The targeting peptide P8 could effectively conjugate with the drug carrier PAM, as detected by SDS-PAGE (Figure S1). CDDP, being smaller in size, can quickly be removed from the kidneys by filtration,38 while PAMCP, of nanometer size, could avoid filtration, maintain a stable blood concentration, enhance therapeutic efficacy, and improve the patients’ compliance.
 | Figure 1 The size characterization of PAMCP. |
PAMCP achieved TFR-mediated cell targeting
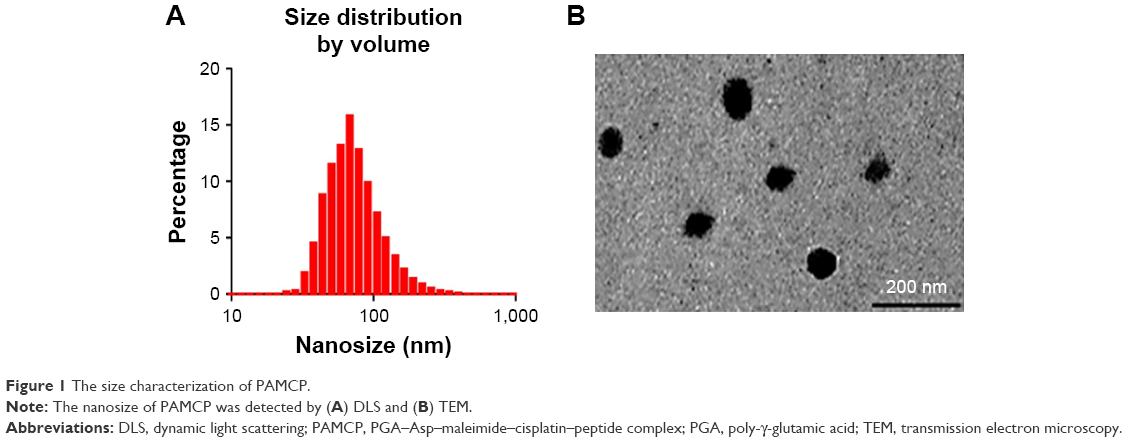
Previous reports and western blotting assay (Figure S2) have confirmed that HeLa cell is a TFR-overexpressed cell line39 and TFR is expressed in low levels in HUVECs. When HeLa cells were incubated with P8 or PAMCP, stronger green fluorescence signal could be detected (Figure 2A). However, when HeLa cells were incubated with the negative control peptide P8X or when HUVECs were incubated with P8, almost no cells with green fluorescence were observed. This phenomenon could be interpreted as the affinity effects between TFR and targeting peptides.
 | Figure 2 In vitro targeting of PAMCP. |
The flow cytometry method (FCM) was further carried out to confirm the TFR-mediated cell targeting. The lowest fluorescence intensity (4.21) was detected when HeLa cells were incubated with PBS (Figure 2B-a; Table 1). When HeLa cells were incubated with P8X, the mean fluorescence intensity (26.95) was almost consistent with that of the PBS-treatment group (Figure 2B-b and Table 1). When HUVECs were incubated with P8, the mean fluorescence intensity was 37.35 (Figure S3). Stronger fluorescence signals (99.7) were detected in PAMCP-treated HeLa cells, compared with cells treated with PBS, P8X, or the HUVEC (Figure S3) treatment groups (Figure 2B-c and Table 1). The strongest fluorescence signal (149.53) was detected when HeLa cells were incubated with the P8 peptide (Figure 2B-d and Table 1). The in vitro cell-targeting test indicated that PAMP could achieve cell targeting through TFR-mediated targeting.
 | Table 1 The mean fluorescence intensity of HeLa cells |
PAMCP reduced the in vitro toxicity
HeLa cells were cultured with CDDP, PAMC, or PAMCP and then, the MTT assay was used to test the cell survival rates. The results indicated that CDDP, PAMC, and PAMCP suppressed cell proliferation in a CDDP concentration-dependent manner (Figure 3 and S4). As shown in Table 2, after treatment with PAMCP, PAMC, or CDDP, the IC50 values of the PAMCP group (98.5±18.5 μg/mL, 40.47±16.3 μg/mL) were higher than those of the CDDP group (26.54±6.8 μg/mL, 7.82±4.3 μg/mL) for general toxicity at 24 hours and 48 hours, respectively, and a little lower than those of the PAMC group (107.38±27.7 μg/mL, 68.14±11.2 μg/mL). However, we detected more obvious cytotoxic effects for the specific toxicity studies in the PAMCP group (217.3±79.5 μg/mL, 120.1±17.3 μg/mL) than in the CDDP group (932.2±435.8 μg/mL, 519.1±174.5 μg/mL) or the PAMC group (2,517.3±217.6 μg/mL, 1,652.1±189.6 μg/mL) at 24 hours and 48 hours, respectively, on removing the drugs after 10 minutes of incubation. This indicated that there was a specific binding effect of PAMCP on HeLa cells and the binding effects enhanced drug cytotoxicity on the target cells. The results revealed that with the aid of TFR-targeted drug carrier, CDDP could achieve target cytotoxicity in vitro.
