Back to Journals » Journal of Experimental Pharmacology » Volume 13
Experimental Pharmacological Management of Psoriasis
Authors Campione E, Cosio T ![]() , Di Prete M
, Di Prete M ![]() , Lanna C
, Lanna C ![]() , Dattola A, Bianchi L
, Dattola A, Bianchi L
Received 19 April 2021
Accepted for publication 30 June 2021
Published 26 July 2021 Volume 2021:13 Pages 725—737
DOI https://doi.org/10.2147/JEP.S265632
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Paola Rogliani
Elena Campione,1 Terenzio Cosio,1 Monia Di Prete,2 Caterina Lanna,1 Annunziata Dattola,1 Luca Bianchi1
1Dermatologic Unit, Department of Systems Medicine, University of Rome Tor Vergata, Rome, 00133, Italy; 2Anatomic Pathology, University of Rome Tor Vergata, Rome, 00133, Italy
Correspondence: Elena Campione
Dermatologic Unit, Department of Systems Medicine, University of Rome Tor Vergata, Via Montpellier,1, Rome, 00133, Italy
Tel +39.06.20900252
Email [email protected]
Abstract: Psoriasis is a chronic, relapsing, immune-mediated systemic disease. Its pathogenesis is complex and not fully understood yet. Genetic and epigenetic factors interact with molecular pathways involving TNF-α, IL-23/IL-17 axis, and peculiar cytokines, as IL-36 or phosphodiesterase 4. This review discusses the mechanisms involved in the development of the disease, as well as the therapeutic options proposed following the investigation of the inflammatory psoriatic pathways. We performed a comprehensive search using the words “psoriasis” and the newest molecules currently under investigation and approval. From these data, a new scenario in psoriasis is occurring to personalize the therapies - especially systemic ones and those using small molecules – and avoid topical and injectable drugs. We reported the newest therapeutic opportunities, including the inhibitors of Janus kinase/tyrosine kinase 2, phosphodiesterase-4 and IL-36 receptor. Today, more than 20 molecules are under investigation for the treatment of cutaneous psoriasis. Most of them are constituted by small molecules or biologic therapies. This underlines how psoriasis needs systemic therapies, due to its complex pathogenesis and multisystemic involvement.
Keywords: psoriasis, janus kinase inhibitors, tyrosine kinase 2 inhibitors, phosphodiesterase 4 inhibitors, IL-36 receptors inhibitors
Introduction
Psoriasis is a chronic, relapsing, immune-mediated disease, with a prevalence of 2–3% worldwide. It is characterized by well-circumscribed erythematous plaques, covered by a squamous scale, generally located on the skin of extensor surfaces of the body. 20% of patients with psoriasis may develop a seronegative polyarthropathy - often associated with nail involvement - with a severe quality of life impairment.1 There are several clinical variants of psoriasis. Besides the psoriasis vulgaris, also called plaque-type psoriasis, we classify guttate, erythrodermic, sebopsoriasis, generalized pustular (GPP) and palm-plantar forms, and, rarely, psoriasis of the mucous membranes.1 Actually, these clinical variants show different cytokines involved in their pathogenesis and different response to therapies. As psoriasis is a dynamic process, microscopic features vary along the evolution of cutaneous lesions. The earliest lesions could be misinterpreted as they consist in superficial dermis capillary vessels dilatation and mild perivascular lymphocytic inflammatory infiltration.2 It is not possible to predict the duration of this phase, but it is inevitably followed by the development of the classic psoriatic plaque, which corresponds histologically to confluent parakeratosis with neutrophilic exocytosis in the epidermis. These changes are accompanied by attenuation to complete loss of granular cell layer, expression of the accelerated keratinocytes migration process, from the basal to the corneum layer, without complete maturation.2 Moreover, basal cell mitotic activity is moderately increased with a consequent characteristic form of epidermal hyperplasia – known as “psoriasiform hyperplasia”. All these changes are associated with dilated and tortuous capillary vessels in the papillary dermis with neutrophils content in their lumen and moderate-to-severe lymphocytic perivascular inflammation. With the progression of the disease, lots of neutrophils migrate in the epidermis reaching the more superficial layers and forming intra-corneal collections, known as Munro’s microabscesses2 (Figure 1). The pathogenesis of psoriasis is complex and not fully understood yet. Many genes and environmental factors, such as stress, injuries, infections, and certain medications, may be responsible for its onset. Understanding the mechanisms behind psoriasis is crucial for the development of new therapies and address tailored approaches for the patients’ conditions.3 Immune system impairment is considered a key trait. The IL-23/Th17/IL-17 axis seems to have a key role in the onset of the inflammation in psoriasis. Although there is currently no definitive cure, various treatment strategies allow prolonged control of the disease. The recently approved new kinase inhibitors act on IL-23/Th17/IL-17 axis in a more selective way, resulting in a reduction in adverse events (AEs) and a chance of therapy for patients with difficult-to-treat psoriasis.4 Alongside the common disease modifying anti-rheumatic drugs, several classes of biologicals and small molecules are now used in the systemic treatment of psoriasis.5,6 Phosphodiesterase (PDE) 4 inhibitors are largely known to improve both skin and joint disease, modulating, at the same time, metabolic biomarkers in diabetic psoriatic patients, with the chance to be employed in psoriatic patients with cardio-metabolic comorbidities.7,8 Moreover, the small molecules showed strong efficacy also in nail psoriasis, which is known as a difficult-to-treat form of the disease. The pathogenetic mechanisms in this form are not completely overlapping those of cutaneous psoriasis. For this reason, PDE4 inhibitors, restoring the nail unit IL-10 levels and the immune privilege site, are particularly effective.9 Advancement in molecular knowledge leads to understand psoriasis better, influencing the development of efficient treatment modalities. However, even with the availability of various therapeutic options, most of the efficient treatment modalities are expensive and are not effective in all subjects. Due to the complexity of psoriasis pathogenesis and the paramount influence of epigenetic factors, new drugs need to be introduced. This review discusses the newest therapeutic strategies in the management of psoriasis, starting from its pathogenesis, highlighting their mechanism of actions - intracellular or on inflammatory factors - and current trials. In this view, also tyrosine kinase (TYK) 2 inhibitors – currently in their approval phase - have been presented.10
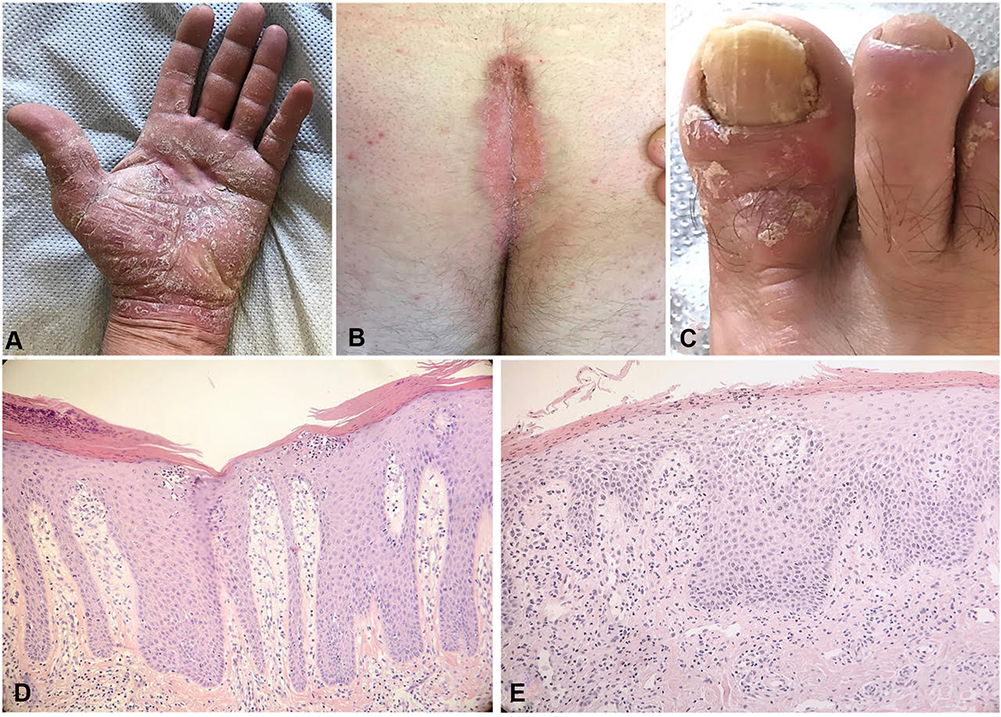 |
Figure 1 (A) Clinical manifestation of palmar psoriasis. (B) Clinical manifestation of sacral psoriasis. (C) Clinical manifestation of nail psoriasis. All the three clinical images report the difficult-to-treat areas, recognized as unmet needs by patients and physician. (D) Histopathological features of plaque psoriasis. The classic epidermal psoriasiform hyperplasia is associated with hyperparakeratosis and loss of granular layer. Collections of neutrophils are observed in the upper stratum spinosum (spongiosiform pustules of Kogoj). In the dermis, thin capillary vessels reach almost the top of the dermal papillae and are surrounded by a chronic lymphocytic infiltrate with sparse neutrophils (Haematoxylin-eosin. Original magnification: 100x). (E) Histopathological features of genital psoriasis. The classic features observed in plaque psoriasis are attenuated in the genital area. The epidermal hyperplasia is associated with less extensive hyperparakeratosis, which is accompanied by mounds of neutrophils in the stratum corneum (Munro’s microabscesses). In the chorion, thin capillary vessels are surrounded by a chronic lymphocytic infiltrate, with sparse neutrophils (Haematoxylin-eosin. Original magnification: 200x). |
Materials and Methods
Search Strategy
The authors performed a comprehensive search using the words “psoriasis” and the newest molecules under investigations or just approved by EMA or FDA, as “piclidenoson/CF101”, “namilumab/AMG203”, “imsidolimab/ANB019”, “spesolimab/BI655130”, “orismilast/LEO 32731”, “hemay005”, “GSK2981278”, “vimirogant/AGN 242428/VTP 43742”, “AUR101”, “ABBV-157”, “SCD-044”, “ponesimod/ACT-128800”, “ND-2158”, “ND-2110”, “belumosudil/KD025/SLx-2119”, “tapinarof/benvitimod/WB-1001/GSK-2894512/DMVT-505”, “deucravacitinib/BMS-986165”. The following databases were evaluated: Cochrane Central Register of Controlled Trials; MEDLINE; Embase; US National Institutes of Health Ongoing Trials Register; NIHR Clinical Research Network Portfolio Database; and the World Health Organization International Clinical Trials Registry Platform. Reference lists and published systematic review articles, starting from year 2018, were studied.
