Back to Journals » Journal of Experimental Pharmacology » Volume 13
Experimental Therapeutic Solutions for Behcet’s Disease
Received 11 December 2020
Accepted for publication 19 January 2021
Published 12 February 2021 Volume 2021:13 Pages 127—145
DOI https://doi.org/10.2147/JEP.S265645
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Bal Lokeshwar
Burçin Cansu Bozca, Erkan Alpsoy
Akdeniz University School of Medicine, Department of Dermatology and Venereology, Antalya, Turkey
Correspondence: Erkan Alpsoy
Akdeniz University School of Medicine, Department of Dermatology and Venereology, Antalya, 07070, Turkey
Tel +90 242 249 67 06
Fax +90 242 249 60 40
Email [email protected]
Abstract: Behcet’s disease (BD) is a chronic systemic vasculitis with inflammation attacks that involve multiple organs. In addition to numerous mucocutaneous symptoms, notably recurrent oral and genital ulcers, ocular, articular, vascular, gastrointestinal, cardiac, and neurological system involvement can be observed. Mucocutaneous lesions are the primary symptom of the disease in most patients, and they usually occur before major organ involvement and other symptoms of the disease. Recognizing the disease’s mucocutaneous lesions is very important to diagnose at an early stage, control with appropriate treatment and close follow-up, and prevent major organ involvement. Genome-wide association studies (GWAS) in recent years have confirmed that HLA-B*51 is the most significant genetic predisposing factor. The majority of gene polymorphisms have been detected in molecules that respond to microorganisms and genes encoding cytokines and adhesion molecules. The infectious agent S. sanguinis -commonly found in the oral mucosa of patients with BD- or the differences in the salivary or intestinal microbiome composition can trigger innate immune-mediated inflammation sustained by acquired or adaptive immune responses. In antigen-presenting cells (APCs), epistatic interactions between HLA-B*51 and endoplasmic reticulum aminopeptidase 1 (ERAP1) variants lead to the disruption of T-cell homeostasis, especially the activation of Type1 T-helper and Th17 pathway and suppression of regulatory T-cells. Recent developments to clarify the disease’s etiopathogenesis provided us with a better understanding of the mechanism of action of the relatively old drugs while opening a way for many new treatment methods. Apremilast has become an important option in the treatment of mucocutaneous symptoms with its high efficacy and safety. The disease increases the mortality rate, especially in young male patients. New treatments, especially anti-TNF-α agents, have provided significant progress and decreased the mortality rates with their rapid effect and high efficacy in patients with severe organ involvement and resistance to traditional immunosuppressive and immunomodulatory therapies. The use of IL-1, IL-6, IL-17, IL-12/IL-23 antagonists in different organ involvement has gradually increased, and the quality of life has significantly improved in many patients.
Keywords: Behcet’s syndrome, algorithms, etiology, therapeutics, morbidity, mortality
Introduction
Behcet’s disease (BD) is a systemic vasculitis distinguished by oral ulcers (OU), genital ulcers (GU), other mucocutaneous lesions (erythema nodosum [EN]-like lesions, papulopustular lesions [PPL], superficial thrombophlebitis, etc.), ocular, vascular, articular, gastrointestinal, neurological, and cardiac involvement. The disease of unknown etiology follows a chronic course with inflammatory attacks. BD usually occurs in the third or fourth decade.1 The disease, which is observed worldwide, is more frequent in the ancient “Silk Road,” extending from Japan to the Middle East and Mediterranean countries. The disease’s prevalence in countries located on the ancient “Silk Road” has been reported as 14–20/100,000. Until now, the highest prevalence was reported from Turkey (420/100,000).2 Population-based studies with large series conducted in the last 30 years show that the disease occurs at a similar rate in both genders.1,3,4
The disease’s course is severe when it occurs at a young age, in male patients and those without regular treatment and follow-up.4,5 BD adversely affects patients’ quality of life and can lead to workday loss. In a multi-center study, Mumcu et al reported that male gender, early onset of illness, smoking, using immunosuppressive agents, ocular and vascular involvement are the most important factors increasing workday loss.6 On the other hand, the disease increases the mortality rate, especially in young male patients. Large vessel involvement, neurological involvement, gastrointestinal system involvement, and cardiac involvement are the most important causes of mortality.7 However, in most patients, BD starts with relatively mild symptoms (mucocutaneous involvement). Severe organ involvement occurs in the advanced stage of the disease.8 Therefore, by providing early diagnosis, appropriate treatment, and regular follow-up, severe organ involvement can be prevented in a significant number of patients.
There is no definitive diagnostic laboratory test for the disease, and diagnosis is based on clinical findings. The common feature of the various diagnostic criteria used so far in the disease is that the diagnoses are based on mucocutaneous lesions, especially OU, GU, cutaneous lesions, and pathergy test positivity. The International Study Group’s criteria for Behçet’s Disease (ISBD) is the most widely used in diagnosis.9
The treatment’s main goal should be to suppress new inflammatory attacks to prevent irreversible organ damage, especially in the early and active phases of the disease. It should be kept in mind that the management of patients with BD requires a multidisciplinary approach. Due to the increasing knowledge about BD etiopathogenesis, many new treatment options have become a part of the standard treatment in recent years. In this review, in the light of recent developments, the clinical spectrum and etiopathogenesis of the disease will be summarized, and we will focus primarily on the treatment. In addition to long-term and generally accepted treatment modalities in BD, new treatment approaches will be examined in detail.
Etiopathogenesis
The etiopathogenesis of the disease is not fully understood yet. There is a consensus that infectious agents can trigger BD.10,11 The heat shock proteins (HSP) of Herpes simplex virus-1 and some streptococci strains, especially S. sanguinis, show significant similarity to human HSP; antibodies against HSPs of these microorganisms in individuals with a genetic predisposition are thought to initiate an immune response in humans by cross-reaction.10,12,13 The fact that BD begins at the oral mucosa,14 dental procedures, and surgical operations for chronic tonsillitis exacerbates the disease,15,16 oral antimicrobial treatments are used successfully in BD, and oral hygiene is worse in BD patients compared to healthy people17–19 suggest that oral microbial flora may play a role in disease pathogenesis. Recent studies suggest that differences in salivary or gut microbiome composition may also play a role in pathogenesis.20,21 The salivary microbiota has been reported to have less diversity in BD patients than in healthy subjects.20 The Major Histocompatibility Complex (MHC) profile seen in BD has been speculated to lead to the formation of a different intestinal bacterial profile and activation of the natural mucosal immune system.22
Shimizu et al23 found that the Lactobacillus family and the genus Bifidobacterium increased gut microbiota in patients with BD compared to the control group. In another study of the same authors,24 Megamonas hypermegale and Butyrivibrio species were found to be decreased and, consequently, the production of butyrate and propionate short-chain fatty acids in the intestine was reduced when compared to normal individuals. It has been emphasized that this situation may lead to a decrease in regulatory T cell response and activation of immunopathological effector T cell responses. An increase in short-chain fatty acid production by the gut microbiota or oral delivery of short-chain fatty acids may be one of the treatment targets to ameliorate skewed T cell differentiation in BD patients.24 Even so, microbial factors and/or microbiome changes are not considered solely responsible for the pathogenesis. However, it should not be ignored that these may play a role in the disease’s progression by causing immune system dysfunction in the presence of the appropriate genetic background.25
The most remarkable studies conducted for BD etiopathogenesis are elucidating the genetic aspects of the disease. BD’s strongest genetic susceptibility factor is found in the MHC class I region, including HLA-B*51. Individuals carrying the HLA-B*51/B5 allele had 5.78 times increased risk for developing BD than those without this allele.26 Although the importance of the HLA-B*51 is already known, it is found to be positive in about 60% of patients. The frequency of HLA-B*51 also varies among populations. The role of HLA-B*51 in the genetic predisposition to the disease has been estimated around 12–19%.27 To develop new hypotheses about the pathogenesis of complex immune system diseases in which genetic factors and environmental factors both play a role, Genome-Wide Association Studies (GWAS), which examine the representation status of thousands of genes, have been applied. GWAS studies have confirmed that HLA-B*51 is the strongest genetic susceptibility factor in BD.
On the other hand, new and non-HLA genes, including endoplasmic reticulum aminopeptidase 1 (ERAP1), IL-23 receptor (IL-23R), IL-23R/IL-12RB2, IL-10, and signal transducer and activator of transcription proteins (STATs) have also been identified.28–32 Generally, gene polymorphisms related to BD have been detected in molecules that respond to microorganisms and genes encoding cytokines and adhesion molecules. Polymorphisms in these genes can affect the functions of the gene and also cause disease susceptibility. Identifying these new genes may play an essential role in determining the disease’s genetic burden and perhaps defining new treatment targets in the future.
In recent years, the diseases in which innate immunity and subsequently acquired immunity are activated due to impaired barrier functions in tissues in contact with environmental factors (skin, oral mucosa, and gastrointestinal system) are called MHC-1 related diseases (MHC-1-opathy). BD overlaps with MHC-1-opathy group diseases in many aspects.33 MHC-Class I alleles present exogenous peptides to CD8 + T cells either through endogenous peptides or through the mechanism called cross-presentation on the surface of APCs. The antigens presented by these molecules and their mechanisms are not fully understood. In recent years, the relationship detected between HLA-B*51 and ERAP1 has given some clues to understand this possible pathological mechanism. ERAP1 polymorphism is a significant genetic susceptibility factor frequently found among patients with BD in GWAS studies.34,35 ERAP1 encodes an enzyme, amino-peptidase, which is essential in the processing of MHC-class I related peptides. This enzyme trims the N-terminal of the peptides and ensures them to reach an ideal length. ERAP1 polymorphisms have been identified as risk factors in patients with BD who are HLA-B*51 positive. It is inherited recessively in patients with BD who are only HLA-B*51 positive. The ERAP1 rs17482078 (p.Arg725Gln) polymorphism affects this enzyme’s peptide specificity by altering the enzyme activity. Thus, the peptide loading onto the antigen-binding groove of HLA-B*51 is affected.36–38 In a recent study, Giza et al showed that the presentation of these modified peptides with HLA-B*51 has a vital role in disease pathogenesis.39
BD starts with the triggering of innate immunity and continues with the activation of acquired immunity.25 The “danger model” proposed by Matzinger is characterized by an excessive immune response against external stimuli. In this model, innate immunity is primarily activated, and the production of Th-1 and Th-17 related acquired immune response cytokines from innate immune cells such as macrophages and dendritic cells is triggered.40,41 BD is also considered a neutrophilic vasculitis, and the pathogenic roles of neutrophils, a member of innate immunity, are well-known.42 The fact that neutrophils probably being hyperactive through HLA-B*51 and form perivascular infiltration may contribute to the tissue damage seen in BD.43,44 Also, activation of neutrophils can lead to oxidative stress and the release of Th-1 related cytokines.44 Testosterone may also contribute to the activation of neutrophils and Th-1 cells.45 This situation may explain why BD is more severe in male patients. Natural killer (NK) cells may also play a role by increasing CD4+ Th-1 cell responses, especially in the active phase of the disease.46,47
Immune system dysregulation, altered T cell balance, and particularly the suppression of T regulatory cells’ activity by activation of the Th1/Th17 pathway are thought to play an essential role in the pathogenesis of BD.42 The number of Th-17 cells has increased in the cutaneous lesions of BD.48 Patients with BD in the active stages of uveitis, OU, GU, and articular symptoms had significantly higher IL-17 levels than patients in the inactive phases of the same symptoms.49,50 Under Th-17 stimulating conditions, IFN-γ release from T cells increases along with IL-17, and this situation further triggers neutrophil activation and the delayed immune response.40 In the acute attack phase of BD, while Th-17 and IL-17 pathways were active, decreased Treg and IL-10 levels were reported.46,51 Th-1 pathway and Th-1 pathway-related cytokines (IL-2, IL-12, IL-18 and IFN-γ) are increased during the active stages of the disease.52 TNF-α, a pro-inflammatory cytokine, plays a central role in the autoimmune response, induction and maintenance of inflammation.52 It contributes to the systemic inflammatory response seen in BD by being released from both innate immune cells, and acquired immune cells, such as Th-1 and Th-17 and it becomes a critical target molecule in the treatment of the disease.53
Consequently, environmental factors (S. sanguinis, etc.) or differences in salivary or gut microbiome composition can trigger innate immune system-mediated inflammation sustained by acquired or adaptive immune responses. Epistatic interactions between HLA-B*51 and ERAP1 variants disrupt T cell homeostasis. In particular, activation of the Th 1 and Th17 pathways and suppression of Treg cells are observed. This leads to the activation of neutrophils and intense neutrophil infiltration in the affected organs in the early inflammation stage.
