Back to Journals » Journal of Experimental Pharmacology » Volume 13
Experimental GABA A Receptor Agonists and Allosteric Modulators for the Treatment of Focal Epilepsy
Authors Janković SM ![]() , Dješević M, Janković SV
, Dješević M, Janković SV
Received 11 December 2020
Accepted for publication 23 February 2021
Published 8 March 2021 Volume 2021:13 Pages 235—244
DOI https://doi.org/10.2147/JEP.S242964
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Bal Lokeshwar
Slobodan M Janković,1 Miralem Dješević,2 Snežana V Janković1
1Faculty of Medical Sciences, University of Kragujevac, Kragujevac, Serbia; 2Cardiology Department, Private Policlinic Center Eurofarm, Sarajevo, Bosnia and Hercegovina
Correspondence: Slobodan M Janković
University of Kragujevac, Faculty of Medical Sciences, Svetozara Markovića 69 Street, Kragujevac, 34000, Serbia
Email [email protected]
Abstract: GABA A receptors are ubiquitous in the central nervous system and there is a huge diversity of receptor subtypes in almost all regions of the brain. However, the expression of GABA A receptor subtypes is altered in both the gray and white matter of patients with focal epilepsy. Although there is a number of anticonvulsants with marketing authorization for the treatment of focal epilepsy which act through GABA A receptors, potentiating the inhibitory effects of GABA, it is necessary to develop more potent and more specific GABAergic anticonvulsants that are effective in drug-resistant patients with focal epilepsy. There are three orthosteric and at least seven allosteric agonist binding sites at the GABA A receptor. In experimental and clinical studies, full agonists of GABA A receptors showed a tendency to cause desensitization of the receptors, tolerance, and physical dependence; therefore, partial orthosteric agonists and positive allosteric modulators of GABA A receptors were further developed. Preclinical studies demonstrated the anticonvulsant efficacy of positive allosteric modulators with selective action on GABA A receptors with α2/α3 subunits, but only a handful of them were further tested in clinical trials. The best results were obtained for clobazam (already marketed), ganaxolone (in phase III trials), CVL-865 (in phase II trials), and padsevonil (in phase III trials). Several compounds with more selective action on GABA A receptors, perhaps only in certain brain regions, have the potential to become effective drugs against specific subtypes of focal-onset epilepsy. However, their development needs time, and in the near future we can expect only one or two new GABA A agonists to obtain marketing authorization for focal epilepsy, an advance that would be of use for just a fraction of patients with drug-resistant epilepsy.
Keywords: GABA A receptors, positive allosteric modulators, focal-onset epilepsy
Introduction
Gamma-aminobutyric acid (GABA) is one of a few key amino acid neurotransmitters in the brain, formed within the neurons from another amino acid, glutamate, by decarboxylation. After release from the endings of presynaptic nerve fibers, it binds to one of three types of membrane receptor: A, B, or C.1 The A and C receptors for GABA are ion channels, which therefore rapidly respond to binding with GABA, whereas the B receptor is a G-protein coupled receptor, reacting somewhat more slowly. GABA is transported from the synaptic cleft back to presynaptic neurons by specific transporters, of which four types are currently known, and then degraded to succinic semialdehyde by the enzyme GABA transaminase.2
Following recognition of the role of GABA A receptors in epileptogenesis, research has linked specific mutations of the α, β, γ, and δ subunits of these pentameric receptors with decreased inhibition of neurons and epilepsy.3 There is a number of anticonvulsants with marketing authorization which act through the GABA A receptor, potentiating the inhibitory effects of GABA: phenobarbital, valproic acid, benzodiazepines (clobazam, clonazepam), felbamate, topiramate, cannabidiol, and stiripentol; for some of these this is not the only mechanism of action. Two other anticonvulsants which have been approved for the treatment of epilepsy enhance transmission through GABA A receptors indirectly, increasing the availability of GABA at the receptors: vigabatrin inhibits GABA transaminase, and therefore degradation of GABA, and tiagabine inhibits the reuptake of GABA through presynaptic membranes.4
Despite the availability of quite a number of anticonvulsants acting through GABA A receptors, almost 22.5% of patients with epilepsy remain drug resistant.5 There is a large unmet need for more potent and more specific anticonvulsants that will be active in drug-resistant patients with focal epilepsy, which is more prevalent (46.2%) than the primarily generalized type, and some of them may act through GABA A receptors.6 The aim of this article was to give an overview of new substances with GABA A agonist properties, which showed promising activity in preclinical models of focal epilepsy and have or may become candidates for testing in clinical trials if their safety is confirmed.
GABA A Receptor
Structure
GABA A receptors are pentameric proteins with five subunits, and are considered to be a member of the Cys loop ligand-gated ion channel superfamily, sharing a common sequence in the amino acid chain where two cysteines are separated by 13 other amino acids.7 The human GABA A receptor is very complex, since there are 19 different types of subunit, each coded by a separate gene: six α, three β, three γ, and three ρ subunits, and one each of ϵ, δ, θ, and π subunits. Each of the subunits is composed of about 450 amino acids; the N-terminal hydrophilic segment of a subunit is placed extracellularly, and then continues in four transmembrane α-helical segments (designated M1–M4), which end with an extracellular C-terminal segment. Between the M3 and M4 segments, there is a large intracellular loop.8
It is believed that more than 500 different subtypes of GABA A receptor exist in the human brain, each with a different combination of the five subunits, but clear experimental proof of activity is only available for 26 subtypes. However, the most prevalent subtypes have α, β, and γ subunits; within the less prevalent subtypes, ϵ, δ, and π subunits replace the γ, and θ may replace the β subunit.9 Receptor subtypes with α, β, and γ subunits are usually composed of two α and two β subunits, and one γ subunit, which connects the other four subunits. All subtypes of GABA A receptor are chloride ion channels that open after binding to GABA.10 GABA A receptors are found at both synaptic and extrasynaptic locations.
