Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Experimental design and optimization of raloxifene hydrochloride loaded nanotransfersomes for transdermal application
Authors Mahmood S, Taher M, Mandal UK
Received 2 April 2014
Accepted for publication 2 June 2014
Published 12 September 2014 Volume 2014:9(1) Pages 4331—4346
DOI https://doi.org/10.2147/IJN.S65408
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Syed Mahmood, Muhammad Taher, Uttam Kumar Mandal
Department of Pharmaceutical Technology, Kulliyyah of Pharmacy, International Islamic University Malaysia (IIUM), Pahang Darul Makmur, Malaysia
Abstract: Raloxifene hydrochloride, a highly effective drug for the treatment of invasive breast cancer and osteoporosis in post-menopausal women, shows poor oral bioavailability of 2%. The aim of this study was to develop, statistically optimize, and characterize raloxifene hydrochloride-loaded transfersomes for transdermal delivery, in order to overcome the poor bioavailability issue with the drug. A response surface methodology experimental design was applied for the optimization of transfersomes, using Box-Behnken experimental design. Phospholipon® 90G, sodium deoxycholate, and sonication time, each at three levels, were selected as independent variables, while entrapment efficiency, vesicle size, and transdermal flux were identified as dependent variables. The formulation was characterized by surface morphology and shape, particle size, and zeta potential. Ex vivo transdermal flux was determined using a Hanson diffusion cell assembly, with rat skin as a barrier medium. Transfersomes from the optimized formulation were found to have spherical, unilamellar structures, with a homogeneous distribution and low polydispersity index (0.08). They had a particle size of 134±9 nM, with an entrapment efficiency of 91.00%±4.90%, and transdermal flux of 6.5±1.1 µg/cm2/hour. Raloxifene hydrochloride-loaded transfersomes proved significantly superior in terms of amount of drug permeated and deposited in the skin, with enhancement ratios of 6.25±1.50 and 9.25±2.40, respectively, when compared with drug-loaded conventional liposomes, and an ethanolic phosphate buffer saline. Differential scanning calorimetry study revealed a greater change in skin structure, compared with a control sample, during the ex vivo drug diffusion study. Further, confocal laser scanning microscopy proved an enhanced permeation of coumarin-6-loaded transfersomes, to a depth of approximately160 µM, as compared with rigid liposomes. These ex vivo findings proved that a raloxifene hydrochloride-loaded transfersome formulation could be a superior alternative to oral delivery of the drug.
Keywords: transfersomes, raloxifene hydrochloride, RSM, Box-Behnken, transdermal flux
Introduction
Orally administered drugs experience a hostile environment in the gastrointestinal tract, where most drugs are degraded in variable pH conditions, or face solubility issues, and (most importantly) first-pass metabolism.1 A transdermal drug delivery system overcomes many of the problems associated with oral drug delivery, and offers several potential advantages, like avoidance of first-pass metabolism, predictable and extended drug action with minimum side effects, improved physiological and pharmacological responses, reduced frequency of drug administration, avoidance of plasma drug level fluctuation, etc.2,3 To deliver the drug through stratum corneum (SC), overcoming its barrier properties to achieve high drug permeability and therapeutic action, lipid-based nanovesicles, which are composed of phospholipid (as a backbone), offer an excellent mode for transdermal delivery, owing to their biocompatibility with skin lipids.4 Liposomes, with phospholipid and cholesterol as structural components, were the first lipid-based carriers to prove increased drug deposition in epidermis as well as dermis, but failed to deliver drugs at therapeutic concentrations into systemic circulation.5 For increased and better systemic absorption of drugs, extensive research in this field has led to the development of a new, lipid-based carrier, transfersomes, which are basically made of phospholipid and surfactant. Because of their ultradeformable and elastic nature, transfersomes result in an increased penetration of drugs through SC, when they are applied under nonoccluded conditions.6 Biocompatible surfactant, also called edge activator, destabilizes the lipid bilayer and causes an increase in its fluidity and elasticity, allowing dramatically improved delivery of the encapsulated agents to and through the skin.7,8 Surfactant is solely responsible for the flexibility of the transfersomes and imparts better hydration properties and extraordinary skin penetration ability. Cevc and Blume reported the permeation mechanism of transfersomes as hydrotaxis (xerophobia) which is moisture-seeking tendency of transfersomes towards the deeper part of skin instead of outer dry surroundings caused by evaporation of moisture from the transfersomes formulation when it is applied to the skin.9 Many authors confirm ex vivo and in vivo success of transfersomes over conventional liposomes, with a wide range of drugs.10
Osteoporosis, commonly known as a “silent disease”, is a progressive medical condition, in which bone density slowly decreases with advancing age. As per report of a WHO Scientific Group, Geneva, World Health Organization, 2003 (WHO Technical Report Series, No. 921), osteoporosis-related fractures have been identified as a major health problem to the older population. With an aging population, the burden of osteoporosis is expected to continue to rise worldwide. With higher socioeconomic status and increasing urbanization in Asian countries, it is estimated that by 2050, more than 50% of the world’s hip fractures will occur in Asia. According to a report, worldwide osteoporosis causes more than 8.9 million fractures annually, resulting in an osteoporotic fracture every 3 seconds.11 According to a 2007 WHO technical report, osteoporosis is estimated to affect 200 million women worldwide, mostly those older than 60.12 Thus, prevention of osteoporosis and its associated fractures will be important in controlling health costs in the future. Apart from the burden of osteoporosis, according to the latest report (12 December 2013, Press release No 223) from the International Agency for Research on Cancer (IARC), the specialized cancer agency of the World Health Organization, breast cancer is the most frequently-diagnosed cancer, and the most common cause of death among women. According to the American Cancer Society’s report, around 232,340 new cases of invasive breast cancer in women were identified in the United States in 2013.13 Based upon a careful risk assessment, health care providers sometimes recommend therapy with one of several medications to reduce the chance of developing breast cancer, in women with increased risk.
Raloxifene hydrochloride, a second-generation selective estrogen receptor modulator, is used in the prevention and treatment of osteoporosis and invasive breast cancer in postmenopausal women. Being an estrogen agonist/antagonist, it mainly increases bone mineral density and decreases low-density lipoprotein cholesterol. It provides an antiestrogen effect on the breast, and reduces the risk of invasive breast cancer in postmenopausal women.14,15 Additionally, it serves as a substitute for long-term female hormone replacement therapy. It is mostly supplied as 60 mg tablets for daily dose. However, the major obstacle for oral delivery of raloxifene is its poor systemic exposure, with only 2% absolute bioavailability, because of its poor solubility in aqueous fluids and its extensive first-pass metabolism.16 Additionally, the commonly-reported adverse events associated with high and regular oral dosing of raloxifene hydrochloride are: hot flushes (flashes), leg cramps, and peripheral edema. Thromboembolic events and increased risk of fatal stroke are rarely-occurring events.16 So, there is huge scope to improve the bioavailability and avoid the commonly-occurring adverse events associated with oral medication, by delivering it through a transdermal route, as this might reduce dose without compromising therapeutic efficacy.
Thus, the aim of the present study was to develop and optimize a nanotransfersome formulation of raloxifene hydrochloride, by response surface methodology, in order to enhance percutaneous permeation and bypass the issue of poor oral bioavailability.
Materials and methods
Materials
Raloxifene hydrochloride was purchased from Apollo Health Care Limited (People’s Republic of China). Phospholipon® 90G (PC90G) was obtained as a generous gift sample from Lipiod GmbH, (Ludwigshafen, Germany). Sodium deoxycholate (SDC) (99.8%) and coumarin-6 were bought from Sigma-Aldrich (St Louis, MO, USA) and Thermo Fisher Scientific (Waltham, MA, USA), respectively. Chloroform, methanol, ethanol, and Triton® X-100 were received from Merck KGaA (Darmstadt, Germany). Acetonitrile, for high-performance liquid chromatography analysis, was obtained from Thermo Fisher Scientific. Polycarbonate membrane (Titan3™), of 0.45 μM and 0.22 μM size, was supplied by Thermo Fisher Scientific. All other reagents used in this study were of analytical grade. Ultrapure distilled water (Kulliyyah of Pharmacy, International Islamic University Malaysia, Pahang, Malaysia) was used throughout the experiment.
Formulation of drug loaded transfersomes and conventional liposomes
Raloxifene hydrochloride-loaded transfersomal vesicles (RTVs) were prepared using the conventional thin-layer rotary evaporation method, with slight modification in order to suit the existing instrumental facility.17 Briefly, PC90G, drug, and SDC were dissolved in a round-bottom flask with a mixture of chloroform and methanol (2:1, by volume). The organic solvent mixture was evaporated by rotary evaporator (Rotavapor® R-205; Buchi Labortechnik AG, Flawil, Switzerland) under reduced pressure at 40°C. The round-bottom flask was kept under vacuum overnight, to remove remaining solvent trace. The lipid film layer deposited onto the inner surface of the flask after solvent evaporation was allowed to hydrate for 1 hour in a saline phosphate buffer (PBS) (pH 6.5) at 41°C–44°C and 120 rpm. The suspended vesicles were furthered allowed to swell for 2–3 hours at room temperature, which resulted in large multilamellar vesicles. These were sonicated for 20–30 minutes using an ULTRASonic 28X (Neytech, Yucaipa, CA, USA), at 325 W power, to reduce their size further. These small vesicles were passed through 450 nM and 220 nM pore size polycarbonate membranes (Titan3™), to obtain final drug-loaded transfersomal vesicles, which were then stored under refrigeration (2°C–8°C) for further characterization. The drug-loaded liposomal formulation used as control for transdermal flux was prepared using the method described by Touitou et al.18 Similarly, coumarin-6 (0.03%, weight to volume) loaded transfersomes and liposomes were formulated to visualize drug permeation by confocal laser scanning microscopy.