Inclusion Criteria
Only articles written in English were included in the search, which was restricted to human studies, with no restrictions on age, sex, ethnicity, or type of study. For Phase I molecules, preclinical results have been considered in the Tables S1-S20, and just the name and the mechanism of actions has been reported. Case reports and case series were included if they described the role of drugs in psoriasis, not present in reviews or trials.
Exclusion Criteria
The target intervention excluded pathologies not of dermatologic interest. Phase I trials have not been included in tables and discussion. Analyses excluded articles not written in English.
Results
We identified 142 manuscripts regarding new therapeutic options against psoriasis. Twenty-six were excluded following the exclusion criteria, while 43 were excluded after evaluating the trials. A total of 72 studies have been included in this comprehensive review, counting clinical trials, published articles, and congress posters (Figure 1S).
Janus Kinases Inhibitors
In order to obtain a more personalized approach according to the clinical and immunological characteristics of each patient, inhibitors of the Janus kinases (JAK) 1, 2,and 3 and TYK2 have emerged as a promising strategy in psoriasis in recent years. The JAK-signal transducer and activator of transcription (JAK-STAT) pathway plays a crucial role in the communication between cytokine signals outside the cell and the cell nucleus, with activation of specific genes transcription in response.11 Cytokines binding to cell membrane receptors cause their dimerization. The JAKs associated with these receptors phosphorylate each other, increasing their kinase domains activity. The consequence is the creation of a SH2 domain-binding site on the receptors, which bind STATs.11 At this point, STATs are phosphorylated and activated by JAKs, dissociate from the receptors and dimerize with other activated STATs. STATs dimers are then translocated to the nucleus starting the transcription.12 JAKs inhibitors (JAKsI) are currently being tested in clinical trials for the treatment of psoriasis,13 particularly those included in the IL-23 mechanism of action. Indeed, IL-23 binding to IL-23 receptor causes phosphorylation and activation of JAK-STAT signaling molecules – JAK2, TYK2, and STAT1, 3, 4, and 5,14 which directly bind to IL-17 and IL-17F genes promoters.15 STAT3 also up-regulates the expression of retinoic acid-related orphan receptor (ROR)-γ, a Th17 specific transcriptional regulator that is crucial for IL-17 and IL-17F expression.16 Among JAKsI, deucravacitinib (BMS-986165) has the unique ability to selectively bind the pseudo-kinase domain of TYK2, inhibiting its functions, involving IL-12, IL-23, and type I and III interferons (IFN), through an allosteric mechanism17 (Table 1; Figure 2; Table S1). Selective TYK2 inhibitors are of great interest for increasing the target specificity and reducing AEs observed with other types of JAKsI.10,18 Pembrocitinib (PF-06700841) is an oral, dual selective TYK2/JAK1 inhibitor, targeting multiple cytokines involved in the signaling (IFN, IL-6, IL-12, IL-21, IL-22, and IL-23), tested to treat patients with moderate-to-severe plaque psoriasis. From oral formulation, a topical cream 0.1% has been produced and tested in a Phase II trial (Table 1; Figure 2; Table S2). Another JAKsI, delgocitinib, has shown in in vitro studies to inhibit JAK1, 2, and 3 and TYK2 in an adenosine triphosphate (ATP)-competitive manner, thus blocking IL-2, IL-6, IL-23, granulocyte-macrophage colony-stimulating factor (GM-CSF) and IFN-α signaling.19 Moreover, delgocitinib inhibits inflammatory cells activation, such as T cells, B cells, monocytes and mast cells, and Th1-, Th2- and Th17-type cytokines production and secretion, by both T and non-T cells.20 It improves skin barrier function and enhances keratinocyte differentiation, as indicated by increased levels of profilaggrin, filaggrin and loricrin.21 On the other hand, as delgocitinib is the agent with a less specific target on the JAK-STAT pathway, more AEs are expected (Table 1; Figure 2).
