Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 14
Extensive Type V Aplasia Cutis Congenita Without Fetus Papyraceus or Placental Infarction: A Rare Case
Authors Effendi RMRA ![]() , Nuraeni L
, Nuraeni L ![]() , Diana IA, Gondokaryono SP
, Diana IA, Gondokaryono SP ![]() , Gunawan H
, Gunawan H ![]() , Dwiyana RF
, Dwiyana RF ![]()
Received 25 July 2021
Accepted for publication 16 September 2021
Published 5 October 2021 Volume 2021:14 Pages 1413—1418
DOI https://doi.org/10.2147/CCID.S330160
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Raden Mohamad Rendy Ariezal Effendi, Lita Nuraeni, Inne Arline Diana, Srie Prihianti Gondokaryono, Hendra Gunawan, Reiva Farah Dwiyana
Department of Dermatology and Venereology, Faculty of Medicine Universitas Padjadjaran - Dr. Hasan Sadikin Hospital, Bandung, Indonesia
Correspondence: Raden Mohamad Rendy Ariezal Effendi
Department of Dermatology and Venereology, Faculty of Medicine, Universitas Padjadjaran - Dr. Hasan Sadikin Hospital, Jl. Pasteur 38, Bandung, 40161, West Java, Indonesia
Email [email protected]
Abstract: Aplasia cutis congenita (ACC) is a congenital disorder characterized by the absence of epidermis, dermis, and sometimes subcutaneous tissue and bone. There are nine types of ACC based on the number and location of the lesions as well as the presence or absence of associated deformities, with type I ACC being the most common. Type V affects the trunk with a characteristic pattern resembling the “H” letter of the alphabet, generally accompanied by fetus papyraceus (death of one of the twin fetuses) or placental infarction. Type V ACC without fetus papyraceus and placental infarction is a rare case. We reported a case of type V ACC in a 3-day-old baby girl, with clinical manifestation of extensive ulcers on the scalp, back, buttocks, and both lower extremities, in an “H”-shaped pattern, with history of placenta accreta, but no fetus papyraceus or placental infarction was found. The patient received dialkyl carbamoyl chloride hydrophobic swab with hydrogel (Sorbact® gel dressing-BSN Medical) and hydrocolloid wound dressing with good response, as indicated by the wounds becoming dried up, leaving an atrophic scar. Complete epithelialization was achieved in 72 days. The cause of type V ACC remains not completely understood. The diagnosis of ACC can be established based on clinical manifestations. Further examination is required if extracutaneous involvement is suspected. Early identification of the etiology of the lesions and appropriate investigation and treatment, including conservative wound management, are needed so that aplastic lesions can heal successfully, although cosmetic repair may be required at a later stage.
Keywords: aplasia cutis congenita, fetus papyraceus, placenta accreta
Introduction
Aplasia cutis congenita (ACC) is a congenital disorder characterized by localized areas where the skin is absent,1,2 and this condition sometimes affects the underlying subcutaneous tissue and bone.1 The most common form of ACC is an oval, sharply marginated atrophic patch on the posterior scalp. Erosion and ulceration with a granulating base or thin shiny membranous covering can be present.1,3,4 The lesions can occur on any surface of the body, although localized scalp lesion is the most frequent pattern.1,5 In 1986, Frieden classified ACC into nine types, with type I being the most common.2,5 Type V is rare,2 associated with fetus papyraceus or placental infarction,2,5 and characterized by stellate lesions in a symmetrical distribution over the trunk and extremities,2 differing from other types of ACC, which are typically localized to the scalp (70–85% of cases) or extremities. Unlike the scalp involvement pattern, there are little epidemiologic data delineating the frequency of other distributions.2 Here, we reported a rare presentation of type V ACC in a newborn with extensive and symmetrical absence of the skin on the scalp, trunk, and limbs without fetus papyraceus and placental infarction.