Clinical Picture
OU, GU, and the other cutaneous lesions and ocular lesions, and arthropathy are the most frequently reported clinical symptoms of BD in all countries. Mucocutaneous lesions are the most common causes for a patient to consult a doctor, and they occur before major organ involvement in most of the patients. For this reason, good knowledge of the mucocutaneous lesions can enable physicians dealing with BD to diagnose the disease early and change the prognosis of the disease positively.3,8,54 OU and GU are characterized by recurrent, painful ulcerations of the oral mucosa, and genital skin/mucosa. Recurrent and bipolar OU and GU strongly indicate BD when any other reason does not explain them. OU seen in BD is similar to recurrent aphthous stomatitis (RAS) in terms of clinical appearance and course; however, it recurs more frequently, the number of lesions is higher in its attacks, and is located more frequently in different anatomical areas of the mouth (Figure 1). Also, major ulcers (> 1 cm) are more frequent in BD than in RAS patients.55 Although the appearance and course of GU are similar to OU, they are deeper and recur less frequently. Deeply located ulcers mostly heal with scarring (Figure 2). Therefore, patients with suspected BD should be investigated for scars from previous lesions, even if there is no active GU at the time of admission.56 PPL is found in at least 3/4 of the cases. It is distinguished by folliculitis or acne-like sterile papulopustular lesions on an erythematous background. EN-like lesions are seen in up to 40% of patients, and it is more frequent in female patients. They are characterized by painful, tender, oval-rounded, erythematous nodules on the anterior and lateral surfaces of the tibia. Superficial thrombophlebitis is observed in roughly one-quarter of the cases. It is more common in male patients and is distinguished by an often erythematous induration that can be palpated throughout the course of a vein, especially in the legs.57,58
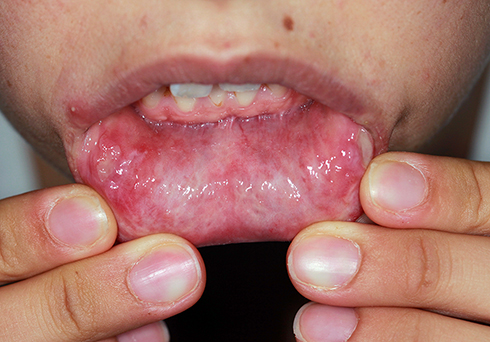 |
Figure 1 Oral ulcers on the lower lip mucosa. |
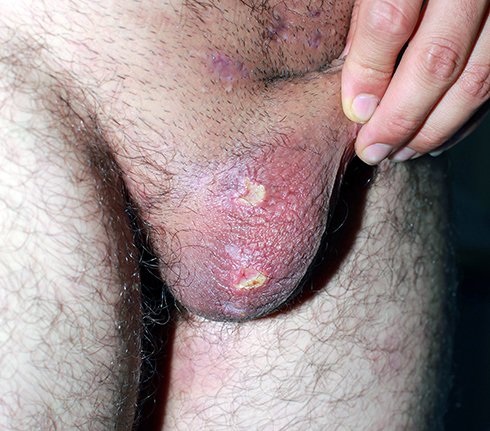 |
Figure 2 Genital ulcers and their scars on the scrotum. |
Pathergy test, one of the disease’s diagnostic criteria, refers to the hypersensitivity reaction that develops in the area where a needle was inserted. Similar to the spontaneously occurring PPL of BD, it is associated with a papule or pustule on an erythematous base at the needle-prick site 48 hours after applying a sterile needle penetrated to the corium of an avascular site on the forearm. While the average test positivity is 50% in Japan and Mediterranean countries, this rate is lower in western countries, which decreases the diagnostic value of the test.57,58
Ocular involvement, seen in about half of the patients, is one of disease’s most serious complications. It is more common and more severe in male patients and can cause visual impairment in approximately %15 patients with recurrent inflammatory attacks.59,60 Ocular involvement can occur as anterior uveitis, intermediate uveitis, posterior uveitis, and panuveitis, affecting both eyes. Posterior uveitis or panuveitis occurs in most patients. Articular involvement is seen in approximately half of the patients. Frequently, it occurs with a monoarticular or oligoarticular pattern. The attacks last a few weeks but usually do not cause deformity. The most frequently affected joint is the knee, followed by the ankle, wrist, and elbow, respectively.61 Being a systemic vasculitis, BD can affect all vessels regardless of diameter. The venous system is the main area of involvement, and superficial thrombophlebitis is the most common type of venous involvement. Inferior and superior vena cava thrombosis, dural venous sinus thrombosis, and Budd-Chiari syndrome can be seen, and these symptoms are associated with poor prognosis. The pulmonary artery aneurysm is rare, but it is the most important cause of mortality.5,62 Neurological involvement is relatively rare, but it is one of the most serious complications of the disease due to its severe prognosis. Neurological involvement is seen more frequently in male patients. Most patients have brainstem involvement, hemispheric symptoms, and parenchymal involvement, including spinal cord lesions and meningoencephalitis.63 Gastrointestinal involvement is characterized by volcano-shaped or punched-out mucosal ulcers localized mainly in the ileocecal region, although they can be seen throughout the gastrointestinal tract.64 In the course of BD, there may be endocarditis, myocarditis, pericarditis, intracardiac thrombus, endomyocardial fibrosis, coronary arthritis, myocardial infarction, and valve diseases.65–67 Arterial and venous involvement has been reported more frequently in patients with cardiac involvement than in patients without cardiac involvement.68
Treatment
The treatment is determined by many variables such as the organ involved, the severity of the involvement, the frequency of the attacks, and the patient’s age and gender. The success of treatment increases with early diagnosis and early treatment of the disease. Irreversible organ damage can be prevented with effective treatment, especially in the disease’s early and active stages. Controlled studies on BD are mostly limited to mucocutaneous, articular, and ocular involvement. There are no controlled studies in treating severe organ involvement because of the rarity of these conditions and ethical concerns. In this review, treatment suggestions have been developed based on the symptom/s that bring the patient to the physician. Although the recommendations are mainly based on controlled studies, it also includes important studies, guidelines, reviews of experts in this field and finally, our personal experience in clinical practice. From a holistic point of view, a symptom-based algorithmic approach has been proposed to manage BD. The “Oxford System” has been used in determining the level of evidence and the strength of the recommendation.69 Table 1 summarizes the spectrum of action of topical and systemic treatments used in BD.
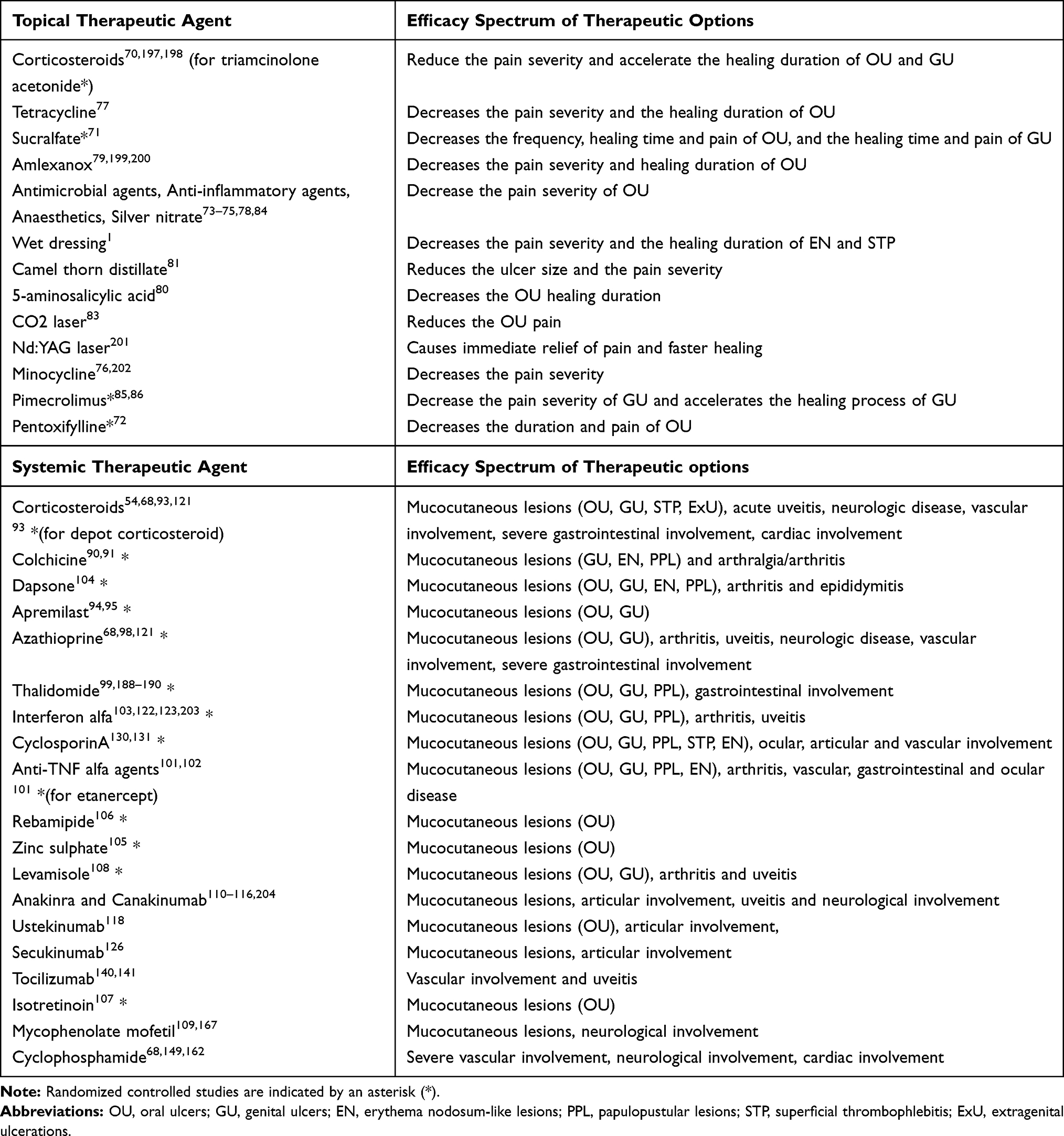 |
Table 1 Effectiveness of Therapeutic Agents Used in the Treatment of Behçet’s Disease |
Mucocutaneous Involvement
Topical Treatment
The number of controlled studies on the use of topical treatments in BD is limited. As we mentioned above, OU seen in BD are clinically identical to RAS, and there are more controlled studies with a large series of RAS in the literature. Therefore, in clinical practice, RAS-related treatment approaches can be used in the OU treatment of BD.1
Triamcinolone acetonide,70 sucralfate,71 and pentoxifylline72 are effective in OU of BD in randomized controlled studies. Triamcinolone acetonide 0.1% ointment (3 times a day) was more effective on OU than phenytoin syrup (2 teaspoons of syrup in half a glass of warm water as a mouthwash for 4–5 minutes, 3 times a day) at the end of one week.70 Sucralfate, which protects the mucous membranes by covering like a barrier, accelerates wound healing and reduces mucosal ulcers’ frequency and pain more than placebo when used 4 times a day for 3 months.71 When it is applied in 4 divided doses and used together with colchicine, pentoxifylline 1000 mg/day, which increases erythrocyte elasticity, decreases blood viscosity and improves microcirculatory flow and tissue perfusion, provided a significant reduction in the duration and pain of OU when compared to the use of colchicine alone, at the end of 2 weeks.72
Antimicrobial agents73 such as ListerineR (Johnson & Johnson, New Brunswick, NJ, USA), chlorhexidine gel,74 penicillin G potassium troches,75 minocycline,76 tetracycline suspension,77 triclosan mouth rinse,78 amlexanox,79 5-aminosalicylate,80 camel thorn distillate,81 diclofenac,82 lasers,83 and silver nitrate84 are other treatments shown to be effective in RAS patients and can also be used in BD patients.
Pimecrolimus cream, twice a day, accelerated the healing time of GU significantly in patients with BD compared to placebo.85 The combination of topical pimecrolimus and colchicine 1–2 mg/day, used twice a day, provided a significant reduction in the pain severity of GU compared to colchicine alone.86 Another option commonly used in clinical practice in GU treatment is corticosteroids. Although there are no controlled studies, potent corticosteroid ointments have been successfully used in GU treatment in the early stages of lesions, alone or in combination with antiseptics.87
EN-like lesions and superficial thrombophlebitis may benefit from wet dressing with 3–5% aluminum subacetate (Burrow) solution.1 Bed rest should be recommended in these patients. For PPL, however, local treatment expectancy is relatively rare; antiseptic containing corticosteroids or antibiotics may be used for this purpose.54
The effects of topical agents are probably limited to the application area. They are often used in the treatment of BD as an adjunct to systemic treatment. However, there may be a subgroup of patients in which a topical treatment approach alone can be used, such as patients without major organ involvement and no new attacks for a long time, female or elderly patients without severe organ involvement.
Systemic Treatment
In the absence of severe organ involvement, systemic therapy is usually based on the severity and clinical spectrum of mucocutaneous symptoms. Colchicine, 0.5 mg, 2–4 times a day, is usually preferred as the first-line treatment in managing OU, GU, EN-like lesions, and PPL.88 It suppresses the adhesion and recruitment of neutrophils by preventing microtubule polymerization. In the first placebo-controlled study of 35 patients,89 colchicine was effective in reducing the number of EN-like lesions. In the second placebo-controlled study involving 116 patients, colchicine reduced the frequency of GU and EN-like lesions in female patients.90 In the most recent, randomized, double-blind, crossover study involving 169 patients, significant improvement was found in the disease activity index and OU, GU, EN-like lesions, and PPL.91 The main difference between the last study and the previous ones is the difference in the medication’s effectiveness on OU and PPL. Open studies and our clinical observations confirm that colchicine is also effective on OU and PPL besides GU, EN-like lesions, especially in those with mild severity of the disease. Once a month, 1.2 million U benzathine penicillin can be added to colchicine treatment for patients for whom colchicine is not adequate alone. Combination therapy decreases OU and EN-like lesions’ duration and the frequency of GU compared to those receiving colchicine alone.92
Systemic corticosteroids can be used to control acute, severe mucocutaneous attacks rapidly. Prednisolone is usually used in major OU attacks, large and deep GU, and extensive and severe EN-like lesions and/or superficial thrombophlebitis. Prednisolone is usually started at a dose of 40–60 mg/day and tapered off within 4 to 6 weeks. Long-term use of corticosteroids should be avoided because of their side effects and their inability to prevent new attacks. In patients, who will be using corticosteroids, it is recommended to start treatment with an agent such as colchicine. In patients with active mucocutaneous lesions without ocular or major organ involvement, low-dose depot corticosteroid (40 mg methylprednisolone acetate every 3 weeks) is useful in the control of EN-like lesions, especially in female patients.93 However, this result does not mean that this compound will not affect other disease symptoms when used daily and at higher doses.