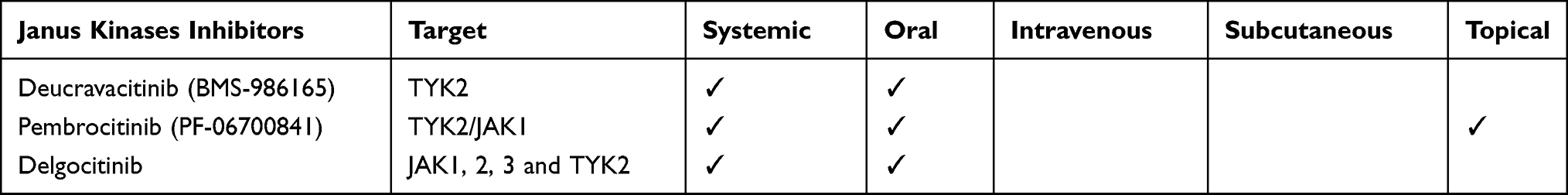 |
Table 1 The Table Shows the Molecules Belonging to the Class of Jak Kinase Inhibitor Under Investigation for Psoriasis |
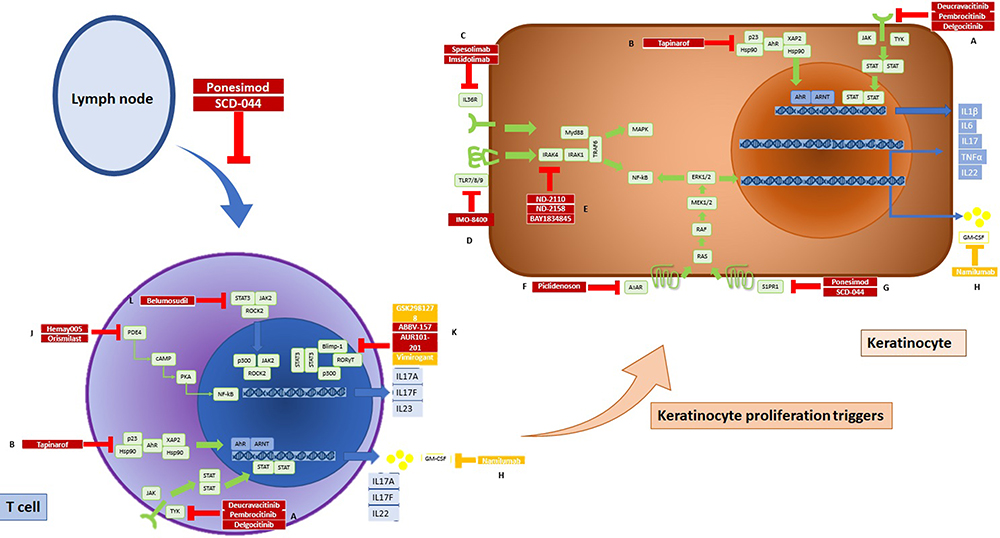 |
Figure 2 Summary of drugs under investigation or with preliminary efficacy in psoriasis disease (red), and drugs with no data reporting efficacy (orange). A) The JAK inhibitor blocks the JAK-STAT pathway signaling in Th17 cells. Specifically, deucravacitinib inhibits TYK2. B) Tapinarof, an AhR agonist, reduces IL-17 and IL-22 in both Th17 cells and keratinocytes. C) Spesolimab and imsidolimab inhibit IL-36R. D) Imo-8400 inhibits TLR-7/8/9. E) BAY1834345, ND2110 and ND2158 inhibit IRAK4. F) Piclidenoson inhibits A3AR in keratinocytes. G) Ponesimod, a selective S1PR1 agonist, induces sequestration of lymphocytes into lymph nodes and decreases peripheral lymphocyte counts and tracking of lymphocytes to peripheral tissues. H) Namilumab has not shown efficacy in psoriasis. J) PDE4 inhibitors act on lymphocytes. K) Among RORγt, just ABVV-157 and AUR-101 have shown efficacy in psoriasis. L) KD025, a ROCK2 inhibitor, reduces IL-17 secretion in Th17 cells. Abbreviations: JAK, Janus kinase; STAT, signal transducer and activator of transcription; S1PR1, sphingosine-1-phosphate receptor 1; ROCK2, Rho-associated kinase 2; IL, interleukin; AhR, aryl hydrocarbon receptor; TLR, toll-like receptor; IRAK, interleukin 1 receptor-associated kinase. |
Aryl Hydrocarbon Receptor Agonist
Aryl hydrocarbon receptor (AhR) is a cytosolic ligand-activated receptor and transcription factor, which is commonly expressed in the skin cells and binds endogenous and exogenous molecules and dioxins.22,23 AhR activation induces oxidative stress through cytochromes P450 and neutralizes oxidative stress through the nuclear factor-erythroid 2-related factor-2 (NRF2) transcription factor.24 Moreover, AhR regulates the Th17/22 axis, which is involved in psoriasis.24 In addition, AhR in cutaneous vascular endothelial cells (VECs) seems to be involved in the arising of psoriasis. Zhu et al have discovered that AhR in cutaneous VECs down-regulates neutrophil recruitment through adhesion molecule ICAM-1 in psoriasis, using a AhR knockout mice.25 AhR agonist reduced IL-23 receptor, Th17 master transcription factor, RORC, and the number of Th17 cells.26 Tapinarof, also known as benvitimod (WB-1001, GSK-2894512, and DMVT-505), is a non-steroidal anti-inflammatory drug in study for the treatment of psoriasis.27,28 In imiquimod-treated mice, tapinarof demonstrated less inflammation, epidermal thickening, and reduced pro-inflammatory cytokines, such as IL-17, IL-19, IL-22, IL-23, and IL-1β. In addition, tapinarof has an antioxidant activity based on the inhibition of reactive oxygen species through the NRF2 pathway.27 In ex vivo human skin, tapinarof reduced Th17 cytokines such as IL-17A, IL-17F, and IL-2227 (Table 2; Figure 2; Table S3).
 |
Table 2 The Table Shows the Molecules Belonging to the Class of Aryl Hydrocarbon Receptor Agonist Under Investigation for Psoriasis |
Retinoic Acid-Related Orphan Receptor-γt Inverse Agonists
RORC2 is a crucial transcription factor for Th17 cell differentiation.29 Inhibiting RORC activity is considered a promising strategy for the treatment of psoriasis.29 Two receptor isoforms are produced from the same RORC gene:30 RORγ (also referred to as RORγ1;31 and RORγt (also known as RORγ2).32 RORγt is the most studied of the two isoforms. The transcription factor is essential for lymph nodes and Peyer’s patches organogenesis.33,34 It inhibits undifferentiated T cells apoptosis, supporting their Th17 differentiation, though the regulation of Fas ligand and IL-2.30 RORγt is a validated drug target for the treatment of cutaneous inflammatory disorders such as psoriasis. Among the RORγt inverse agonist, ABBV-157 is a small molecule currently employed in a clinical trial on the treatment of chronic plaque psoriasis, still in the recruitment phase (Table 3; Figure 2; Table S4). AUR101 is a potent oral RORγt inverse agonist, with high selectivity among other ROR isoforms, showing IL-17 modulation with an acceptable safety profile, which has recently completed a phase I human study. AUR-101 has demonstrated inhibition of IL-17A in whole blood from psoriasis patients and very significant reduction in histopathology scores in two separate pre-clinical psoriasis models. In addition, AUR-101 has been found to be safe in preclinical toxicology evaluations, at several fold of anticipated efficacious doses in humans (Table 3; Figure 2; Table S5). To data, other two RORγt inverse agonists have been studied, vimirogant (also known as AGN 242428 or VTP 43742) and GSK2981278, but no efficacy in cutaneous psoriasis has been demonstrated (Tables S6, S18).
 |
Table 3 The Table Shows the Molecules Belonging to the Class of Retinoic Acid-Related Orphan Receptor γt Inverse Agonists Under Investigation for Psoriasis. Vimirogant and GSK2981278 are Reported in the Table S19 Due to Any Efficacy in the Pathology |
Sphingosine-1-Phosphate Receptors Agonists
Sphingosine is an 18-carbon amino alcohol, which forms a primary part of sphingolipids, a class of cell membrane lipids created from ceramide, through ceramidase.35 The Sphingosine-1-phosphate (S1P) is a lipid mediator associated with cellular proliferation, survival, migration, inflammation, immune cell trafficking, angiogenesis, vascular integrity, and adhesion in the immune and vascular systems.36 S1P plays on five specific G protein-coupled receptors named S1P receptors (S1PR) 1–5.36 S1PR1 is expressed on lymphocytes and controls their way out from thymus and secondary lymphoid organs.37,38 S1PR1 modulators induce internalization of this receptor and segregation of most circulating lymphocytes in lymph nodes, decreasing peripheral lymphocytes count and trafficking to peripheral tissues.39,40 Moreover, it has been described that S1P hinders the growth of keratinocytes and demonstrates anti-proliferative and anti-inflammatory effects in mouse models of psoriasis.41–45 It results by keratinocytes entrance in G0 phase of the cell cycle and is not due to a cytotoxic effect.45 Moreover, S1P induces calcium intracellular content elevation, and this is a strong signal for keratinocytes differentiation.35,42 Ponesimod (ACT-128800) is an oral selective S1PR1 agonist, which blocks the egress of T cells from lymphoid organs (Table 4; Figure 2; Table S7).46,47 SCD-044 is a novel oral S1PR1 agonist that is going to be tested for the treatment of cutaneous inflammatory diseases such as atopic dermatitis and psoriasis (Table 4; Figure 2; Table S8).