Function
There are three types of orthosteric GABA binding site on the GABA A receptor, which are well characterized by specific ligands: muscimol, 4,5,6,7-tetrahydroisoxazolo(5,4-c)pyridin-3-ol (THIP), and bicuculline.11 The orthosteric GABA binding sites are located on the extracellular domain of the GABA A receptor, at the interface of the α and β subunits (two per receptor).12
Allosteric binding sites are also present on GABA A receptors, and were named according to the main ligands that were first discovered to bind to them: benzodiazepine binding site, picrotoxinin binding site, binding site for anesthetics, alcohol (ethanol) binding site, cannabinoid binding site, neurosteroid binding site, avermectin B1a binding site, and binding sites of ions (Table 1).13 After GABA binds to an orthosteric site, an ion channel within the GABA A receptor opens, chloride anions enter inside the cell through the ion channel, and the membrane is then hyperpolarized. Two types of transmembrane current are produced after the opening of GABA A receptors: synaptic (phasic) and tonic.14 Benzodiazepine and ethanol binding sites are located on the extracellular domains of the GABA A receptor, while the other allosteric binding sites are located within the transmembrane domains (the picrotoxinin binding site is located within the chloride channel itself, sopicrotoxinin causes closure of the chloride channel, producing convulsions in experimental animals).15 After binding their ligands, allosteric binding sites undergo conformational change, which further causes blooming and twisting of peptide chains, thus reorganizing the interfaces between the GABA A receptor subunits; since orthosteric binding sites are located at these interfaces, they are also conformationally changed, and their affinity for orthosteric ligands becomes different, as well as their capability to induce intracellular activity, leading to a cellular response. Allosteric ligands that enhance the effects of GABA and other orthosteric ligands after binding are called positive allosteric modulators (PAMs), whereas negative allosteric ligands reduce the effects of GABA.16
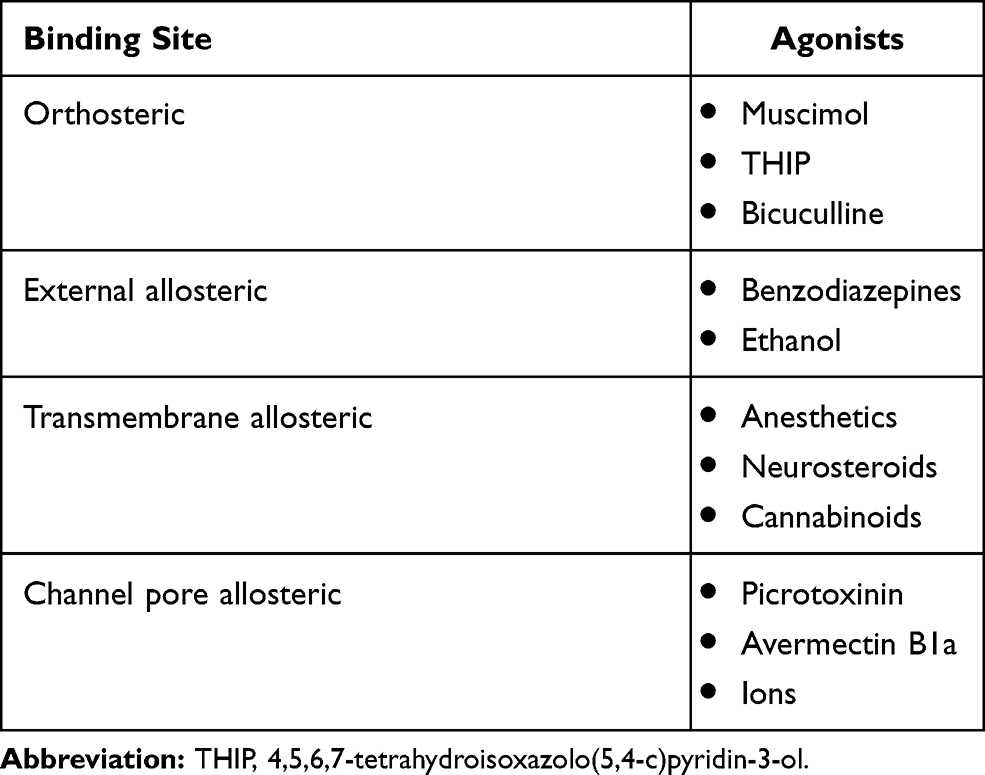 |
Table 1 Agonists and Their Binding Sites at GABA A Receptors |
GABA A Receptor Agonists, Antagonists, and Allosteric Modulators
Orthosteric Agonists and Antagonists
The orthosteric GABA binding site at the GABA A receptor is structurally complex, and therefore few compounds besides GABA itself can be recognized and cause activation of the receptor.17 Two compounds (muscimol and THIP) behaving as GABA A receptor agonists were discovered early on, and then modified in a number of ways to produce other activators of the receptor with different characteristics. Muscimol, naturally produced by the Amanita muscaria mushroom, was the first substance with proven full agonistic action on GABA A receptors, and has been widely tested in experimental studies. It was used as a lead for further synthesis of a number of other full agonists, which are more or less potent, as well as of other substances which may affect GABAergic transmission in various ways. Some of the derived full agonists are listed here in decreasing order of potency: dihydromuscimol, thiomuscimol, isoguvacine, and isonipecotic acid.18
In experimental and clinical studies, full agonists of GABA A receptors showed a tendency to cause desensitization of the receptors, which was translated into tolerance and physical dependence, with withdrawal symptoms in the case of abrupt cessation of therapy. This was one reason why partial agonists of GABA A receptors began to be sought. Piperidine-4-sulfonic acid (P4S) is a partial agonist that shows between 15% and 75% of full agonist activity, depending on the subtype of the GABA A receptor.19 THIP (synonym: gaboxadol), thio-THIP, and 1,2,3,6-tetrahydropyridine-4-sulfonic acid (DH-P4S) are also partial agonists.
