Back to Journals » Journal of Experimental Pharmacology » Volume 13
Experimental and Investigational Targeted Therapies for the Management of Fibrosis in NASH: An Update
Authors Huisman TM, Dieterich DT, Friedman SL ![]()
Received 8 January 2021
Accepted for publication 23 February 2021
Published 18 March 2021 Volume 2021:13 Pages 329—338
DOI https://doi.org/10.2147/JEP.S265286
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Bal Lokeshwar
Tsipora M Huisman, Douglas T Dieterich, Scott L Friedman
Division of Liver Diseases, Icahn School of Medicine at Mount Sinai, New York, NY, USA
Correspondence: Scott L Friedman
Icahn School of Medicine at Mount Sinai, Box 1123, 1425 Madison Ave, Room 1170C, New York, NY, 10025, USA
Email [email protected]
Abstract: There have been major advances in the treatment of HBV and HCV with anti-viral treatments, which is reducing the prevalence of fibrosis due to these viruses and obviating the need for anti-fibrotic therapies in these diseases. At the same time, however, the prevalence of non-alcoholic fatty liver disease (NAFLD) has been increasing, of which a substantial fraction of patients have non-alcoholic steatohepatitis (NASH), which may progress to cirrhosis. Accordingly, NASH is emerging as the leading indication for liver transplantation in North America and Europe. Progress in uncovering pathogenic determinants of fibrosis in NASH include metabolic dysregulation in hepatocytes that induce inflammation and cytokine secretion leading to cell injury and apoptosis, among others. These pathogenic events converge upon hepatic stellate cells, which are the primary fibrogenic cell in liver, and represent a target of new therapeutic candidates that are currently being evaluated in animal models and clinical trials. This review highlights key experimental and investigational therapies for NASH fibrosis, whose evaluation will be accelerated as new non-invasive markers of fibrosis are established. While no drugs are approved yet for NASH fibrosis, there is growing optimism that new pharmacotherapies are likely to emerge within the next 3 years that will favorably alter the natural history of disease.
Keywords: liver fibrosis, novel treatments, NASH, pharmacotherapies, antifibrotic therapy, cirrhosis, hepatic stellate cell
Introduction
Background
For several decades hepatitis B and C have been the leading causes of cirrhosis and the need for liver transplantation. However, with major advances in their treatment leading to either long-term suppression of HBV and cure of HCV, these etiologies have been replaced by NAFLD as the most common etiology of cirrhosis and indication for liver transplantation.1,2 Other causes of cirrhosis also include chronic alcohol use, as well as autoimmune hepatitis, PBC, and PSC. Chronic inflammation, fibrosis, and cirrhosis of any cause, not only lead to end-stage liver disease and the need for liver transplantation but also increase the risk of HCC, and thus efforts to prevent fibrosis or promote its regression are critically needed to reduce the rising incidence of HCC.
Fibrosis or scarring is the accumulation of extracellular matrix (ECM) in chronically injured tissues, including liver, and is the key determinant of clinical outcomes, especially in NASH. The pathogenesis of fibrosis has been extensively elucidated in the past 3 decades, revealing many cellular and molecular responses that promote damage to hepatocytes, which in turn provokes inflammation, secretion of cytokines, and hepatocyte apoptosis. These and other convergent drivers lead to activation of hepatic stellate cells (HSCs), a resident pericytic non-parenchymal cell that is the primary source of ECM accumulation.3–6
As noted, many circulating cytokines, mediators and growth factors are implicated in fibrogenesis. These include TGFβ1,7–9 metalloproteinases,10 as well as growth factors such as PDGF11 and VEGF,12 among others. Intracellular signaling networks within both hepatocytes and HSCs may attempt to restore normal liver homeostasis, including nuclear receptors, for example FXR, PPAR, vitamin D receptor.4,13,14 Mediators of hepatocellular damage are increasingly implicated, as signals directly from damaged or dying hepatocytes may provoke HSC activation.15 In addition, direct toxic effects from free cholesterol and increased levels of insulin can be a trigger for HSC activation.16 These and other elements in the cascade of liver injury and fibrosis each represent potential targets of antifibrotic therapies.30
Mode of Diagnosis