Optimization of transfersomes formulation
A three-factor, three-level, Box-Behnken design (BBD) was applied, to optimize transfersome formulation, using response surface methodology (RSM). The amount of phospholipid (A), surfactant (B), and sonication time (C) were selected as independent variables, considering their overall effects on the formulation’s stability and transdermal flux, as reported by many researchers.19–21 Accordingly, entrapment efficiency (EE) (Y1), vesicle size (Y2 ), and transdermal flux (Y3 ) were selected as dependent variables for the optimization process. After a significant number of preliminary studies, the amount of PC90G (A), SDC (B), and sonication time (C) were selected in the range of 100–300 mg, 15–55 mg, and 15–35 minutes, respectively, as independent variables for raloxifene hydrochloride loaded transfersomes formulation optimization by RSM (Table 1). Seventeen trial formulations of various factor combinations, including five center points, were prepared according to the BBD, as shown in Table 2. All other formulation and processing variables were kept constant throughout the study.
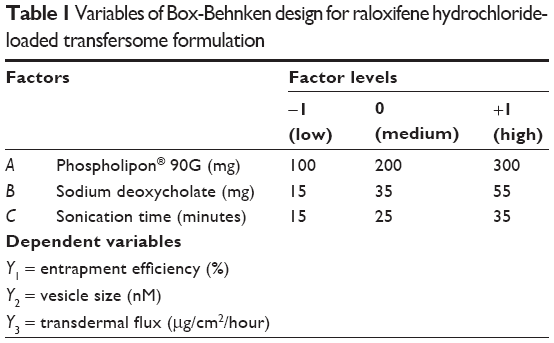 | Table 1 Variables of Box-Behnken design for raloxifene hydrochloride-loaded transfersome formulation |
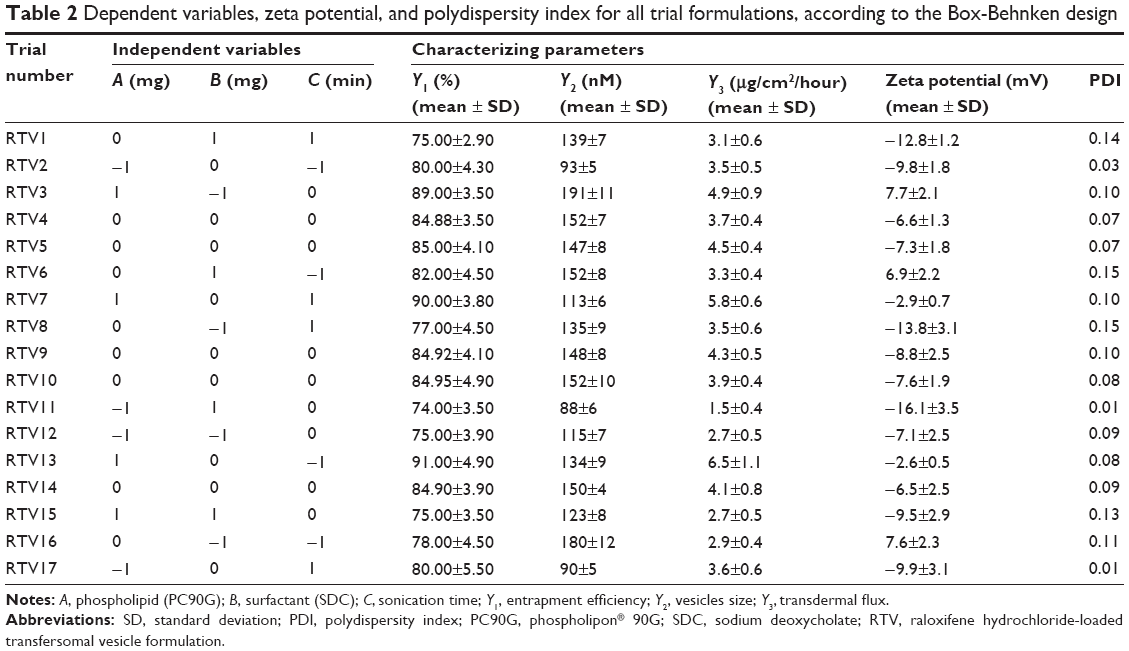 | Table 2 Dependent variables, zeta potential, and polydispersity index for all trial formulations, according to the Box-Behnken design |
Data analysis and validation of RSM model
A three-factor, three-level BBD was adopted for the present RSM technique of formulation optimization. Various RSM computations were performed using Design-Expert® version 8.0.7.1 software (Stat-Ease Inc, Minneapolis, MN, USA). Polynomial models, including interaction and quadratic terms, were generated for all dependent variables using a multiple linear regression analysis (MLRA) approach. The general form of the MLRA model is represented by the following equation,
| (1) |
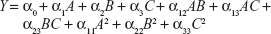
where, Y is the measured response associated with each factor level combination; α0 is a constant value that represents the arithmetic average of all quantitative outcomes of 17 experimental trials; α1, α2, and α3 are linear coefficients computed from the observed experimental response values of Y; α12, α13, α23 are interaction coefficients between two factors; and α11, α22, α33 are their quadratic coefficients.22 Three-dimensional response surface graphs and two-dimensional contour plots were generated for individual response variables, in order to evaluate the effects of individual independent variables on response variables. The developed model was further validated, based on theoretical versus predicted values and their corresponding residual plot. Finally, the best formulation was selected using the point prediction method of the software, utilizing constrains of response values. The optimized formulation was further characterized by vesicular size, size distribution, shape, surface morphology, zeta potential, differential scanning calorimetry (DSC) study, and confocal laser scanning microscopy (CLSM) study.
Vesicular size, zeta potential, and size distribution
Vesicle size and size distribution are important parameters for drug permeation through SC. Measurement of size and zeta potential, for all the trial formulations, was carried out in three replicates, using a particle size analyser (Zetamaster ZEM 5002 and Zetasizer Nano-Z; Malvern Instruments, Malvern, UK), applying dynamic light scattering techniques. The vesicular formulations were mixed with deionized water, in a ratio of 0.4 mL in 10 mL of deionized water, before taking the measurement using the particle size analyser in a multimodal mode.18,23 The polydispersity index (PDI) was used as a measure of size distribution, which was measured in triplicate. A PDI value of less than 0.1 was considered as a homogeneous distribution of vesicles, whereas a PDI value of greater than 0.3 was of higher heterogeneity.21
Vesicular shape and surface morphology
Transmission electron microscopy (TEM) was used for visualization of transfersomes (Libra® 120 Plus; Carl Zeiss Microscopy LLC, Thornwood, NY, USA). Vesicles were examined by the negative staining method. A drop of the vesicular system was placed on to a film-coated copper grid of 300 mesh size, with a diameter of 3.05 mm, to form a thin film, before the specimen was dried on the grid. Then, the film was negatively stained with a drop of 1% phosphotungstic acid. Excess solution was removed with filter paper. The grid was allowed to air-dry thoroughly and was then viewed through TEM at 20–80 K-fold enlargement, at an accelerating voltage of 120 kV.23
Morphology of the vesicles was visualized by scanning electron microscopy (SEM). One drop of transfersomal preparation was spread onto a glass slide and was allowed to dry. Once dried, the specimen was gold-coated using a cool sputter coater (Leica EM SCD005, Leica Microsystems, Wetzlar, Germany), and viewed under a scanning electron microscope (EVO series, Carl Zeiss Microscopy) at an accelerating voltage of 10 KV.
Content of raloxifene hydrochloride by high-performance liquid chromatography (HPLC)
The amount of raloxifene present in the transfersome formulation was determined by an HPLC method, as earlier reported by Trontelj et al with slight modification.24 Transfersome vesicles from 15 mL of formulation were separated by high-speed cold centrifugation. After that, the obtained vesicles were disrupted with 50 mL of 50% methanol, in order to release the entrapped drug into the solvent. The sample was then filtered through a 0.45 μM membrane filter, followed by 20-times dilution with mobile phase, and injection into the HPLC system (Agilent 1200 Compact LC; Agilent Technologies, Santa Clara, CA, USA), which was equipped with an ultraviolet detector. An Agilent Eclipse XDB-C18 column (150 mM ×4.6 mM; 5 μM particle size) (Agilent Technologies) was used for chromatographic separation. The mobile phase comprised of an isocratic mixture of acetonitrile and pH 3.0 buffer solution of orthophosphoric acid (33:67, by volume), delivered at a flow rate of 1 mL per minute. The buffer solution was prepared daily, filtered through a 0.45 μM membrane filter, and degassed before use. Analysis was carried out at 287 nM at 25°C. The injection volume was 10 μL, and the relative retention time of raloxifene was found to be 4.68±0.5 minutes. A six-point standard curve in the range of 0.125–5.0 μg/mL was prepared, taking the peak area of the drug versus the concentration (μg/mL), using a weighted linear least-squares regression (1/concentration2) as the mathematical model. The prepared standard curve was linear, and the equation was y =76.082×+8.466, with a correlation coefficient of 0.999.