The naturally occurring plant alkaloid bicuculline is a competitive antagonist at the orthosteric GABA binding site of the GABA A receptor.20 Bicuculline is a highly unstable compound which, in solution, is slowly converted to bicucine, a much less active GABA antagonist. Bicuculline is equally potent at all subtypes of GABA A receptor, which is a characteristic of all GABA A-binding antagonists in general. Besides bicuculline, several other GABA antagonists have been discovered in experimental studies (mostly in vitro), with high affinity for orthosteric binding sites at the GABA A receptor: gabazine, RU5135 (a steroid derivative with convulsant properties), and 4-(3-biphenyl)-5-(4-piperidyl)-3-isoxazolol.20
Allosteric Modulators
Benzodiazepines bind with at least three allosteric sites; the most abundant sites, site 1 and site 2, are located on the extracellular domain of the GABA A receptor, while site 3 is located on the transmembrane domain. There is substantial heterogeneity in the activity of benzodiazepines at these sites; for example, some of the sites 1 (having α4/α6 subunits) and sites 2 and 3 are insensitive to diazepam and alprazolam, but react with ethyl alcohol.12 On the other hand, bretazenil and flumazenil bind with all sites 1, regardless of the subunit composition.21
Picrotoxinin binds with another allosteric site at the GABA A receptor, which is located on the transmembrane part of the chloride channel itself. It could also be considered as a chloride channel blocker; there is a plethora of various substances, some of which are poisonous to mammals, that bind to the picrotoxinin binding site, block chloride channels, and cause excitation or even convulsions: t-butyl bicyclophosphorothionate (TBPS), thujone, organochlorine insecticides (lindane, dieldrin, and fipronil), pitrazepin, and tetramethylenedisulfotetramine (TETS). Some substances with general anesthetic properties (eg, propanidid) also bind to the picrotoxinin binding site, but behave as agonists, as they increase the flux of chloride ions.22
The antiparasitic compounds avermectin B1a and ivermectin bind to yet another site within the chloride channel of the GABA A receptor, acting as channel openers; that is, facilitating the action of GABA.23
Barbiturates, intravenous anesthetics (propofol, etomidate), neuroactive steroids (allopregnanolone and tetrahydrodeoxycorticosterone), volatile anesthetics, and ethanol in high doses bind to a variety of sites at the transmembrane interfaces of GABA A receptor subunits, facilitating the opening of chloride channels. These sites can be grouped into three distinct entities, called “sites”, according to the preferential binding of some substances with general anesthetic properties: etomidate binding (site 1), R-mTFD-MPAB (mephobarbital derivative) binding (site 2), and neurosteroid binding (site 3).24
Some cannabinoid CB1 inverse agonists (rimonabant and AM251) also bind to specific allosteric sites in GABA A receptors, potentiating the inhibitory action of GABA.25 The endocannabinoid 2-arachidonoyl glycerol (2-AG) also binds to the same site, which is located on the transmembrane segment of the GABA A receptor, having two β subunits out of the five subunits.26
As already mentioned, ethanol in high concentrations binds to allosteric sites on GABA A receptors specific for general anesthetics, but there is also a high-affinity site for binding ethanol located on the extracellular part of the receptor.15 The high-affinity site is present on both synaptic and extrasynaptic GABA A receptors, and is responsible for the majority of the signs and symptoms of ethanol intoxication.
Pathophysiology of Focal Epilepsy
Focal epilepsy is characterized by a range of seizure types, limited to one part of the brain, or in one hemisphere at most, the origin of which could be unifocal or multifocal.27 A variety of pathophysiological mechanisms could be behind focal seizures, leading to the abnormal discharge and spread of electrical activity. Details on the pathophysiology of mesial temporal lobe epilepsy, one of the most frequent forms of focal epilepsy in adults, were reviewed by Avoli et al,28 giving insights into the role of certain brain structures and neurotransmitter systems. The entorhinal cortex plays a primary role in the generation of seizures in this specific type of focal epilepsy, and glutamatergic neurotransmission through ionotropic NMDA receptors is a key factor in maintaining the ictal discharges. Although GABA A receptors normally mediate inhibition, under special circumstances their activation may lead to the depolarization of some neuronal groups in the entorhinal cortex, and thus contribute to the initiation of seizures. Dysfunction of hippocampal CA3 neurons is a necessary further element that enables the development and maintenance of seizures initiated in the entorhinal cortex, because their inhibitory action on CA1 and subiculum is lost.28
Morphological studies have shown that hippocampal sclerosis is a characteristic finding in patients with mesial temporal lobe epilepsy. Within the framework of sclerosis, neurons are lost in the hilus of the dentate gyrus and the cornu ammonis (CA), and replaced by glia. There is abundant experimental evidence that several factors in the damaged hippocampus contribute to the maintenance of seizures initiated in the entorhinal cortex: loss of both principal neurons and interneurons, neurogenesis with sprouting of nerve fibers, activation of microglia and astrocytes, neuroinflammation, and loss of the blood–brain barrier.29
Role of GABA A Receptors in Control of Focal Epilepsy
The main mechanism of action of GABA in the human brain is the induction of inhibitory postsynaptic potentials (IPSPs), predominantly at presynaptic nerve fibers. GABA A receptors (being rapid-acting chloride ion channels) are responsible for the early phase of the IPSP, and GABA B receptors (G-protein coupled) for the late phase. There are numerous studies on animal and human material showing reduced numbers of GABA A receptors in brain areas that were the origin of focal epilepsies with or without secondary generalization.30 Mesial temporal lobe epilepsy (the most frequent form among resistant focal epilepsies) has been linked to a significant loss of benzodiazepine binding sites,31 and the activation of GABA A receptors by various allosteric ligands is crucial for the prevention of complex focal seizures.32 Several substances that either increase the activity of GABA A receptors through binding to allosteric sites (benzodiazepines, barbiturates, cannabidiol) or raise the concentration of GABA in the proximity of its receptors (vigabatrin, which blocks GABA catabolism, and tiagabine, which blocks reuptake) are already established anticonvulsants in clinical practice with marketing authorization for focal epilepsy, among other types. However, owing to the diversity of allosteric sites at GABA A receptors and the great variety of subunit composition, with regional specificities, there are great opportunities to find and develop drugs with even higher efficacy against focal seizuresand a lower incidence of adverse effects.
GABA A Receptor Agonists and Allosteric Modulators in Preclinical Stages of Development
A number of substances may act as GABA A receptor agonists and have the potential to become effective (and safe) antiepileptic drugs if appropriately tested in preclinical and clinical trials. An in silico study using structure-guided virtual screening found 19 compounds capable of activating GABA A receptors and having beneficial ADMET (absorption, distribution, metabolism, elimination, and toxicity) properties similar to already approved antiepileptic drugs.33 However, the screened compounds have not yet been synthesized, or tested in vitro or in animal experiments, so the destiny of these substances is not known.