The diagnosis of fibrosis has traditionally been made by liver biopsy; however, biopsy is being reconsidered as the gold standard because it is prone to sampling variability and cannot be performed more than 2–3 times during the course of a clinical trial.17 Additionally, liver biopsy is invasive, not without risks, and therefore, non-invasive methods of diagnosis are urgently needed and rapidly developing.18 For example, blood tests widely used include the ELF test, the NAFLD fibrosis score19 and the Fibrosis-4 (FIB-4) Index for Liver Fibrosis,20,21 which incorporates the patient’s age, ALT, AST, and platelet count, with a high negative predictive value for excluding advanced fibrosis. Transient elastography (TE) to assess liver stiffness, which utilizes a bedside device, has a high sensitivity, but is less accurate in obese patients.22–24 Other non-invasive modalities include imaging such as MR elastography and corrected T1 imaging, among others.25 MR elastography has a higher specificity than TE, and can be useful to rule out fibrosis.26 MRI-PDFF has great promise as a diagnostic modality to assess fat content.26 Noninvasive modalities are urgently needed because they are objective and reduce the need for invasive procedures. However, they are not yet sufficiently validated to enable them to replace liver biopsy in Phase 3 clinical trials seeking regulatory approval.24,27,28
Therapeutic Targets
At present, there are no drugs approved to specifically treat hepatic fibrosis in NASH or any chronic liver disease. Based on NASH’s rising prevalence and the significant advances in anti-viral drugs to treat viral hepatitis, most efforts to develop an antifibrotic drug are now focused on NASH. Interestingly, effective antiviral therapy in HBV and HCV has a significant anti-fibrotic impact by restoring hepatocellular health and promoting fibrosis regression, indicating that the healing liver has endogenous anti-fibrotic pathways that are not well understood yet. The process of fibrosis in NASH, and the identified targets that induce ECM formation could be targets to reverse the fibrosis by interfering with the pathways leading to fibrogenesis and thereby allowing the liver to regenerate.29 Despite the lack of an approved drug for fibrosis, significant advances in understanding the pathogenesis of this disease have unearthed many therapeutic targets that are currently being evaluated in clinical trials. Moreover, although several drugs being tested to treat NASH fibrosis may not be directly antifibrotic, but may nonetheless improve fibrosis by attenuating the upstream cellular injury and metabolic drivers of fibrogenesis by HSCs. At this time, lifestyle modifications and treatment of underlying co-morbidities are the mainstay treatment for NASH and its associated fibrosis. However, while there are no approved pharmaceutical treatments, there are many targets that are currently being studied, with some promising stage 3 trials. Here we review several of the key targets and drugs in clinical development to treat fibrosis associated with NASH, divided in agents targeting metabolic processes and agents targeting the inflammatory response and the development of fibrosis, as well as emphasize the mode of diagnosis used in the different studies.
Therapeutic Targets and Novel Emerging Therapies (See Table 1 and Figure 1)
Metabolic Agents
The nuclear receptor, Farnesoid X Receptor (FXR), regulates many intracellular pathways including cholesterol metabolism,31 and is downregulated during the development of fibrosis. Based on this rationale, there are several FXR agonists in clinical development,32 led by obeticholic acid (OCA), which has been studied in Phase 2 and 3 clinical trials.33,34 While in the phase 3 trial OCA doubled the percentage of patients who had reduced fibrosis from 11.9% to 23.1% (NCT02548351), it did not statistically improve the likelihood of resolution of NASH and has not yet been approved for use in NASH. In contrast, OCA is already approved and effective in primary biliary cholangitis, where it significantly delays disease progression.35 There is an ongoing phase 3 trial of OCA in cirrhotic patients with NASH, the REVERSE (NCT03439254) trial, which is a double-blind, randomized, and placebo-controlled, seeking evidence of fibrosis improvement. Yet another FXR agonist EDP-305, is under evaluation in a phase 2, randomized double-blind, placebo-controlled study (NCT03421431) based on its ability to reduce ALT in patients with biopsy-proven NASH. The FXR agonist tropifexor is currently being evaluated in combination with a small molecule, cenicriviroc, which blocks the chemokine receptors CCR2 and CCR5,36 while cilifexor is another FXR agonist is under evaluation combined with a drug that blocks the enzymes acetyl CoA carboxylase 1 and 2,37 which reduce liver fat content.