Drug entrapment efficiency
The drug entrapment efficiency of the transfersome formulation was determined by an indirect method. The transfersome formulation was centrifuged at 22,000 rpm for 30 minutes at a temperature of 4°C, to separate the vesicles from the unentrapped drug. Separated vesicles were washed again with phosphate buffer, to remove further any unentrapped drug attached to the vesicles. The resulting supernatant liquid was then analysed by the above-described HPLC method. Drug entrapment efficiency was calculated from the following equation,
| (2) |
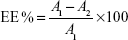
where, A1 is the amount of drug added initially, A2 is the amount of drug determined in the supernatant, and A1−A2 is the amount of drug entrapped in the formulation.
DSC study of transfersomal system treated and untreated skin
Freshly scarified rat skin was used for this study. The skin sample, with the SC facing up, was incubated in a petri dish over a Whatman® filter paper saturated with 0.1% (weight to volume) trypsin in 0.5% (weight to volume) sodium bicarbonate solution at 37°C±1°C for 3 hours.25 The SC was detached from the rest of the skin, thoroughly washed, and kept for drying in a vacuum dessicator. Around 20 mg of dried SC sample was taken and allowed to hydrate in saturated sodium sulphate solution for 5 days. After a water gain of approximately 20%–25%, the SC was treated with optimized transfersomal formulation for 12 hours. A control sample was prepared in a similar way, without any formulation treatment. The SC sample was washed with distilled water, and excess water was removed with tissue paper. A sample SC section of 5 mg was cut, sealed in aluminum hermetic pans and scanned using DSC (DSC1; Mettler-Toledo LLC, Columbus, OH, USA) at a scanning rate of 5°C per minute over the range of 30°C–150°C. The changes in structure of the SC were assessed from the obtained thermograms.25
| (3) |

Ex vivo permeation and deposition study
Albino Wistar rats (80–120 gm) were obtained from the Faculty of Pharmacy, International Islamic University, Kuantan, Malaysia. Hair on the outer skin surface was removed with dissecting scissors, as close as possible to the skin, without harming the SC. The majority of subcutaneous tissue was removed with scissors. The adhering fatty layer and connective tissue layer were cleaned with isopropyl alcohol. The skin was washed with 0.9% sodium chloride solution, wrapped in aluminium foil, and stored in a deep freezer at −40°C, for a maximum of 1 month, for further use.
A Hanson diffusion cell assembly, with an effective permeation area of 1.5 cm2, receptor volume of 6 mL, and rat skin as a barrier medium, was utilized to measure in vitro drug release and its permeation from the transfersomal formulation. The receptor chamber was filled with a receptor medium composed of ethanol and PBS (40:60, by volume) (pH 7.4), maintained at 37°C±1°C, and stirred at 100 rpm. The skin sample was checked thoroughly, to ensure that it was free from any kind of defect that might influence the results of the study. On the day of the experiment, the skin sample was taken out from deep freeze and thawed at room temperature. Prior to the study, the skin sample was equilibrated for 30 minutes in 0.9% sodium chloride solution.
The skin sample was fixed over the diffusion cell so that the dermis side faced the receptor compartment, in contact with receptor medium, while the SC side faced the donor compartment, for formulation application. An amount of therapeutic transdermal daily dose was calculated and an amount of formulation equivalent to 1.2 mg of raloxifene hydrochloride was administered in the donor compartment. Samples of 200 μL were withdrawn from the receptor compartment, via the sample port, at 0.5, 1, 2, 4, and 6 hours. Immediately after sampling, the receptor compartment was compensated with an equal volume of fresh receptor medium, to maintain a sink condition throughout the study period. The permeation sample was analyzed by HPLC. A similar procedure was performed with the conventional liposome formulation, and with an ethanolic PBS of pure raloxifene hydrochloride. Once the permeation study was completed, the skin sample attached to the diffusion cell was removed and washed thoroughly in running tap water. The skin sample was cleaned, if required, with tissue paper, to remove any trace of the formulation attached to the skin. The skin was then cut into small pieces and sonicated with 10 mL of methanol for 30 minutes, to extract the drug retained in the skin. The formulation thus obtained was centrifuged for 10 minutes at 4,000 rpm. The supernatant liquid was separated, passed through a 0.45 μM nylon membrane filter, and analyzed using HPLC. The transdermal flux of tested formulations was calculated and further used to determine the enhancement ratio (ER), which is defined as the extent of enhancement of transdermal flux of the tested formulations, compared with that of the control formulation.
| (4) |

CLSM study
CLSM can be used to visualize improved skin delivery and skin structure. Furthermore, it allows the skin structure to be studied in three dimensions with very high accuracy.8 Three types of drug-loaded formulation (optimized transfersome formulation, conventional liposome, and ethanolic PBS solution), each of them tagged with coumarin-6 as a fluorescent marker, were used for a comparative study. Each sample was subjected to ex vivo study, nonocclusively, on a Hanson diffusion cell assembly (Hanson Research Corporation, Chatsworth, CA, USA) for 8 hours. At the end of the diffusion study, the skin samples were removed from the experimental setup and washed in running tap water, to remove any sample deposit. The skin samples – specifically the treatment area – were cut and sliced in sections of 5–10 μM thickness, and CLSM was used to optically scan through the z-axis of a confocal laser microscope (Leica TCS SPE, Leica Microsystems), using an argon beam at an excitation wavelength of 488 nM and emission wavelength of 502 nM. Relative fluorescence intensity of the probe in the skin samples was detected using Leica LAS AF advanced fluorescence software.
Results and discussion
Preparation of raloxifene hydrochloride-loaded transfersomes
The conventional rotary evaporation sonication method was used to prepare the transfersomes. The method was selected because the formation of the thin film takes place on a large surface area, with a complete hydration of the vesicles; this leads to improvement in entrapment efficiency.17 The basic composition of the transfersome formulations comprised of a phospholipid (PC90G), as a membrane forming lipid, and a surfactant (SDC), as an edge activator, to impart elasticity to the vesicle membrane. As per the BBD design, 17 RTV formulations were prepared using different concentrations of PC90G, surfactant, and different sonication times (Table 2). The therapeutic transdermal daily dose of the drug was calculated according to the equation given by Vaddi et al26
| (5) |
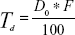
where, Td = therapeutic transdermal daily dose, F = bioavailability (as a percentage, after oral administration), and D0 = oral daily dose. Considering a 60 mg oral dose of raloxifene hydrochloride tablets, 2% oral bioavailability and 10%–15% transdermal absorption capacity of the applied dose, 15 mg of drug was loaded for all 17 formulations.
Working ranges of the independent variables
Preliminary trials were carried out (data not shown) to determine the probable ranges of the independent variables, which provide extreme effects on various dependent variables selected for this study. With respect to the formation of lipid vesicles, most of the available literature reported concentration ranges of phospholipid to surfactant in ratios of 75:25, 80:20, 85:15, 90:10, etc.17,18,20,24 However, the ratio of phospholipid and surfactant used in this study was optimized, based on the initial experiments and their observations (data not shown). PC90G concentrations less than 100 mg produced transfersomes with poor values of transdermal flux, whereas PC90G concentrations greater than 300 mg resulted in larger size vesicles, due to aggregation.20 In the case of the surfactant, it was observed that too low or too high a concentration of SDC was unsuitable for proper vesicle formation.19 In both cases, less than 15 mg or more than 55 mg of surfactant resulted in very low entrapment efficiency, as well as low transdermal flux. Vesicle size, which depends significantly on the sonication time, affects transdermal flux and skin deposition, as studied by Verma et al.21 In line with that report, we found that sonication time of less than 15 minutes was not sufficient for formation of transfersome vesicles with high concentration of phospholipid (about 300 mg) and the desired size was not achieved, due to aggregation of vesicles. Thirty-five minutes of sonication was too long; in one of the studies, a PC90G concentration of 100 mg and a surfactant concentration of 55 mg resulted in leaking of vesicles, with very low entrapment efficiency.
Mathematical modelling for RSM optimization
In order to optimize the raloxifene-loaded transfersome formulation, relationships among independent variables, such as PC90G (A), SDC (B), and sonication time (C), at three levels (−1, 0, +1), with dependent variables, such as entrapment efficiency (Y1), vesicles size (Y2), and transdermal flux (Y3), were evaluated by BBD, using Design Expert® software. The obtained response values of the individual trial formulations were fitted into the model matrix, in order to find the best-fitted model. After due consideration, the best model for all three dependent variables was found to be quadratic. Mathematical relationships generated using MLRA for the studied dependent variables are expressed in the following equations (6–8).
| (6) |
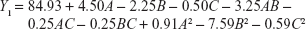
|
|
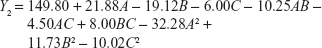
| (8) |
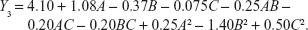
For estimation of the significance of the model, analysis of variance was performed using Design Expert® software (Table 4). Using a 5% significance level, a model is considered significant if the P-value (significance probability value) is less than 0.05.27 Most of the terms in Table 4, including the individual model, were found to be significant (P<0.05), except for a few terms (shown in bold). A positive coefficient represents a synergistic effect of the factor on the response, while a negative coefficient indicates an antagonistic effect. From the above equations (6–8), it is evident that independent variable A (first-order) and intercept have positive effects for all three responses. It is sugested that concentration of phospholipid (A) has a prominent effect on individual dependent variables. On the other hand, the other first-order main terms (B and C), interaction terms and, higher-order terms (except A2, in equation 6, B2 and BC, in equation 7, and A2 and C2, in equation 8) have negative effects on all of the dependent variables.