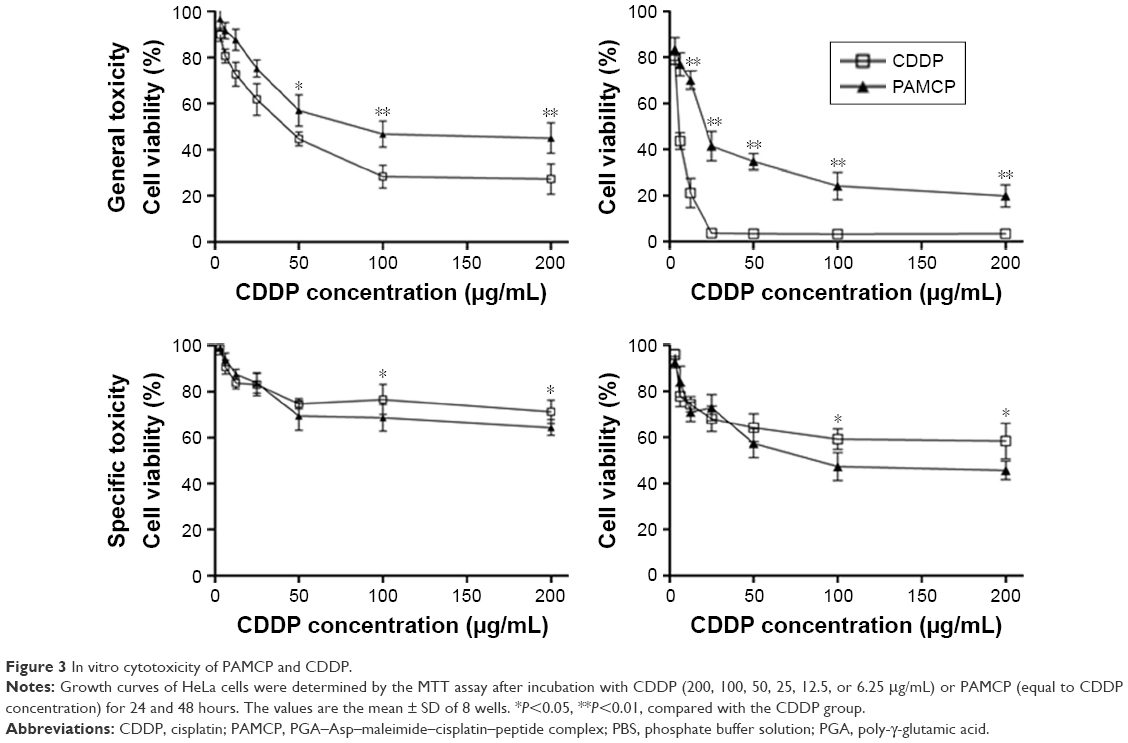 | Figure 3 In vitro cytotoxicity of PAMCP and CDDP. |
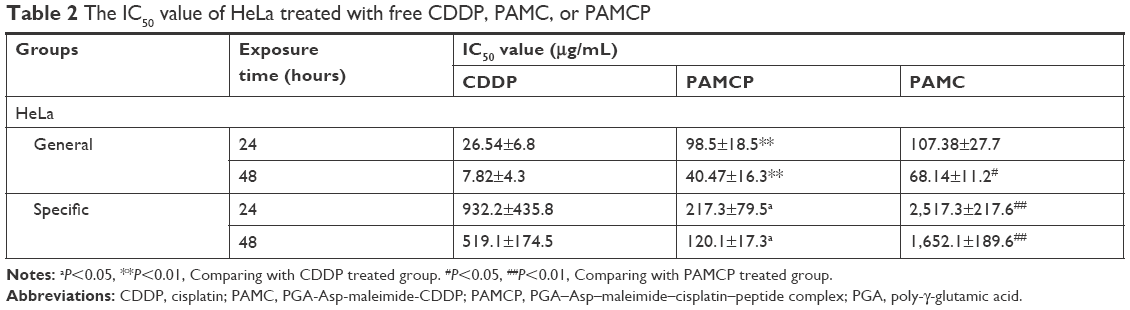 | Table 2 The IC50 value of HeLa treated with free CDDP, PAMC, or PAMCP |
PAMCP reduced the in vivo toxicity
The KM mice were used to evaluate the in vivo toxicity. The changes in survival rate and body weight were indicators of systemic toxicity of PAMCP. The whole experimental period was 14 days. The survival rate of the mice was 100% when administered with PAMCP (4 mg/kg CDDP); while 50% survival rates were noticed when the mice received 4 mg/kg CDDP (Figure 4A). No death appeared when the mice received the empty drug carrier (vehicle) PAMP. Body weight loss was another index to evaluate in vivo toxicity. When the mice were injected with CDDP, the body weight loss was about 17% (P<0.001) in the end, and spleens of the mice significantly decreased. Compared with the PBS or PAMP treatment group, there were no obvious body weight changes or spleen decreases observed in the PAMCP group (Figure 4B–D). Compared with the PBS or PAMP group, there were no obvious changes in terms of decrease in body weight or the WBC/RBC counts in the PAMCP group (Table 3). However, in the CDDP treatment group, the WBC and RBC counts in mice significantly decreased (P<0.01). BUN and CRE are two classic renal function detection indexes, and ALT and AST are standards to estimate liver function. Analysis of serum urea, CRE, ALT, and AST levels indicated that only the free CDDP treatment group brought about obvious kidney function and liver function injury.
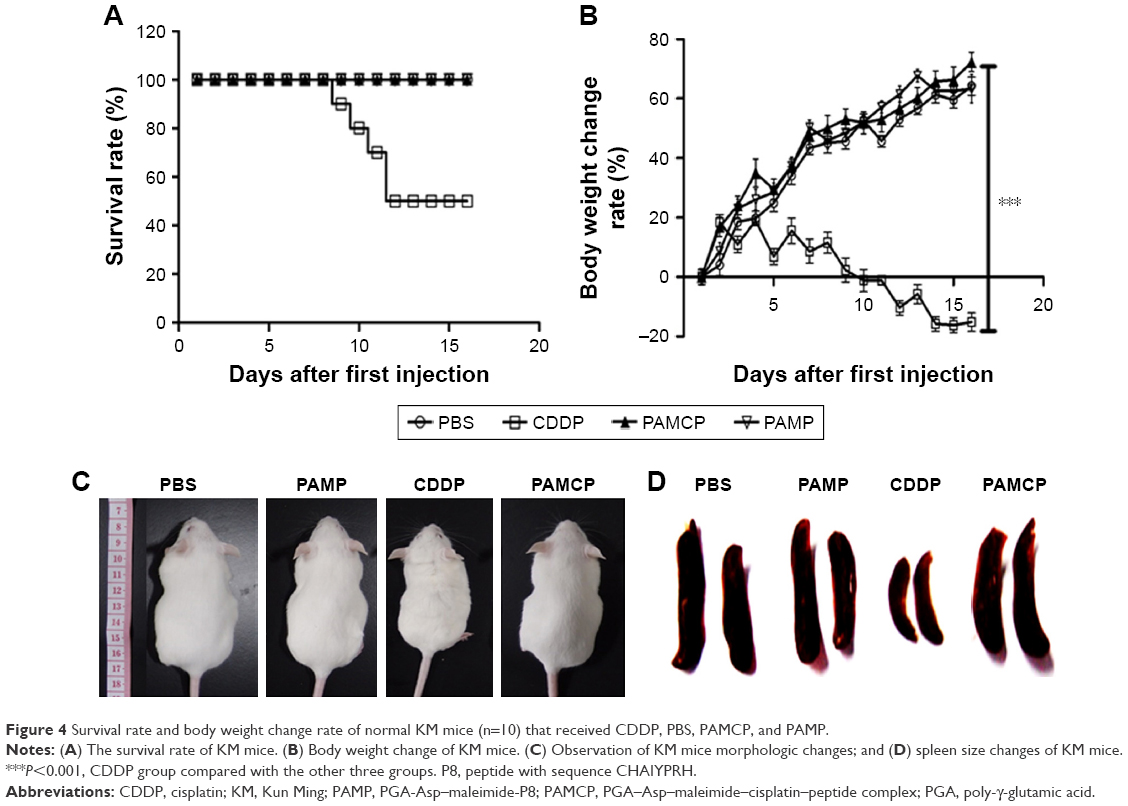 | Figure 4 Survival rate and body weight change rate of normal KM mice (n=10) that received CDDP, PBS, PAMCP, and PAMP. |
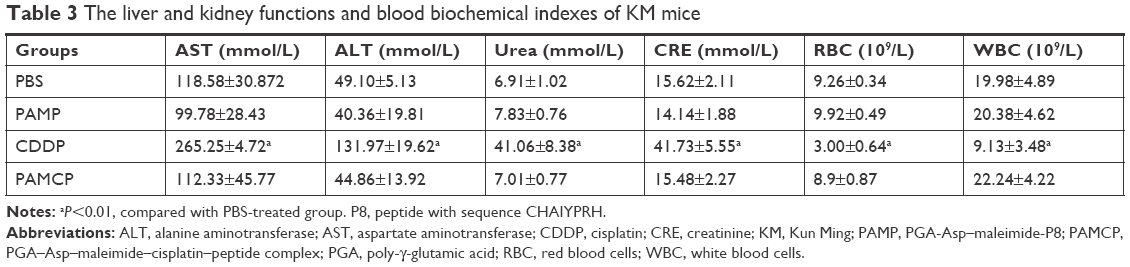 | Table 3 The liver and kidney functions and blood biochemical indexes of KM mice |
There were no significant differences (P>0.05) among the remaining groups, which indicated that PAMCP reduced the liver and kidney toxicity induced by CDDP. The TUNEL assay was used to detect the apoptosis of the liver, kidney, and spleen tissues. The apoptotic cells were stained with green fluorescence. When CDDP was administered, significant toxicity was detected as a number of positive, stained regions in the liver, spleen, and kidney (Figure 5). Compared with the free CDDP treatment group, the number of apoptotic cells in the PAMCP-treated groups was significantly decreased, and no obvious apoptosis was detected in the PBS- or PAMP-treated group.