Apremilast, an oral phosphodiesterase-4 inhibitor, is one of the most exciting treatment choices for BD in recent years. In the Phase 2 study, apremilast (30 mg, twice a day) decreased the number of OU and GU and was effective in reducing pain due to OU.94 In a randomized, placebo-controlled, double-blind Phase 3 study, in patients with BD with active OU, without major organ involvement, apremilast significantly reduced the OU count and OU pain when compared to placebo, and this effect was observed as early as 1 week. Normally, the response seen at 12 weeks was prolonged to 28 weeks. In double-blind studies, the side effect profile was similar between apremilast and placebo. Diarrhea, nausea, and headache caused by apremilast are generally mild to moderate.95 Following this study, apremilast was approved by the FDA in patients with BD with active OU who had been previously treated with at least one nonbiological agent. Recently, real-life data on the use of apremilast in BD have also been published. Lopalco et al noted that apremilast decreased the number of OU and GU at the end of 3 months significantly compared to baseline and the number of active OU and OU attacks was still lower than the baseline at the end of 6 months.96 At the end of the 3rd month of treatment, the pain score was found to be significantly lower than the baseline, and this effect continued in the 6th month. Also, the disease activity score was significantly lower than the baseline at the end of the 3rd and 6th months. In the study of De Luca et al, at the end of the 12th week, a significant decrease was found in the number of OU and GU, disease activity score, and pain score using apremilast in patients with BD who were resistant or intolerant to conventional treatment. This clinical improvement enabled corticosteroid dose reduction and corticosteroid discontinuation. There was a significant improvement in the quality of life of the patients compared to the baseline. In 4 of 12 patients, the treatment was discontinued mostly due to diarrhea.97 In conclusion, apremilast is a candidate to be an important treatment option in patients with colchicine-resistant mucocutaneous symptoms, with a relatively lower side effect profile than immunosuppressive treatments. It can be evaluated as an important alternative in the first step in patients who do not respond to previous treatments (Table 2).
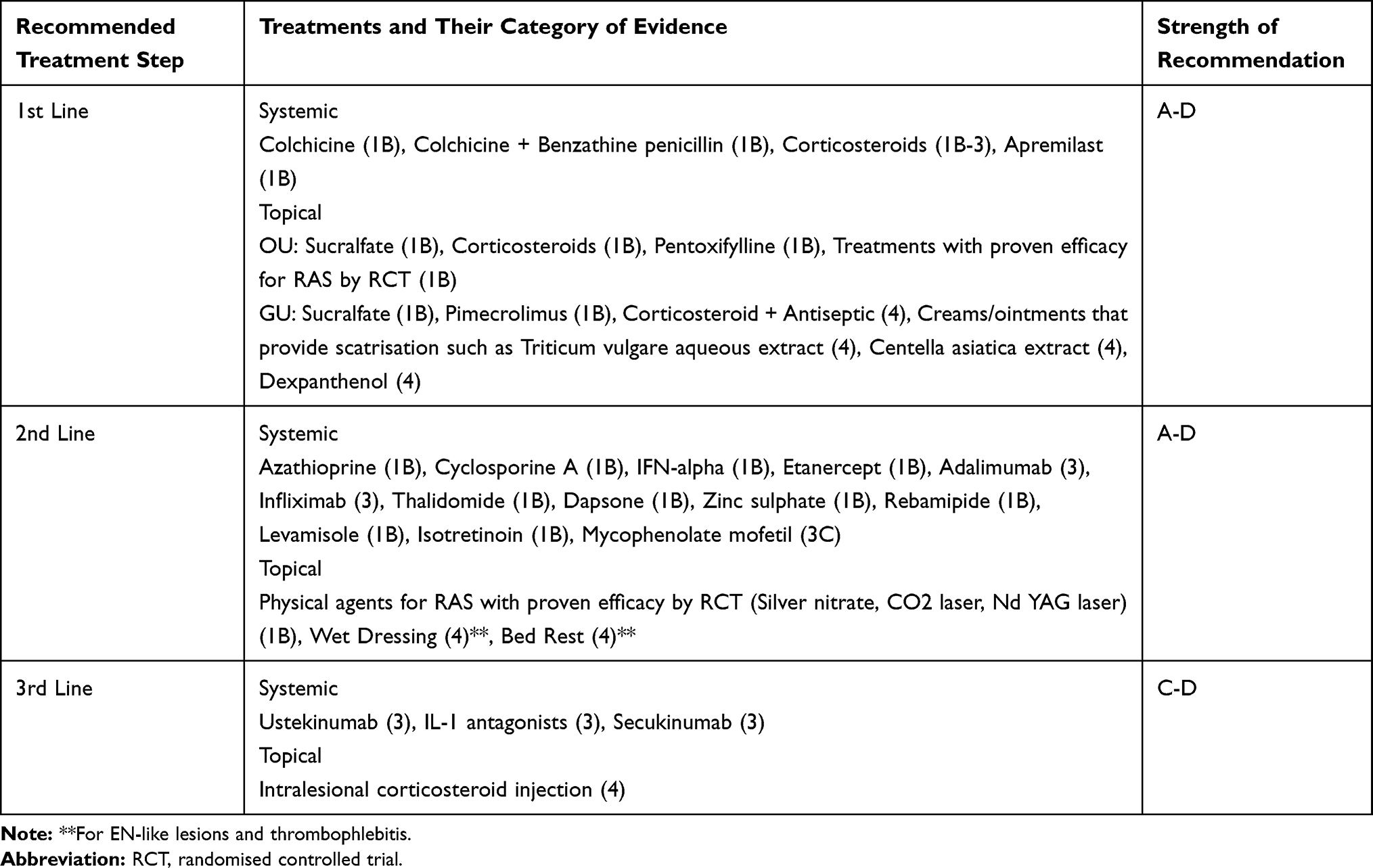 |
Table 2 Evidence-Based Algorithmic Treatment for Mucocutaneous Behcet’s Disease |
In the randomized, placebo-controlled, double-blind study of Yazici et al,98 azathioprine was used at a dose of 2.5 mg/kg/day and decreased the frequency of OU and GU when compared to placebo. Anti-TNF-α agents may also be recommended as the second-line treatment in refractory cases. When used at a dose of 25 mg, 2 days a week in a placebo-controlled study, etanercept had a rapid effect, especially on GU and EN-like lesions, and significantly reduced the numbers of OU, EN-like lesions, and PPL.99 In multiple and open studies with large series, infliximab and adalimumab were found to be effective at a rate of 88% in mucocutaneous lesions in severe cases and/or cases resistant to immunosuppressive agents, with no significant difference in effect.100 Interferon-alpha 2a treatment (3 times a week, 6 million U) significantly decreased the healing time and pain of OU and the frequency of GU and PPL at the end of 3 months in a placebo-controlled study.101 Cyclosporine-A used at a dose of 5 mg/kg/day was found to be more effective than conventional treatment (prednisolone, azathioprine) on OU, GU, superficial thrombophlebitis, and cutaneous lesions in a randomized controlled study.102 In a randomized, placebo-controlled, double-blind study conducted by Hamuryudan et al,103 thalidomide used at a dose of 100–300 mg/day provided long-term remission in OU, GU, and PPL. However, this agent should be used only in selected patients with care because of its potential side effects. It should also be kept in mind that nodular lesions may increase during thalidomide treatment.103
In a double-blind, placebo-controlled study, dapsone has been shown to reduce the number, healing time, and frequency of OU and the number of GU, EN-like lesions, and PPL when compared to placebo.104 Dapsone, an antineutrophilic activity like colchicine, can be an alternative to colchicine in treating mucocutaneous manifestations of BD in regions without glucose-6-phosphate dehydrogenase deficiency. It has also been reported that the use of oral zinc sulfate (300 mg/day, 6 months) provided improvement in mucocutaneous symptoms without any significant side effects.105 Rebamipide (300 mg/day, 6 months), a gastroprotective drug, significantly reduced the OU count and pain level compared to placebo.106 In the study of Sharrquie et al,107 isotretinoin, used for 12 weeks at a dose of 20 mg/day, provided a significant improvement in the clinical symptom index and OU and skin manifestation parameters compared to placebo. Levamisole (3x50 mg, 2 days/w) was found more effective on OU and GU than placebo.108 Dapsone, zinc sulfate, rebamipide, isotretinoin, and levamisole might have been considered in the treatment’s previous steps because of their controlled studies showing promising results. However, these treatments were not included in the previous steps because of the reason(s) summarized below. Relatively few patients were included in these publications; there have not been new publications about these treatments; new and more effective treatments have emerged in recent years; effectiveness is limited to OU (rebamipide). Enteric-coated mycophenolate mofetil (6 months, 720 mg twice a day) is another treatment that can be preferred in patients with mucocutaneous symptoms, resistant to at least one of the standard treatment methods as a second-line treatment.109
There is a growing body of research on the use of IL-1 antagonists in BD. The IL-1 receptor antagonist anakinra (ANA) and the full human anti-IL-1β antibody canakinumab (CAN) had good clinical results in patients with BD.110–115 In a multicenter retrospective study,116 90% of patients received ANA (100 mg/day) and 10% CAN (150 mg every 6–8 weeks) as the initial treatment. Complete remission (CR) was achieved in all 13 patients on anti-IL-1 treatment for 12 months. In 6 of these patients they switched from ANA to CAN during the treatment. During the 12-month follow-up, 8 of 30 patients were switched to non-anti IL-1 antagonist treatment due to ineffectiveness or loss of effect. During the study process, no serious adverse effect was developed. A mild local cutaneous reaction developed in 15% of the group receiving ANA, and no adverse effects developed in the group receiving CAN. These data indicate that anti-IL-1β antagonists are effective and safe in BD.
There are two studies on the use of ustekinumab, a monoclonal antibody developed against IL-12 and IL-23 p40 subunit.117,118 In the first study,117 14 BD patients with colchicine-resistant active OU were included in the study. 45 or 90 mg subcutaneous ustekinumab injection was administered on the first day, the first month, and every 3 months. At the end of the 12th week, 64% CR (defined as no OU), 21% partial response (PR) was obtained, while no response was obtained for 14%. Ustekinumab reduced the corticosteroid dose, decreased disease activity score, and treatment was discontinued in one patient due to headache. In another multicenter, prospective, open-label study,118 30 BD patients with colchicine-resistant active OU were included to study. CR (defined as no OU) was achieved in 60% of patients at the end of 12 weeks and 88.9% of patients at the end of 24 weeks by injecting 90 mg subcutaneous ustekinumab on day 1, the first month, and every 3 months. Serious adverse effect has not been observed. Ustekinumab appears to be a promising and safe treatment for colchicine-resistant OU.
As an IL-17 antagonist, secukinumab has been used in psoriatic arthritis, and ankylosing spondylitis and its efficacy in BD are being investigated due to similarities of its pathogenesis and clinical characteristics to seronegative arthritis. On 5 BD patients with OU and articular symptoms resistant to colchicine, conventional disease-modifying anti-rheumatic drugs (DMARDs), and at least one anti-TNF-α agent, secukinumab 150 mg (4 patients) or 300 mg (1 patient) were used once a month. The patient receiving 300 mg reached CR (50% reduction in OU count) within 3 months. Half of the 4 patients who received 150 mg reached CR in the 6th month, but relapse was observed in one patient who also reached CR. CR was achieved using 300 mg in patients with relapse and in whom CR could not be reached.119 On the other hand, two BD cases have been reported to be triggered by the use of secukinumab.120 Table 2 shows evidence-based algorithmic treatment for mucocutaneous BD.