 |
Table 4 The Table Shows the Molecules Belonging to the Class of Sphingosine-1-Phosphate Receptor 1 Agonist Under Investigation for Psoriasis |
Rho-Associated Kinases Inhibitor
Rho family kinases, consisting of Rho-associated coiled-coil kinase (ROCK) 1 and 2, are serine-threonine kinases activated by Rho GTPases and mediate the phosphorylation of downstream targets in cells.48,49 ROCK2 down-regulates autoimmune responses binding phosphorylated-STAT3 and controlling the formation of ROCK2/STAT3/JAK2 complex and optimal STAT3 phosphorylation in human CD4+ T cells during Th17 skewing.50 Thus, ROCK2 seems to be a promising target in the treatment of immune-mediated and inflammatory condition, as psoriasis, due to the role on T cells. Belumosudil (KD025 or SLx-2119) is an oral ROCK inhibitor Table 5; Figure 2; Table S9). Recent studies have showed that oral belumosudil administration in healthy subjects, without psoriasis, decreases IL-17 and IL-21 secretion, induced by ex vivo stimulation.49 Moreover, ROCK2 inhibition shifted the balance between pro-inflammatory and immunosuppressive T-cell subsets through concurrent regulation of STAT3/STAT5 phosphorylation. In a phase II clinical trial, KD025 significantly reduced both IL-17 and IL-23 levels. Moreover, epidermal thickness, K16 expression, and T-cell cutaneous infiltration also decreased in skin biopsies of treated patients.51 In addition, the PASI50 response to KD025 (200 mg twice daily) at week 12 was 71%.61 Finally, KD025 significantly increased levels of the immune-modulant cytokine IL-10, but tumor necrosis factor (TNF)-α and IL-6 levels were not changed.51
 |
Table 5 The Table Shows the Molecules Belonging to the Class of Rho-Associated Coiled-Coil Kinase 2 Inhibitor Under Investigation for Psoriasis |
Interleukin-1 Receptor Associated Kinase 4 Inhibitors
Interleukin-1 receptor associated kinase (IRAK)-4, a member of serine/threonine kinase family, is a crucial downstream signaling factor of both toll-like receptors (TLRs) and IL-1 receptor pathways of innate immune responses. The main expression of IRAK-4 seems to be on peripheral blood mononuclear cells and CD8+ cells.52,53 IRAK-4 achieved great attention when results from a poster at the Annual Scientific Meeting of American College of Rheumatology reported the beneficial potentials of selective and potent IRAK-4 inhibitors in the pre-clinical evaluation for various inflammatory- and immune-related conditions, as psoriasis.54 Experimental reports have demonstrated that IRAK-4 activity reduction stops cytokine production, and consequently, appear to be promising in inflammatory- and immune-related disorders.55,56 The oral administration of IRAK-4 inhibitors (ND-2110, ND-2158) has demonstrated efficacy in a mouse model of imiquimod-induced psoriasis.55 BAY18344845 is an orally bioavailable small molecule IRAK-4 inhibitor. It blocks nuclear factor-kappa of B cell (NF-κB)-mediated secretion of TNF-α, IL-1, and IL-6 in peripheral blood mononuclear cells. BAY1834845 is currently under investigation for rheumatoid arthritis, other autoimmune conditions and hematological malignancies. To data, just one phase I trial is evaluating safety and tolerability of multiple oral doses in healthy male subjects and in patients with psoriasis (Table 6; Figure 2; Table S17).
 |
Table 6 The Table Shows the Molecules Belonging to the Class of Interleukin-1 Receptor Associated Kinase 4 Inhibitor Under Investigation for Psoriasis |
Phosphodiesterase-4 Inhibitors
PDE4 is an intracellular non-receptor enzyme modulating inflammation and epithelial integrity, mainly present in immune, epithelial, and brain cells.57 Inhibition of PDE4 has several effects via the elevation of the cyclic adenosine monophosphate (cAMP) levels. It has been identified that PDE4 is a promising therapeutic target for the treatment of diverse pulmonary, dermatological, and severe neurological diseases.58 In the skin, PDE4 is primarily expressed in keratinocytes, neutrophils, Langerhans and T cells, which contribute to the psoriatic plaque formation.59 Previous studies demonstrated that PDE4 mRNA level in peripheral blood was higher in psoriatic patients compared to normal individuals.59,60 Hemay005 is a novel small molecule inhibiting PDE4, developed for the treatment of psoriasis (Table 7; Figure 2; Table S10).61 Oral orismilast (LEO 32731) was developed as a candidate for psoriasis treatment. It has been demonstrated to be superior over placebo in randomized, double-blinded, placebo-controlled clinical studies for this indication (Table 7; Figure 2; Table S11).
 |
Table 7 The Table Shows the Molecules Belonging to the Class of Phosphodiesterase-4 Inhibitors Under Investigation for Psoriasis |
A3 Adenosine Receptor Agonist
A3 adenosine receptor (A3AR) is expressed on all types of immune cells with a broad distribution. A direct correlation has been found between A3AR expression level and disease progression in inflammatory and cancer diseases, in both experimental animal models and humans.62 Boukamp et al demonstrated A3AR over-expression in skin lesions and peripheral blood of psoriatic patients with moderate-to-severe disease.63 Targeting the receptor with the highly specific agonist piclidenoson (CF101), it was induced the in vitro inhibition of cell proliferation.63 Moreover, piclidenoson reduces PI3K, p-AKT, NF-κB, TNF-α, IL-17, and IL-23 expression, all known to act as potent inflammatory mediators in psoriasis (Table 8; Figure 2; Table S12).64,65
 |
Table 8 The Table Shows the Molecules Belonging to the Class of A3 Adenosine Receptor Agonist Under Investigation for Psoriasis |
Toll-Like Receptors Antagonist
TLRs are a class of proteins that plays a key role in the innate immune system. They are single-pass membrane-spanning receptors usually expressed on antigen presenting cells, as macrophages and dendritic cells, that recognize structurally conserved molecules derived from microbes. Aberrant TLR-7, 8, and 9 activation by self-nucleic acids is involved in immune-mediated inflammatory diseases, such as psoriasis. IMO-8400 is a first-in-class antagonist of TLR-7, 8, and 9. Balak et al evaluated the short-term safety and proof-of-concept for efficacy of IMO-8400 in a phase II trial66 (Table 9; Figure 2; Table S13).
 |
Table 9 The Table Shows the Molecules Belonging to the Class of Toll-Like Receptor Antagonist Under Investigation for Psoriasis |
IL-36 Receptor Inhibitors
The IL-36 receptor (IL-36R) axis has been identified as the involved pathways in GPP and palmo-plantar pustulosis (PPP). IL-36 is an inflammatory cytokine, member of the IL-1 superfamily, secreted by T cells and acting on keratinocytes, which express IL-36R. IL-36 is composed of three agonists, IL-36α, IL-36β, and IL-36γ (previously called IL-1F6, IL-1F8, and IL-1F9) and an antagonist, IL-36 receptor antagonist (IL-36Ra, formerly known as IL-1F5).67 Spesolimab (BI 655130), is a monoclonal antibody that blocks IL-36R, significantly improving GPP symptoms in moderate and severe disease flares.68 In vivo studies highlighted the capability of spesolimab to block skin inflammation. Inhibiting the IL-36R pathway results in significant attenuation of skin thickening and psoriasis-relevant gene expression. These data suggest a crosstalk between IL-36R and the IL-23/Th17 axis both in PPP and GPP69 (Table 10; Figure 2; Table S14). Imsidolimab (ANB019) is an antibody inhibiting the IL-36R function, which has been initially developed as a therapy in GPP, PPP, EGFR inhibitors-mediated skin toxicity, and ichthyosis (Table 10; Figure 2; Table S15).
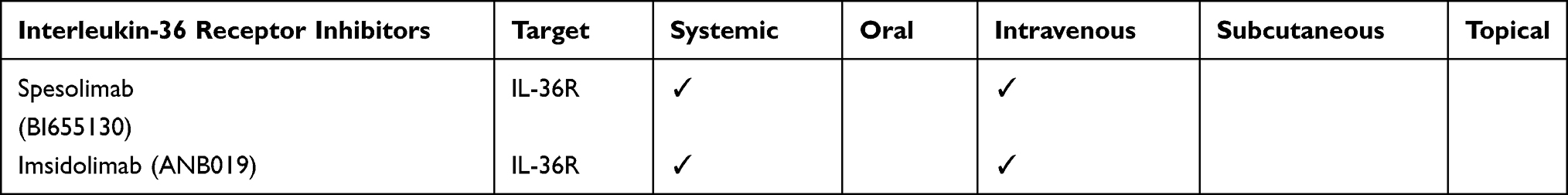 |
Table 10 The Table Shows the Molecules Belonging to the Class of Interleukin-36 Receptor Inhibitors Under Investigation for Psoriasis |
Granulocyte-Macrophage Colony-Stimulating Factor
As a major immune modulator, GM-CSF regulation may be of potential interest in psoriasis treatment. Within the skin, GM-CSF is produced by activated T lymphocytes, myeloid cells, endothelial cells, macrophages, fibroblasts and keratinocytes. It is detectable in psoriasis related skin blister fluid and in the serum of patients with psoriasis.70 Namilumab (AMG203) is a human IgG1 monoclonal antibody potently and specifically neutralizing human and macaque circulating GM-CSF (Table S16). Both in vivo and in vitro studies supported a hypothetical use of GM-CSF inhibitors in psoriasis.71,72 Data from the NEPTUNE trial have highlighted all benefits from the clinical usage of namilumab in moderate-to-severe plaque psoriasis (Tables S14, S20).