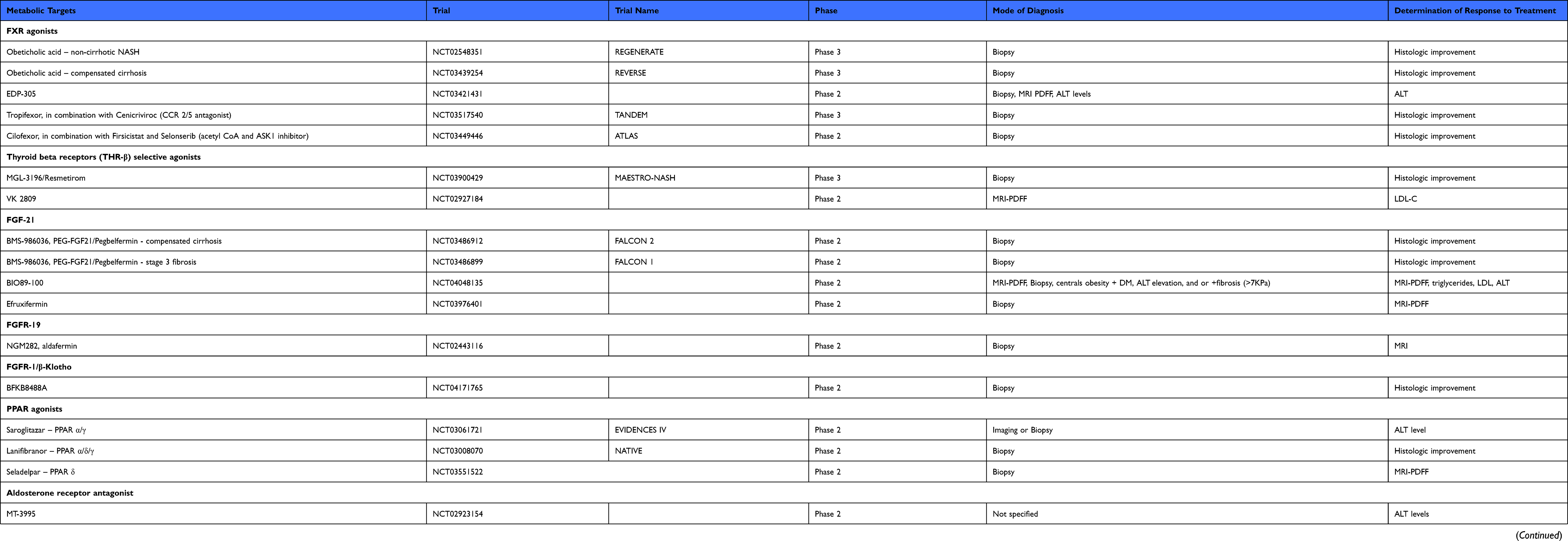 |
Table 1 Summary of Therapeutic Targets and Novel Emerging Therapies |
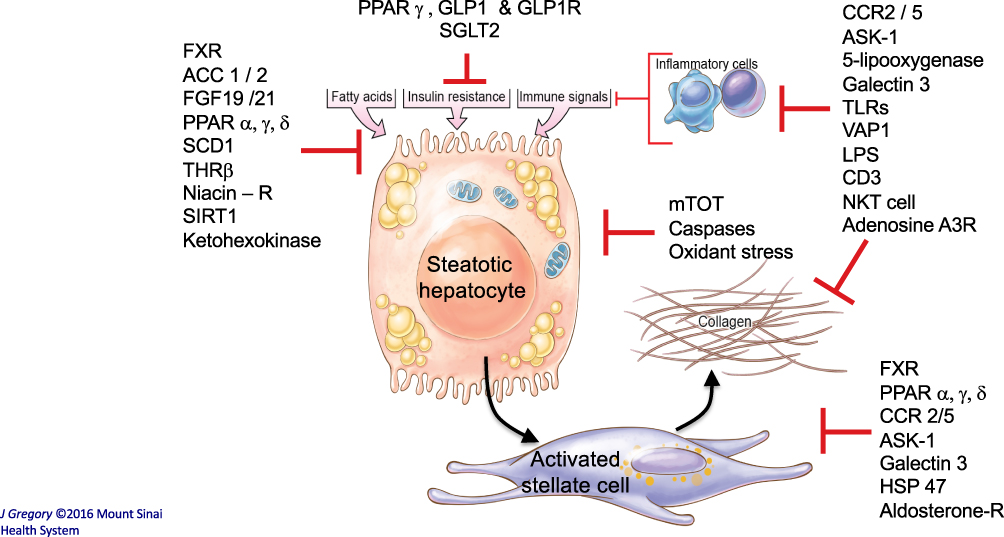 |
Figure 1 Key liver-related NASH targets in phase 2 and 3 clinical trials. This diagram illustrates the key cellular targets of many drugs currently in clinical trials for treatment of NASH. They include targets whose modulation is intended to reduce liver fat, improve insulin resistance and glucose homeostasis, reduce inflammation, improve mitochondrial function of hepatocytes and/or directly block fibrosis by hepatic stellate cells. Note that some targets, for example FXR and ASK-1, may contribute to responses in more than one cell type. Image illustration by Jill K Gregory, CMI. Printed with permission from Mount Sinai Health System, licensed under CC BY-NC. Ownership of original artwork, copyright, and all rights not specifically transferred herein remain the exclusive property of Mount Sinai Health System. |
Selective agonism of thyroid beta receptors (THR-β) is attracting interest.38 This mechanism of action has the relative advantage of improving cardiovascular risk factors while also potentially attenuating the severity of NASH. The MAESTRO-NASH trial is a double-blind placebo-controlled randomized phase 3 trial, using a THR-β (thyroid hormone receptor) agonist Resmetirom/MGL-3196 (NCT03900429), which is thought to improve NASH by increasing hepatic fat metabolism.39 The primary endpoint of this trial is biopsy-proven resolution of NASH in non-cirrhotic stage 2 or 3 fibrosis patients. VK 2809, another drug of the same class,40,41 is being studied in a phase 2 trial (NCT02927184) in patients with NAFLD and primary hypercholesterolemia, measuring a change in LDL-C as well, with secondary outcome using fat content reduction on MRI-PDFF. Other THR-β agonists are under development as well.