Case
A 3-day-old healthy baby girl with a birth weight of 2.8 kg and a length of 48 cm, delivered full term through vaginal delivery from a multigravida mother, presented with congenital absence of the skin on the scalp, back, buttocks, and upper limbs with an atrophic scar on the chest and abdomen. The mother was currently 40 years old and had four children from a previous pregnancy. During antenatal care (ANC), the mother had no history of illness, trauma, or taking medicine other than vitamins prescribed by the doctor. Ultrasonography was performed at the third trimester and showed a single live fetus with no obvious congenital malformations. Soon after delivery, the mother had vaginal bleeding due to retained placenta. The bleeding did not stop despite subsequent curettage, and a hysterectomy was performed. Histopathological examination of the uterus showed placenta accreta with no signs of infarction. All the previous pregnancies were full-term, with no complications, and all her children were delivered vaginally. A history of miscarriages was denied. The first child was a male, died at the age of 2 months due to diarrhea, the second is a female, currently 16 years old, the third is a boy – 14 years old, and the fourth is a male – 7 years old. There was no family history with a similar skin condition. Upon physical examination, the lesions were well demarcated, symmetrical, and stellate, covered with a thin, gelatinous, shiny membrane. The lesions on the trunk were distributed in an H-shaped pattern measuring 20×7 cm, while lesion on the scalp was 6×4 cm. Atrophic scar, 14×5 cm in size, was found in the chest and abdomen (Figure 1). There were no embryological malformations such as meningomyeloceles, spinal dysgraphia, cranial stenosis, congenital midline porencephaly, leptomeningeal angiomatosis, ectopia of the ear, omphalocele and gastroschisis, blistering, nail deformity, or malformation syndrome. Laboratory examination revealed hypoalbuminemia (2.26 g/dL) and elevated C-reactive protein (6.73 mg/dL), while other parameters were within the normal limit. Head X-ray showed irregularity of the parietal lobe in the vertex region, while radiological examination of the trunk showed no abnormalities. Magnetic resonance imaging (MRI) with contrast in the head revealed no subcutaneous defect. The patient was consulted to otolaryngologist, ophthalmologist, neuropediatrician, and cardiologist; consultation results indicated no abnormalities other than dermatological abnormality. Initially, lesions were treated with Sorbact® gel dressing-BSN Medical for autolytic debridement, and then covered