The amino acid taurine is a weak agonist of GABA A receptors, but its internal activity depends on the subtype of GABA A receptor; that is, on the subunit composition of the receptor. Knowing that there is great regional variability in GABA A receptor subtypes in both animal and human brains, the activity of taurine could be quite specific; this creates an opportunity to use taurine or some of its analogues for the treatment of focal or other types of epilepsy.34 Effective anticonvulsant doses of taurine ranged from 20 to 80 mg/kg when administered intraperitoneally in rats.35 The equivalent human dose would be more than 5 g for an adult person, which may produce toxic effects (decreased blood glucose, somnolence); however, the lethal dose that kills 50% of experimental rodents is as high as 7 g/kg.36 Taurine is present in many energy drinks (about 1 g per serving), but it is unlikely that such a dose could counteract the proconvulsive effects of caffeine (up to 320 mg per serving).37 Whether taurine could become an anticonvulsant is just a hypothesis for now, and elaborate experimental studies are necessary to either confirm or reject it.
It has been confirmed in a number of experimental studies that GABA A receptors that have α1 subunits mediate sedative/ataxic effects of substances, while those that have α2/α3 subunits mediate anticonvulsant effects, primarily owing to the slower decay of inhibitory postsynaptic currents. Following on from this knowledge, many substances with relatively selective modulatory action on GABA A receptors with α2/α3 subunits were synthesized and tested in vitro or in animal studies. The substance designated as HZ-166 was found to be an α2/α3-selective GABA A agonist in vitro, but its low bioavailability after oral or intraperitoneal administration precluded testing of its activity in vivo. However, replacement of the ester radical at the imidazole ring with bioisosteres led to analogues with good bioavailability and unchanged selectivity for receptors with α2/α3 subunits. The analogue KRM-II-81 showed excellent anticonvulsant activity in several animal models of epilepsy (electroshock-induced convulsions in mice, 6 Hz seizure model in mice, pentylenetetrazole (PTZ)-induced seizures in rats, and basolateral amygdala kindling in rats), while its motor-impairing effect was less than that of benzodiazepines.38
Other substances with positive allosteric modulatory action selective for GABA A receptors with α2/α3 subunits that showed anticonvulsant effects in animal models of epilepsy are: AZD7325 (4-amino-8-(2-fluoro-6-methoxy-phenyl)-N-propyl-cinnoline-3-carboxamide), which increased the threshold for hyperthermia-induced seizures in Scn1a+/− mice,39 and PF-06372865 (7-ethyl-4-[3-(4-ethylsulfonyl-2-methoxyphenyl)-4-fluorophenyl]imidazo[4,5-c]pyridazine), which efficiently suppressed spike-and-wave discharges in rats genetically prone to absence seizures (Genetic Absence Epilepsy Rats from Strasbourg [GAERS] strain).40 However, only the latter found its way into clinical trials, and is now known under the name CVL-865.
To avoid some key adverse effects of classic benzodiazepines, such as tolerance and dependence, several partial agonists of benzodiazepine binding sites at GABA A receptors were designed, one of which was successfully developed throughout the preclinical studies for the treatment of focal epilepsy: imepitoin (ELB138). However, when the pharmacokinetics of the drug was tested in phase I studies on humans, the interindividual variability of elimination parameters was unacceptably high, especially when compared between smokers and non-smokers; the company that developed imepitoin decided to proceed with its clinical development in veterinary medicine, for the treatment of canine epilepsy.41 Imepitoin proved to be effective in both newly diagnosed canine epilepsy, as monotherapy, and in chronic canine epilepsy, as add-on therapy.42
Steroid hormones from the circulation easily penetrate the blood–brain barrier and are transformed in the brain to metabolites with positive allosteric activity on GABA A receptors. Allopregnanolone (5α-pregnan-3α-ol-20-one) is derived from progesterone, allotetrahydrodeoxycorticosterone (5α-pregnane-3α,21-diol-20-one) comes from deoxycorticosterone, and androstanediol (5α-androstan-3α,17β-diol) is derived from testosterone; since these substances are synthesized in the brain, they are called “neurosteroids”. Neurosteroids bind preferentially to GABA A receptors that contain the δ subunit, and enhance the action of GABA, behaving as positive allosteric modulators. The anticonvulsant activity of neurosteroids has been demonstrated in a number of animal models of epilepsy: androstanediol protects against seizures elicited by the hippocampus kindling model of epilepsy in mice,43 while allopregnanolone and allotetrahydrodeoxycorticosterone inhibit convulsions induced in animal models by pentylenetetrazol, pilocarpine, bicuculline, or electroshock.44 Lucchi et alshowed that allopregnanolone and pregnanolone levels are reduced in the hippocampus of epileptic rats, but only levels of allopregnanolone correlated significantly with the frequency of seizures.45 After reviewing the role of neurosteroids in epilepsy, Lévesque et al46 concluded that they can modulate both the emergence of seizures and the interictal activity of epileptic foci (frequency of spikes and oscillations); specifically, the onset of spontaneous seizures in mesial temporal lobe epilepsy is delayed after the administration of neurosteroids, as shown in animal models.
There is also clinical evidence of the anticonvulsant action of neurosteroids. Pugnaghi et al reported the case of a woman with temporal lobe epilepsy, who lost control over her seizures after starting therapy for concomitant hirsutism with finasteride, a well-known inhibitor of neurosteroid synthesis; after stopping finasteride, the patient regained seizure control with the original anticonvulsant therapy.47 A case series of 12 patients with protocadherin 19 (PCDH19)-female limited epilepsy (PCDH19-FE) confirmed significantly decreased blood levels of the neurosteroids allopregnanolone, pregnanolone, and pregnenolone sulfate.48 In a controlled study with 41 patients and 41 controls, either non-detectable or significantly decreased allopregnanolone levels were found in cerebrospinal fluid obtained from patients with status epilepticus.49 However, a synthetic neurosteroid derivative, ganaxolone, was more effective than naturally occurring neurosteroids in animal models of epilepsy and had superior pharmacokinetic properties (minor biotransformation); these features have helped its transition to the clinical development phase.