Discussion
The involvement of specific cytokine-driven signaling pathways in the pathogenesis of psoriasis has been the key observation for the development of target therapies, some of which are currently used in the clinical setting with excellent results in terms of induction of disease remission.13,68,73 Antimicrobial peptides (AMPs) play important roles in host protection, leading to kill several microorganisms, including bacteria, protozoa, fungi, and viruses.74 In psoriasis, certain AMPs, including β-defensins and cathelicidin, are highly expressed and produced by keratinocytes, neutrophils, and macrophages in response to injury and cytokine secretion.75 They stimulate TLRs, especially TLR-9 on plasmacytoid dendritic cells, which, in turn, produce IFN-α and β, promoting myeloid dendritic cells (mDCs) activation.76 Activated mDCs migrate to lymph nodes and secrete IL-12 e IL-23, which modulate the differentiation and proliferation of Th1 and Th17 lymphocytes subsets respectively.75 Th17 cytokines (IL-17, IL-21, and IL-22) induce keratinocytes proliferation causing epidermal hyperplasia and thickening. Intracellular pathways implied in the signal transduction of these molecules include NF-kB overexpression, with increased TNF-α and IL-6 production, and JAK-STAT pathway activation.77 In this context, JAKsI may be employed in the treatment of psoriasis as they oppose to the immune-mediated mechanisms underlying psoriasis.69 In particular, as JAK-STAT pathway activation, induced by IL-23, upregulates STAT3,5 JAKsI down-regulate IL-17 and IL-17F transcription, which are critical for Th17 lymphocytes development.16 Among JAKsI, daucravacitinib, with its unique ability to selectively inhibit TYK2 promises to target specific cytokine pathways, involving IL-12, IL-23, and IFN, arousing great interest for increasing target specificity and reducing AEs.10,17,78 An increased infections occurrence in patients treated with JAKsI was expected since TYK2, in particular, is required for antiviral defense controlled by type I IFN.17 However, from real life data, the incidence of infections was not significant when compared to placebo or other small molecules groups. The most common infective events reported were pharyngitis and upper respiratory tract infections, but they were mild-to-moderate and did not lead to treatment discontinuation.18 The different clinical subtypes of psoriasis could be related to different cytokines involved in the pathogenesis. For instance, recessive mutations in the antagonist of the IL-36R, belonging to the IL-1 pro-inflammatory cytokine family, have been linked to GPP.67 This mutation is also found in PPP and acrodermatitis continua of Hallopeau. In patients with pre-existing plaque-type psoriasis, the gain-of-function mutation in CARD14, p.Asp176His, was found to be a predisposing factor for developing GPP.67 In this context, IL-36R inhibitors conquer a central role in the rational treatment of GPP and PPP. Improvement in understanding psoriasis pathogenesis reveals that different clinical psoriatic phenotypes underly various involved cytokines and diverse genetic substrate. Cumulative evidences implicate a substantial role of genetic factors in psoriasis susceptibility and manifestations. The genetic basis of psoriasis is supported by family-based investigations, population based epidemiological studies, association studies with human leukocyte antigens (HLAs), genome-wide linkage scans, and candidate gene studies within and outside the major histocompatibility complex region.79 The genetic landscape of psoriasis is dominated by mutations in the psoriasis susceptibility locus 1 (PSORS1), which comprises genes in the HLA complex.80 The strongest HLA-related associations in psoriasis is mapped on HLA-C*06, particularly in patients with early-onset and more severe disease who have a positive family history.80 However, regression analyses have identified at least seven independent HLA genetic signatures for psoriasis based on single-nucleotide polymorphism typing, which have been mapped by imputation to HLA-C, HLA-B, HLA-A, and HLA-DRA.81 Confirmed associations include mutations in the PSORS2 region (17q24-q25), hosting the CARD14 gene;82,83 PSORS4 in the epidermal differentiation complex;84 PSORS6, on chromosome 19p13, spanning the TYK2 locus;85 and PSORS7 on chromosome 1p, in which the IL23R locus is located.86 Studies suggested that CARD14 gene mutations enhance NF-κB activation, resulting in abnormal inflammation. NF-κB activation in keratinocytes should be the target in CARD14-related psoriasis. Following what was explained before, in the era of small molecules, these patients could benefit of PDE4 and IL-36R inhibitors.57,67 More than genetics, epigenetics paved the way to understand the interactions between gene and environment. Epigenetic modifications are able to induce chromatin changes without modifying the DNA sequence.87 Epigenetic mechanisms may operate at transcriptional (methylation and histone modifications) and post-transcriptional (microRNAs and long non-coding RNAs) level. Non-genetic factors (including smoking, alcohol, stress, drugs assumption, pollution, UV radiation, and diet) can induce epigenetic responses and, ultimately, modulate individual gene expression profiles and susceptibility to disease trait.88–90 Small molecules, as PDE4 inhibitors, permit to cure those patients with difficult-to-treat or moderate-severe disease. They are of particular help in the clinical practice, as oral administration and high security profile in fragile and elderly patients are very appreciated features in a chronically administrated therapy. In this view, PDE4 inhibitors resolve the unmet needs not yet under control with the current biologic therapies. In the scenario of new targeted therapies, finding other pathogenetic molecular target in a disease as psoriasis - where the pathogenetic mechanism are not yet fully understood - is a valid therapeutic option. Based on such paradigm shift, next generation therapeutic targets might be identified. It is recognized today that psoriatic patients present a number of important comorbidities, observed more often than expected, based on their respective prevalence in general population. These include psoriatic arthritis, metabolic syndrome, cardiovascular disorders, as well as numerous other diseases, such as anxiety/depression, non-alcoholic fatty liver disease, Crohn’s disease, or lymphoma. High prevalence, chronicity, disfiguration, disability, and associated comorbidity make it a challenge for clinicians of multiple specialties.86
Conclusions
Several clinical trials in experimental phase may provide new therapeutic options to satisfy the unmet needs of patients with a resistant psoriatic disease to the last biologic therapies. Although the concept of “psoriatic disease” comprises extra-cutaneous manifestations also, we had focused only on cutaneous disease, to a reason of major clarity. Several new classes of anti-psoriatic drugs are currently undergoing clinical development. These new therapies promise earlier and higher-level responses that are durable and more specific, as they target cytokines involved directly in psoriatic inflammation. Additionally, based on results from clinical trials evaluating these new agents, it may be possible to find predictive markers identifying classes of patients who are treated with better results with certain drug classes,91 those prone to lose response to treatments, and those who may discontinue treatment maintaining remission. It remains to be determined whether the promising results seen in early studies of therapies in development for psoriasis will translate into actual improvements over currently available treatment options. Numerous small molecules are under development for psoriasis, although their place in the anti-psoriatic treatment schemes has not yet been established. Nevertheless, advantage in oral administration, reducing subcutaneous or intravenous therapies, lead JAKsI and PDE4 inhibitors as the more under development molecules. In terms of future research, randomized trials comparing directly active agents are necessary once high-quality evidence of benefit against placebo is established, including head-to-head trials amongst and between conventional systemic therapies, biological agents and the newest small molecules.
Acknowledgments
We would like to thank Denis Mariano, who kindly revised the English language.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lanna C, Mancini M, Gaziano R, et al. Skin Immunity and Its Dysregulation in Psoriasis. Cell Cycle. 2019;18:2581–2589. doi:10.1080/15384101.2019.1653099.