Effect of independent factors on entrapment efficiency (Y1)
The effects of the independent variables on entrapment efficiency are represented by three-dimensional response surface graphs and their corresponding contour plots (Figure 1). As is evident from these graphs and plots, entrapment efficiency of the drug increased significantly with increase of SDC concentration from 15 mg to 35 mg (by weight). However, further increase of SDC, from 35 mg to 55 mg (by weight), resulted in decreased entrapment efficiency. The maximum entrapment efficiency obtained was 91%±4.9%, for RTV13, while the minimum value was 74%±3.5%, for RTV11 (Table 2). PC90G and SDC at 300:35 ratio (by weight) showed maximum entrapment efficiency, while a 100:55 ratio of the same showed minimum entrapment efficiency. According to Lopez et al the entrapment of drug occurs both in the bilayer and in the aqueous compartment of the vesicle, for which both surfactant and lipids are responsible.28 When the lipid compartment and the aqueous phase become saturated with the drug, the vesicle provides limited entrapment capacity.29 SDC, with a steroidal structure and two OH groups, was used as a surfactant in this study. It is an anionic molecule with a hydrophilic/lipophilic balance value of 16.7. This value gives a measure of the physiological properties of a surfactant, in terms of its affinity for water or lipids.30 Upon incorporation of SDC in low concentration, formation of vesicles occurred, whereas a further increase in SDC concentration may have led to the formation of pores in the bilayers of the vesicles.31 When the concentration of SDC was at the highest level (+1), the entrapment efficiency of the drug was at minimum, due to the possible coexistence of mixed micelles and vesicles. Another possible explanation for lower drug permeation through SC at a high SDC concentration may be that high concentration of SDC disrupts the lipid membrane, which becomes leaky to the entrapped drug.32
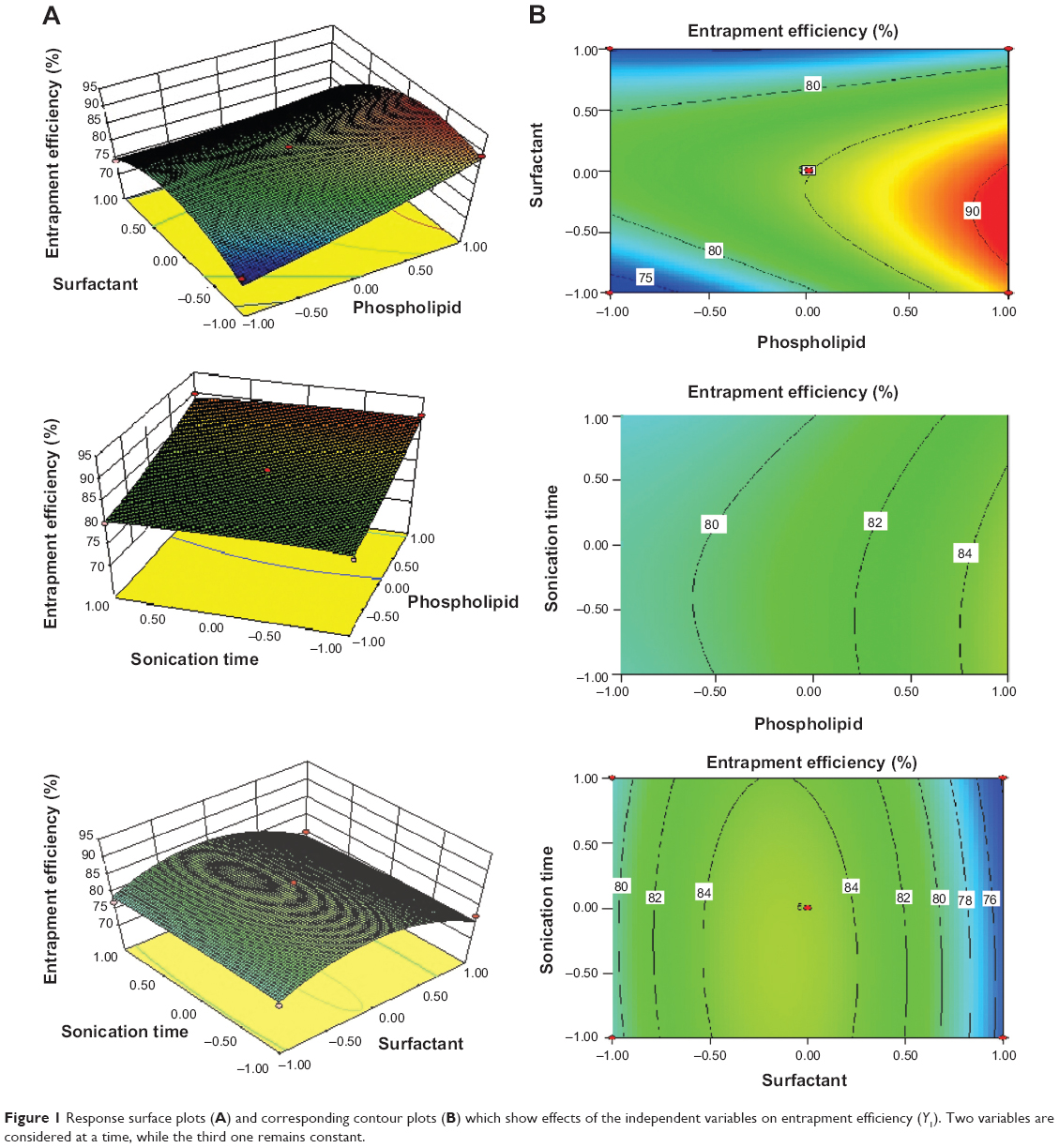 | Figure 1 Response surface plots (A) and corresponding contour plots (B) which show effects of the independent variables on entrapment efficiency (Y1). Two variables are considered at a time, while the third one remains constant. |
Effect of independent factors on vesicle size (Y2)
The mean vesicle sizes of the RTV formulations are shown in Table 2. The smallest mean vesicle size was observed for RTV17 (90±5 nM), while the maximum vesicle size was obtained for formulation RTV3 (191±11 nM). The sizes of the RTVs were less than 200 nM, which is the optimum range of these nano-sized vesicles for effective transdermal application.33 The sizes were smaller than the conventional liposome (226±20 nM) prepared by the same rotary evaporation sonication method. These variations in vesicle size were statistically significant (P<0.05). The PDI values of the RTV formulations were less than 0.3, indicating narrow size distribution and good homogeneity of distribution, as shown in Table 3. The incorporation of SDC, an anionic surfactant, showed a reasonable reduction in size, as evidenced by three-dimensional response surface graphs and their corresponding contour plots (Figure 2). The observations of particle size showed that the average vesicle size of the RTVs decreased with an increasing amount of SDC (from 15 mg to 35 mg). However, a further increase in the concentration of SDC (from 35 mg to 55 mg) led to a reduced average size of the RTVs. This might be due to the formation of micelles (instead of vesicles) that are relatively smaller in size, as discussed earlier. Another possible explanation of such increase or decrease in the size of the vesicles might arise from the duration of sonication time, involved at the last stage of transfersome preparation. High concentration of SDC and low concentration of PC90G resulted in small-size vesicles, probably due to breakage of the vesicles. On the other hand, high concentration of PC90G and low concentration SDC resulted in large-size vesicles, as the bilayer was probably thick enough to obtain reduction in size. Du Plessis et al showed that intermediate particle size results in both the highest reservoir in the deeper skin layers, as well as highest drug concentration in the reservoir, confirming that topical delivery is influenced by the size of vesicles.33 This data was further confirmed by Verma et al.21