FGF-21 analogues are also being developed by multiple companies because of their ability to improve glucose homeostasis and insulin sensitivity, in addition to potential antifibrotic activity.42–44 Pegbelfermin, a pegylated FGF-21 also known as PEG-FGF21, is being tested in patients with NASH in a multicenter, randomized, double-blind, placebo-controlled, parallel, phase 2 study.45 Patients with a BMI > 25 and biopsy-proven NASH, a fibrosis stage 1–3, with MRI-PDFF score of ≥10% were included, and treated with either a placebo, 10 mg pegbelfermin, and 20 mg pegbelfermin, measuring a change in the fat fraction on MRI-PDFF as a readout of treatment response. The drug elicited a significant improvement in MRI-PDFF absolute fat fraction compared to placebo of −6.8% versus −1.3%, p=0.0004 in the 10 mg pegbelmerfin group, and −5.2% versus −1.3%, p=0.008 in the 20 mg pegbelmerfin group, with the expectation of a phase 3 trial to follow to further assess response in liver histology and safety. Pegbelmerfin is additionally being evaluated in the FALCON 2 study, a phase 2 randomized, double-blind, placebo-controlled study, in patients with NASH or compensated cirrhosis, with the primary outcome being a reduction equal or greater than 1 stage of fibrosis on biopsy.45 The FALCON 1 study is a phase 2B randomized double-blind placebo-controlled study looking at patients with NASH and stage 3 fibrosis, also with the primary outcome of a reduction of equal or greater than 1 stage of fibrosis on biopsy. FGF-21 agent BIO89-100, which has a different structure than pegbelfermin, is currently in a phase 2, randomized, double-blind, placebo-controlled study (NCT04048135). The study focuses on patients with steatosis on fibroscan® (transient hepatic elastography) or MRI-PDFF, as well as patients with biopsy-proven NASH, central obesity with DM or with elevated ALT. Efruxifermin is a third FGF-21 agent currently being evaluated in a phase 2, randomized, double-blind, placebo-controlled study (NCT03976401), seeking a reduction in fat on MRI-PDFF in patients with biopsy-proven NASH.
FGFR19 is a fibroblast growth factor that promotes hepatocyte proliferation but also has antifibrotic and beneficial metabolic activity. To remove the risk of carcinogenesis, the parent molecule was engineered to eliminate its growth-promoting activity while preserving its beneficial metabolic mechanism of action. The resulting molecule, aldafermin, is currently under evaluation and showed striking antifibrotic activity within 4 months in a phase 2 trial.46 A related approach to FGF19 is to use an antibody to activate the FGFR19 receptor, FGFR1/β-Klotho; this too shows promise in animal studies47 and is currently being evaluated in a phase 2 study. This study is seeking evidence of histologic resolution of NASH as the primary endpoint (NCT04171765).
The peroxisome proliferator-activated receptors (PPAR) are nuclear receptors regulating cholesterol metabolism and dyslipidemia.38,48–50 There are three subgroups: PPARα is involved in ketogenesis, PPARγ in lipid uptake, and PPARδ in the catabolism of cholesterol. The EVIDENCES IV trial (NCT03061721) is a phase 2 trial that focuses on Saroglitazar, which is PPAR α/γ agonist,51 powered to demonstrate a reduction in ALT in NASH/NAFLD patients diagnosed by either biopsy or imaging. Lanifibranor, a PPARα/γ/δ agonist (NCT03008070)52 is being evaluated in a biopsy-based phase 2 study as well, assessing a response in the SAF (Steatosis, Activity, Fibrosis) score, with a response defined as a decrease of at least 2 points of the SAF score. NCT03551522 is a phase 2 trial of Seladelpar, a PPAR δ agonist,53,54 using MRI-PDFF to assess reduction in hepatic fat content. One recent notable failure was RESOLVE-IT, a phase 3 study, using a PPAR α/δ agonist, Elafibranor55 which was evaluated in patients with NASH seeking histological improvement, however the primary endpoint was not met.