 | Figure 5 The detection of liver, spleen, and kidney apoptosis. |
PAMCP exhibited pronounced tumor suppression in vivo
The in vivo tumor suppression experiment was conducted on HeLa cell-bearing nude mice. Five mice died when 6 mg/kg CDDP was administered and no death was noted in the remaining groups (Figure S5). Significant tumor volume decrease was seen when mice were exposed to CDDP or PAMCP compared with the remaining groups (Figure 6A). There were no significant differences in terms of tumor volumes changes between the CDDP or PAMCP treatment groups until 7 days after injection. PAMCP exhibited a more pronounced tumor suppression effect than free CDDP (Figure 6B and C). The inhibition of tumor proliferation by PAMCP could be attributed to the EPR effect40–42 and the targeting ability through affinity of PAMCP to TFR.
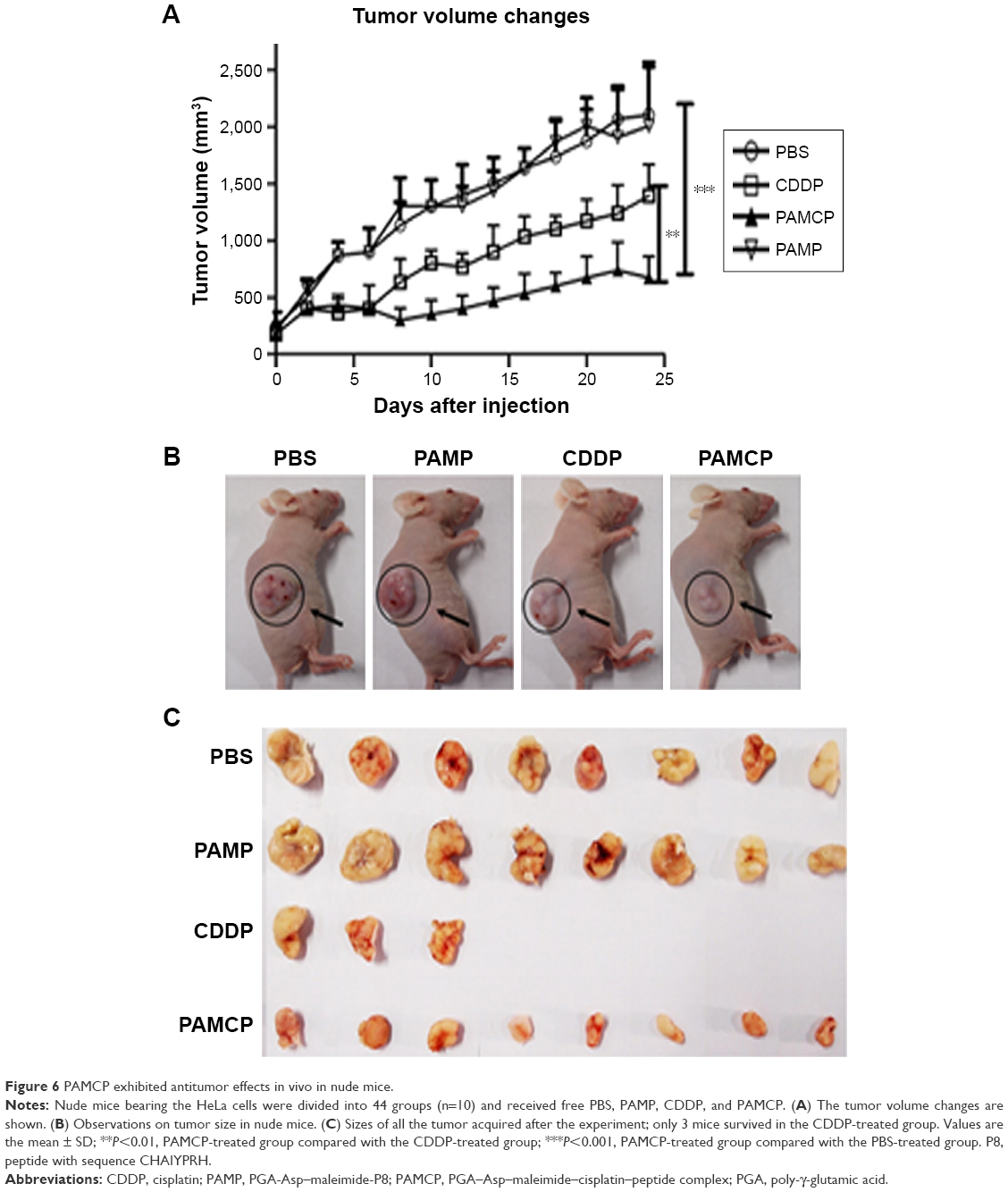 | Figure 6 PAMCP exhibited antitumor effects in vivo in nude mice. |
PAMCP achieved enriched tumor distribution in HeLa-bearing mice
Distributions of PAMCP were tested in HeLa tumor-bearing mice. The tumor-bearing mice were injected with PAMCP and P8X and killed at 24 hours. Different tissue sections were imaged and analyzed by Carestream MI SE to generate the corresponding fluorescence intensities. There was a certain distribution of P8X in different organs, but we could not detect obvious enhancement in tumor tissue. However, compared with the P8X treatment group, more conspicuous signals were detected after intravenous injection of PAMCP (Figure 7). In the PAMCP treatment group, the fluorescence intensity in different organs showed that the tumor tissue gained 5–10 folds higher fluorescence intensity than other tissues (Figure 8). These results suggested that PAMCP could prolong retention in tumor tissues, which could be due to the TFR-mediated cell targeting.