GABA A Receptor Agonists in the Clinical Stage of Development
The benzodiazepine clobazam was first approved for the treatment of severe anxiety, and then repurposed for the adjunctive treatment of resistant focal epilepsy.50 However, only recently have several clinical trials been conducted, aiming to investigate the efficacy and safety of clobazam when used as monotherapy in patients with focal epilepsy. The active metabolite of clobazam, N-desmethyl clobazam, shows partial selectivity for GABA A receptors that contain α2/α3 subunits,39 thus acting as a positive allosteric modulator. A Cochrane review analyzed its effectiveness and safety when used as monotherapy for focal or generalized seizures.51 Only three clinical trials with 206 patients in total fulfilled the minimum requirements for methodological quality to be included in the review. The available evidence failed to show advantages of clobazam over carbamazepine, but it was slightly more effective and safer than phenytoin: the retention rate after 12 months of therapy was higher (relative risk 1.43 [95% CI 1.08–1.90]) and the discontinuation rate due to adverse effects lower (relative risk 0.1 [95% CI 0.01–1.65]).52 The most frequent adverse effects of clobazam are dose-dependent sedation, dizziness, and ataxia; however, it is less sedating than other benzodiazepines, including clonazepam. Serious adverse effects of clonazepam are extremely rare, below 1 per 100,000 patient-years.53
When clobazam was tried as monotherapy in a study with 45 children with Rolandic epilepsy, and compared with carbamazepine, it led to freedom from seizures in 94.1% of patients, while carbamazepine was effective in all investigated patients.54 Clobazam achieved freedom from seizures earlier than carbamazepine (after 33.3 days on average versus 48.2 days with carbamazepine) and had fewer side effects. However, two of the children treated with clobazam experienced worsening of their seizures, and the drug caused disturbances of cognition and behavior.
Ganaxolone is a derivative of the endogenous neurosteroid allopregnanolone, created by methylation of its A-ring at position 3. The methylation prevents biotransformation of the drug and avoids the induction of tolerance, which is a characteristic feature of the parent compound.55 It is an agonist of allosteric neurosteroid binding sites on GABA A receptors (both synaptic and extrasynaptic receptors), which facilitates the inhibitory action of GABA (positive allosteric modulator of GABA). Ganaxolone was the most efficient drug in clinical trials as add-on therapy against partial seizures with or without secondary generalization in adult patients,56 as well as for the treatment of refractory status epilepticus.57 Although it was tested in children with infantile spasms, its beneficial effects were modest and did not reach clinical significance. However, ganaxolone is a remarkably safe drug: the most prominent adverse effect is somnolence, and the vast majority of recorded adverse effects are mild (dizziness, fatigue, headache, impaired coordination), followed by full recovery after discontinuation of ganaxolone; arthralgia and convulsions were recorded very rarely as serious adverse effects in clinical trials and postmarketing studies.58,59
CVL-865 (previously PF-06372865) is a positive allosteric modulator of GABA A receptors, which binds selectively to the receptor subtypes that contain α2, α3, and α5 subunits, while the subtypes with α1 subunits remain completely unaffected.60 Based on its mechanism of action, which is more selective than that of marketed benzodiazepines, CVL-865 is expected to have strong anticonvulsive action with less pronounced sedation and cognitive impairment. It was first tried in adult patients with generalized anxiety disorder (GAD), but its effect in a phase II clinical trial was not differentiated sufficiently from that of placebo, and the trial was ended prematurely.61 The strong anticonvulsant action of CVL-865 is strongly believed in by its developers, especially following the completion of a phase II trial in patients with photosensitive epilepsy. In this small pilot trial, with only seven participants, CVL-865 strongly inhibited the photoparoxysmal response to intermittent photic stimulation (complete inhibition in six out of seven patients), without significant adverse effects.62 However, whether CVL-865 will be of use in patients with focal epilepsy, and what its true safety profile is, are questions that can be answered only after future clinical trials. In a study investigating the analgesic potential of CVL-865, lower doses caused dizziness (39%), fatigue (33%), and bradyphrenia (28%), and higher doses dizziness (53%), somnolence (32%), bradyphrenia (32%), fatigue (26%), balance disorder (26%), and feeling abnormal (26%).63
Padsevonil is a new antiepileptic drug with two mechanisms of action: it binds with high affinity to synaptic vesicle 2 (SV2) proteins and behaves as a partial agonist at the benzodiazepine binding site on GABA A receptors, where it is bound with low to moderate affinity.64 Padsevonil has been tested as add-on therapy in a clinical trial on adult patients with drug-resistant focal epilepsy (four or more focal seizures/week, unresponsive to at least four different antiepileptic drugs). The patients received padsevonil or placebo for 3 weeks in hospital, followed by an 8-week open-label study period when all patients received padsevonil. At discharge from the hospital, 30.8% of patients taking padsevonil were ≥75% responders (the frequency of seizures was reduced by 53.7%), compared to 11.1% taking placebo (the frequency of seizures was reduced by 12.5%). After the treatment outside the hospital ended, 31.4% of patients were ≥75% responders and the frequency of seizures was decreased by 55.2%. This remarkable efficacy was associated with mild adverse effects: somnolence, headache, and dizziness.65 Thanks to the beneficial ratio of efficacy and safety in a subpopulation of difficult-to-treat patients with epilepsy, padsevonil is currently one of the few promising positive allosteric modulators of GABA A receptors that are entering phase III clinical trials, which should reveal its true place in the treatment of focal epilepsy.66
Market Projections
Having been repurposed for the treatment of epilepsy, clobazam was the first among new GABA A agonists to reach the market. It currently has marketing authorization in the UK for add-on treatment of all types of epilepsy (both focal and generalized) in children over 2 years of age and adults.67 Clobazam is also approved in the USA “for the adjunctive treatment of seizures associated with Lennox–Gastaut syndrome (LGS) in patients 2 years of age or older”.68 In the near future, clobazam could be expected to receive marketing authorization for the treatment of epilepsy in the European Union and other large markets, owing to its wide spectrum of action and excellent safety profile.