2. Picciani BLS, Domingos TA, Teixeira-Souza T, Santos VDCBD. Geographic Tongue and Psoriasis: clinical, Histopathological, Immunohistochemical and Genetic Correlation - a Literature Review. An Bras Dermatol. 2016;91:410–421. doi:10.1590/abd1806-4841.20164288.
3. Rønholt K, Iversen L. Old and New Biological Therapies for Psoriasis. Int J Mol Sci. 2017;18(11):2297. doi:10.3390/ijms18112297.
4. Sun C, Xia J. Treatment of Psoriasis: janus Kinases Inhibitors and Biologics for the Interleukin-23/Th17 Axis. Minerva Med. 2020;111:254–265. doi:10.23736/S0026-4806.20.06460-5.
5. Balato A, Campione E, Cirillo T, Malara G, Trifirò C, Bianchi L. Long-Term Efficacy and Safety of Apremilast in Psoriatic Arthritis: focus on Skin Manifestations and Special Populations. Dermatol Ther. 2020;33:e13440. doi:10.1111/dth.13440.
6. Lanna C, Cesaroni GM, Mazzilli S, et al. Apremilast as a Target Therapy for Nail Psoriasis: a Real-Life Observational Study Proving Its Efficacy in Restoring the Nail Unit. J Dermatolog Treat. 2020;1–5. doi:10.1080/09546634.2020.1801976.
7. Lanna C, Cesaroni GM, Mazzilli S, Bianchi L, Campione E Small Molecules, Big Promises: improvement of Psoriasis Severity and Glucidic Markers with Apremilast: a Case Report. Available from: https://www.dovepress.com/small-molecules-big-promises-improvement-of-psoriasis-severity-and-glu-peer-reviewed-article-DMSO.
8. Mazzilli S, Lanna C, Chiaramonte C, et al. Real Life Experience of Apremilast in Psoriasis and Arthritis Psoriatic Patients: preliminary Results on Metabolic Biomarkers. J Dermatol. 2020. doi:10.1111/1346-8138.15293.
9. Lanna C, Cesaroni GM, Mazzilli S, et al. Nails as Immune-Privileged Sites: a Case of Disabling Acrodermatitis Continua of Hallopeau Successfully Treated with Apremilast. Dermatol Ther. 2019;32:e12946. doi:10.1111/dth.12946.
10. Nogueira M, Puig L, Torres T. JAK Inhibitors for Treatment of Psoriasis: focus on Selective TYK2 Inhibitors. Drugs. 2020;80:341–352. doi:10.1007/s40265-020-01261-8.
11. Kan S-H, Mancini G, Gallagher G. Identification and Characterization of Multiple Splice Forms of the Human Interleukin-23 Receptor Alpha Chain in Mitogen-Activated Leukocytes. Genes Immun. 2008;9:631–639. doi:10.1038/gene.2008.64.
12. Roskoski R. Janus Kinase (JAK) Inhibitors in the Treatment of Inflammatory and Neoplastic Diseases. Pharmacol Res. 2016;111:784–803. doi:10.1016/j.phrs.2016.07.038.
13. Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJJAK. Inhibition as a Therapeutic Strategy for Immune and Inflammatory Diseases. Nat Rev Drug Discov. 2017;16:843–862. doi:10.1038/nrd.2017.201.
14. O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, The LA. JAK-STAT Pathway: impact on Human Disease and Therapeutic Intervention. Annu Rev Med. 2015;66:311–328. doi:10.1146/annurev-med-051113-024537.
15. Chen Z, Laurence A, Kanno Y, et al. Selective Regulatory Function of Socs3 in the Formation of IL-17-Secreting T Cells. Proc Natl Acad Sci U S A. 2006;103:8137–8142. doi:10.1073/pnas.0600666103.
16. Yang XO, Panopoulos AD, Nurieva R, et al. STAT3 Regulates Cytokine-Mediated Generation of Inflammatory Helper T Cells. J Biol Chem. 2007;282:9358–9363. doi:10.1074/jbc.C600321200.
17. Papp K, Gordon K, Thaçi D, et al. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N Engl J Med. 2018;379:1313–1321. doi:10.1056/NEJMoa1806382.
18. Chang Y, Xu S, Ding K. Tyrosine Kinase 2 (TYK2) Allosteric Inhibitors To Treat Autoimmune Diseases. J Med Chem. 2019;62:8951–8952. doi:10.1021/acs.jmedchem.9b01612.
19. Tanimoto A, Ogawa Y, Oki C, et al. Pharmacological Properties of JTE-052: a Novel Potent JAK Inhibitor That Suppresses Various Inflammatory Responses in Vitro and in Vivo. Inflamm Res. 2015;64:41–51. doi:10.1007/s00011-014-0782-9.
20. Tanimoto A, Shinozaki Y, Yamamoto Y, et al. A Novel JAK Inhibitor JTE-052 Reduces Skin Inflammation and Ameliorates Chronic Dermatitis in Rodent Models: comparison with Conventional Therapeutic Agents. Exp Dermatol. 2018;27:22–29. doi:10.1111/exd.13370.
21. Amano W, Nakajima S, Kunugi H, et al. The Janus Kinase Inhibitor JTE-052 Improves Skin Barrier Function through Suppressing Signal Transducer and Activator of Transcription 3 Signaling. J Allergy Clin Immunol. 2015;136:667–677.e7. doi:10.1016/j.jaci.2015.03.051.
22. Denison MS, Nagy SR. Activation of the Aryl Hydrocarbon Receptor by Structurally Diverse Exogenous and Endogenous Chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi:10.1146/annurev.pharmtox.43.100901.135828.
23. Stockinger B, Di Meglio P, Gialitakis M, Duarte JH. The Aryl Hydrocarbon Receptor: multitasking in the Immune System. Annu Rev Immunol. 2014;32:403–432. doi:10.1146/annurev-immunol-032713-120245.
24. Furue M, Hashimoto-Hachiya A, Tsuji G. Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis. Int J Mol Sci. 2019;20(21):5424. doi:10.3390/ijms20215424.
25. Zhu Z, Chen J, Lin Y, et al. Aryl Hydrocarbon Receptor in Cutaneous Vascular Endothelial Cells Restricts Psoriasis Development by Negatively Regulating Neutrophil Recruitment. J Invest Dermatol. 2020;140:1233–1243.e9. doi:10.1016/j.jid.2019.11.022.
26. Ramirez J-M, Brembilla NC, Sorg O, et al. Activation of the Aryl Hydrocarbon Receptor Reveals Distinct Requirements for IL-22 and IL-17 Production by Human T Helper Cells. Eur J Immunol. 2010;40:2450–2459. doi:10.1002/eji.201040461.
27. Smith SH, Jayawickreme C, Rickard DJ, et al. Tapinarof Is a Natural AhR Agonist That Resolves Skin Inflammation in Mice and Humans. J Invest Dermatol. 2017;137:2110–2119. doi:10.1016/j.jid.2017.05.004.
28. Richardson WH, Schmidt TM, Nealson KH. Identification of an Anthraquinone Pigment and a Hydroxystilbene Antibiotic from Xenorhabdus Luminescens. Appl Environ Microbiol. 1988;54:1602–1605. doi:10.1128/aem.54.6.1602-1605.1988
29. Tang L, Yang X, Liang Y, Xie H, Dai Z, Zheng G. Transcription Factor Retinoid-Related Orphan Receptor Γt: a Promising Target for the Treatment of Psoriasis. Front Immunol. 2018;9. doi:10.3389/fimmu.2018.01210
30. He Y-W, Deftos ML, Ojala EW, Bevan MJ. RORγt, a Novel Isoform of an Orphan Receptor, Negatively Regulates Fas Ligand Expression and IL-2 Production in T Cells. Immunity. 1998;9:797–806. doi:10.1016/S1074-7613(00)80645-7
31. Medvedev A, Chistokhina A, Hirose T, Jetten AM. Genomic Structure and Chromosomal Mapping of the Nuclear Orphan Receptor ROR Gamma (RORC) Gene. Genomics. 1997;46:93–102. doi:10.1006/geno.1997.4980.
32. Villey I, de Chasseval R, de Villartay JP. RORgammaT, a Thymus-Specific Isoform of the Orphan Nuclear Receptor RORgamma/TOR, Is up-Regulated by Signaling through the Pre-T Cell Receptor and Binds to the TEA Promoter. Eur J Immunol. 1999;29:4072–4080. doi:10.1002/(SICI)1521-4141(199912)29:12<4072::aid-immu4072>3.0.CO;2-E.