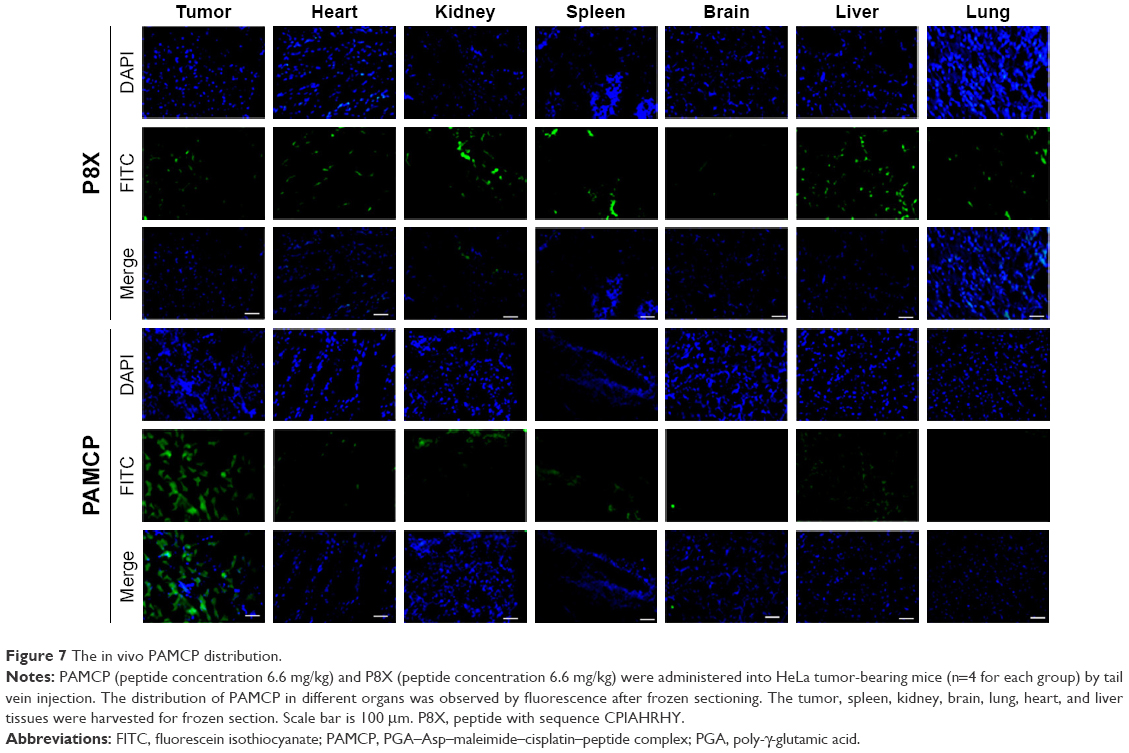 | Figure 7 The in vivo PAMCP distribution. |
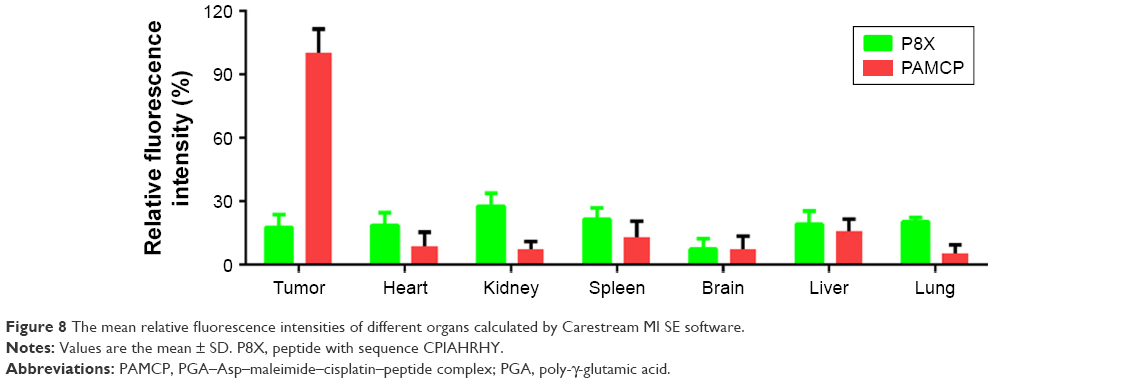 | Figure 8 The mean relative fluorescence intensities of different organs calculated by Carestream MI SE software. |
Discussion
The efficacies of major antitumor chemical drugs in the clinic were hindered due to the short half-life, nonselective delivery approaches, and side effects.43–47 Enormous challenges have been encountered in tumor treatment, and one of these is enhancement of the efficacy by site-specific drug delivery. The usage of targeting drug carrier for cancer treatment based on specific protein markers presenting in tumor tissues is one of the research highlights. Our group is focused on improving the chemotherapeutic efficacy and selectivity of drug delivery methods and reducing drug toxicity by coupling the drug to biosynthesized PGA. PAMCP was designed based on a carrier named GAP460, synthesized by an amidation reaction between PGA and Asp. GAP460 appeared to be sensitive to high temperatures and extreme pH but was quite stable at room temperature and neutral pH.48 PGA–Asp complex can overcome the disadvantages of free CDDP, such as low solubility in water and instability under physiological conditions. Biomacromolecules with relatively greater molecular weights could overcome the renal filtration clearance, have a longer plasma half-life, and have prolonged circulating time in vivo. Previously, the PGA–CDDP complex was mixed with plasma and dialyzed against 0.9% normal saline, and the results showed that the release of CDDP in PGA–CDDP complex was slow in plasma, and the drug release rate was only 55% at 100 hours.49 In this research, we also find PAMCP showing sustained CDDP release properties, compared with the free CDDP (Figure S6). Compared with the neutral environment, PAMCP presented faster CDDP release efficiency under relatively acidic conditions. The tumor-targeting peptide, with low molecular weight and high affinity, conjugated with the drug delivery system conferred obvious therapeutic advantage compared with mAbs. In our research, we used a peptide screened from an in vivo phage display method to implement TFR targeting of the drug carrier. The delivery system improved the systemic efficiency and decreased the side effects of CDDP. As a small molecule, CDDP could rapidly implement the drug efficacy, but it was hard to maintain stable blood drug concentration.50 The CDDP encapsulation rate of PAMCP reached 37%±6%, which is a high encapsulation efficiency. PAMCP was a slow-release system and was present for a long time in the blood circulation. The CDDP emulsification method using the self-assembly approach displayed a favorable character of high encapsulation efficiency, low energy consumption, and mild preparation conditions, which are promising for development of more number of hydrophobic substances. The MTT study suggested that PAMCP reduced the cytotoxicity of CDDP. These results suggest that PAMCP is stable and suitable for the EPR effect and can target TFR-positive tumor cells in vitro.
The capillary endothelium in malignant tumors tends to be impaired, and the aperture is probably >200 nm (mostly, in the range of 1–100 nm). The disorder of capillary endothelia enables the extravasation and retention of macromolecules and nanocarriers at pathological sites. The size of PAMCP was determined with dynamic light scattering (DLS) (89±18 nm) and TEM (110±28 nm). The size detected by DLS in aqueous phase (smaller than 100 nm) was similar to that in the physiological state. The TEM and DLS results indicate that the particle size of our newly-formed PAMCP was in the range for EPR effects for tumor tissue,41 and our in vivo experiment suggested that PAMCP was efficaciously accumulated for tumor targeting.