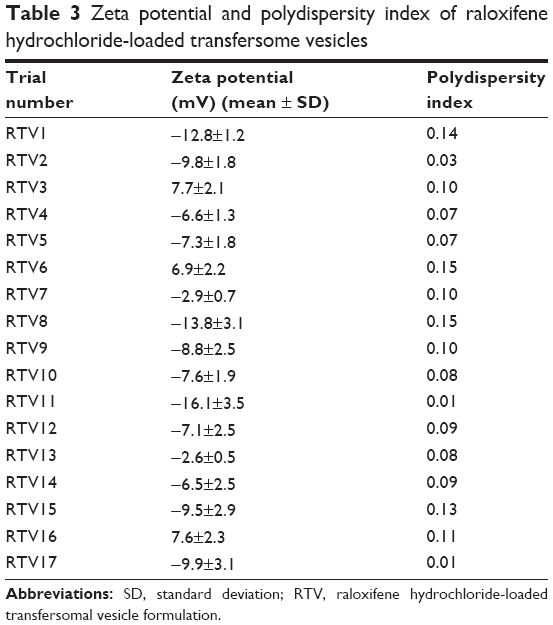 | Table 3 Zeta potential and polydispersity index of raloxifene hydrochloride-loaded transfersome vesicles |
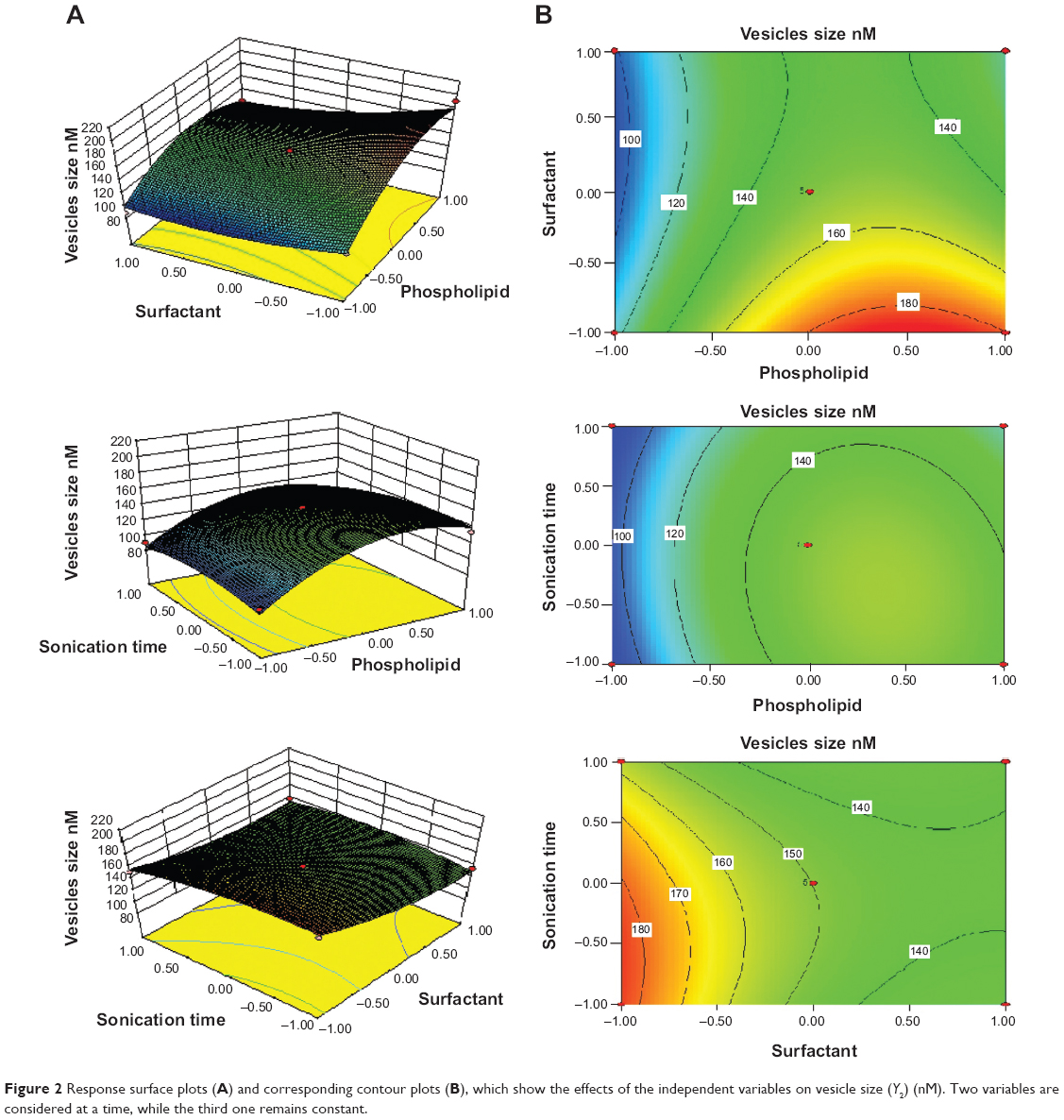 | Figure 2 Response surface plots (A) and corresponding contour plots (B), which show the effects of the independent variables on vesicle size (Y2) (nM). Two variables are considered at a time, while the third one remains constant. |
Effect of independent factors on transdermal flux (Y3)
The results of ex vivo skin permeation studies are shown in Table 2. RTV13, with a high concentration of PC90G and a medium concentration of SDC, showed maximum flux (ie, 6.5±1.1 μg/cm2/hour), whereas the minimum flux value was from RTV11 (1.5±0.4 μg/cm2/hour), with low concentration of PC90G and high concentration of SDC. The flux values for the liposomes and for the ethanolic PBS solution were 1.0±0.3 μg/cm2/hour and 0.7±0.4 μg/cm2/hour, respectively; the ER values shown by transfersomal formulation, compared against conventional liposomes and the ethanolic PBS solution, were 6.50 and 9.25, respectively. This enhancement ratio of transdermal flux in the transfersomal system is comparable to a previous finding of Mishra et al.20 A possible explanation for the better performance of the transfersome formulation, compared with conventional liposomes, may be the difference in their flexibility. Being more flexible in their nature, the transfersome vesicles can squeeze themselves through pores much smaller than their own diameters during permeation through the SC.34 The unique flexibility and deformability of the transfersome membrane are achieved by combining at least two lipophilic/amphiphilic components (phospholipid and biosurfactant) with sufficiently different packing characteristics into a single bilayer. Furthermore, these vesicles are highly stress-dependent and, as a result, they minimize the risk of complete vesicle rupture in the skin. Effects of the independent variables on transdermal flux are shown in three-dimensional response surface graphs and their corresponding contour plots (Figure 3). It was observed from these graphs and plots that after 6 hours of permeation studies, with rat skin as a barrier membrane, the transdermal flux from the RTV formulations initially increased with an increase of SDC concentration (from 15 mg to 35 mg), then decreased (Table 2). It is evident that too low or too high a concentration of SDC is unable to deliver the drug through viable skin. These findings are in agreement with the studies conducted by Cevc and Blume.9 In their study, transdermal flux was also found to be affected by the ratio of SDC to PC90G (determinant of drug entrapment efficiency).
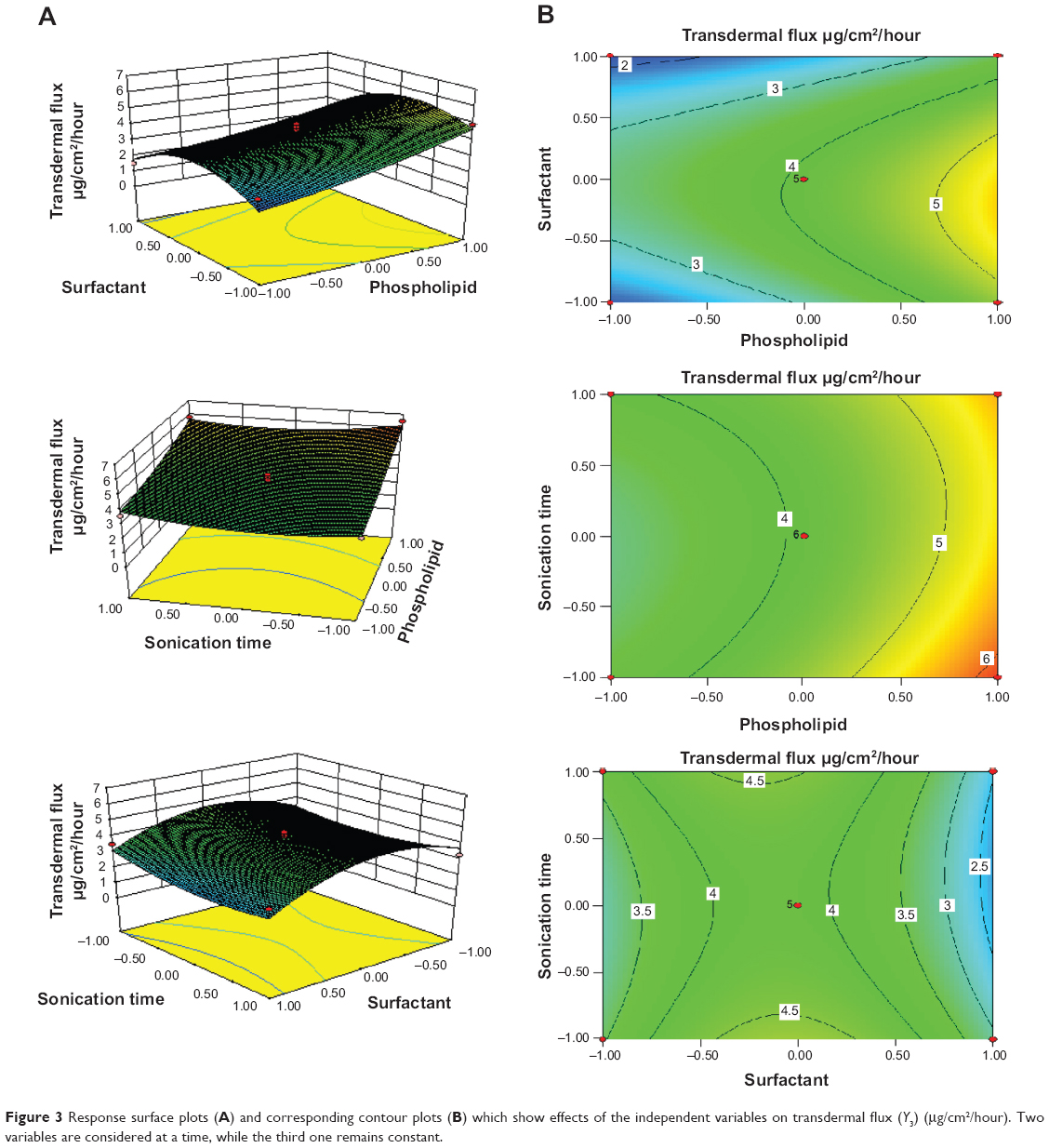 | Figure 3 Response surface plots (A) and corresponding contour plots (B) which show effects of the independent variables on transdermal flux (Y3) (μg/cm2/hour). Two variables are considered at a time, while the third one remains constant. |
When the concentration of SDC was at the highest level (+1), transdermal flux of the drug was minimal, due to the possible coexistence of mixed micelles and vesicles, and showed heterogeneity in the distribution of the vesicles (PDI >0.3). Micelles are less effective in transdermal delivery, compared against nano-size vesicles, as reported by Cevc et al.7 According to that research group, micelles, being less than 10 nm in size, are much less sensitive to the water activity gradient and drag force associated with transdermal permeation of lipid vesicles. Micelles are also less deformable and have less skin permeability, compared with transfersomes, as reported by Jain et al.17 Another possible explanation for the lower drug permeation of transfersomes through the SC at a high concentration of SDC may be that SDC at high concentration decreases drug entrapment efficiency and disrupts the lipid membrane, so that it becomes more leaky to the entrapped drug, as described earlier by Lasch et al.32
Validation of the developed RSM model
The theoretical values of Y1, Y2, and Y3, for all RTVs, were obtained by substituting their corresponding values of A, B, and C in the respective equations generated by Design Expert® software. Figure 4 quantitatively compares the resultant experimental values of the responses with those of predicted values. The theoretical (predicted) values and the observed values were in reasonably good agreement (data not shown). These results proved the validity of the RSM model developed.