Ganaxolone is currently in phase III clinical trials for the treatment of early infantile epileptic encephalopathy 2 and status epilepticus, and the developer plans to apply for marketing authorization from the Food and Drug Administration (FDA) and European Medicines Agency (EMA) in the first half of 2021.69 It is also being tested in a phase II trial on protocadherin-19-related childhood epilepsy, where a much better response rate was achieved in patients with decreased plasma allopregnanolone sulfate levels, which may serve as a biomarker if confirmed when the study is completed.70 On the other hand, CVL-865 is still in phase II of clinical development, as phase II clinical trials in patients with focal-onset epilepsy started only in 2020;71 therefore, its marketing authorization cannot be expected within the next 2–3 years.
A phase III clinical trial with padsevonil in adult patients with focal-onset epilepsy was completed in late 2020, but the results are still awaited.72 The developer of padsevonil is not planning to apply for marketing authorization in 2021,73 and further actions in that direction have not yet been announced.
Conclusions
The GABA A receptor is ubiquitous in the central nervous system and there is a huge diversity of receptor subtypes in almost all regions of the brain. Some of newer GABA A receptor agonists are more or less selective for receptor subtypes that contain α2/α3 subunits, which makes them more effective as anticonvulsants and less disadvantageous in regard to sedation and motor impairment. There are several compounds with more selective action on GABA A receptors, perhaps only in certain brain regions, that have the potential to become effective drugs against specific subtypes of focal-onset epilepsy. However, their development needs time, and in the near future we can expect only one or two new GABA A agonists with marketing authorization for focal epilepsy, an advance that would be of use for just a fraction of patients with drug-resistant epilepsy.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chuang S-H, Reddy DS. Genetic and molecular regulation of extrasynaptic GABA-A receptors in the brain: therapeutic insights for epilepsy. J Pharmacol Exp Ther. 2018;364(2):180–197. doi:10.1124/jpet.117.244673
2. Vargas RA. The GABAergic system: an overview of physiology, physiopathology and therapeutics. Int J Clin Pharmacol Pharmacother. 2018;4:143.
3. Hernandez CC, Macdonald RL. A structural look at GABAA receptor mutations linked to epilepsy syndromes. Brain Res. 2019;1714:234–247. doi:10.1016/j.brainres.2019.03.004
4. Abou-Khalil BW. Update on antiepileptic drugs 2019. Continuum. 2019;25(2):508–536. doi:10.1212/CON.0000000000000715
5. Laxer KD, Trinka E, Hirsch LJ, et al. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014;37:59–70. doi:10.1016/j.yebeh.2014.05.031
6. Oller LFV. The prevalence of different types of epilepsy in clinical practice. Rev Neurol. 2002;34(6):526–531.
7. Sigel E, Steinmann ME. Structure, function, and modulation of GABA(A) receptors. J Biol Chem. 2012;287(48):40224–40231. doi:10.1074/jbc.R112.386664
8. Olsen RW, Sieghart W. International union of pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Pharmacol Rev. 2008;60(3):243–260. doi:10.1124/pr.108.00505
9. Sieghart W. Structure, pharmacology, and function of GABAA receptor subtypes. Adv Pharmacol. 2006;54:231–263.
10. Zhu S, Noviello CM, Teng J, et al. Structure of a human synaptic GABAA receptor. Nature. 2018;559(7712):67–72. doi:10.1038/s41586-018-0255-3
11. Chua HC, Chebib M. GABAA receptors and the diversity in their structure and pharmacology. Adv Pharmacol. 2017;79:1–34.
12. Olsen RW. GABAA receptor: positive and negative allosteric modulators. Neuropharmacology. 2018;136(Pt A):10–22.
13. Sieghart W. Chapter three - allosteric modulation of GABAA receptors via multiple drug-binding sites. In: Rudolph U, editor. Advances in Pharmacology [Internet]. Academic Press; 2015: 53–96. (Diversity and functions of GABA receptors: a tribute to Hanns Möhler, part A; vol. 72). Available from: http://www.sciencedirect.com/science/article/pii/S1054358914000374.
14. Lorenz-Guertin JM, Jacob TC. GABA type a receptor trafficking and the architecture of synaptic inhibition. Dev Neurobiol. 2018;78(3):238–270.
15. Olsen RW. Allosteric ligands and their binding sites define γ-aminobutyric acid (GABA) type A receptor subtypes. Adv Pharmacol. 2015;73:167–202.
16. Changeux J-P, Christopoulos A. Allosteric modulation as a unifying mechanism for receptor function and regulation. Diabetes Obes Metab. 2017;19(Suppl 1):4–21. doi:10.1111/dom.12959
17. Krogsgaard-Larsen P, Frølund B, Liljefors T. Specific GABA(A) agonists and partial agonists. Chem Rec. 2002;2(6):419–430. doi:10.1002/tcr.10040
18. Falch E, Jacobsen P, Krogsgaard-Larsen P, Curtis DR. GABA-mimetic activity and effects on diazepam binding of aminosulphonic acids structurally related to piperidine-4-sulphonic acid. J Neurochem. 1985;44(1):68–75. doi:10.1111/j.1471-4159.1985.tb07114.x
19. Hansen SL, Ebert B, Fjalland B, Kristiansen U. Effects of GABA(A) receptor partial agonists in primary cultures of cerebellar granule neurons and cerebral cortical neurons reflect different receptor subunit compositions. Br J Pharmacol. 2001;133(4):539–549. doi:10.1038/sj.bjp.0704121
20. Johnston GAR. Advantages of an antagonist: bicuculline and other GABA antagonists. Br J Pharmacol. 2013;169(2):328–336. doi:10.1111/bph.12127
21. Masiulis S, Desai R, Uchański T, et al. GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature. 2019;565(7740):454–459. doi:10.1038/s41586-018-0832-5
22. Kirkness EF, Turner AJ. The gamma-aminobutyrate/benzodiazepine receptor from pig brain. Enhancement of gamma-aminobutyrate-receptor binding by the anaesthetic propanidid. Biochem J. 1986;233(1):259–264. doi:10.1042/bj2330259
23. Dawson GR, Wafford KA, Smith A, et al. Anticonvulsant and adverse effects of avermectin analogs in mice are mediated through the gamma-aminobutyric acid(A) receptor. J Pharmacol Exp Ther. 2000;295(3):1051–1060.