Our group is focused on improving the chemotherapeutic efficacy and reducing the drug toxicity by coupling the drug to biosynthesized PGA. In the present study, the in vivo toxicity – such as bone marrow suppression, acute nephrotoxicity, and hepatotoxicity – restricted the clinical application of CDDP. The in vivo results suggested that the PAMCP could prolong the retention in tumor tissues, which could be due to the TFR-mediated endocytosis. In the current study, PAMCP reduced the death of mice treated with CDDP and decreased the toxicity in vivo, which suggested that PAMCP was safe for in vivo usage. PAMCP could inhibit the malignant tumor proliferation and reduce the cytotoxic effect against normal cells. TFR, aiding cells in transferring iron ions, was expressed in low amounts in normal cells. In most tumor cells, however, more iron ions were obtained to facilitate cell growth. TFR was expressed in different tissues, and PAMCP could bind with the normal tissues. Although a large number of PAMCP molecules were distributed in normal tissues within 24 hours after injection, the TFR expression in tumor tissue was much higher than in normal tissues. High enrichment of PAMCP in tumor tissue was detected by FITC fluorescence intensities. The drug delivery system was capable of improving therapeutic agent concentration in the tumor tissue, limiting systemic toxicity, and extending agent half-life in blood relative to the free agents. PAMCP was not only highly enriched in tumor tissues but it also significantly reduced the side effects on liver and kidney, as well as prominently decreased the mortality induced by free CDDP. Therefore, PAMCP represents a very effective drug candidate for tumor therapy, with low adverse side effects.
Conclusion
A novel TFR-targeted drug carrier with loaded CDDP was fabricated, and the targeting carrier could improve the curative effect of CDDP and reduce the side effects. PAMCP could serve as an agent for developing novel antitumor therapy and promises to extend the clinical application of CDDP.
Acknowledgments
This research work was supported by the National Natural Science Foundation of China (31170920, 81771306), Scientific Research Project of Shanghai Municipal Commission of Health and Family Planning (20174Y0108), and the National Key Research and Development Program of China (2016YFC1200400).
Author contributions
The authors alone are responsible for the content and writing of the paper. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Xu Z, Chan HM, Li C, et al. Synthesis, structure, and cytotoxicity of oxaliplatin-based platinum(IV) anticancer prodrugs bearing one axial fluoride. Inorg Chem. 2018;57(14):8227–8235. | ||
Tikoo K, Ali IY, Gupta J, Gupta C. 5-Azacytidine prevents cisplatin induced nephrotoxicity and potentiates anticancer activity of cisplatin by involving inhibition of metallothionein, pAKT and DNMT1 expression in chemical induced cancer rats. Toxicol Lett. 2009;191(2–3):158–166. | ||
Gad AM, El-Raouf OMA, El-Sayeh BM, Fawzy HM, Abdallah DM. Renoprotective effects of montelukast in an experimental model of cisplatin nephrotoxicity in rats. J Biochem Mol Toxicol. 2017;31(12):e21979. | ||