Articular Involvement
The effect of colchicine on arthritis and arthralgia has been shown with randomized placebo-controlled studies.89,91 Therefore, colchicine is recommended as a first-line treatment in patients with arthritis.121 Calguneri et al92 showed that when benzathine penicillin (1.2 million U, every 3 weeks) was combined with colchicine treatment, the number of arthritis attacks was decreased, and the duration of the attack-free period was prolonged compared to the colchicine group alone. Azathioprine, used for 2 years at the dose of 2.5 mg/kg/day, significantly reduced arthritis incidence compared to placebo.98 Azathioprine may be preferred in patients with recurrent arthritis and/or refractory disease. IFN-alpha 2a (3 days a week, 3–12 million U) can alleviate the symptoms of arthritis and arthralgia.7 Studies have shown that IFN-alpha 2a (3 days a week, 6–9 million U) and IFN-alpha 2b (3 days a week, 5 million U) treatments reduce the number of arthritis attacks and shorten the duration of arthritis.122–124 Infliximab and adalimumab were effective in 70% of patients with severe articular involvement resistant to immunosuppressive agents (azathioprine, cyclophosphamide, methotrexate, mycophenolate mofetil, cyclosporine-A) and IFN-alpha.100 Although systemic corticosteroids and nonsteroidal anti-inflammatory drugs are widely used to treat symptoms associated with arthritis, the results from controlled trials with azapropazone or intramuscular methylprednisolone acetate have been disappointing.93,125 In patients with mono-arthritis, intra-articular corticosteroid injections may be considered besides systemic treatment.121
In a multicenter, prospective, open-label study, with ustekinumab treatment (90 mg at inclusion, at week 4, and then every 12 weeks), no arthritis was detected in any of the patients during the treatment process, and there was a decrease in articular involvement compared to baseline at the end of 12, 24, 36 and 48 weeks. At the end of the 4-year follow-up, arthralgia continued in only 15% of the patients.118
In a multicenter, retrospective study, with secukinumab treatment (150 mg/month, 300 mg/month in polyarticular involvement), 66.7% of the patients showed a significant improvement in articular symptoms within 3 months, and sustained remission was achieved in 84.6% of the patients.126 In another multicenter, retrospective study, complete improvement in articular symptoms (CR) was achieved in all patients after 12 months of treatment with IL-1 antagonists (100 mg/day ANA and 150 mg CAN every 6–8 weeks).116 Ustekinumab, secukinumab, and IL-1 antagonists can be used as an alternative treatment option in treatment-resistant cases. Table 3 summarizes the evidence-based algorithmic treatment for articular BD.
 |
Table 3 Evidence-Based Algorithmic Treatment for Articular Behcet’s Disease |
Ocular Involvement
To prevent irreversible losses in visual acuity, it is crucial to suppress inflammatory attacks quickly and prevent relapses.127129 In acute attacks, when the anterior part of the eye is involved (anterior uveitis), topically applied corticosteroid eye drops combined with mydriatics or cycloplegic agents can control the disease. Subconjunctival or posterior sub-Tenon corticosteroid injections according to the severity of eye involvement, and intravitreal corticosteroid injections in acute attacks limited to one eye can be administered in suitable patients. However, these treatments are recommended to be combined with systemic treatments.121
Systemic corticosteroids can be used in unresponsive and/or acute inflammatory ocular attacks of posterior uveitis, panuveitis, and retinal vasculitis.121 Systemic corticosteroid therapy may not be sufficient when administered alone. It is generally recommended to be used in conjunction with systemic immunosuppressive treatment.121 The first randomized controlled trial of an immunosuppressive agent in ocular involvement is related to azathioprine. Azathioprine treatment was effective in uveitis compared to placebo and significantly reduced patients’ corticosteroid requirement and improved visual acuity. Also, the development of new ocular diseases was significantly reduced in azathioprine users.98 Cyclosporine-A has been found superior to both colchicine130 and conventional therapies (prednisolone, chlorambucil) in reducing the frequency and severity of ocular attacks.130,131 Some experts suggest that although there is no controlled study of combining azathioprine or cyclosporin-A with anti-TNF-α agents, combined therapy will increase success.121 Anti-TNF-α agents can also be used alone or in combination with these agents in severe and/or resistant cases to cyclosporine A, azathioprine, systemic corticosteroids. In a placebo-controlled phase 3 study, including patients with BD, adalimumab (a loading dose of 80 mg followed by a dose of 40 mg every 2 weeks) was associated with a lower risk of uveitis exacerbation or visual impairment in non-infectious active intermediate, posterior uveitis and panuveitis.132 Recent, multicenter and large series studies have shown that adalimumab (40 mg/every 2 weeks subcutaneously) and infliximab (3–5 mg/kg iv at 0, 2, 6 days, and then every 4–8 weeks) are effective and relatively safe options for ocular involvement resistant to conventional treatments.133,134 In a multicenter study involving 177 patients, these two agents were compared with each other. After 1 year of treatment, a significant improvement was observed in all ocular parameters in both groups. However, improvement in some parameters (improvement in anterior chamber inflammation, vitritis, and best-corrected visual acuity) was more pronounced in patients using adalimumab than in infliximab.135 Switching between agents seems possible in patients with unresponsiveness to any of these drugs or patients with adverse events.121 IFN-alpha 2a is another alternative in patients resistant to corticosteroids and traditional immunosuppressive agents, and its effectiveness has been shown in controlled102 and open studies.122,136 This compound is effective and safe in the long-term treatment of severe uveitis (3 million units thrice a week).122,136 IFN-alpha decreases the need for corticosteroids and immunosuppressive agents and increases the quality of life.137
Randomized, placebo-controlled studies on gevokizumab, an IL-1β antagonist, and secukinumab, show that these two agents are far from achieving their goals.138,139 Monoclonal antibodies targeting these two cytokines play an important role in etiopathogenesis. The eye has a highly protected microenvironment; this may be one reason why eye involvement is relatively resistant to these treatments. On the other hand, studies show that tocilizumab, an IL-6 blocker, can be an effective treatment option in uveitis resistant to immunosuppressive agents.140,141 Table 4 shows evidence-based algorithmic treatment for ocular BD.
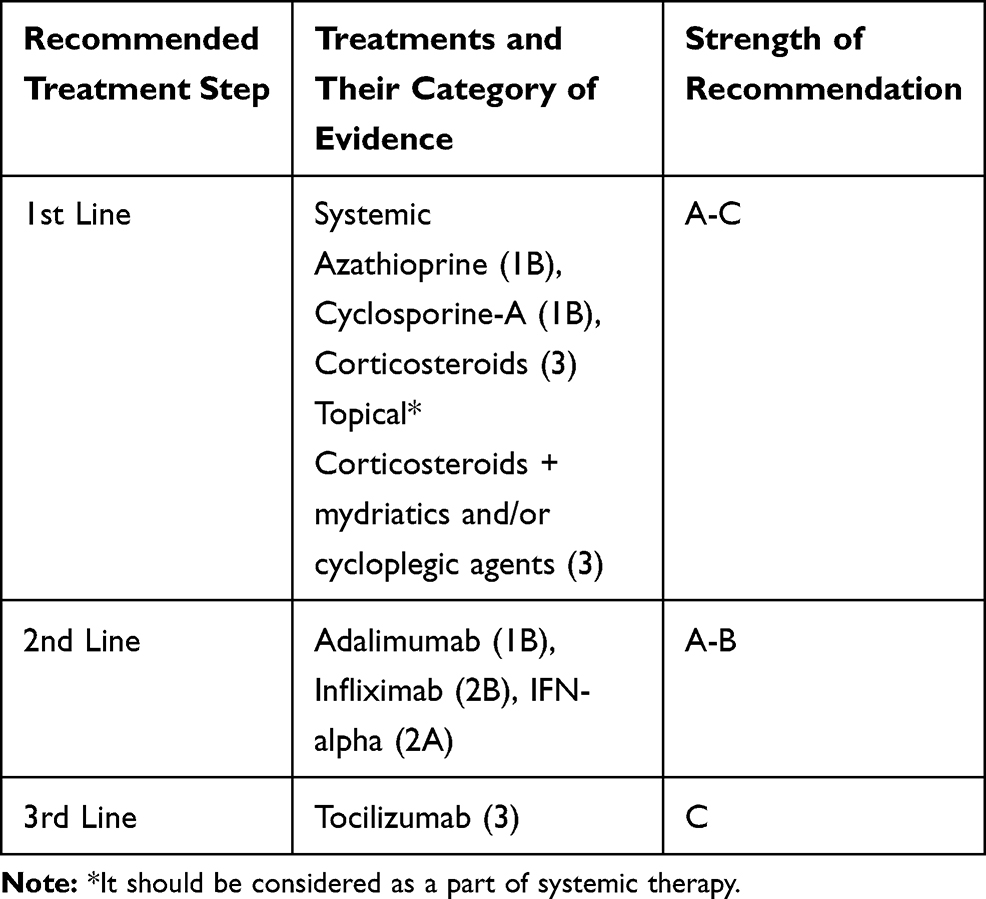 |
Table 4 Evidence-Based Algorithmic Treatment for Ocular Behcet’s Disease |
Vascular Involvement
The treatment of vascular BD symptoms varies according to the affected vascular area and the nature of this involvement. Inflammation of vascular structures in BD profoundly affects the treatment approach.
Venous Involvement
Acute deep vein thrombosis (DVT) treatment is based on systemic corticosteroids and immunosuppressive agents such as azathioprine, cyclosporine-A, cyclophosphamide.121,142,143 Immunosuppressive treatment is crucial to prevent relapse and reduce the risk of post-thrombotic syndrome.142 There is no evidence that one immunosuppressive agent should be preferred over others.121 However, cyclophosphamide may be preserved for patients with extensive thrombosis in larger vessels such as the vena cava due to its potential severe side effects.121 The use of anticoagulants in DVT is still controversial.142
In a meta-analysis, the combined use of immunosuppressive agents and anticoagulants was found to be more effective in preventing the relapse risk of DVT than using anticoagulants alone. However, treatment with anticoagulants and immunosuppressive agents did not provide a significant benefit in preventing relapses compared to immunosuppressive agents alone.121,144-146 On the other hand, a retrospective study shows that not using anticoagulants increases the risk of post-thrombotic syndrome.147
Anti-TNF-α agents may be considered in patients with resistant DVT.121 Anti-TNF-α agents can also be used in combination with conventional DMARDs148,149 or interferon-alpha.150
Arterial Involvement
Primary management of pulmonary artery aneurysms and thrombosis is carried out with high-dose corticosteroids and cyclophosphamide.121,149,150 Anti-TNF-α agents, particularly infliximab, can be life-saving in treatment-resistant cases.121,151 The mortality rate is high in patients undergoing surgical treatment, and surgery should not be preferred except for life-threatening situations.150,152,153 Embolization may be necessary for patients with a high risk of bleeding.149,153,154
If peripheral arterial aneurysms are small and asymptomatic and have a low risk of rupture, medical treatment with high-dose corticosteroids and cyclophosphamide may be sufficient.154–156 Surgery or stenting is required to treat all peripheral artery aneurysms in those who do not have these features.
In a retrospective study,157 tocilizumab was used at a dose of 8 mg/kg every 4 weeks in 7 patients with resistant arterial involvement who were poorly controlled with corticosteroids and immunosuppressive agents. After a mean follow-up of 19 months, all clinical symptoms and blood markers of inflammation improved, and no new-onset, arterial or venous lesions were reported during follow-up. Therefore, tocilizumab with immunosuppressive agents and corticosteroids may be a good alternative in managing active refractory vasculo-BD. Table 5 summarizes the evidence-based algorithmic treatment for vasculo-BD.
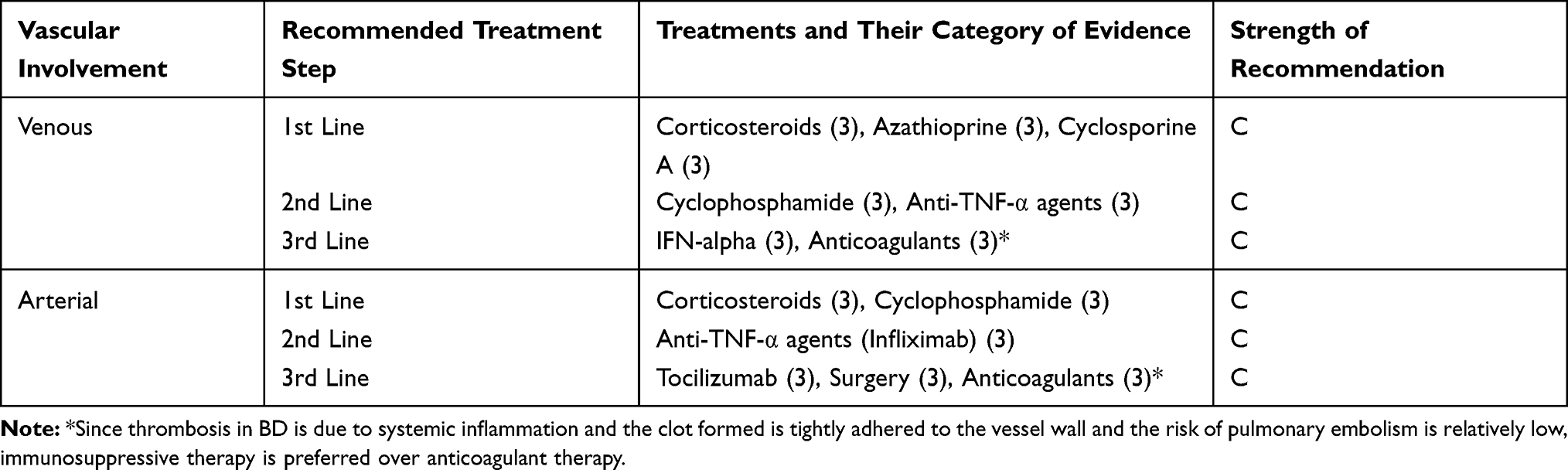 |
Table 5 Evidence-Based Algorithmic Treatment for Vasculo-Behcet’s Disease |
Neurological Involvement
Acute parenchymal attacks should be treated with high doses of corticosteroids. Corticosteroid treatment usually begins with pulse intravenous methylprednisolone at a dose of 1 gr/day, which can be given for up to 7 days, and continues with oral prednisolone or prednisone given at a dose of 1 mg/kg/day for 1 month and this treatment is reduced by 5–10 mg in every 10–15 days. Azathioprine (2–2.5 mg/kg/day) is added to this treatment. In patients with progressive neurological involvement who persist or relapse despite corticosteroids and/or azathioprine, anti-TNF-α agents may be preferred as effective first-line therapy.158–161 Anti-TNF-α agents, particularly infliximab, have been associated with a high response rate of 80%. Anti-TNF-α agents can prevent the risk of relapse and disability.100,121,158–161 Cyclophosphamide may be an alternative, and it can be preferred in patients with parenchymal involvement having severe and poor prognostic factors. Cyclophosphamide can be administered orally (1–3 mg/kg/day) or pulse intravenously (500–1000 mg/m2 every month for 6–9 months). In a retrospective study comparing three different therapeutic regimes (corticosteroids alone, azathioprine + corticosteroids, cyclophosphamide + corticosteroids), no significant difference in long-term outcomes was reported. However, a longer event-free survival was achieved in patients with a severe disability who were initially treated with high dose corticosteroids + intravenous cyclophosphamide.162 Tocilizumab (8 mg/kg every 4 weeks for a mean period of 24 months) provided partial remission in two patients and near-complete remission in one patient of three patients with neuro-BD, who were resistant to immunosuppressive and anti-TNF-α agents, and these effects could be achieved in a relatively short time, such as months.163 ANA110,116 and CAN110 have also been reported to be effective in neuro-BD resistant to other immunosuppressive therapies. Since limited data are available for IFN-alpha,164,165 methotrexate,166 and mycophenolate mofetil,167 their use is recommended in selected cases.