24. Kent DE, Savechenkov PY, Bruzik KS, Miller KW. Binding site location on GABAA receptors determines whether mixtures of intravenous general anaesthetics interact synergistically or additively in vivo. Br J Pharmacol. 2019;176(24):4760–4772. doi:10.1111/bph.14843
25. Baur R, Gertsch J, Sigel E. The cannabinoid CB1 receptor antagonists rimonabant (SR141716) and AM251 directly potentiate GABA(A) receptors. Br J Pharmacol. 2012;165(8):2479–2484. doi:10.1111/j.1476-5381.2011.01405.x
26. Sigel E, Baur R, Rácz I, et al. The major central endocannabinoid directly acts at GABA(A) receptors. Proc Natl Acad Sci U S A. 2011;108(44):18150–18155. doi:10.1073/pnas.1113444108
27. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58(4):512–521. doi:10.1111/epi.13709
28. Avoli M, D’Antuono M, Louvel J, et al. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog Neurobiol. 2002;68(3):167–207. doi:10.1016/S0301-0082(02)00077-1
29. Curia G, Lucchi C, Vinet J, et al. Pathophysiogenesis of mesial temporal lobe epilepsy: is prevention of damage antiepileptogenic? Curr Med Chem. 2014;21(6):663–688. doi:10.2174/0929867320666131119152201
30. Treiman DM. GABAergic mechanisms in epilepsy. Epilepsia. 2001;42(Suppl 3):8–12. doi:10.1046/j.1528-1157.2001.042suppl.3008.x
31. Fritschy JM, Kiener T, Bouilleret V, Loup F. GABAergic neurons and GABA(A)-receptors in temporal lobe epilepsy. Neurochem Int. 1999;34(5):435–445. doi:10.1016/S0197-0186(99)00040-6
32. Stamboulian-Platel S, Legendre A, Chabrol T, et al. Activation of GABAA receptors controls mesiotemporal lobe epilepsy despite changes in chloride transporters expression: in vivo and in silico approach. Exp Neurol. 2016;284(Pt A):11–28. doi:10.1016/j.expneurol.2016.07.009
33. Mehta P, Srivastava S, Sharma M, et al. Identification of chemically diverse GABAA agonists as potential anti-epileptic agents using structure-guided virtual screening, ADMET, quantum mechanics and clinical validation through off-target analysis. Int J Biol Macromol. 2018;119:1113–1128. doi:10.1016/j.ijbiomac.2018.08.032
34. Ochoa-de la Paz L, Zenteno E, Gulias-Cañizo R, Quiroz-Mercado H. Taurine and GABA neurotransmitter receptors, a relationship with therapeutic potential? Expert Rev Neurother. 2019;19(4):289–291. doi:10.1080/14737175.2019.1593827
35. Li Q, Guo J-C, Jin H-B, et al. Involvement of taurine in penicillin-induced epilepsy and anti-convulsion of acupuncture: a preliminary report. Acupunct Electrother Res. 2005;30(1–2):1–14. doi:10.3727/036012905815901325
36. Taranukhin AG, Saransaari P, Kiianmaa K, et al. Comparison of toxicity of taurine and GABA in combination with alcohol in 7-day-old mice. Adv Exp Med Biol. 2017;975(Pt 2):1021–1033.
37. Curran CP, Marczinski CA. Taurine, caffeine, and energy drinks: reviewing the risks to the adolescent brain. Birth Defects Res. 2017;109(20):1640–1648. doi:10.1002/bdr2.1177
38. Witkin JM, Smith JL, Ping X, et al. Bioisosteres of ethyl 8-ethynyl-6-(pyridin-2-yl)-4H-benzo[f]imidazo [1,5-a][1,4]diazepine-3-carboxylate (HZ-166) as novel alpha 2,3 selective potentiators of GABAA receptors: improved bioavailability enhances anticonvulsant efficacy. Neuropharmacology. 2018;137:332–343. doi:10.1016/j.neuropharm.2018.05.006
39. Nomura T, Hawkins NA, Kearney JA, et al. Potentiating α2 subunit containing perisomatic GABAA receptors protects against seizures in a mouse model of Dravet syndrome. J Physiol. 2019;597(16):4293–4307. doi:10.1113/JP277651
40. Duveau V, Buhl DL, Evrard A, et al. Pronounced antiepileptic activity of the subtype-selective GABAA -positive allosteric modulator PF-06372865 in the GAERS absence epilepsy model. CNS Neurosci Ther. 2019;25(2):255–260. doi:10.1111/cns.13046
41. Rundfeldt C, Löscher W. The pharmacology of imepitoin: the first partial benzodiazepine receptor agonist developed for the treatment of epilepsy. CNS Drugs. 2014;28(1):29–43. doi:10.1007/s40263-013-0129-z
42. Löscher W, Potschka H, Rieck S, et al. Anticonvulsant efficacy of the low-affinity partial benzodiazepine receptor agonist ELB 138 in a dog seizure model and in epileptic dogs with spontaneously recurrent seizures. Epilepsia. 2004;45(10):1228–1239. doi:10.1111/j.0013-9580.2004.21204.x
43. Reddy DS, Jian K. The testosterone-derived neurosteroid androstanediol is a positive allosteric modulator of GABAA receptors. J Pharmacol Exp Ther. 2010;334(3):1031–1041. doi:10.1124/jpet.110.169854
44. Miziak B, Chrościńska-Krawczyk M, Czuczwar SJ. Neurosteroids and seizure activity. Front Endocrinol. 2020;11:541802. doi:10.3389/fendo.2020.541802
45. Lucchi C, Costa AM, Rustichelli C, Biagini G. Allopregnanolone and pregnanolone are reduced in the hippocampus of epileptic rats, but only allopregnanolone correlates with the seizure frequency. Neuroendocrinology. 2020. doi:10.1159/000509093
46. Lévesque M, Biagini G, Avoli M. Neurosteroids and focal epileptic disorders. Int J Mol Sci. 2020;21(24):9391. doi:10.3390/ijms21249391
47. Pugnaghi M, Monti G, Biagini G, Meletti S. Temporal lobe epilepsy exacerbation during pharmacological inhibition of endogenous neurosteroid synthesis. BMJ Case Rep. 2013;2013(feb19 1):bcr2012008204. doi:10.1136/bcr-2012-008204
48. Trivisano M, Lucchi C, Rustichelli C, et al. Reduced steroidogenesis in patients with PCDH19-female limited epilepsy. Epilepsia. 2017;58(6):e91–5. doi:10.1111/epi.13772
49. Meletti S, Lucchi C, Monti G, et al. Decreased allopregnanolone levels in cerebrospinal fluid obtained during status epilepticus. Epilepsia. 2017;58(2):e16–20. doi:10.1111/epi.13625
50. Pernea M, Sutcliffe AG. Clobazam and its use in epilepsy. Pediatr Rep. 2016;8(2):6516. doi:10.4081/pr.2016.6516
51. Arya R, Giridharan N, Anand V, Garg SK. Clobazam monotherapy for focal or generalized seizures. Cochrane Database Syst Rev. 2018;11(7):CD009258.