Hsieh AC, Liu Y, Edlind MP, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485(7396):55–61. | ||
Cho H, Du X, Rizzi JP, et al. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature. 2016;539(7627):107–111. | ||
Chen W, Hill H, Christie A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature. 2016;539(7627):112–117. | ||
Ahmed M, Cheung NK. Engineering anti-GD2 monoclonal antibodies for cancer immunotherapy. FEBS Lett. 2014;588(2):288–297. | ||
Mohit E, Hashemi A, Allahyari M. Breast cancer immunotherapy: monoclonal antibodies and peptide-based vaccines. Expert Rev Clin Immunol. 2014;10(7):927–961. | ||
Wang L, Shi C, Wright FA, et al. Multifunctional telodendrimer nanocarriers restore synergy of bortezomib and doxorubicin in ovarian cancer treatment. Cancer Res. 2017;77(12):3293–3305. | ||
Chang KH, Kim MS, Hong GW, Shin YN, Kim SH. Conversion of a murine monoclonal antibody A13 targeting epidermal growth factor receptor to a human monoclonal antibody by guided selection. Exp Mol Med. 2012;44(1):52–59. | ||
Shin TH, Sung ES, Kim YJ, et al. Enhancement of the tumor penetration of monoclonal antibody by fusion of a neuropilin-targeting peptide improves the antitumor efficacy. Mol Cancer Ther. 2014;13(3):651–661. | ||
Rudnick SI, Lou J, Shaller CC, et al. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res. 2011;71(6):2250–2259. | ||
Petrenko VA, Jayanna PK. Phage protein-targeted cancer nanomedicines. FEBS Lett. 2014;588(2):341–349. | ||
Li M, Fan X, Liu J, Hu Y, Huang H. Selection by phage display of nanobodies directed against hypoxia inducible factor-1α (HIF-1α). Biotechnol Appl Biochem. 2015;62(6):738–745. | ||
Nogueira-Librelotto DR, Codevilla CF, Farooqi A, Rolim CM. Transferrin-Conjugated Nanocarriers as Active-Targeted Drug Delivery Platforms for Cancer Therapy. Curr Pharm Des. 2017;23(3):454–466. | ||
Tortorella S, Karagiannis TC. The significance of transferrin receptors in oncology: the development of functional nano-based drug delivery systems. Curr Drug Deliv. 2014;11(4):427–443. | ||
Qian ZM, Li H, Sun H, Ho K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev. 2002;54(4):561–587. | ||
Li H, Sun H, Qian ZM. The role of the transferrin-transferrin-receptor system in drug delivery and targeting. Trends Pharmacol Sci. 2002;23(5):206–209. | ||
Han L, Li J, Huang S, et al. Peptide-conjugated polyamidoamine dendrimer as a nanoscale tumor-targeted T1 magnetic resonance imaging contrast agent. Biomaterials. 2011;32(11):2989–2998. | ||
Oh S, Kim BJ, Singh NP, Lai H, Sasaki T. Synthesis and anti-cancer activity of covalent conjugates of artemisinin and a transferrin-receptor targeting peptide. Cancer Lett. 2009;274(1):33–39. | ||
Liu S, Guo Y, Huang R, et al. Gene and doxorubicin co-delivery system for targeting therapy of glioma. Biomaterials. 2012;33(19):4907–4916. | ||
Gao LY, Liu XY, Chen CJ, et al. Core-shell type lipid/rPAA-Chol polymer hybrid nanoparticles for in vivo siRNA delivery. Biomaterials. 2014;35(6):2066–2078. | ||