A meta-analysis of observational studies with cyclosporine-A showed that the risk of nervous system involvement is increased in patients using this agent.168–171 Therefore, cyclosporine-A should be avoided in BD patients with neurological involvement, even when the nervous system involvement is no longer active.121
In the acute attack treatment of cerebral venous thrombosis, corticosteroids are started at high doses, and the dose is reduced according to the response. Short-term anticoagulants can be used, especially in patients with prothrombotic tendency. It has not been demonstrated that adding immunosuppressive agents to these treatments has any benefits. In the first phase, the use of immunosuppressive agents may not be recommended as relapses are uncommon.121 However, in resistant and relapsed cases, azathioprine, cyclophosphamide, and anti-TNF-α agents can be used.172,173 Table 6 summarizes the evidence-based algorithmic treatment for neuro-BD.
 |
Table 6 Evidence-Based Algorithmic Treatment for Neuro-Behcet’s Disease |
Gastrointestinal Involvement
The treatment approach in gastrointestinal system involvement is generally determined according to the severity of the involvement. In mild cases, it would be wise to start treatment with five aminosalicylate derivatives and salazosulfapyridine.174 Azathioprine can be considered in unresponsive or severe cases.121 In unresponsive cases, oral or intravenous high-dose corticosteroids can be used.175,176 Corticosteroids are thought to accelerate the healing of ulcers in acute exacerbations. However, since high doses of corticosteroids have the potential to trigger perforation, they should be used with a well-calculated benefit-to-harm ratio.57 The current evidence on the efficacy of corticosteroids in gastrointestinal involvement is insufficient to recommend their routine use in clinical practice.176 Occasionally, colchicine is administered empirically, but there is insufficient evidence regarding its use.174 Colchicine should not be used alone in mucosal inflammation and ulcers.174
The absolute indications of surgical treatments are intestinal perforation, severe stricture, large abscesses, and massive gastrointestinal bleeding.174 Immunosuppressive agents reduce the risk of postoperative recurrence and complications in such patients.121 Relative indications of surgery are; resistance to medical treatment and intestinal complications affecting the quality of life.174 Enteral nutrition therapy with elemental diets may be useful in inducing remission and is indicated for patients resistant to drug therapy and those with severe intestinal disorders such as stenosis.174
In recent years, the most crucial development in this field is anti-TNF-α agents in many cases with high success rates. Therefore, it can be considered as a suitable alternative for those with severe symptoms resistant to azathioprine.177,178 Infliximab is useful in the rapid induction and maintenance of remission in intestinal BD.179–181 Adalimumab also shows a similar efficacy profile to infliximab in patients with resistant or severe intestinal BD.182–184 Etanercept,185 golimumab,186 and certolizumab187 are other anti-TNF-α agents found to be effective in intestinal BD.
Thalidomide can be administered successfully in cases with gastrointestinal involvement resistant to immunosuppressive agents such as corticosteroids and azathioprine.188–190 It has been suggested that methotrexate may be useful in intestinal BD as combination therapy with infliximab.178 IL-1 antagonists, ANA and CAN110,114,116 and IL-6 antagonist tocilizumab140 have also been studied in patients with BD, including, but not limited to, intestinal BD. These agents may be useful in intestinal BD. Ustekinumab has been approved for treating Crohn’s disease, which shows some similarities with intestinal BD in terms of genetic background, clinical features, and treatment. This treatment has not yet been studied in intestinal BD.191 Table 7 shows evidence-based algorithmic treatment for intestinal BD.
 |
Table 7 Evidence-Based Algorithmic Treatment for Gastrointestinal Behcet’s Disease |
Cardiac Involvement
Cardiac complications are one of the leading causes of death in patients with BD. Therefore, early diagnosis and treatment of these complications are of great importance. Since cardiac involvement is rare, there are no large series and controlled studies in the literature. There is no standard protocol for remission and maintenance treatment.
Pericarditis can be treated with aspirin, colchicine, and/or immunosuppressive agents.68 The main therapeutic approach in acute myocardial infarction is based on revascularization (surgical or percutaneous).68,192 Corticosteroids, immunosuppressive agents, surgery, or anticoagulant therapy, may be used in the intracardiac thrombus.68,192 The surgical approach may be preferred in addition to immunosuppressive therapy in endomyocardial fibrosis and coronary aneurysm.68,192–194 In cardiac failure, high dose corticosteroids and conventional heart failure therapy may improve cardiac performance.192,195
Kwon et al196 reported that anticoagulants such as coumadin could cause aneurysm formation and enlargement in BD. Intracardiac thrombotic events are caused by blood pooling due to endothelial or myocardial damage secondary to inflammation. Therefore, some authors do not recommend anticoagulants, as they may cause bleeding in patients with thrombotic complications.
Funding
There is no funding to report.
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Alpsoy E. Behçet’s disease: A comprehensive review with a focus on epidemiology, etiology and clinical features, and management of mucocutaneous lesions. J Dermatol. 2016;43(6):620–632. doi:10.1111/1346-8138.13381
2. Azizlerli G, Kose AA, Sarica R, et al. Prevalence of Behcet’s disease in Istanbul, Turkey. Int J Dermatol. 2003;42:803–806.
3. Alpsoy E, Zouboulis CC, Ehrlich CE. Mucocutaneous lesions of Behcet’s disease. Yonsei Med J. 2007;48:573–585.
4. Alpsoy E, Donmez L, Onder M, et al. Clinical features and natural course of Behcet’s disease in 661 cases: a multicentre study. Br J Dermatol. 2007;157:901–906.
5. Kural-Seyahi E, Fresko I, Seyahi N, et al. The long-term mortality and morbidity of Behcet syndrome: a 2-decade outcome survey of 387 patients followed at a dedicated center. Medicine. 2003;82:60–76.
6. Mumcu G, Yay M, Aksoy A, et al. Predictive factors for work-day loss in Behçet’s syndrome: A multi-center study. Int J Rheum Dis. 2020;23:240–246.
7. Yazici H, Esen F. Mortality in Behçets syndrome. Clin Exp Rheumatol. 2008;26(5 Suppl 51):S138.
8. Ugurlu N, Bozkurt S, Bacanli A, Akman Karakas A, Uzun S, Alpsoy E. The natural course and factors affecting severity of Behçet’s disease: a single-center cohort of 368 patients. Rheumatol Int. 2015;35:2103–2107.
9. International Study Group for Behcet’s Disease. Criteria for diagnosis of Behçet’s disease. Lancet. 1990;335:1078–1080.
10. Sallakci N, Bacanli A, Coskun M, Yavuzer U, Alpsoy E, Yegin O. CTLA-4 gene 49A/G polymorphism in Turkish patients with Behcet’s disease. Clin Exp Dermatol. 2005;30(5):546–550. doi:10.1111/j.1365-2230.2005.01846.x
11. Behçet H. Uber rezidivierende aphthose, durch ein Virus verursachte Geschwure, am Mund, am Auge, und an den Genitalien. Dermatol Wochenschr. 1937;105:1152–1157.
12. Lehner T. The role of heat shock protein, microbial and autoimmune agents in the aetiology of Behget’s disease. Int Rev Immunol. 1997;14(1):21–32. doi:10.3109/08830189709116842
13. Studd M, McCance DJ, Lehner T. Detection of HSV-1 DNA in patients with Behcet’s syndrome and in patients with recurrent oral ulcers by the polymerase chain reaction. J Med Microbiol. 1991;34(1):39–43. doi:10.1099/00222615-34-1-39
14. Mumcu G, et al. Oral health is impaired in Behcet’s disease and is associated with disease severity. Rheumatology. 2004;43(8):1028–1033. doi:10.1093/rheumatology/keh236
15. Kaneko F, Oyama N, Nishibu A. Streptococcal infection in the pathogenesis of BehÇet’s disease and clinical effects of minocycline on the disease symptoms. Yonsei Med J. 1997;38(6):444–454. doi:10.3349/ymj.1997.38.6.444
16. Mizushima Y, Matsuda T, Hoshi K, Ohno S. Induction of Behçet’s disease symptoms after dental treatment and streptococcal antigen skin test.. J Rheumatol. 1988;15(6):1029–1030.
17. Alpsoy E. Behçet’s disease: treatment of mucocutaneous lesions.. Clin Exp Rheumatol. 2005;23(4):532–539.
18. Akman A, Kacaroglu H, Donmez L, Bacanli A, Alpsoy E. Relationship between periodontal findings and Behçet’s disease: a controlled study. J Clin Periodontol. 2007;34:485–491.
19. Celenligil-Nazliel H, Kansu E, Ebersole J. Periodontal findings and systemic antibody responses to oral microorganisms in Behçet’s disease. J Periodontol. 1999;70:1449–1456.
20. Coit P, Mumcu G, Ture-Ozdemir F, et al. Sequencing of 16S rRNA reveals a distinct salivary microbiome signature in Behcet’s disease. Clin Immunol. 2016;169:28–35.
21. Consolandi C, Turroni S, Emmi G, et al. Behcet’s syndrome patients exhibit specific microbiome signature. Autoimmun Rev. 2015;14:269–276.
22. Oezguen N, Yalcinkaya N, Kücükali CI, et al. Microbiota stratification identifies disease-specific alterations in neuro-Behçet’s disease and multiple sclerosis. Clin Exp Rheumatol. 2019;37 Suppl 121(6):58–66.
23. Shimizu J, Kubota T, Takada E, et al. Bifidobacteria abundance-featured gut microbiota compositional change in patients with Behcet’s disease. PLoS One. 2016;11:e0153746.
24. Shimizu J, Kubota T, Takada E, et al. Relative abundance of Megamonas hypermegale and Butyrivibrio species decreased in the intestine and its possible association with the T cell aberration by metabolite alteration in patients with Behcet’s disease. Clin Rheumatol. 2019;38:1437–1445.
25. Leccese P, Alpsoy E. Behçet’s Disease: an Overview of Etiopathogenesis. Front Immunol. 2019;10(10):1067. doi:10.3389/fimmu.2019.01067
26. de Menthon M, Lavalley MP, Maldini C, Guillevin L. Mahr A. HLA-B51/B5 and the risk of Behçet’s disease: a systematic review and meta-analysis of case-control genetic association studies. Arthritis Rheum. 2009;61:1287–1296.
27. Gul A. Genetics of Behçet’s disease: lessons learned from genomewide association studies. Curr Opin Rheumatol. 2014;26:56–63.
28. Hughes T, Coit P, Adler A, et al. Identification of multiple independent susceptibility loci in the HLA region in Behcet’s disease. Nat Genet. 2013;45:319–324.
29. Takeuchi M, Mizuki N, Meguro A, et al. Dense genotyping of immune-related loci implicates host responses to microbial exposure in Behçet’s disease susceptibility. Nat Genet. 2017;49:438–443.
30. Hou S, Yang Z, Du L, et al. Identification of a susceptibility locus in STAT4 for Behçet‘s disease in Han Chinese in a genome-wide association study. Arthritis Rheum. 2012;64:4104–4113.
31. Mizuki N, Meguro A, Ota M, et al. Genome-wide association studies identify IL23R-IL12RB2 and IL10 as Behçet’s disease susceptibility loci. Nat Genet. 2010;42:703–706.
32. Salmaninejad A, Gowhari A, Hosseini S, et al. Genetics and immunodysfunction underlying Behçet’s disease and immunomodulant treatment approaches. J Immunotoxicol. 2017;14:137–151.
33. McGonagle D, Aydin SZ, Gül A, Mahr A, Direskeneli H. ‘MHC-I-opathy’ — unified concept for spondyloarthritis and Behçet disease. Nat Rev Rheumatol. 2015;11:731–740.
34. Remmers EF, Cosan F, Kirino Y, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behçet’s disease. Nat Genet. 2010;42:698–702.
35. Kirino Y, Bertsias G, Ishigatsubo Y, et al. Genome-wide association analysis identifies new susceptibility loci for Behçet’s disease and epistasis between HLA-B-51 and ERAP1. Nat Genet. 2013;45:202–207.