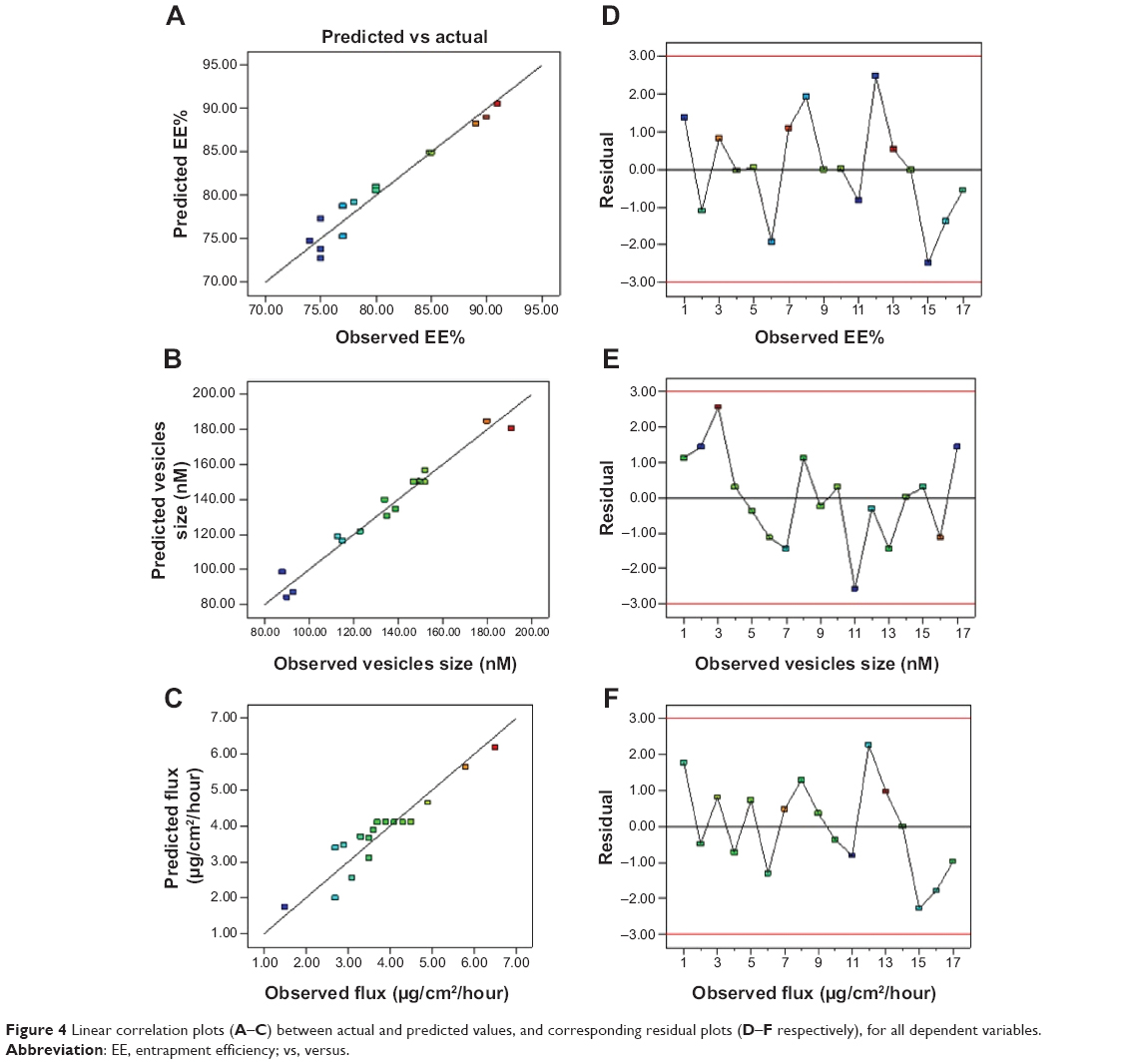 | Figure 4 Linear correlation plots (A–C) between actual and predicted values, and corresponding residual plots (D–F respectively), for all dependent variables. |
Selection of the optimized formulation
The point prediction method of Design Expert® software was utilized to select the optimized formulation from 17 trial formulations, according to the BBD experimental design. This selection was based on the criteria of attaining maximum values of entrapment efficiency and transdermal flux, and a minimum value of vesicle size. After thorough evaluation by Design Expert® software, it was found that formulation RTV13, with the combination of 300 mg PC90G, 35 mg SDC, and 15 minutes sonication time, fulfilled the requisites of an optimum formulation. The optimized formulation showed 91.00%±4.90% entrapment efficiency, 134±9 nM vesicles size, and 6.5±1.1 μg/cm2/hour transdermal flux across rat skin.
Visualization of vesicles from the optimized formulation by electron microscopy
The optimized transfersomal preparation (RTV13) was initially evaluated for shape and lamellarity using TEM (Figure 5A). Transfersomes were examined by negative staining with 0.1% phosphotungstic acid. The vesicles appeared as unilamellar and spherical in shape. The surface morphology and three-dimensional nature of the transfersomes were further visualized by SEM (Figure 5B).
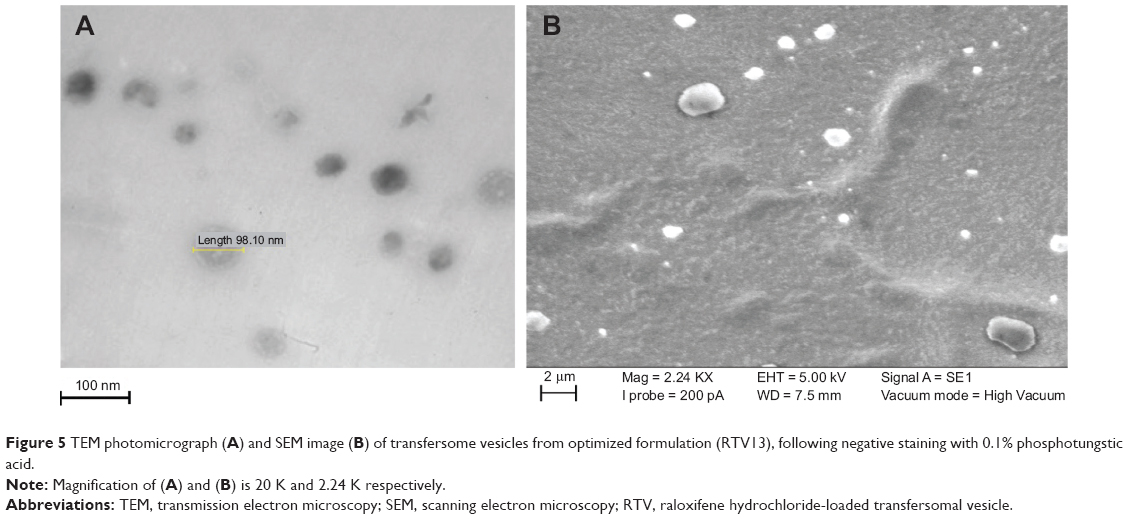 | Figure 5 TEM photomicrograph (A) and SEM image (B) of transfersome vesicles from optimized formulation (RTV13), following negative staining with 0.1% phosphotungstic acid. |
Size and charge of the transfersome vesicles from the optimized formulation
Vesicle size plays an important role in entrapment efficiency and transdermal permeation of RTVs, as shown in Figure 6. The vesicular size of the optimized formulation was found to be 134±9 nM. These drug-loaded, nano-size vesicles resulted in a better skin permeation profile (6.5±1.1 μg/cm2/hour). Subsequently for the formulation RTV16, vesicles’ size was 180±12 nM and transdermal flux was found to be 2.9±0.4 μg/cm2/hour. A similar report on the importance of vesicular size towards the permeation of drug-loaded vesicles has been published by Verma et al.21 However, larger and smaller size vesicles, as observed in formulations RTV3 (191±11 nM) and RTV7 (113±6 nM), produced a similar transdermal flux. This contradiction may arise due to other influencing parameters, like vesicle charges, entrapment efficiency, PDI, etc. The vesicle charge of all trial formulations varied as the concentration of the phospholipid and anionic surfactant changed (Table 4). The values of the charges varied within a wide range of −16.1±3.5 mV to 7.7±2.1 mV. In addition to their nano-size ranges, a narrow and homogeneous size distribution of the optimized formulation (PDI =0.08) helped better drug permeation through the SC. In contrast, nano-size liposomes (226±20 nM) were found to produce minimum transdermal flux (1.0±0.3 μg/cm2/hour). As reported by Sinico et al charge is an important parameter that can affect the stability of a vesicular system, as well as skin–vesicle interaction.35 The charge of the optimized formulation was found to be −2.61±0.5 mV. Being negatively charged, transfersomal vesicles strongly improved skin permeation of the drug.