33. Eberl G, Littman DR. The Role of the Nuclear Hormone Receptor RORgammat in the Development of Lymph Nodes and Peyer’s Patches. Immunol Rev. 2003;195:81–90. doi:10.1034/j.1600-065x.2003.00074.x.
34. Ivanov II, McKenzie BS, Zhou L, et al. The Orphan Nuclear Receptor RORgammat Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell. 2006;126:1121–1133. doi:10.1016/j.cell.2006.07.035.
35. Borodzicz S, Rudnicka L, Mirowska-Guzel D, Cudnoch-Jedrzejewska A. The Role of Epidermal Sphingolipids in Dermatologic Diseases. Lipids Health Dis. 2016;15:13. doi:10.1186/s12944-016-0178-7.
36. Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the Sphingosine-1-Phosphate Axis in Cancer, Inflammation and Beyond. Nat Rev Drug Discov. 2013;12:688–702. doi:10.1038/nrd4099.
37. Krause A, D’Ambrosio D, Dingemanse J. Modeling Clinical Efficacy of the S1P Receptor Modulator Ponesimod in Psoriasis. J Dermatol Sci. 2018;89:136–145. doi:10.1016/j.jdermsci.2017.11.003.
38. Cyster JG, Schwab SR. Sphingosine-1-Phosphate and Lymphocyte Egress from Lymphoid Organs. Annu Rev Immunol. 2012;30:69–94. doi:10.1146/annurev-immunol-020711-075011.
39. Chew WS, Wang W, Herr DR. To Fingolimod and beyond: the Rich Pipeline of Drug Candidates That Target S1P Signaling. Pharmacol Res. 2016;113:521–532. doi:10.1016/j.phrs.2016.09.025.
40. Piali L, Froidevaux S, Hess P, et al. The Selective Sphingosine 1-Phosphate Receptor 1 Agonist Ponesimod Protects against Lymphocyte-Mediated Tissue Inflammation. J Pharmacol Exp Ther. 2011;337:547–556. doi:10.1124/jpet.110.176487.
41. Ji M, Xue N, Lai F, et al. Validating a Selective S1P1 Receptor Modulator Syl930 for Psoriasis Treatment. Biol Pharm Bull. 2018;41:592–596. doi:10.1248/bpb.b17-00939.
42. Vogler R, Sauer B, Kim D-S, Schäfer-Korting M, Kleuser B. Sphingosine-1-Phosphate and Its Potentially Paradoxical Effects on Critical Parameters of Cutaneous Wound Healing. J Invest Dermatol. 2003;120:693–700. doi:10.1046/j.1523-1747.2003.12096.x.
43. Schüppel M, Kürschner U, Kleuser U, Schäfer-Korting M. Sphingosine 1-Phosphate Restrains Insulin-Mediated Keratinocyte Proliferation via Inhibition of Akt through the S1P2 Receptor Subtype. J Invest Dermatol. 2008;128:1747–1756. doi:10.1038/sj.jid.5701259.
44. Schaper K, Dickhaut J, Japtok L, et al. Sphingosine-1-Phosphate Exhibits Anti-Proliferative and Anti-Inflammatory Effects in Mouse Models of Psoriasis. J Dermatol Sci. 2013;71:29–36. doi:10.1016/j.jdermsci.2013.03.006.
45. Manggau M, Kim DS, Ruwisch L, et al. 1Alpha,25-Dihydroxyvitamin D3 Protects Human Keratinocytes from Apoptosis by the Formation of Sphingosine-1-Phosphate. J Invest Dermatol. 2001;117:1241–1249. doi:10.1046/j.0022-202x.2001.01496.x.
46. Park S-J, Im D-S. Sphingosine 1-Phosphate Receptor Modulators and Drug Discovery. Biomol Ther (Seoul). 2017;25:80–90. doi:10.4062/biomolther.2016.160.
47. Bolli MH, Abele S, Binkert C, et al. 2-Imino-Thiazolidin-4-One Derivatives as Potent, Orally Active S1P1 Receptor Agonists. J Med Chem. 2010;53:4198–4211. doi:10.1021/jm100181s.
48. Riento K, Ridley AJ. Rocks: multifunctional Kinases in Cell Behaviour. Nat Rev Mol Cell Biol. 2003;4:446–456. doi:10.1038/nrm1128.
49. Zanin-Zhorov A, Weiss JM, Nyuydzefe MS, et al. Selective Oral ROCK2 Inhibitor Down-Regulates IL-21 and IL-17 Secretion in Human T Cells via STAT3-Dependent Mechanism. Proc Natl Acad Sci U S A. 2014;111:16814–16819. doi:10.1073/pnas.1414189111.
50. Chen W, Nyuydzefe MS, Weiss JM, Zhang J, Waksal SD, Zanin-Zhorov A. ROCK2, but Not ROCK1 Interacts with Phosphorylated STAT3 and Co-Occupies TH17/TFH Gene Promoters in TH17-Activated Human T Cells. Sci Rep. 2018;8:16636. doi:10.1038/s41598-018-35109-9.
51. Zanin-Zhorov A, Weiss JM, Trzeciak A, et al. Cutting Edge: selective Oral ROCK2 Inhibitor Reduces Clinical Scores in Patients with Psoriasis Vulgaris and Normalizes Skin Pathology via Concurrent Regulation of IL-17 and IL-10. J Immunol. 2017;198:3809–3814. doi:10.4049/jimmunol.1602142.
52. Li S, Strelow A, Fontana EJ, Wesche H. IRAK-4: a Novel Member of the IRAK Family with the Properties of an IRAK-Kinase. Proc Natl Acad Sci U S A. 2002;99:5567–5572. doi:10.1073/pnas.082100399.
53. Kim TW, Staschke K, Bulek K, et al. A Critical Role for IRAK4 Kinase Activity in Toll-like Receptor–Mediated Innate Immunity. J Exp Med. 2007;204:1025–1036. doi:10.1084/jem.20061825.
54. Croasdell G. A Report from the American College of Rheumatology/Association of Rheumatology Health Professionals (ACR/ARHP) - 2012 Annual Meeting (November 9–14, 2012 - Washington, D.C., USA). Drugs Today (Barc). 2013;49:153–155. doi:10.1358/dot.2013.49.2.1937427.
55. Koziczak-Holbro M, Littlewood-Evans A, Pöllinger B, et al. The Critical Role of Kinase Activity of Interleukin-1 Receptor-Associated Kinase 4 in Animal Models of Joint Inflammation. Arthritis Rheum. 2009;60:1661–1671. doi:10.1002/art.24552.
56. Staschke KA, Dong S, Saha J, et al. IRAK4 Kinase Activity Is Required for Th17 Differentiation and Th17-Mediated Disease. J Immunol. 2009;183:568–577. doi:10.4049/jimmunol.0802361.
57. Sanz MJ, Cortijo J, Morcillo EJ. PDE4 Inhibitors as New Anti-Inflammatory Drugs: effects on Cell Trafficking and Cell Adhesion Molecules Expression. Pharmacol Ther. 2005;106:269–297. doi:10.1016/j.pharmthera.2004.12.001.
58. Li H, Zuo J, Tang W. Phosphodiesterase-4 Inhibitors for the Treatment of Inflammatory Diseases. Front Pharmacol. 2018;9:1048. doi:10.3389/fphar.2018.01048.
59. Chiricozzi A, Caposiena D, Garofalo V, et al. Therapeutic for the Treatment of Moderate-to-Severe Plaque Psoriasis: apremilast. Expert Rev Clin Immunol. 2016;12:237–249. doi:10.1586/1744666X.2016.1134319.
60. Schafer PH, Adams M, Horan G, Truzzi F, Marconi A, Pincelli C. Apremilast Normalizes Gene Expression of Inflammatory Mediators in Human Keratinocytes and Reduces Antigen-Induced Atopic Dermatitis in Mice. Drugs R D. 2019;19:329–338. doi:10.1007/s40268-019-00284-1.
61. Liu X, Chen R, Zeng G, et al. Determination of a PDE4 Inhibitor Hemay005 in Human Plasma and Urine by UPLC-MS/ MS and Its Application to a PK Study. Bioanalysis. 2018;10:863–875. doi:10.4155/bio-2018-0004.