with sterile gauze until the gelatinous-shiny-membrane disappeared. Following that, we used hydrocolloid wound dressing (WD) until complete epithelialization was achieved (Figure 2). Prophylactic antibiotic was given with intravenous injection of ampicillin sulbactam and gentamicin. We also gave 20% albumin due to hypoalbuminemia until the normal level was reached. Bacterial culture from wound base was positive for Klebsiella pneumoniae with resistance to ampicillin and gentamicin. Therefore, antibiotic treatment was modified to intravenous injection of meropenem. After 26 days, the patient was discharged from neonatal intensive care unit (NICU) when more than 70% of the lesion showed epithelialization. Following daily local treatment, full epithelialization was achieved at day 72, leaving a smooth, hairless, papery scar (Figure 3). Until the age of five months, no medical problems were apparent, and the infant showed completely normal motor skills development.
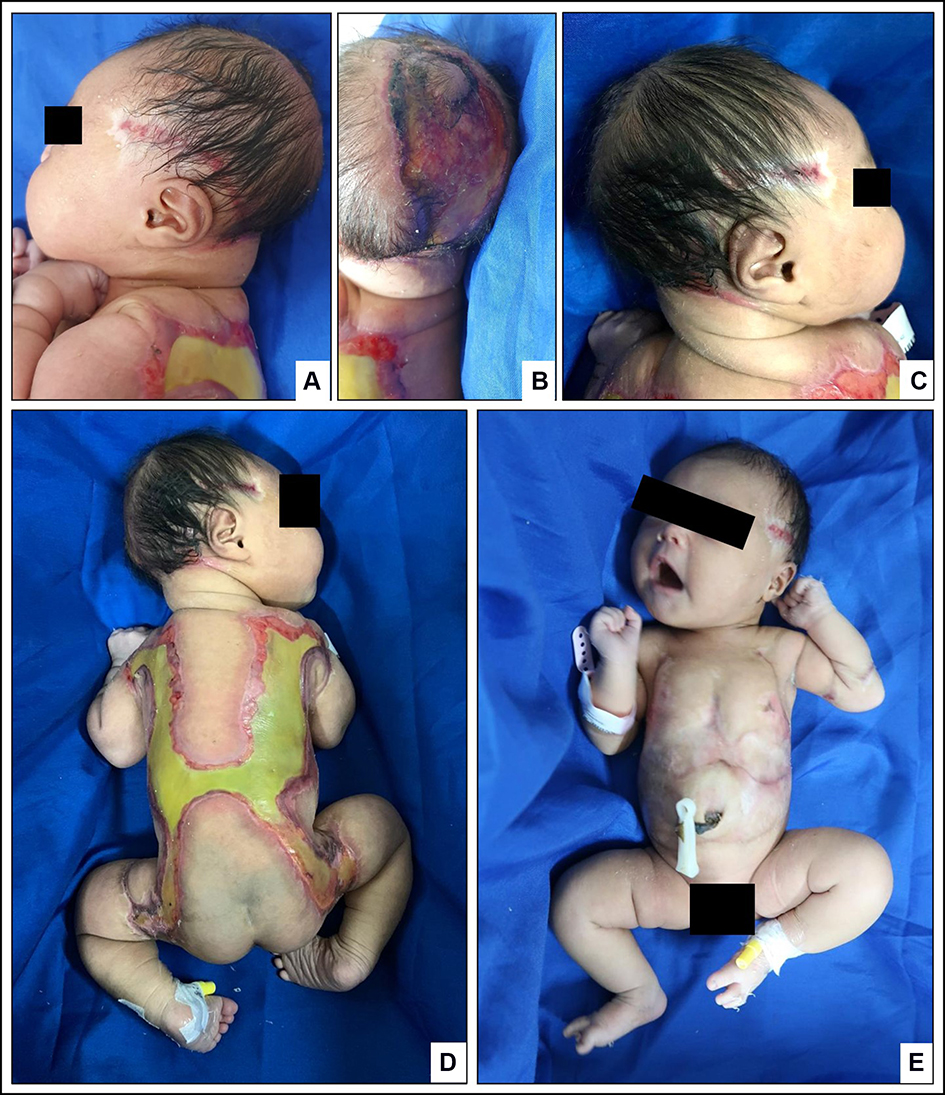 |
Figure 1 Clinical manifestation of ACC, absence of the skin on scalp (A–C), back, buttocks, and upper limbs (D) with an atrophic scar on the chest and abdomen (E). |
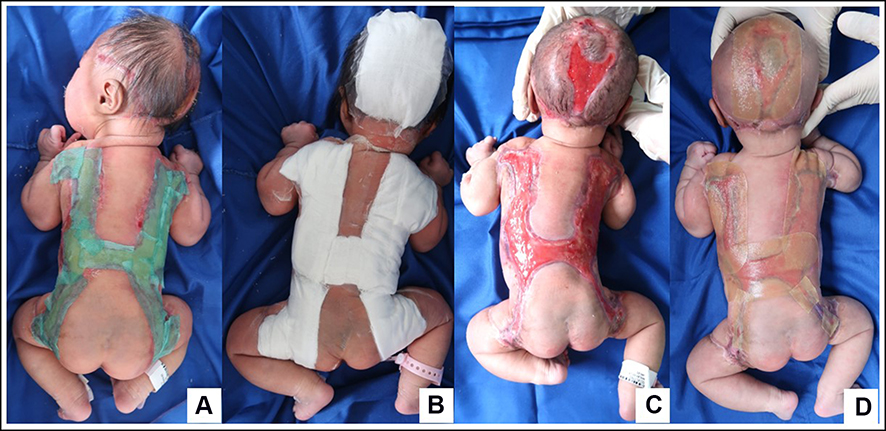 |
Figure 2 WD used on the patient. Initially, lesions were treated with Sorbact® gel dressing-BSN Medical and then covered with sterile gauze (A and B), after the gelatinous-shiny-membrane disappeared (C) hydrocolloid WD was used until complete epithelialization was achieved (D). |
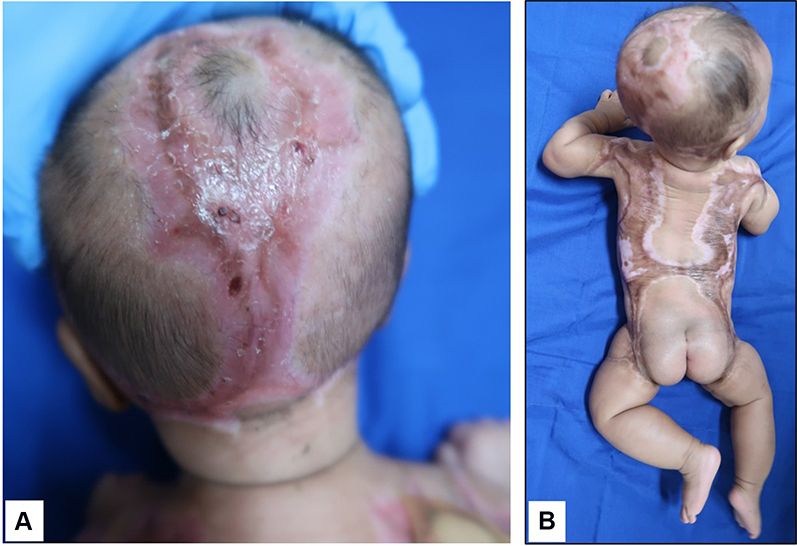 |
Figure 3 Complete epithelialization on day 72, on scalp (A) and trunk (B). |
Discussion
The term aplasia cutis congenita is used for a heterogenous group of disorders in which the skin and sometimes the underlying subcutaneous tissues and bones are absent at birth,1,4 or a congenital atrophic scar is present.4,6 In most cases, this condition is localized and isolated on the scalp.2,4 There are multiple factors that might be contributing to the development of ACC, including chromosomal abnormalities, trauma, amniotic irregularities, vascular disorders, teratogenic substances (misoprostol, benzodiazepines, valproic acid, cocaine, methotrexate, angiotensin-converting enzyme inhibitors, methimazole), fetus papyraceus,5 and congenital infections (herpes simplex virus and varicella virus).6 The pathogenesis of ACC is unknown;5 however, it is thought to occur due to two mechanisms: the first is a disruption to the development of the overlying skin layer,5,6 and the second is destruction of the skin that had been previously formed.6 Based on the physical examination, the patient in this case report had absence of skin on her scalp, back, buttocks, and upper limbs, with an atrophic scar on the chest and abdomen at birth, which is consistent with the clinical features of ACC.