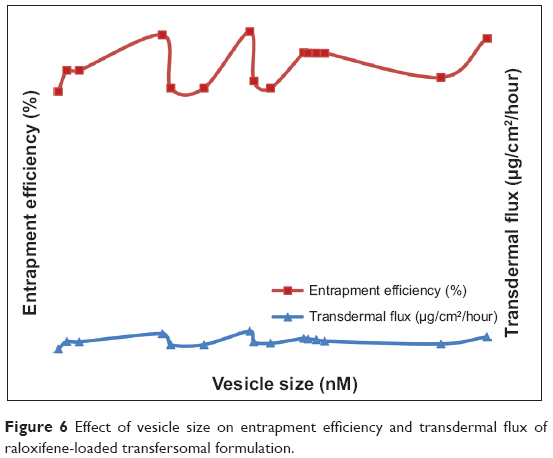 | Figure 6 Effect of vesicle size on entrapment efficiency and transdermal flux of raloxifene-loaded transfersomal formulation. |
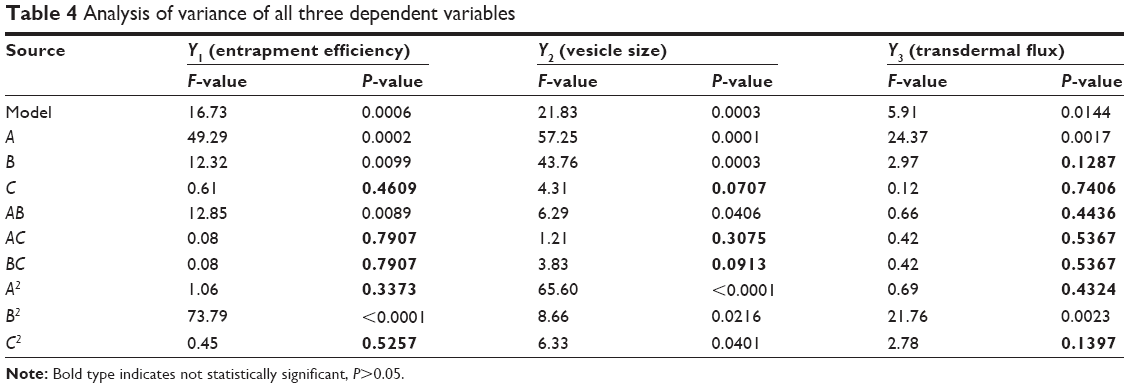 | Table 4 Analysis of variance of all three dependent variables |
CLSM study of the optimized formulation
The ability of the transfersomes to deliver raloxifene to deeper layers of the skin, and its extent of penetration, were investigated by CLSM study. Fluorescent images of the drug-loaded, optimized transfersomal formulation (RTV13), the conventional liposomes, and the ethanolic PBS solution are shown in Figure 7. In each case, 0.03% of 6-coumarin was tagged with the formulations. Images of the optimized transfersomal formulation showed greater intensity and uniform distribution of the fluorescence throughout the SC, viable epidermis, and dermis, compared with the other two samples. 6-coumarin-loaded transfersomes penetrated the skin to a depth of approximately 160 μM, giving a fluorescence intensity of 165 AU, compared with liposomes, which showed a penetration depth of 60 μM and a maximum fluorescence intensity of 40 AU. Ethanolic PBS was found to penetrate the skin up to 140 μM. However, its fluorescence intensity was found to be only 60 AU (Figures 6). Similar results have been reported by many researchers.17,18,21
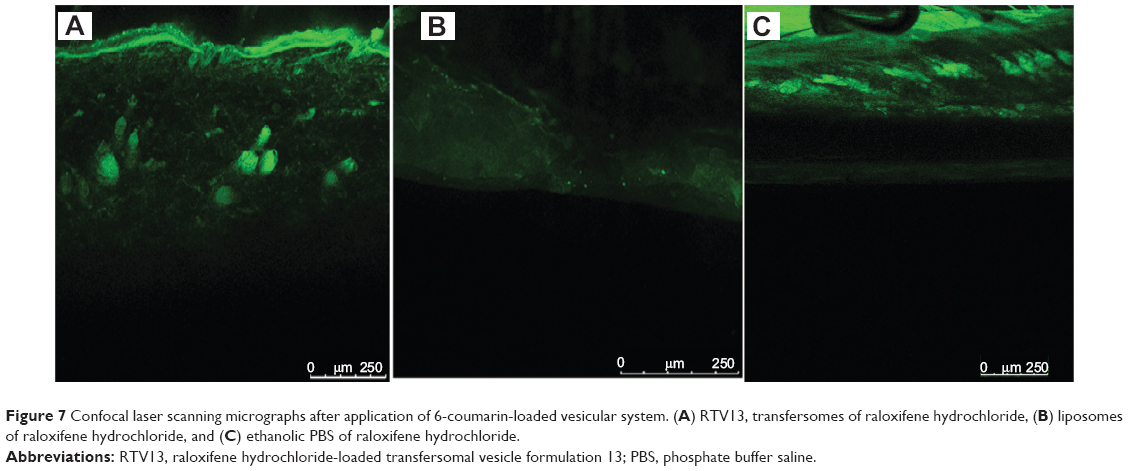 | Figure 7 Confocal laser scanning micrographs after application of 6-coumarin-loaded vesicular system. (A) RTV13, transfersomes of raloxifene hydrochloride, (B) liposomes of raloxifene hydrochloride, and (C) ethanolic PBS of raloxifene hydrochloride. |
DSC study on the optimized formulation
The permeation-enhancing effect of transfersomes was further confirmed by DSC study of the transfersomal system, with treated and untreated skin. This study involved ex vivo permeation study of the optimized transfersomal formulation for 8 hours, and evaluation of its effects on SC structure, compared with untreated skin, based on thermal properties.36 Three expected transitions were observed in the untreated (control) SC sample, as shown in Figure 8: T1, at around 67°C, T2, at around 79°C, and T3, at around 105°C. The first two peaks (T1 and T2) were due to melting of the SC lipids; the third peak (T3) was due to denaturation of SC protein, as reported by Yamane et al.37 However, both T1 and T2 endotherms disappeared in the thermogram of the treated skin sample (Figure 8). It is expected that T1 melting or shifting happened, due to the lipid bilayer melting, T2 melting took place due to the melting of the lipids associated with keratin, or due to disruption of the polar head groups of the lipids.38 The T3 peak was shifted to a lower melting point (around 84°C), suggesting keratin denaturation and a possible intracellular permeation mechanism.
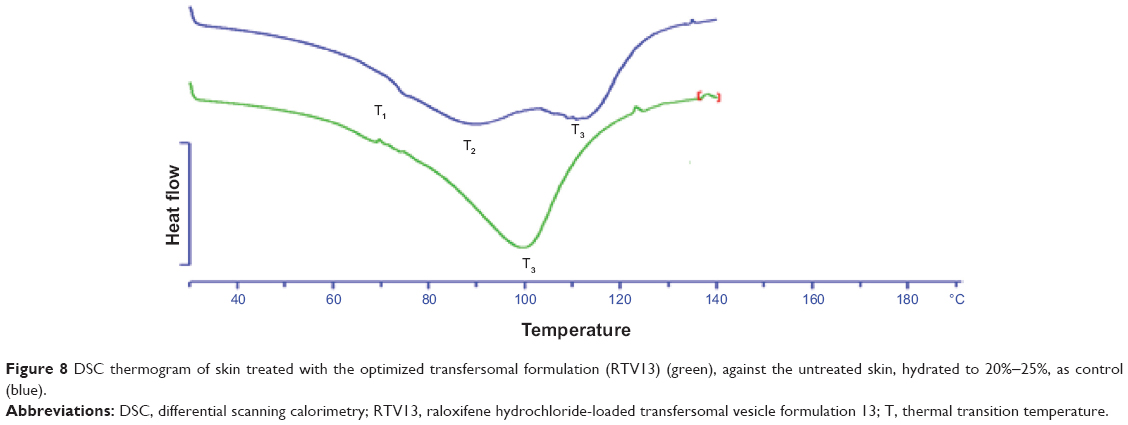 | Figure 8 DSC thermogram of skin treated with the optimized transfersomal formulation (RTV13) (green), against the untreated skin, hydrated to 20%−25%, as control (blue). |
The result of the DSC investigation showed that problems associated with transdermal delivery of raloxifene hydrochloride could be overcome by incorporating it into transfersomes, the new “ultraflexible drug carrier”. These lipid vesicles differ from conventional liposomes in their characteristic fluid membrane with high elasticity, which makes them special in responding to an external stress by rapid shape transformation. The elasticity of these vesicles is attributed to the simultaneous presence of different stabilizing (phospholipid) and destabilizing (surfactant) molecules and their tendency to redistribute in bilayers, which contributes to forming a flexible bilayer that can be deformed by itself.