Han L, Huang R, Liu S, Huang S, Jiang C. Peptide-conjugated PAMAM for targeted doxorubicin delivery to transferrin receptor overexpressed tumors. Mol Pharm. 2010;7(6):2156–2165. | ||
Xiong Y, Jiang W, Shen Y, et al. A poly(γ, L-glutamic acid)-citric acid based nanoconjugate for cisplatin delivery. Biomaterials. 2012;33(29):7182–7193. | ||
Zhang L, Chang J, Zhao Y, et al. Fabrication of a triptolide-loaded and poly-γ-glutamic acid-based amphiphilic nanoparticle for the treatment of rheumatoid arthritis. Int J Nanomedicine. 2018;13:2051–2064. | ||
Khalil IR, Burns AT, Radecka I, et al. Bacterial-Derived Polymer Poly-y-Glutamic Acid (y-PGA)-based micro/nanoparticles as a delivery system for antimicrobials and other biomedical applications. Int J Mol Sci. 2017;18(2):E313. | ||
Ogunleye A, Bhat A, Irorere VU, Hill D, Williams C, Radecka I. Poly-γ-glutamic acid: production, properties and applications. Microbiology. 2015;161(Pt 1):1–17. | ||
Luo Z, Guo Y, Liu J, et al. Microbial synthesis of poly-γ-glutamic acid: current progress, challenges, and future perspectives. Biotechnol Biofuels. 2016;9:134. | ||
Zhang L, Wang T, Li Q, et al. Fabrication of novel vesicles of triptolide for antirheumatoid activity with reduced toxicity in vitro and in vivo. Int J Nanomedicine. 2016;11:2663–2673. | ||
Gabriel CM, Keener M, Gallou F, Lipshutz BH. Amide and peptide bond formation in water at room temperature. Org Lett. 2015;17(16):3968–3971. | ||
Geng X, Ye H, Feng Z, et al. Synthesis and characterization of cisplatin-loaded, EGFR-targeted biopolymer and in vitro evaluation for targeted delivery. J Biomed Mater Res A. 2012;100(10):2839–2848. | ||
Zhang L, Geng X, Zhou J, et al. Fabrication of poly(γ-glutamic acid)-based biopolymer as the targeted drug delivery system with enhanced cytotoxicity to APN/CD13 over-expressed cells. J Drug Target. 2015;23(5):453–461. | ||
Feng Z, Li W, Xue X, et al. A novel GAP460 biopolymer for use as a carrier in drug-delivery applications. J Biomater Sci Polym Ed. 2011;22(15):2023–2040. | ||
Desale SS, Cohen SM, Zhao Y, Kabanov AV, Bronich TK. Biodegradable hybrid polymer micelles for combination drug therapy in ovarian cancer. J Control Release. 2013;171(3):339–348. | ||
Levina TN, Sarycheva TG, Kniazeva ES. Experience in the use of flow scintillator Cobas Micros-18 OT in hematological practice. Klin Lab Diagn. 1997;10(10):18–20. Russian. | ||
Maji R, Dey NS, Satapathy BS, Mukherjee B, Mondal S. Preparation and characterization of Tamoxifen citrate loaded nanoparticles for breast cancer therapy. Int J Nanomedicine. 2014;9:3107–3118. | ||
Yan F, Zhang C, Zheng Y, et al. The effect of poloxamer 188 on nanoparticle morphology, size, cancer cell uptake, and cytotoxicity. Nanomedicine. 2010;6(1):170–178. | ||
Miao Z, Ren G, Liu H, Qi S, Wu S, Cheng Z. PET of EGFR expression with an 18F-labeled affibody molecule. J Nucl Med. 2012;53(7):1110–1118. | ||
Citores L, Ferreras JM, Muñoz R, Benítez J, Jiménez P, Girbés T. Targeting cancer cells with transferrin conjugates containing the non-toxic type 2 ribosome-inactivating proteins nigrin b or ebulin l. Cancer Lett. 2002;184(1):29–35. | ||