In 1986, Frieden classified ACC into nine types, based on the location, pattern of skin disorders, inheritance, associated malformations, teratogens, and involved syndromes.5,7,8 Some types of ACC were inherited in an autosomal dominant or recessive manner.8 Type I ACC, the most common type, is localized on the scalp with no other associated anomalies. Type II is scalp involvement associated with limb abnormalities; type III is scalp involvement associated with epidermal nevus, neurological, and ophthalmic abnormalities; type IV is ACC accompanied by embryological deformities, can affect the scalp, abdomen, lumbar, and any other sites; type V is ACC associated with fetus papyraceus or placental infarction with the extended absence of the skin on the trunk or limbs; type VI ACC is divided into localized on extremities or widespread on extremities and torso accompanied by epidermolysis bullosa (EB); type VII is ACC on extremities without EB; type VIII is ACC associated with teratogens or varicella and herpes simplex infections, can affect scalp locally or any other areas; type IX is ACC associated with malformation syndromes.5,7
Based on Frieden's classification, type IV, V, VI, and VIII are ACC that can affect the trunk. Type IV ACC is accompanied by embryological malformations, such as meningomyelocele, spinal dystrophy, cranial stenosis, omphalocele, and gastroschisis. The body areas affected in type VI are the abdomen, lumbar, scalp, and other body parts associated with EB. In this type, blisters are found with abnormalities in the nails, with pyloric or duodenal atresia, abnormalities in the ears and nose, and urethral stenosis. In this patient, there were no blisters, no abnormalities in the ears, nose, gastrointestinal tract, or urinary tract. Therefore, differential diagnosis with type VI can be excluded. The mother’s pregnancy history revealed neither a history of chicken pox (varicella) or herpes simplex infection nor a history of consuming medicines other than vitamins prescribed by doctor during pregnancy. Therefore, differential diagnosis with type VIII can be excluded. The absence of skin formation in the body can also be differentially diagnosed with iatrogenic trauma caused by forceps.3,4,8 However, the patient was delivered vaginally without forceps, ruling out the possibility of iatrogenic trauma disorders. There was no family history with similar skin condition.