36. Ombrello MJ, Kirino Y, de Bakker PI, Gül A, Kastner DL, Remmers EF. Behcet disease-associated MHC class I residues implicate antigen binding and regulation of cell-mediated cytotoxicity. Proc Natl Acad Sci. 2014;111:8867–8872.
37. Guasp P, Alvarez-Navarro C, Gomez-Molina P, et al. The Peptidome of Behçet’s Disease-Associated HLA-B*51:01 Includes Two Subpeptidomes Differentially Shaped by Endoplasmic Reticulum Aminopeptidase 1. Arthritis Rheumatol. 2016;68:505–515.
38. Padula MC, Leccese P, Pellizzieri E, et al. Distribution of rs17482078 and rs27044 ERAP1 polymorphisms in a group of Italian Behçet’s syndrome patients: a preliminary case-control study. Intern Emerg Med. 2019;14:713–718.
39. Giza M, Koftori D, Chen L, Bowness P. Is Behçet’s disease a ‘class 1-opathy’? The role of HLA-B*51 in the pathogenesis of Behçet’s disease. Clin Exp Immunol. 2018;191:11–18.
40. Deniz R, Tulunay-Virlan A, Ture Ozdemir F, et al. Th17-Inducing Conditions Lead to in vitro Activation of Both Th17 and Th1 Responses in Behcet’s Disease. Immunol Invest. 2017;46:518–525.
41. Alpsoy E, Kodelja V, Goerdt S, Orfanos CE, Zouboulis CC. Serum of patients with Behçet’s disease induces classical (pro-inflammatory) activation of human macrophages in vitro. Dermatology. 2003;206(3):225–232.
42. Greco A, De Virgilio A, Ralli M, et al. Behçet’s disease: new insights into pathophysiology, clinical features and treatment options. Autoimmun Rev. 2018;17:567–575.
43. Eksioglu-Demiralp E, Direskeneli H, Kibaroglu A, Yavuz S, Ergun T, Akoglu T. Neutrophil activation in Behçet’s disease. Clin Exp Rheumatol. 2001;19(5Suppl. 24):S19–S24.
44. Becatti M, Emmi G, Silvestri E, et al. Neutrophil activation promotes fibrinogen oxidation and thrombus formation in Behçet disease. Circulation. 2016;33:302–311.
45. Yavuz S, Akdeniz T, Hancer V, Bicakcigil M, Can M, Yanikkaya-Demirel G. Dual effects of testosterone in Behçet’s disease: implications for a role in disease pathogenesis. Genes Immun. 2016;17:335–341.
46. Hasan MS, Ryan PL, Bergmeier LA, Circulating FF. NK cells and their subsets in Behçet’s disease. Clin Exp Immunol. 2017;188:311–322.
47. Yamaguchi Y, Takahashi H, Satoh T, et al. Natural killer cells control a T-helper 1 response in patients with Behçet’s disease. Arthritis Res Ther. 2010;12:R80.
48. Shimizu J, Takai K, Fujiwara N, et al. Excessive CD4+ T cells co-expressing interleukin-17 and interferon-γ in patients with Behçet’s disease. Clin Exp Immunol. 2012;168:68–74.
49. Chi W, Zhu X, Yang P, et al. Upregulated IL-23 and IL-17 in Behçet patients with active uveitis. Invest Ophthalmol Vis Sci. 2008;49:3058–3064.
50. Ekinci NS, Alpsoy E, Karakas AA, Yilmaz SB, Yegin O. IL-17A has an important role in the acute attacks of Behçet’s disease. J Invest Dermatol. 2010;130:2136–2138.
51. Touzot M, Cacoub P, Bodaghi B, Soumelis V, Saadoun D. IFNa induces IL-10 production and tilt the balance between Th1 and Th17 in Behçet disease. Autoimmun Rev. 2015;14:370–375.
52. Lee YJ, Horie Y, Wallace GR, et al. Genome-wide association study identifies GIMAP as a novel susceptibility locus for Behçet’s disease. Ann Rheum Dis. 2013;72:1510–1516.
53. Tong B, Liu X, Xiao J, Su G. Immunopathogenesis of Behcet’s Disease. Front Immunol. 2019;29(10):665.
54. Alpsoy E, Akman A. Behçet’s disease: an algorithmic approach to its treatment. Arch Dermatol Res. 2009;301:693–702.
55. Oh SH, Han EC, Lee JH. Comparison of the clinical features of recurrent aphthous stomatitis and behcet’s disease. clin exp dermatol. 2009; 34: 208-12.nakamura k, iwata y, asai j, kawakami t, tsunemi y, takeuchi m, et al; members of the consensus conference on treatment of skin and mucosal lesions (committee of guideline for the diagnosis and treatment of mucocutaneous lesions of behçet’s disease). guidelines for the treatment of skin and mucosal lesions in behçet’s disease: A secondary publication. J Dermatol. 2020;47:223–235.
56. Mat C, Goksugur N, Ergin B, Yurdakul S, Yazici H. The frequency of scarring after genital ulcers in Behcet’s syndrome: a prospective study. Int J Dermatol. 2006;45:554–556.
57. Esatoglu SN, Hatemi G. Update on the treatment of Behçet’s syndrome. Intern Emerg Med. 2019;14(5):661–675.
58. Alpsoy E, Donmez L, Bacanli A, Apaydin C, Butun B. Review of the clinical manifestations’ chronology in 60 patients with Behçet’s disease. Dermatology. 2003;117:354–356.
59. Inaba G. Clinical features of neuro-Behçet’s syndrome. In: Lehner T, Barnes CG, editors. Recent Advances in Behçet’s Disease. London: Royal Society of Medicine Services; 1986:235–246.
60. Verity DH, Wallace GR, Seed PT, et al. Soluble adhesion molecules in Behcet’s disease. Ocul Immunol InXamm. 1998;6:81–92.
61. Yurdakul S, Hamuryudan V, Yazici H. Behcet syndrome. Curr Opin Rheumatol. 2004;16:38–42.
62. Saadoun D, Wechsler B, Desseaux K, et al. Mortality in Behçet’s disease. Arthritis Rheum. 2010;62:2806–2812.
63. Siva A, Altintas A, Saip S. Behçet’s syndrome and the nervous system. Curr Opin Neurol. 2004;17:347–357.
64. Sakane T, Takeno M, Suziki N, Inaba G. Behcet’s disease. N Engl J Med. 1999;341:1284–1291.
65. Heper G, Polat M, Yetkin E, Senen K. Cardiac findings in Behçet’s patients. Int J Dermatol. 2010;49(5):574–578.
66. Gurgun C, Ercan E, Ceyhan C, et al. Cardiovascular involvement in Behcet’s disease. Jpn Heart J. 2002;43:389–398.
67. Aslam F, Bandeali SJ, Crowson C, Alam M. Cardiac function and diastolic dysfunction in Behcet's disease: A systematic review and meta-analysis. Int J Rheumatol. 2016;2016:9837184.
68. Geri G, Wechsler B. Thi Huong du L, Isnard R, Piette JC, Amoura Z, et al. Spectrum of cardiac lesions in behçet disease: a series of 52 patients and review of the literature. Medicine. 2012;91:25–34.
69. van der Heijde D, Aletaha D, Carmona L, et al. Update of the EULAR standardised operating procedures for EULAR-endorsed recommendations. Ann Rheum Dis. 2014;2015(74):8–13.
70. Fani MM, Ebrahimi H, Pourshahidi S, Aflaki E, Shafiee Sarvestani S. Comparing the effect of phenytoin syrup and triamcinolone acetonide ointment on aphthous ulcers in patients with Behcet’s syndrome. Iran Red Crescent Med J. 2012;14:75–78.
71. Alpsoy E, Er H, Durusoy C, Yilmaz E. The use of sucralfate suspension in the treatment of oral and genital ulcerations of Behcet’s disease: a randomised, placebo-controlled and double-blind study. Arch Dermatol. 1999;135:529–532.
72. Hatemi G, Yurttas B, Kutlubay Z, et al. Pentoxifylline Gel for Oral Ulcers in Patients with Behçet’s Syndrome [abstract]. Arthritis Rheumatol. 2019;71(supl):10.
73. Meiller TF, Kutcher MJ, Overholser CD, Niehaus C, DePaola LG, Siegel MA. Effect of an antimicrobial mouthrinse on recurrent aphthous ulcerations. Oral Surg Oral Med Oral Pathol. 1991;72:425–429.
74. Addy M, Carpenter R, Roberts WR. Management of recurrent aphthous ulceration. A trial of chlorhexidine gel. Br Dent J. 1976;141:118–120.
75. Kerr AR, Drexel CA, Spielman AI. The efficacy and safety of 50 mg penicillin G potassium troches for recurrent aphthous ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;96:685–694.
76. Gorsky M, Epstein J, Raviv A, Yaniv R, Truelove E. Topical minocycline for managing symptoms of recurrent aphthous stomatitis. Spec Care Dentist. 2008;28:27–31.
77. Graykowski EA, Kingman A. Double-blind trial of tetracycline in recurrent aphthous ulceration. J Oral Pathol. 1978;7:376–382.
78. Skaare AB, Herlofson BB, Barkvoll P. Mouthrinses containing triclosan reduce the incidence of recurrent aphthous ulcers (RAU). J Clin Periodontol. 1996;23:778–781.
79. Liu J, Zeng X, Chen Q, et al. An evaluation on the efficacy and safety of amlexanox oral adhesive tablets in the treatment of recurrent minor aphthous ulceration in a Chinese cohort: a randomized, double-blind, vehicle-controlled, unparallel multicenter clinical trial. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;102:475–481.
80. Collier PM, Neill SM, Copeman PW. Topical 5-aminosalicylic acid: a treatment for aphthous ulcers. Br J Dermatol. 1992;126:185–188.
81. Pourahmad M, Rahiminejad M, Fadaei S, Kashafi H. Effects of camel thorn distillate on recurrent oral aphthous lesions. J Dtsch Dermatol Ges. 2010;8:348–352.
82. Saxen MA, Ambrosius WT. al Rehemtula KF, Russell AL, Eckert GJ. Sustained relief of oral aphthous ulcer pain from topical diclofenac in hyaluronan: a randomized, double-blind clinical trial. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:356–361.
83. Zand N, Ataie-Fashtami L, Djavid GE, et al. Relieving pain in minor aphthous stomatitis by a single session of non-thermal carbon dioxide laser irradiation. Lasers Med Sci. 2009;24:515–520.
84. Alidaee MR, Taheri A, Mansoori P, Ghodsi SZ. Silver nitrate cautery in aphthous stomatitis: a randomized controlled trial. Br J Dermatol. 2005;153:521–525.
85. Chams-Davatchi C, Barikbin B, Shahram F, et al. Pimecrolimus versus placebo in genital aphthous ulcers of Behcet’s disease: a randomized double-blind controlled trial. Int J Rheum Dis. 2010;13:253–258.
86. Kose O, Dinç A, Simsek I. Randomized trial of pimecrolimus cream plus colchicine tablets versus colchicine tablets in the treatment of genital ulcers in Behcet’s disease. Dermatology. 2009;218:140–145.
87. Alpsoy E. New evidence-based treatment approach in Behcet’s disease. Patholog Res Int. 2012;2012:871019.
88. Angelidis C, Kotsialou Z, Kossyvakis C, et al. Colchicine Pharmacokinetics and Mechanism of Action. Curr Pharm Des. 2018;24:659–663.
89. Aktulga E, Altaç M, Müftüoglu A, et al. A double blind study of colchicine in Behçet’s disease. Haematologica. 1980;65:399–402.
90. Yurdakul S, Mat C, Tuzun Y, et al. A double-blind trial of colchicine in Behçet’s syndrome. Arthritis Rheum. 2001;44:2686–2692.
91. Davatchi F, Sadeghi Abdollahi B, Tehrani Banihashemi A, et al. Colchicine versus placebo in Behcet’s disease: randomized, double-blind, controlled crossover trial. Mod Rheumatol. 2009;19:542–549.
92. Calguneri M, Ertenli I, Kiraz S, Erman M, Celik I. Effect of prophylactic benzathine penicillin on mucocutaneous symptoms of Behcet’s disease. Dermatology. 1996;192:125–128.
93. Mat C, Yurdakul S, Ozyazgan Y, Uysal S, Uysal O, Yazıcı H. A double-blind trial of depot corticosteroids in Behçets syndrome. Rheumatology. 2006;45:348–352.
94. Hatemi G, Melikoglu M, Tunc R, et al. Apremilast for Behcet’s syndrome – a phase 2, placebo-controlled study. N Engl J Med. 2015;16:1510–1518.
95. Hatemi G, Mahr A, Ishigatsubo Y, et al. Trial of Apremilast for Oral Ulcers in Behçet’s Syndrome. N Engl J Med. 2019;381:1918–1928.
96. Lopalco G, Venerito V, Leccese P, et al. Real-world effectiveness of apremilast in multirefractory mucosal involvement of Behçet’s disease. Ann Rheum Dis. 2019;78:1736–1737.
97. De Luca G, Cariddi A, Campochiaro C, et al. Efficacy and safety of apremilast for Behçet’s syndrome: a real-life single-centre Italian experience. Rheumatology. 2020;59:171–175.