Greish K. Enhanced permeability and retention of macromolecular drugs in solid tumors: a royal gate for targeted anticancer nanomedicines. J Drug Target. 2007;15(7–8):457–464. | ||
Maeda H, Sawa T, Konno T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Control Release. 2001;74(1–3):47–61. | ||
Greish K, Fang J, Inutsuka T, Nagamitsu A, Maeda H. Macromolecular therapeutics: advantages and prospects with special emphasis on solid tumour targeting. Clin Pharmacokinet. 2003;42(13):1089–1105. | ||
Khare P, Jain A, Gulbake A, Soni V, Jain NK, Jain SK. Bioconjugates: harnessing potential for effective therapeutics. Crit Rev Ther Drug Carrier Syst. 2009;26(2):119–155. | ||
Ramasamy T, Ruttala HB, Gupta B, et al. Smart chemistry-based nanosized drug delivery systems for systemic applications: A comprehensive review. J Control Release. 2017;258:226–253. | ||
Ramasamy T, Ruttala HB, Chitrapriya N, et al. Engineering of cell microenvironment-responsive polypeptide nanovehicle co-encapsulating a synergistic combination of small molecules for effective chemotherapy in solid tumors. Acta Biomater. 2017;48:131–143. | ||
Ramasamy T, Haidar ZS, Tran TH, et al. Layer-by-layer assembly of liposomal nanoparticles with PEGylated polyelectrolytes enhances systemic delivery of multiple anticancer drugs. Acta Biomater. 2014;10(12):5116–5127. | ||
Fang Y, Tan J, Lim S, Soh S. Rupturing cancer cells by the expansion of functionalized stimuli-responsive hydrogels. NPG Asia Mater. 2018;10(2):e465. | ||
Feng Z, Li W, Xue X, et al. A novel GAP460 biopolymer for use as a carrier in drug-delivery applications. J Biomater Sci Polym Ed. 2011;22(15):2023–2040. | ||
Feng Z, Lai Y, Ye H, Huang J, Xi XG, Wu Z. Poly (γ, L-glutamic acid)-cisplatin bioconjugate exhibits potent antitumor activity with low toxicity: a comparative study with clinically used platinum derivatives. Cancer Sci. 2010;101(11):2476–2482. | ||
Morris TT, Ruan Y, Lewis VA, Narendran A, Gailer J. Fortification of blood plasma from cancer patients with human serum albumin decreases the concentration of cisplatin-derived toxic hydrolysis products in vitro. Metallomics. 2014;6(11):2034–2041. |
Supplementary materials
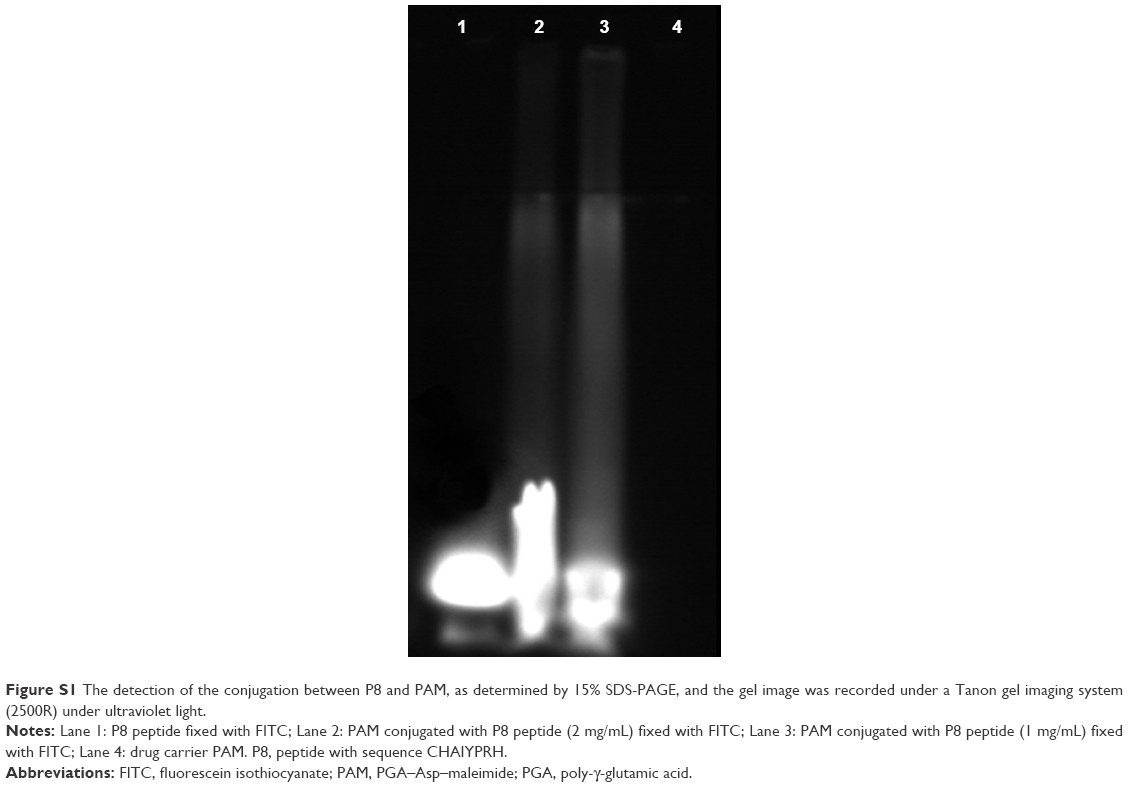 | Figure S1 The detection of the conjugation between P8 and PAM, as determined by 15% SDS-PAGE, and the gel image was recorded under a Tanon gel imaging system (2500R) under ultraviolet light. |
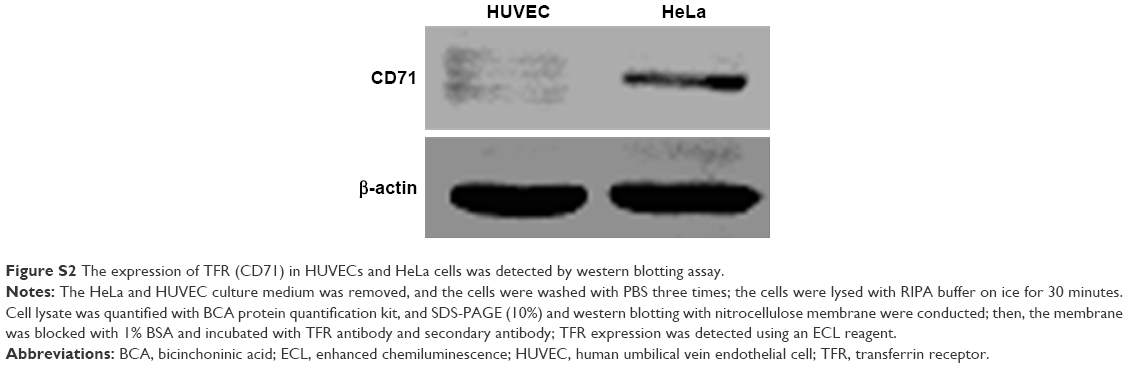 | Figure S2 The expression of TFR (CD71) in HUVECs and HeLa cells was detected by western blotting assay. |
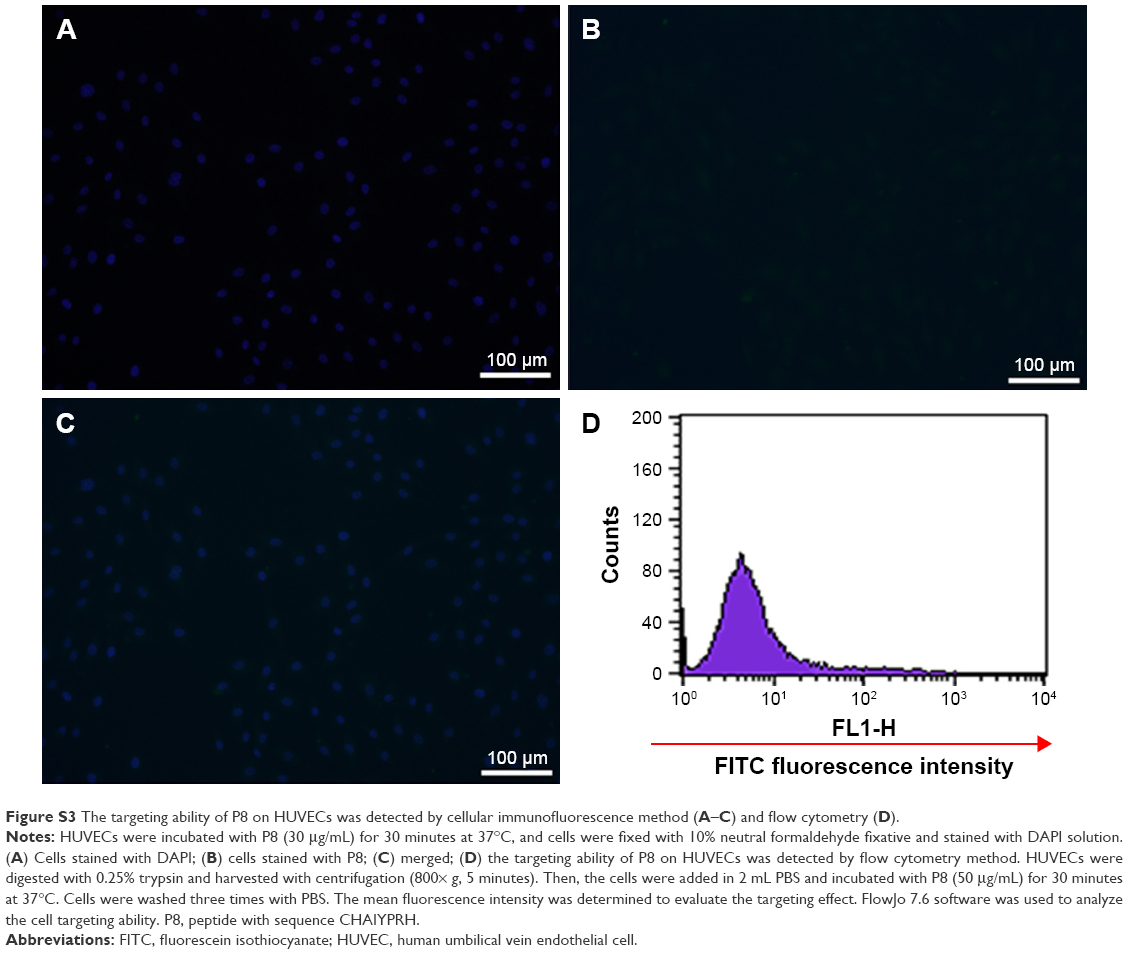 | Figure S3 The targeting ability of P8 on HUVECs was detected by cellular immunofluorescence method (A–C) and flow cytometry (D). |
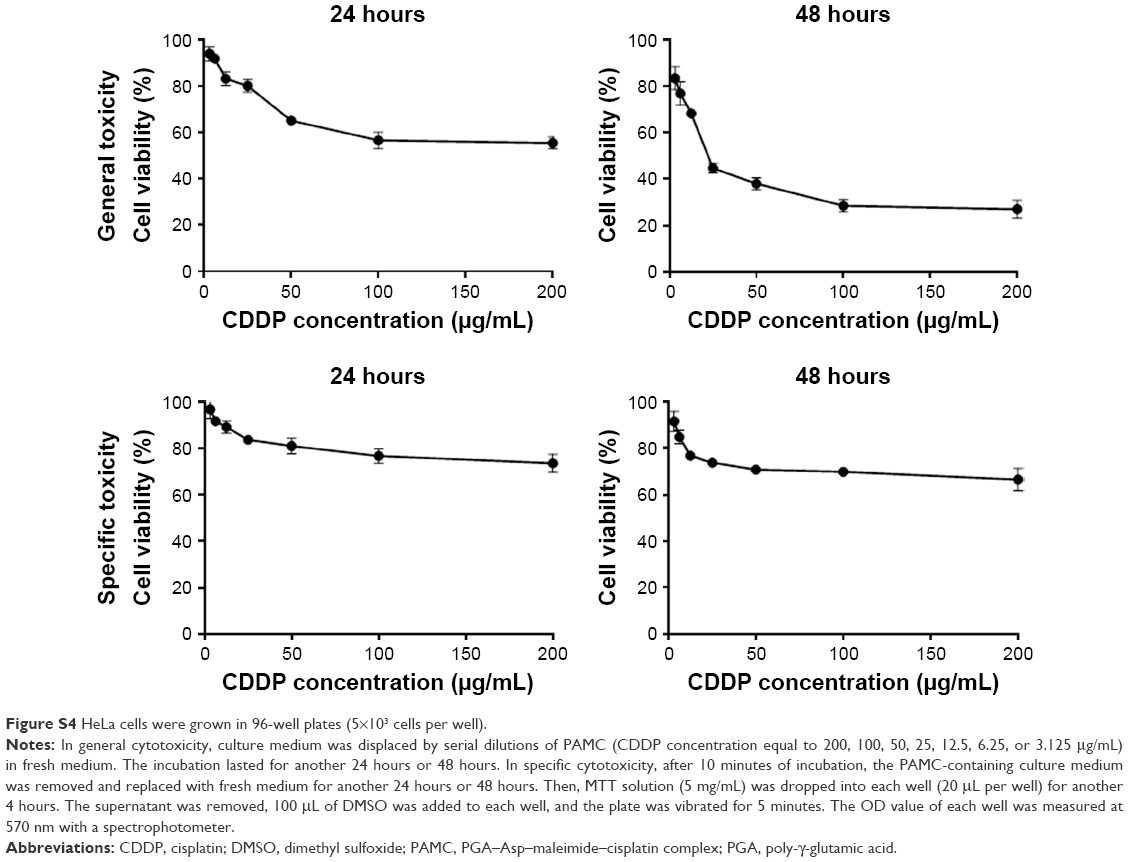 | Figure S4 HeLa cells were grown in 96-well plates (5×103 cells per well). |
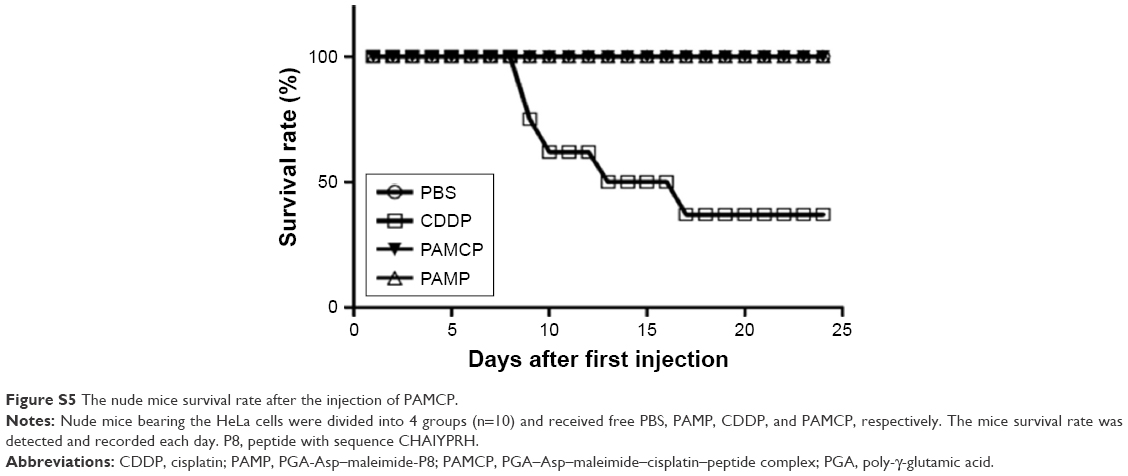 | Figure S5 The nude mice survival rate after the injection of PAMCP. |
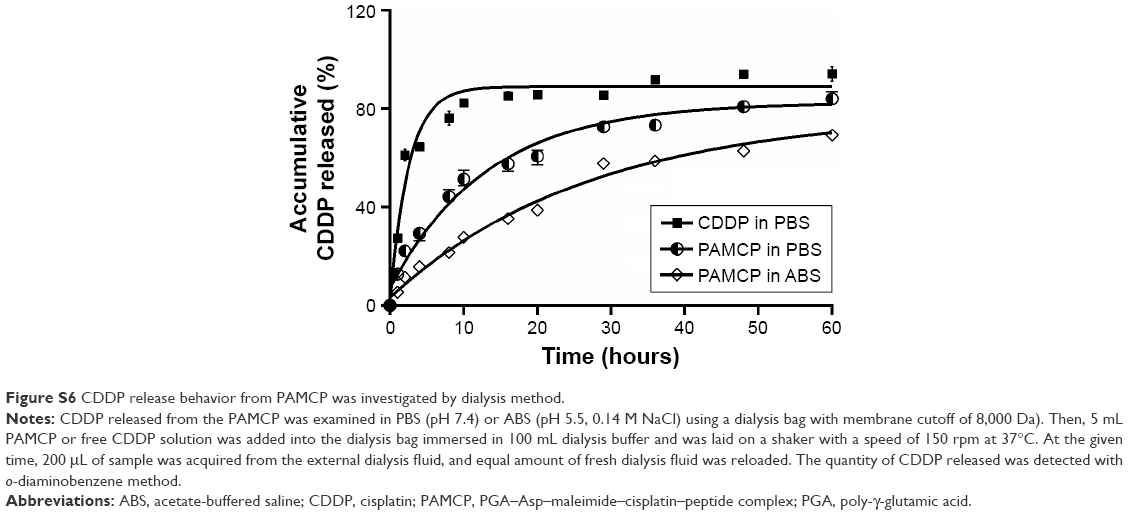 | Figure S6 CDDP release behavior from PAMCP was investigated by dialysis method. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.