Bullous dermolysis of the newborn (BDN) or transient bullous dermolysis is an extremely rare clinical type of dystrophic EB, characterized by subepidermal blistering at birth or shortly thereafter.9 Skin lesions in BDN are found in friction areas and the extremities at birth,10,11 and new blisters can appear due to local trauma followed by rapid improvement with pigmentation or minimal scarring.9,10 In this patient, no blisters were found and there were no new skin lesions following local trauma.
The precise incidence of ACC is unknown, but thought to be around 0.5–1 in 10,000 births,6 with a significantly lower incidence observed in type V.7 The cause of type V ACC is more often associated with fetus papyraceus or placental infarction.2 In 1995, Boente et al reported one case of type V ACC without fetus papyraceus or placental infarction,12 then in 2016 Kaur et al reported two similar cases.13 Although the exact etiology has not yet been definitively proven, type V is thought to be the result of a transient vascular process due to fetus papyraceus.7 Type V ACC commonly has extensive lesion. The skin defects are located on the flanks, buttocks/thighs, abdomen, scalp, axillae/arms, and back.14 The lesion had highly characteristic, symmetric distribution pattern that encircled the umbilicus and formed an “H” pattern on the anterior and less often, posterior trunk.6,12,14 Type V lesions formed early in utero may heal with scarring before delivery.7 In this patient, at birth, we found skin absence on the scalp, back, buttocks, and upper limbs, forming an “H” pattern on the posterior trunk, accompanied by an atrophic scar on the chest and abdomen, which was thought to be ACC lesion that healed before delivery. There was no fetus papyraceus or placental infarction, but histopathology examination of samples taken from the mother’s uterus revealed placenta accreta. We believe this is the first case report that showed extensive ACC on the scalp and trunk that formed an “H” pattern on the back associated with placenta accreta. Based on the patient’s history and clinical manifestation, the diagnosis of type V ACC without fetus papyraceus or placental infarction was established.
Although most ACC lesions heal spontaneously, the use of WD is highly recommended for a faster healing process.15 Sorbact® gel dressing-BSN Medical is a modern WD that reduces bacterial load in a wound to support moist wound healing in low exuding or sloughy wounds. This hydrophobic interaction attracts bacteria and binds them irreversibly to the dressing fibers which will prevent them from proliferating, releasing harmful endotoxins, or shedding disrupted cellular content that otherwise might result in delayed wound healing.16,17 The gel formulation of this dressing provides moisture to dry wounds, which is needed to assist with debridement. The moist environment also facilitates the movement of bacteria into the dressing, where it becomes irreversibly bound.16 We used Sorbact® gel dressing-BSN Medical as an antibacterial dressing and for wound debridement.
Hydrocolloid is a modern WD, which has a debridement ability and can absorb exudate.18,19 The gel will form when hydrocolloid contacts with exudate making wound environment damp. Hydrocolloid is recommended for pediatric patients because it does not cause pain when removed.19 We have used hydrocolloid as WD since the 4th day of observation after the gelatinous-shiny-membrane on lesion disappeared. The lesion had completely dried up at day 72, leaving an atrophic scar.
Despite the extensive skin lesions, the defects healed with an appropriate wound care and observation during the early days of life, with no additional surgical procedures. The patient also had a normal development according to her age, implying a good clinical prognosis for the disease.
Conclusion
Type V aplasia cutis congenita is a rare case, usually accompanied by fetus papyraceus and placental infarction, and typically occurs as a large cutaneous defect in the trunk and extremities. Further examination is required if extracutaneous involvement is suspected. With early identification of its etiology and appropriate investigation and treatment, including proper wound management, aplastic lesions can heal successfully, but may require cosmetic repair at a later stage.
Ethical Statement
The publications of images were included in the parents’ consent for publication of the case.
Institutional approval to publish the case details has been obtained.
Consent Statement
The authors certify that they have obtained all appropriate patient consent forms. The parents of the patient signed a consent form for the publication of the case details and images.