98. Yazici H, Pazarli H, Barnes CG, et al. A Controlled Trial of Azathioprine in Behçet’s Syndrome. N Engl J Med. 1990;322(5):281–285. doi:10.1056/NEJM199002013220501
99. Melikoglu M, Fresko I, Mat C, et al. Short-term trial of etanercept in Behçet’s disease: a double blind, placebo controlled study.. J Rheumatol. 2005;32(1):98–105.
100. Vallet H, Riviere S, Sanna A, et al. Efficacy of anti-TNF alpha in severe and/or refractory Behçet’s disease: multicenter study of 124 patients. J Autoimmun. 2015;62:67–74. doi:10.1016/j.jaut.2015.06.005
101. Alpsoy E, Durusoy C, Yilmaz E, et al. Interferon alfa-2a in the treatment of Behçet disease: a randomized placebo-controlled and double-blind study. Arch Dermatol. 2002;138(4):467–471. doi:10.1001/archderm.138.4.467
102. Assaad-Khalil SH. Low-dose cyclosporin in Behçet’s disease: follow-up controlled study with emphasis on extraocular manifestations and neuro-Behçet’s disease. In: Jd O, Kokmen E, editors. Behçet’s Disease: Basic and Clinical Aspects. Vols. 603-612. New York: Marcel Dekker; 1991.
103. Hamuryudan V, et al. Thalidomide in the treatment of the mucocutaneous lesions of the Behcet syndrome. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1998;128(6):443–450. doi:10.7326/0003-4819-128-6-199803150-00004
104. Sharquie KE, Najim RA, Abu-Raghif AR. Dapsone in Behçet’s Disease: A Double-Blind, Placebo-Controlled, Cross-Over Study. J Dermatol. 2002;29(5):267–279. doi:10.1111/j.1346-8138.2002.tb00263.x
105. Sharquie KE, Najim RA, Al-Dori WS, Al-Hayani RK. Oral zinc sulfate in the treatment of Behcet’s disease: A double blind cross-over study. J Dermatol. 2006;33(8):541–546. doi:10.1111/j.1346-8138.2006.00128.x
106. Matsuda T, Ohno S, Hirohata S, et al. Efficacy of Rebamipide as Adjunctive Therapy in the Treatment of Recurrent Oral Aphthous Ulcers in Patients with Beh??et??s Disease. Drugs R D. 2003;4(1):19–28. doi:10.2165/00126839-200304010-00002
107. Sharquie KE, Helmi RM, Noiami AA, Al-Hayani RK, Kadhom MA. The therapeutic role of isotretinoin in the management of Behcet’s disease: a single-blinded, controlled therapeutic study. J Drugs Dermatol. 2013;12:68–73.
108. Lehner T, Wilton JMA. The therapeutic and immunological effects of levamisole in recurrent oral ulcers and Behçet’s syndrome. In: Lehner T, Cb B, editors. Behçet’s Syndrome: Clinical and Immunological Features. New York: Academic Press; 1979:291–305.
109. Kose O, Simsek İ, Pay S. Mycophenolate sodium in the treatment of mucocutaneous Behcet’s diseases. Int J Dermatol. 2011;50(7):895–896. doi:10.1111/j.1365-4632.2010.04505.x
110. Cantarini L, Vitale A, Scalini P, et al. Anakinra treatment in drug-resistant Behcet’s disease: a case series. Clin Rheumatol. 2015;34(7):1293–1301. doi:10.1007/s10067-013-2443-8
111. Emmi G, Silvestri E, Cameli AM, et al. Anakinra for resistant Behçet uveitis: why not? Clin Exp Rheumatol. 2013;31(3 Suppl 77):152–153.
112. Caso F, Rigante D, Vitale A, Lucherini OM, Cantarini L. Efficacy of anakinra in refractory Behçet’s disease sacroiliitis. Clin Exp Rheumatol. 2014;32:S171.
113. Ugurlu S, Ucar D, Seyahi E, Hatemi G, Yurdakul S. Canakinumab in a patient with juvenile Behçet’s syndrome with refractory eye disease: figure 1. Ann Rheum Dis. 2012;71(9):1589–1591. doi:10.1136/annrheumdis-2012-201383
114. Vitale A, Rigante D, Caso F, et al. Inhibition of Interleukin-1 by Canakinumab as a Successful Mono-Drug Strategy for the Treatment of Refractory Behçet’s Disease: A Case Series. Dermatology. 2014;228(3):211–214. doi:10.1159/000358125
115. Cantarini L, Vitale A, Borri M, Galeazzi M, Franceschini R. Successful use of canakinumab in a patient with resistant Behçet’s disease. Clin Exp Rheumatol. 2012;30:S115.
116. Emmi G, Talarico R, Lopalco G, et al. Efficacy and safety profile of anti-interleukin-1 treatment in Behçet’s disease: a multicenter retrospective study. Clin Rheumatol. 2016;35(5):1281–1286. doi:10.1007/s10067-015-3004-0
117. Mirouse A, Barete S, Monfort J-B, et al. Ustekinumab for Behçet’s disease. J Autoimmun. 2017;82:41–46. doi:10.1016/j.jaut.2017.05.002
118. Mirouse A, Barete S, Desbois A-C, et al. Long-Term Outcome of Ustekinumab Therapy for Behçet’s Disease. Arthritis Rheumatol. 2019;71(10):1727–1732. doi:10.1002/art.40912
119. Di Scala G, Bettiol A, Cojan RD, Finocchi M, Silvestri E, Emmi G. Efficacy of the anti-IL 17 secukinumab in refractory Behçet’s syndrome: A preliminary study. J Autoimmun. 2019;97:108–113. doi:10.1016/j.jaut.2018.09.002
120. Dincses E, Yurttas B, Esatoglu SN, Melikoglu M, Hamuryudan V, Seyahi E. Secukinumab induced Behçet’s syndrome: a report of two cases. Oxford Medical Case Reports. 2019;2019(5):omz041. doi:10.1093/omcr/omz041
121. Hatemi G, Christensen R, Bang D, et al. update of the EULAR recommendations for the management of Behçet’s syndrome. Ann Rheum Dis. 2018;2018(77):808–818.
122. Kötter I, Vonthein R, Zierhut M, et al. Differential efficacy of human recombinant interferon-α2a on ocular and extraocular manifestations of behçet disease: results of an open 4-center trial. Semin Arthritis Rheum. 2004;33(5):311–319. doi:10.1016/j.semarthrit.2003.09.005
123. Hamuryudan V, Moral F, Yurdakul S, et al. Systemic interferon alpha 2b treatment in Behçet’s syndrome.. J Rheumatol. 1994;21(6):1098–1100.
124. Calguneri M. Effects of interferon alpha treatment on the clinical course of refractory Behcet’s disease: an open study. Ann Rheum Dis. 2003;62(5):492–493. doi:10.1136/ard.62.5.492
125. Moral F, Hamuryudan V, Yurdakul S, Yazici H. Inefficacy of azapropazone in the acute arthritis of Behçet’s syndrome: a randomized, double blind, placebo controlled study.. Clin Exp Rheumatol. 1995;13(4):493–495.
126. Fagni F, Bettiol A, Talarico R, et al. Long-term effectiveness and safety of secukinumab for treatment of refractory mucosal and articular Behçet’s phenotype: a multicentre study. Ann Rheum Dis. 2020;79(8):1098–1104. doi:10.1136/annrheumdis-2020-217108
127. Tugal-Tutkun I, Onal S, Altan-Yaycioglu R, Huseyin Altunbas H, Urgancioglu M. Uveitis in Behçet disease: an analysis of 880 patients. Am J Ophthalmol. 2004;138(3):373–380. doi:10.1016/j.ajo.2004.03.022
128. Ohno S, Namba K, Takemoto Y. Behcet’ s disease. In: Zierhut M, Pavesio C, Ohno S, Orefice F, Na R, editors. Intraocular Inflammation.,Berlin, Heidelberg, New York, Dordrecht, London: Springer-Verlag; 2016:785–795.
129. Mizuki N, Takeuchi M. Treatment Algorithm of Ocular Behcet’s Disease. Clinical Practice Guidelines for Behcet’s Disease 2020. Tokyo: Shindanto Chiryosha; 2020:56–57.
130. Masuda K, Urayama A, Kogure M, Nakajima A, Nakae K, Inaba G. DOUBLE-MASKED TRIAL OF CYCLOSPORIN VERSUS COLCHICINE AND LONG-TERM OPEN STUDY OF CYCLOSPORIN IN BEHÇET’S DISEASE. Lancet. 1989;333(8647):1093–1096. doi:10.1016/S0140-6736(89)92381-7
131. BenEzra D, Cohen E, Chajek T, et al. Evaluation of conventional therapy versus cyclosporine A in Behçet’s syndrome.. Transplant Proc. 1988;20(3 Suppl 4):136–143.
132. Jaffe GJ, Dick AD, Brézin AP, et al. Adalimumab in patients with active noninfectious uveitis. New England Journal of Medicine. 2016;375(10):932–943. doi:10.1056/NEJMoa1509852
133. Silvestri E, Bitossi A, Bettiol A, et al. Adalimumab effectively controls both anterior and posterior noninfectious uveitis associated with systemic inflammatory diseases: focus on Behçet’s syndrome. Inflammopharmacology. 2020;28(3):711–718. doi:10.1007/s10787-020-00697-4
134. Martín-Varillas JL, Atienza-Mateo B, Calvo-Rio V, et al. Biological Therapy in refractory Uveitis due to Behçet’s Disease Collaborative Group. Long Term Follow-up and Optimization of Infliximab in Refractory Uveitis Due to Behçet’s Disease. National Study of 103 Caucasian Patients. J Rheumatol. 2020;1:200300. doi:10.3899/jrheum.200300
135. Atienza‐Mateo B, Martín‐Varillas JL, Calvo‐Río V, et al. Comparative Study of Infliximab Versus Adalimumab in Refractory Uveitis due to Behçet’s Disease: national Multicenter Study of 177 Cases. Arthritis Rheumatol. 2019;71(12):2081–2089. doi:10.1002/art.41026
136. Sobaci G, Erdem Ü, Durukan AH, et al. Safety and Effectiveness of Interferon Alpha-2a in Treatment of Patients with Behçet’s Uveitis Refractory to Conventional Treatments. Ophthalmology. 2010;117(7):1430–1435. doi:10.1016/j.ophtha.2009.11.022
137. Lightman S, Taylor SR, Bunce C, et al. Pegylated interferon-α-2b reduces corticosteroid requirement in patients with Behçet’s disease with upregulation of circulating regulatory T cells and reduction of Th17. Ann Rheum Dis. 2015;74(6):1138–1144. doi:10.1136/annrheumdis-2014-205571
138. Tugal-Tutkun I, Pavesio C, De Cordoue A, Bernard-Poenaru O, Gül A. Use of gevokizumab in patients with Behçet’s disease uveitis: an international, randomized, double-masked, placebo-controlled study and open-label extension study. Ocul Immunol Inflamm. 2018;25:1–11.
139. Dick AD, Tugal-Tutkun I, Foster S, et al. Secukinumab in the treatment of noninfectious uveitis: results of three randomized, controlled clinical trials. Ophthalmology. 2013;120(4):777–787. doi:10.1016/j.ophtha.2012.09.040
140. Deroux A, Chiquet C, Bouillet L. Tocilizumab in severe and refractory Behcet’s disease: four cases and literature review. Semin Arthritis Rheum. 2016;45(6):733–737. doi:10.1016/j.semarthrit.2015.11.012
141. Atienza-Mateo B, Calvo-Río V, Beltrán E, et al. Anti-interleukin 6 receptor tocilizumab in refractory uveitis associated with Behçet’s disease: multicentre retrospective study. Rheumatology. 2018;57(5):856–864. doi:10.1093/rheumatology/kex480
142. Emmi G, Becatti M, Bettiol A, Hatemi G, Prisco D, Fiorillo C. Behçet’s Syndrome as a Model of Thrombo-Inflammation: the Role of Neutrophils. Front Immunol. 2019;1.
143. Emmi G, Mannucci A, Argento FR, et al. Stem-Cell-Derived Circulating Progenitors Dysfunction in Behçet’s Syndrome Patients Correlates With Oxidative Stress. Front Immunol. 2019;10:2877. doi:10.3389/fimmu.2019.02877
144. Ahn JK, Lee YS, Jeon CH, Koh E-M, Cha H-S. Treatment of venous thrombosis associated with Behcet’s disease: immunosuppressive therapy alone versus immunosuppressive therapy plus anticoagulation. Clin Rheumatol. 2008;27(2):201–205. doi:10.1007/s10067-007-0685-z
145. Desbois AC, Wechsler B, Resche-Rigon M, et al. Immunosuppressants reduce venous thrombosis relapse in Behçet’s disease. Arthritis Rheum. 2012;64(8):2753–2760. doi:10.1002/art.34450
146. Alibaz-Oner F, Karadeniz A, Ylmaz S, et al. Behçet disease with vascular involvement: effects of different therapeutic regimens on the incidence of new relapses. Medicine. 2015;94(6):e494. doi:10.1097/MD.0000000000000494
147. Seyahi E, Cakmak OS, Tutar B, et al. Clinical and ultrasonographic evaluation of lower-extremity vein thrombosis in Behcet syndrome: an observational study. Medicine. 2015;94(44):e1899. doi:10.1097/MD.0000000000001899
148. Tascilar K, Melikoglu M, Ugurlu S, et al. Vascular involvement in Behçet’s syndrome: a retrospective analysis of associations and the time course. Rheumatology. 2014;53(11):2018–2022. doi:10.1093/rheumatology/keu233
149. Hamuryudan V, Er T, Seyahi E, et al. Pulmonary artery aneurysms in Behçet syndrome. Am J Med. 2004;117(11):867–870. doi:10.1016/j.amjmed.2004.05.027
150. Saba S, Saricaoğlu S, Bayram B, et al. Arterielle Läsionen beim Morbus Behçet. Vasa. 2003;32(2):75–81. doi:10.1024/0301-1526.32.2.75
151. Hamuryudan V, Seyahi E, Ugurlu S, et al. Pulmonary artery involvement in Behçet׳s syndrome: effects of anti-Tnf treatment. Semin Arthritis Rheum. 2015;45(3):369–373. doi:10.1016/j.semarthrit.2015.06.008
152. Hamuryudan V, Yurdakul S, Moral F, et al. PULMONARY ARTERIAL ANEURYSMS IN BEHÇET’S SYNDROME: A REPORT OF 24 CASES. Br J Rheumatol. 1994;33(1):48–51. doi:10.1093/rheumatology/33.1.48
153. Seyahi E, Melikoglu M, Akman C, et al. Pulmonary Artery Involvement and Associated Lung Disease in Behçet Disease. Medicine. 2012;91(1):35–48. doi:10.1097/MD.0b013e318242ff37
154. Le Thi HD, Wechsler B, Papo T, et al. Arterial lesions in Behçet’s disease. A study in 25 patients.. J Rheumatol. 1995;22(11):2103–2113.