52. Gimigliano F. Is clobazam monotherapy effective and safe in people with focal or generalized seizures? A cochrane review summary with commentary. Dev Med Child Neurol. 2020;62(6):670–672. doi:10.1111/dmcn.14539
53. Gauthier AC, Mattson RH. Clobazam: a safe, efficacious, and newly rediscovered therapeutic for epilepsy. CNS Neurosci Ther. 2015;21(7):543–548. doi:10.1111/cns.12399
54. Andrade R, García-Espinosa A, Machado-Rojas A, et al. A prospective, open, controlled and randomised study of clobazam versus carbamazepine in patients with frequent episodes of rolandic epilepsy. Rev Neurol. 2009;49(11):581–586.
55. Vidaurre J, Herbst J. New antiepileptic drugs. Medicina (Mex). 2019;79(Suppl 3):48–53.
56. Jankovic S, Lukic S. Antiepileptic potential of ganaxolone. Vojnosanit Pregl. 2016;74:157.
57. Bialer M, Johannessen SI, Koepp MJ, et al. Progress report on new antiepileptic drugs: a summary of the fifteenth eilat conference on new antiepileptic drugs and devices (EILAT XV). II. Drugs in more advanced clinical development. Epilepsia. 2020;61(11):2365–2385. doi:10.1111/epi.16726
58. Ligsay A, Van Dijck A, Nguyen DV, et al. A randomized double-blind, placebo-controlled trial of ganaxolone in children and adolescents with fragile X syndrome. J Neurodev Disord. 2017;9(1):26. doi:10.1186/s11689-017-9207-8
59. Sperling MR, Klein P, Tsai J. Randomized, double-blind, placebo-controlled Phase 2 study of ganaxolone as add-on therapy in adults with uncontrolled partial-onset seizures. Epilepsia. 2017;58(4):558–564. doi:10.1111/epi.13705
60. Boada CM, French JA, Dumanis SB. Proceedings of the 15th antiepileptic drug and device trials meeting: state of the science. Epilepsy Behav. 2020;111:107189. doi:10.1016/j.yebeh.2020.107189
61. Simen A, Whitlock M, Qiu R, et al. An 8-week, randomized, phase 2, double-blind, sequential parallel-group comparison study of two dose levels of the GABAA positive allosteric modulator PF-06372865 compared with placebo as an adjunctive treatment in outpatients with inadequate response to standard of care for generalized anxiety disorder. J Clin Psychopharmacol. 2019;39(1):20–27. doi:10.1097/JCP.0000000000000997
62. Gurrell R, Gorman D, Whitlock M, et al. Photosensitive epilepsy: robust clinical efficacy of a selective GABA potentiator. Neurology. 2019;92(15):e1786–95. doi:10.1212/WNL.0000000000007271
63. van Amerongen G, Siebenga PS, Gurrell R, et al. Analgesic potential of PF-06372865, an α2/α3/α5 subtype-selective GABAA partial agonist, in humans. Br J Anaesth. 2019;123(2):e194–203. doi:10.1016/j.bja.2018.12.006
64. Wood M, Daniels V, Provins L, et al. Pharmacological profile of the novel antiepileptic drug candidate padsevonil: interactions with synaptic vesicle 2 proteins and the GABAA receptor. J Pharmacol Exp Ther. 2020;372(1):1–10. doi:10.1124/jpet.119.261149
65. Muglia P, Hannestad J, Brandt C, et al. Padsevonil randomized phase IIa trial in treatment-resistant focal epilepsy: a translational approach. Brain Commun. 2020;2(2):fcaa183. doi:10.1093/braincomms/fcaa183
66. Mula M. Emerging drugs for focal epilepsy. Expert Opin Emerg Drugs. 2018;23(3):243–249. doi:10.1080/14728214.2018.1527903
67. Clobazam wockhardt 1mg/ml oral suspension - summary of product characteristics (SmPC) - (emc) [Internet]. [
68. Clobazam oral suspension - FDA prescribing information, side effects and uses [Internet]. Drugs.com. [
69. Marinus launches expanded access program for ganaxolone treatment in CDKL5 deficiency disorder [Internet]. [
70. Sullivan J, Specchio N, Chez MG, et al. Preliminary evidence of a predictive clinical biomarker in pcdh19-related epilepsy: significant treatment effect of ganaxolone in biomarker-positive patients. Annual meeting abstracts [Internet]. American Epilepsy Society; 2018 [
71. Cerevel Therapeutics, LLC. A randomized, double-blind, placebo-controlled, parallel group, multicenter trial of CVL-865 as adjunctive therapy in adults with drug-resistant focal onset seizures (REALIZE trial) [Internet]. clinicaltrials.gov; 2020 [
72. UCB Biopharma SRL. A multicenter, randomized, double-blind, placebo-controlled, parallel-group study to evaluate the efficacy and safety of padsevonil as adjunctive treatment of focal-onset seizures in adult subjects with drug-resistant epilepsy [Internet]. clinicaltrials.gov; 2020 [
73. Key events – UCB integrated annual report 2019 [Internet]. Integrated annual report; 2019 [
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.