Acknowledgments
The authors would like to thank the staff of the Department of Dermatology and Venereology, Faculty of Medicine, Universitas Padjadjaran–Dr. Hasan Sadikin General Hospital, Bandung, West Java, Indonesia.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hunt R, Chang MW, Shah KN. Neonatal dermatology. In: Kang S, Amagai M, Bruckner AL, et al, editors. Fitzpatrick’s Dermatology.
2. Snyder ML, Ilyas H. Type V aplasia cutis congenita with fetus papyraceus. JAAD Case Rep. 2019;5(4):303–305. doi:10.1016/j.jdcr.2019.02.015
3. Paller AS, Mancini AJ. Cutaneous Disorders of the Newborn. Hurwitz Clinical Pediatric Dermatology.
4. Wobser M, Hamm H. Developmental anomalies. In: Irvine AD, Hoeger PH, Yan AC, editors. Harper’s Textbook of Pediatric Dermatology.
5. Alexandros B, Dimitrios G, Elias A, et al. Aplasia cutis congenita: two case reports and discussion of the literature. Surg Neurol Int. 2017;8:273.
6. Humphrey SR, Hu X, Adamson K, Schaus A, Jensen JN, Drolet B. A practical approach to the evaluation and treatment of an infant with aplasia cutis congenita. J Perinatol. 2018;38(2):110–117. doi:10.1038/jp.2017.142
7. Perry BM, Maughan CB, Crosby MS, Hadenfeld SD. Aplasia cutis congenita type V: a case report and review of the literature. Int J Dermatol. 2017;56(6):118–121. doi:10.1111/ijd.13611
8. Frieden IJ. Aplasia cutis congenita: a clinical review and proposal for classification. J Am Acad Dermatol. 1986;14(4):646–660. doi:10.1016/S0190-9622(86)70082-0
9. Shi BJ, Zhu XJ, Liu Y, et al. Transient bullous dermolysis of the newborn: a novel de novo mutation in the COL7A1 gene. Int J Dermatol. 2015;54(4):438–442. doi:10.1111/ijd.12704
10. Hashimoto K, Matsumoto M, Iacobelli D. Transient bullous dermolysis of the newborn. Arch Dermatol. 1985;121(11):1429–1438.
11. Mellerio JE, Martinez AE, Has C. Epidermolysis bullosa and kindler syndrome. In: Irvine AD, Hoeger PH, Yan AC, editors. Harper’s Textbook of Pediatric Dermatology.
12. Boente MD, Frontini MD, Acosta MI, Saleme C, Barrionuevo S, Asial R. Extensive symmetric truncal aplasia cutis congenita without fetus papyraceus or macroscopic evidence of placental abnormalities. Pediatr Dermatol. 1995;12(3):228–230. doi:10.1111/j.1525-1470.1995.tb00164.x
13. Kaur S, Sangwan A, Dayal S, Dua I, Jain V. Aplasia cutis congenita, group 5 without fetus papyraceus in two newborns. Indian J Dermatol Venereol Leprol. 2016;82(6):695–697. doi:10.4103/0378-6323.188653
14. Schaffer JV, Popiolek DA, Orlow SJ. Symmetric truncal aplasia cutis congenita following multifetal reduction of a sextuplet pregnancy. J Pediatr. 2008;153(6):860–863. doi:10.1016/j.jpeds.2008.07.009
15. Ahčan U, Janeič T. Management of aplasia cutis congenita in a non-scalp location. Br J Plas Surg. 2002;55(6):530–532. doi:10.1054/bjps.2002.3915
16. Pirie G, Duguid K, Timmons J. Cutimed® Sorbact® gel: a new infection management dressing. Wounds. 2009;5(2):74–78.
17. Cutting K, McGuire J. In vitro and clinical experience of cutimed sorbact: the evidence base. J Wound Care. 2015;24(5 Suppl):S6–S30.
18. Barčot Z, Bašković M, Car A, Jurić M. Aplasia cutis congenita of the scalp: the success of conservative approach in treatment of a large defect. Indian J Dermatol. 2019;20(2):166–168. doi:10.4103/ijpd.IJPD_93_18
19. Dhivya S, Padma VV, Santhini E. Wound dressings–a review. BioMedicine. 2015;5:4. doi:10.7603/s40681-015-0022-9
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.