155. Saadoun D, Asli B, Wechsler B, et al. Long-term outcome of arterial lesions in Behçet disease: a series of 101 patients. Medicine. 2012;91(1):18–24. doi:10.1097/MD.0b013e3182428126
156. Park M-C, Hong B-K, Kwon HM, Hong Y-S. Surgical outcomes and risk factors for postoperative complications in patients with Behcet’s disease. Clin Rheumatol. 2007;26(9):1475–1480. doi:10.1007/s10067-006-0530-9
157. Ding Y, Li C, Liu J, et al. Tocilizumab in the treatment of severe and/or refractory vasculo-Behçet’s disease: a single-centre experience in China.. Rheumatology. 2018;57(11):2057–2059. doi:10.1093/rheumatology/key230
158. Pipitone N, Olivieri I, Padula A, et al. Infliximab for the treatment of Neuro-Behçet’s disease: A case series and review of the literature. Arthritis Rheum. 2008;59(2):285–290. doi:10.1002/art.23345
159. Borhani Haghighi A, Safari A, Nazarinia MA, Habibagahi Z, Shenavandeh S. Infliximab for patients with neuro-Behcet’s disease: case series and literature review. Clin Rheumatol. 2011;30(7):1007–1012. doi:10.1007/s10067-011-1726-1
160. Zeydan B, Uygunoglu U, Saip S, et al. Infliximab is a plausible alternative for neurologic complications of Behçet disease. Neurol Neuroimmunol Neuroinflamm. 2016;3(5):e258. doi:10.1212/NXI.0000000000000258
161. Asa A-A, Saip S. Treatment of NeuroBehcet’s disease with infliximab: an international multi-centre case-series of 18 patients. Clin Exp Rheumatol. 2010;28(Suppl 60):S119.
162. Noel N, Bernard R, Wechsler B, Resche-Rigon M, Depaz R. Long-Term Outcome of Neuro-Behçet’s Disease. Arth Rheumatol. 2014;66(5):1306–1314. doi:10.1002/art.38351
163. Addimanda O, Pipitone N, Pazzola G, Salvarani C. Tocilizumab for severe refractory neuro-Behçet: three cases IL-6 blockade in neuro-Behçet. Semin Arthritis Rheum. 2015;44(4):472–475. doi:10.1016/j.semarthrit.2014.08.004
164. Monastirli A, Chroni E, Georgiou S, et al. Interferon- treatment for acute myelitis and intestinal involvement in severe Behcet’s disease. QJM. 2010;103(10):90. doi:10.1093/qjmed/hcq125
165. Nichols JC, Ince A, Akduman L, Mann ES. Interferon-alpha 2a treatment of neuro-Behcet disease. J Neuroophthalmol. 2001;21(2):109–111. doi:10.1097/00041327-200106000-00011
166. Hirohata S, Kikuchi H, Sawada T, et al. Retrospective analysis of long-term outcome of chronic progressive neurological manifestations in Behcet’s disease. J Neurol Sci. 2015;349(1–2):143–148. doi:10.1016/j.jns.2015.01.005
167. Shugaiv E, Tuzun E, Mutlu M, Kiyat-Atamer A, Kurtuncu M, Akman-Demir G. Mycophenolate mofetil as a novel immunosuppressant in the treatment of neuro-Behçet’s disease with parenchymal involvement: presentation of four cases.. Clin Exp Rheumatol. 2011;29(4 Suppl 67):S64–S67.
168. Kato Y, Numaga J, Kato S, Kaburaki T, Kawashima H, Fujino Y. Central nervous system symptoms in a population of Behçet’s disease patients with refractory uveitis treated with cyclosporine A. Clin Exp Ophthalmol. 2001;29:335–336.
169. Kotake S, Higashi K, Yoshikawa K, Sasamoto Y, Okamoto T, Matsuda H. Central nervous system symptoms in patients with Behçet disease receiving cyclosporine therapy. Ophthalmology. 1999;106(3):586–589. doi:10.1016/S0161-6420(99)90120-3
170. Kötter I, Günaydin I, Batra M, et al. CNS involvement occurs more frequently in patients with Behçet’s disease under cyclosporin A (CSA) than under other medications—results of a retrospective analysis of 117 cases. Clin Rheumatol. 2006;25(4):482–486. doi:10.1007/s10067-005-0070-8
171. Akman-Demir G, Ayranci O, Kurtuncu M, Vanli EN, Mutlu M, Tugal-Tutkun I. Cyclosporine for Behçet’s uveitis: is it associated with an increased risk of neurological involvement? Clin Exp Rheumatol. 2008;26(4 Suppl 50):S84–90.
172. Saadoun D, Wechsler B, Resche-Rigon M, et al. Cerebral venous thrombosis in Behçet’s disease. Arthritis Rheum. 2009;61(4):518–526. doi:10.1002/art.24393
173. Ferro JM, Canhao P, Stam J, Bousser M-G. Prognosis of cerebral vein and dural sinus thrombosis: results of the International Study on Cerebral Vein and Dural Sinus Thrombosis (ISCVT). Stroke. 2004;35(3):664–670. doi:10.1161/01.STR.0000117571.76197.26
174. Watanabe K, Tanida S, Inoue N, et al. Evidence-based diagnosis and clinical practice guidelines for intestinal Behçet’s disease 2020 edited by Intractable Diseases, the Health and Labour Sciences Research Grants. J Gastroenterol. 2020;55(7):679–700. doi:10.1007/s00535-020-01690-y
175. Hisamatsu T, Ueno F, Matsumoto T, et al. The 2nd edition of consensus statements for the diagnosis and management of intestinal Behçet’s disease: indication of anti-TNFα monoclonal antibodies. J Gastroenterol. 2014;49(1):156–162. doi:10.1007/s00535-013-0872-4
176. Hisamatsu T, Hayashida M. Treatment and outcomes: medical and surgical treatment for intestinal Behçet’s disease. Intest Res. 2017;15(3):318–327. doi:10.5217/ir.2017.15.3.318
177. Park YE, Cheon JH. Updated treatment strategies for intestinal Behçet’s disease. Korean J Intern Med. 2018;33:1–19.
178. Iwata S, Saito K, Yamaoka K, et al. Efficacy of combination therapy of anti-TNF-α antibody infliximab and methotrexate in refractory entero-Behçet’s disease. Mod Rheumatol. 2011;21:184–191.
179. Ideguchi H, Suda A, Takeno M, et al. Gastrointestinal manifestations of Behcet’s disease in Japan: a study of 43 patients. Rheumatol Int. 2014;34:851–856.
180. Hibi T, Hirohata S, Kikuchi H, et al. Infliximab therapy for intestinal, neurological, and vascular involvement in Behcet disease: efficacy, safety, and pharmacokinetics in a multicenter, prospective, open-label, single-arm phase 3 study. Medicine. 2016;95:3863.
181. Zou J, Ji DN, Shen Y, Guan JL, Zheng SB. Mucosal healing at 14 weeks predicts better outcome in low-dose infliximab treatment for Chinese patients with active intestinal Behcet’s disease. Ann Clin Lab Sci. 2017;47:171–177.
182. Tanida S, Inoue N, Kobayashi K, et al. Adalimumab for the treatment of Japanese patients with intestinal Behcet’s disease. Clin Gastroenterol Hepatol. 2015;13:940.
183. Inoue N, Kobayashi K, Naganuma M, et al. Long-term safety and efficacy of adalimumab for intestinal Behcet’s disease in the open label study following a phase 3 clinical trial. Intest Res. 2017;15:395–401.
184. Vitale A, Emmi G, Lopalco G, et al. Adalimumab effectiveness in Behcet’s disease: short and longterm data from a multicenter retrospective observational study. Clin Rheumatol. 2017;36:451–455.
185. Ma D, Zhang CJ, Wang RP, Wang L, Yang H. Etanercept in the treatment of intestinal Behcet’s disease. Cell Biochem Biophys. 2014;69:735–739.
186. Vitale A, Emmi G, Lopalco G, et al. Long-term efficacy and safety of golimumab in the treatment of multirefractory Behcet’s disease. Clin Rheumatol. 2017;36:2063–2069.
187. Lopalco G, Emmi G, Gentileschi S, et al. Certolizumab pegol treatment in Behcet’s disease with different organ involvement: a multicenter retrospective observational study. Mod Rheumatol. 2017;27:1031–1035.
188. Sayarlioglu M, Kotan MC, Topcu N, Bayram I, Arslanturk H, Gul A. Treatment of recurrent perforating intestinal ulcers with thalidomide in Behçet’s disease. Ann Pharmacother. 2004;38:808–811.
189. Yasui K, Uchida N, Akazawa Y, et al. Thalidomide for treatment of intestinal involvement of juvenile-onset Behçet disease. Inflamm Bowel Dis. 2008;14:396–400.
190. Hatemi I, Hatemi G, Pamuk ON, Erzin Y, Celik AF. TNF-alpha antagonists and thalidomide for the management of gastrointestinal Behçet’s syndrome refractory to the conventional treatment modalities: a case series and review of the literature. Clin Exp Rheumatol. 2015;33(6 Suppl 94):129–137.
191. Park Y, Cheon JH. Update on the Treatment of Behcet’s Disease of the Small Bowel with Biologic Agents. Curr Gastroenterol Rep. 2020;19(22):24.
192. Demirelli S, Degirmenci H, Inci S, Arisoy A. Cardiac manifestations in Behcet;s disease. Intractable Rare Dis Res. 2015;4:70–75.
193. Ozatli D, Kav T, Haznedaroglu IC, et al. Cardiac and great vessel thrombosis in Behcet's disease. Intern Med. 2001;40:68–72.
194. Ozeren M, Dogan OV, Dogan S, Yucel E. True and pseudo aneurysms of coronary arteries in a patient with Behçet’s disease. Eur J Cardiothorac Surg. 2004;25:465–467.
195. Kaatz M, Görnig M, Bocker T, Zouboulis CC, Wollina U. Late manifestation of a fatal Behcet's disease with cardiac involvement and lethal outcome. Dtsch Med Wochenschr. 1998;123:217–222.
196. Kwon CM, Lee SH, Kim JH, et al. A case of Behcet's disease with pericarditis, thrombotic thrombocytopenic purpura, deep vein thrombosis and coronary artery pseudo aneurysm. Korean J Intern Med. 2006;21:50–56.
197. Al-Na’mah ZM, Carson R, Thanoon IA. Dexamucobase: a novel treatment for oral aphthous ulceration. Quintessence Int. 2009;40:399–404.
198. Ghate J, Jorizzo J. Behcet’s disease and complex aphthosis. J Am Acad Dermatol. 1999;40:1–18.
199. Binnie WH, Curro FA, Khandwala A, Van Inwegan RG. Amlexanox oral paste: a novel treatment that accelerates the healing of aphthous ulcers. Compend Contin Educ Dent. 1997;18:1116–1124.
200. Greer RO, Lindenmuth JE, Juarez T, Khandwala A. A double blind study of topically applied 5% amlexanox in the treatment of aphthous ulcers. J Oral Maxillofac Surg. 1993;51:243–248.
201. Arabaci T, Kara C, Ciçek Y. Relationship between periodontal parameters and Behçet’s disease and evaluation of different treatments for oral recurrent aphthous stomatitis. J Periodontal Res. 2009;44:718–725.
202. Gorsky M, Epstein J, Rabenstein S, Elishoov H, Yarom N. Topical minocycline and tetracycline rinses in treatment of recurrent aphthous stomatitis: a randomized cross-over study. Dermatol Online J. 2007;13:1.
203. Wechsler B, Bodaghi B, Thi Huong DL, et al. Efficacy of interferon alfa-2a in severe and refractory uveitis associated with Behçet’s disease. Ocul Immunol Inflamm. 2000;8:293–301.
204. Fabiani C, Vitale A, Emmi G, et al. Interleukin (IL)-1 inhibition with anakinra and canakinumab in Behçet’s disease-related uveitis: a multicenter retrospective observational study. Clin Rheumatol. 2017;36:191–197.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.