62. Fishman P, Bar-Yehuda S, Liang BT, Jacobson KA. Pharmacological and Therapeutic Effects of A3 Adenosine Receptor Agonists. Drug Discov Today. 2012;17:359–366. doi:10.1016/j.drudis.2011.10.007.
63. Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal Keratinization in a Spontaneously Immortalized Aneuploid Human Keratinocyte Cell Line. J Cell Biol. 1988;106:761–771. doi:10.1083/jcb.106.3.761.
64. Cohen S, Barer F, Itzhak I, Silverman MH, Fishman P. Inhibition of IL-17 and IL-23 in Human Keratinocytes by the A3 Adenosine Receptor Agonist Piclidenoson. J Immunol Res. 2018;2018:2310970. doi:10.1155/2018/2310970.
65. David M, Gospodinov DK, Gheorghe N, et al. Treatment of Plaque-Type Psoriasis With Oral CF101: data from a Phase II/III Multicenter, Randomized, Controlled Trial. J Drugs Dermatol. 2016;15:931–938.
66. Balak DMW, van Doorn MBA, Arbeit RD, et al. IMO-8400, a Toll-like Receptor 7, 8, and 9 Antagonist, Demonstrates Clinical Activity in a Phase 2a, Randomized, Placebo-Controlled Trial in Patients with Moderate-to-Severe Plaque Psoriasis. Clin Immunol. 2017;174:63–72. doi:10.1016/j.clim.2016.09.015.
67. Yuan Z-C, Xu W-D, Liu X-Y, Liu X-Y, Huang A-F, Su L-C. Biology of IL-36 Signaling and Its Role in Systemic Inflammatory Diseases. Front Immunol. 2019;10. doi:10.3389/fimmu.2019.02532
68. Bachelez H, Choon S-E, Marrakchi S, et al. Inhibition of the Interleukin-36 Pathway for the Treatment of Generalized Pustular Psoriasis. N Engl J Med. 2019;380:981–983. doi:10.1056/NEJMc1811317.
69. Su Z, Paulsboe S, Wetter J, et al. IL-36 Receptor Antagonistic Antibodies Inhibit Inflammatory Responses in Preclinical Models of Psoriasiform Dermatitis. Exp Dermatol. 2019;28:113–120. doi:10.1111/exd.13841.
70. Bonifati C, Carducci M, Fei PC, et al. Correlated Increases of Tumour Necrosis Factor-α, Interleukin-6 and Granulocyte Monocyte-Colony Stimulating Factor Levels in Suction Blister Fluids and Sera of Psoriatic Patients Relationships with Disease Severity. Clin Exp Dermatol. 1994;19:383–387. doi:10.1111/j.1365-2230.1994.tb02687.x.
71. Alvarez-Ruiz S, Peñas PF, Fernández-Herrera J, Sánchez-Pérez J, Fraga J. Maculopapular Eruption with Enlarged Macrophages in Eight Patients Receiving G-CSF or GM-CSF. J Eur Acad Dermatol Venereol. 2004;18:310–313. doi:10.1111/j.1468-3083.2004.00872.x.
72. Kelly R, Marsden RA, Bevan D. Exacerbation of Psoriasis with GM-CSF Therapy. Br J Dermatol. 1993;128:468–469. doi:10.1111/j.1365-2133.1993.tb00218.x.
73. Koziczak-Holbro M, Joyce C, Glück A, et al. IRAK-4 Kinase Activity Is Required for Interleukin-1 (IL-1) Receptor- and Toll-like Receptor 7-Mediated Signaling and Gene Expression. J Biol Chem. 2007;282:13552–13560. doi:10.1074/jbc.M700548200.
74. Lai Y, Gallo RL. AMPed Up Immunity: how Antimicrobial Peptides Have Multiple Roles in Immune Defense. Trends Immunol. 2009;30:131–141. doi:10.1016/j.it.2008.12.003.
75. Rendon A, Schäkel K. Psoriasis Pathogenesis and Treatment. Int J Mol Sci. 2019;20. doi:10.3390/ijms20061475
76. Mylonas A, Conrad C. Psoriasis: classical vs. Paradoxical. The Yin-Yang of TNF and Type I Interferon. Front Immunol. 2018;9:2746. doi:10.3389/fimmu.2018.02746.
77. Lowes MA, Suárez-Fariñas M, Krueger JG. Immunology of Psoriasis. Annu Rev Immunol. 2014;32:227–255. doi:10.1146/annurev-immunol-032713-120225.
78. Liu C, Lin J, Langevine C, et al. Discovery of BMS-986202: a Clinical Tyk2 Inhibitor That Binds to Tyk2 JH2. J Med Chem. 2021;64:677–694. doi:10.1021/acs.jmedchem.0c01698.
79. AlShobaili HA, Shahzad M, Al-Marshood A, Khalil A, Settin A, Barrimah I. Genetic Background of Psoriasis. Int J Health Sci (Qassim). 2010;4:23–29.
80. Greb JE, Goldminz AM, Elder JT, et al. Psoriasis. Nat Rev Dis Primers. 2016;2:16082. doi:10.1038/nrdp.2016.82.
81. Baran W, Szepietowski JC, Mazur G, Baran EA. 308 Promoter Polymorphism of Tumor Necrosis Factor Alpha Gene Does Not Associate with the Susceptibility to Psoriasis Vulgaris. No Difference Either between Psoriasis Type I and Type II Patients. Acta Dermatovenerol Alp Pannonica Adriat. 2006;15:113–118.
82. Carroll JM, Romero MR, Watt FM. Suprabasal Integrin Expression in the Epidermis of Transgenic Mice Results in Developmental Defects and a Phenotype Resembling Psoriasis. Cell. 1995;83:957–968. doi:10.1016/0092-8674(95)90211-2.
83. Signa S, Campione E, Rusmini M, et al. Whole Exome Sequencing Approach to Childhood Onset Familial Erythrodermic Psoriasis Unravels a Novel Mutation of CARD14 Requiring Unusual High Doses of Ustekinumab. Pediatr Rheumatol Online J. 2019;17:38. doi:10.1186/s12969-019-0336-3.
84. Hammer RE, Maika SD, Richardson JA, Tang JP, Taurog JD. Spontaneous Inflammatory Disease in Transgenic Rats Expressing HLA-B27 and Human Beta 2m: an Animal Model of HLA-B27-Associated Human Disorders. Cell. 1990;63:1099–1112. doi:10.1016/0092-8674(90)90512-d.
85. Stern RS. Psoriasis. Lancet. 1997;350:349–353. doi:10.1016/S0140-6736(97)05257-4.
86. Boehncke WH, Sterry W, Hainzl A, Scheffold W, Kaufmann R. Psoriasiform Architecture of Murine Epidermis Overlying Human Psoriatic Dermis Transplanted onto SCID Mice. Arch Dermatol Res. 1994;286:325–330. doi:10.1007/BF00402223.
87. Furrow RE, Christiansen FB, Feldman MW. Environment-Sensitive Epigenetics and the Heritability of Complex Diseases. Genetics. 2011;189:1377–1387. doi:10.1534/genetics.111.131912.
88. Fogel O, Richard-Miceli C, Tost J. Epigenetic Changes in Chronic Inflammatory Diseases. Adv Protein Chem Struct Biol. 2017;106:139–189. doi:10.1016/bs.apcsb.2016.09.003.
89. Mervis JS, McGee JS. Epigenetic Therapy and Dermatologic Disease: moving beyond CTCL. J Dermatolog Treat. 2019;30:68–73. doi:10.1080/09546634.2018.1473550.
90. Caputo V, Strafella C, Termine A, et al. Overview of the Molecular Determinants Contributing to the Expression of Psoriasis and Psoriatic Arthritis Phenotypes. J Cell Mol Med. 2020;24:13554–13563. doi:10.1111/jcmm.15742.
91. Campione E, Cosio T, Lanna C, et al. Predictive Role of Vitamin A Serum Concentration in Psoriatic Patients Treated with IL-17 Inhibitors to Prevent Skin and Systemic Fungal Infections. J Pharmacol Sci. 2020;144:52–56. doi:10.1016/j.jphs.2020.06.003.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.