Conclusion
Transfersome vesicles were studied as a possible vehicle for transdermal delivery of raloxifene hydrochloride. The optimized formulation resulted in an appropriate size, reasonable entrapment efficiency, and higher drug permeation capability, compared with conventional liposomes and an ethanolic PBS solution of the drug. CLSM further confirmed the potential of transfersomes, by showing a high fluorescent intensity of 6-coumarin-loaded formulation, compared with conventional liposomes and ethanolic PBS solution of the drug. The results of the DSC study further confirmed that skin treated with transfersomes was more fluidized, compared with untreated skin. Overall, these ex vivo findings indicate that the raloxifene hydrochloride-loaded transfersomal formulation has great potential to deliver the drug through the transdermal route. However, rat skin is very different from human skin. Therefore, the studies should be repeated on human cadaver skin to confirm ex vivo permeation efficacy of raloxifene hydrochloride formulated as transfersomal vesicles. Further investigations are also required on suitable in vivo models to establish the superiority of the developed tranfersomal formulation of raloxifene hydrochloride over existing oral medications of the drug.
Acknowledgments
This work was supported by the Fundamental Research Grant Scheme of the Ministry of Education, Malaysia (Grant number: 13-091-0332). The authors acknowledge the generous gift of phospholipon® 90G by Lipoid GmbH, Ludwigshafen, Germany.
Disclosure
The authors report no conflicts of interest in this work.
References
Langer R. Transdermal drug delivery: past progress, current status, and future prospects. Adv Drug Deliv Rev. 2004;56(5):557–558. | ||
Barry BW. Breaching the skin’s barrier to drugs. Nat Biotecnol. 2004;22(2):165–167. | ||
Honeywell-Nguyen PL, Bouwstra JA. Vesicles as a tool for transdermal and dermal delivery. Drug Discov Today Technol. 2005;2(1):67–74. | ||
Chourasia MK, Kang L, Chan SY. Nanosized ethosomes bearing ketoprofen for improved transdermal delivery. Results Pharma Sci. 2011; 1(1):60–67. | ||
Mezei M, Gulasekharam V. Liposomes-a selective drug delivery system for the topical route of administration I. Lotion dosage form. Life Sci. 1980;26(18):1473–1477. | ||
Ahad A, Aqil M, Kohli K, Sultana Y, Mujeeb M, Ali A. Formulation and optimization of nanotransfersomes using experimental design technique for accentuated transdermal delivery of valsartan. Nanomedicine. 2012; 8(2):237–249. | ||
Cevc G, Blume G, Schätzlein A, Gebauer D, Paul A. The skin: a pathway for systemic treatment with patches and lipid-based agent carriers. Adv Drug Deliv Rev. 1996;18(3):349–378. | ||
Alvarez-Román R, Naik A, Kalia Y, Fessi H, Guy RH. Visualization of skin penetration using confocal laser scanning microscopy. Eur J Pharm Biopharm. 2004;58(2):301–316. | ||
Cevc G, Blume G. New, highly efficient formulation of diclofenac for the topical, transdermal administration in ultradeformable drug carriers, transfersomes. Biochim Biophys Acta. 2001;1514(2):191–205. | ||
Elsayed MM, Abdallah OY, Naggar VF, Khalafallah NM. Lipid vesicles for skin delivery of drugs: reviewing three decades of research. Int J Pharm. 2007;332(1–2):1–16. | ||
Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoperos Int. 2006; 17(12):1726–1733. | ||
Kanis JA. WHO technical report. Sheffield, UK: University of Sheffield; 2007:66. | ||
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | ||
Cummings SR, Eckert S, Krueger KA, et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. JAMA. 1999;281(23):2189–2197. | ||
Delmas PD, Bjarnason NH, Mitlak BH, et al. Effects of raloxifene on bone mineral density, serum cholesterol concentrations, and uterine endometrium in postmenopausal women. N Engl J Med. 1997;337(23): 1641–1647. | ||
Heringa M. Review on raloxifene: profile of a selective estrogen receptor modulator. Int J Clin Pharmacol Ther. 2003;41(8):331–345. | ||
Jain S, Jain P, Umamaheshwari R, Jain N. Transfersomes – a novel vesicular carrier for enhanced transdermal delivery: development, characterization, and performance evaluation. Drug Dev Ind Pharm. 2003;29(9):1013–1026. | ||
Touitou E, Dayan N, Bergelson L, Godin B, Eliaz M. Ethosomes – novel vesicular carriers for enhanced delivery: characterization and skin penetration properties. J Control Release. 2000;65(3):403–418. | ||
El Zaafarany GM, Awad GA, Holayel SM, Mortada ND. Role of edge activators and surface charge in developing ultradeformable vesicles with enhanced skin delivery. Int J Pharm. 2010;397(1–2):164–172. | ||
Mishra D, Garg M, Dubey V, Jain S, Jain NK. Elastic liposomes mediated transdermal delivery of an anti-hypertensive agent: propranolol hydrochloride. J Pharm Sci. 2007;96(1):145–155. | ||
Verma DD, Verma S, Blume G, Fahr A. Particle size of liposomes influences dermal delivery of substances into skin. Int J Pharm. 2003; 258(1–2):141–151. | ||
Khajeh M. Application of Box–Behnken design in the optimization of a magnetic nanoparticle procedure for zinc determination in analytical samples by inductively coupled plasma optical emission spectrometry. J Hazard Mater. 2009;172(1):385–389. | ||
Jain S, Sapre R, Tiwary AK, Jain NK. Proultraflexible lipid vesicles for effective transdermal delivery of levonorgestrel: development, characterization, and performance evaluation. AAPS Pharm Sci Tech. 2005;6(3):E513–E522. | ||
Trontelj J, Vovk T, Bogataj M, Mrhar A. HPLC analysis of raloxifene hydrochloride and its application to drug quality control studies. Pharmacol Res. 2005;52(4):334–339. | ||
Vaddi HK, Ho PC, Chan YW, Chan SY. Oxide terpenes as human skin penetration enhancers of haloperidol from ethanol and propylene glycol and their modes of action on stratum corneum. Biol Pharm Bull. 2003; 26(2):220–228. | ||
Vaddi HK, Ho PC, Chan S. Terpenes in propylene glycol as skin-penetration enhancers: permeation and partition of haloperidol, fourier transform infrared spectroscopy, and differential scanning calorimetry. J Pharm Sci. 2002;91(7):1639–1651. | ||
Mandal U, Gowda V, Ghosh A, Selvan S, Solomon S, Pal TK. Formulation and optimization of sustained release matrix tablet of metformin HCl 500 mg using response surface methodology. Yakugaku Zasshi. 2007;127(9):1281–1290. | ||
López O, de la Maza A, Coderch L, López-Iglesias C, Wehrli E, Parra JL. Direct formation of mixed micelles in the solubilization of phospholipid liposomes by Triton X-100. FEBS Lett. 1998;426(3):314–318. | ||
Ning MY, Guo YZ, Pan HZ, Yu HM, Gu ZW. Preparation and evaluation of proliposomes containing clotrimazole. Chem Pharm Bull (Tokyo). 2005;53(6):620–624. | ||
El Maghraby GM, Williams AC, Barry BW. Oestradiol skin delivery from ultradeformable liposomes: refinement of surfactant concentration. Int J Pharm. 2000;196(1):63–74. | ||
van den Bergh BA, Wertz PW, Junginger HE, Bouwstra JA. Elasticity of vesicles assessed by electron spin resonance, electron microscopy and extrusion measurements. Int J Pharm. 2001;217(1–2):13–24. | ||
Lasch J, Hoffman J, Omelyanenko WG, et al. Interaction of Triton X-100 and octyl glucoside with liposomal membranes at sublytic and lytic concentrations. Spectroscopic studies. Biochim Biophys Acta. 1990;1022(2):171–180. | ||
du Plessis J, Ramachandran C, Weiner N, Müller D. The influence of particle size of liposomes on the deposition of drug into skin. Int J Pharm. 1994;103(3):277–282. | ||
Cevc G, Schätzlein A, Blume G. Transdermal drug carriers: basic properties, optimization and transfer efficiency in the case of epicutaneously applied peptides. J Control Release. 1995;36(1–2):3–16. | ||
Sinico C, Manconi M, Peppi M, Lai F, Valenti D, Fadda AM. Liposomes as carriers for dermal delivery of tretinoin: in vitro evaluation of drug permeation and vesicle–skin interaction. J Control Release. 2005; 103(1):123–136. | ||
Brandts JF, Hu CQ, Lin LN, Mos MT. A simple model for proteins with interacting domains. Applications to scanning calorimetry data. Biochemistry. 1989;28(21):8588–8596. | ||
Yamane MA, Williams AC, Barry BW. Terpene penetration enhancers in propylene glycol/water co-solvent systems: effectiveness and mechanism of action. J Pharm Pharmacol. 1995;47(12A):978–989. | ||
Barry B. Mode of action of penetration enhancers in human skin. J Control Release. 1987;6(1):85–97. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.