A growing list of drugs focus on improving glucose homeostasis as a means to improve NASH. The most prominent of these, Semaglutide, is a GLP-1R (glucagon-like peptide-1 receptor), which promotes insulin secretion from pancreatic beta cells, was studied in a phase 2 trial (NCT02970942). This was a double-blind, randomized, phase 2 trial that included biopsy-confirmed NASH and liver fibrosis (stage 1–3), with NASH resolution without worsening of fibrosis as primary endpoint, combined with a secondary endpoint of improvement in fibrosis stage in patients with stage 2 or 3.56 Three hundred twenty patients were included, who received placebo, 0.1 mg, 0.2 mg, or 0.4 mg each, with a statistically significant dose-dependent response in NASH resolution with no worsening fibrosis (17% of patients in the placebo group, 40% of patients in the 0.1-mg group, 36% in the 0.2-mg group, and 59% in the 0.4-mg group, P<0.001 for 0.4 mg versus placebo). Surprisingly, despite a dramatic benefit in NASH resolution, there was no antifibrotic activity (secondary outcome); a larger phase 3 study is anticipated.
An aldosterone receptor antagonist is currently being evaluated in a phase 2 study (NCT02923154), measuring ALT levels as a surrogate to response to treatment. HTD1801 is a lipid modulator being tested in a phase 2 clinical trial (NCT03656744) with patients with NASH and diabetes mellitus, measuring a decrease in fat content as a primary outcome. NCT03763877, is evaluating PXL 770, which activates AMPK (adenosine monophosphate-activated protein kinase), involved in the regulation of ischemia and stress, as well as energy deprivation; the drug may inhibit hepatic fibrosis.57 This phase 2 study measures the efficacy of the agent through the improvement in liver fat by the use of MRI-PDFF. TVB-2640, a FASN (fatty acid synthase) inhibitor, an essential enzyme in de-novo synthesis of long-chain fatty acids, is being evaluated in a phase 2 trial (NCT03938246), using MRI-PDFF as well to measure a reduction in hepatic fat content. NCT04006145 is a phase 2 study, using Elobixibat, an IBAT (ileal bile acid transporter) inhibitor, looking at the change in serum LDL-C (low-density lipoprotein cholesterol) as a primary outcome, with measure of fibrosis as secondary outcome via the use of surrogates, such as the Fibrosis-4 Index. MSDC-0602K is a MPC (mitochondrial pyruvate carrier) modulator, involved in fat metabolism, similar to the PPAR family mentioned above,58 currently in phase 2 trial, with a primary endpoint of at least 2 points on NAS score on liver histology in patients with NASH fibrosis (NCT02784444).
Icosabutate is a structurally engineered fatty acid, thought to have anti-fibrotic effects, as well as increasing insulin sensitivity and decreasing hepatic lipotoxicity in mice.59,60 A recent study60 evaluated its ability to decrease lipid levels, as well as its antifibrotic and anti-atherogenic effects in mice with NASH. The results showed a decrease in hepatic fibrosis, with an overall decrease in lipid levels, increased lipid metabolism in the liver, as well as downregulated pathways of inflammation. Icosabutate is currently being evaluated in a phase 2 trial (NCT04052516), with histologic confirmation of the resolution of NASH without worsening of fibrosis as primary endpoint, coupled with improvement in the NAFLD activity score as a secondary endpoint. The drug has the potential advantage of treating both NASH and the concurrent metabolic defects associated with increased cardiovascular risk in these patients.
The ARMOR study is a phase 3 clinical trial (NCT04104321), testing the activity of Aramchol, which inhibits SCD1 (Stearoyl-CoA Desaturase-1), which promotes the synthesis of fatty acids and reduces insulin resistance. The drug is well tolerated and shows promise based on animal studies61 and a phase 2 trial.62,63 The phase 2 study was a double-blind, randomized, placebo-controlled trial, including 60 patients with NAFLD confirmed on biopsy, given placebo, or 100 mg, 300mg of Aramchol, with the primary endpoint of a decrease in fat percentage on magnetic resonance spectroscopy. There was a statistically significant decrease in fat content percentage in the patients receiving 300 mg Aramchol versus placebo (−12.57% ± 22.14% versus +6.39% ± 36.27%, P = 0.02). An ongoing phase 3 trial includes overweight and obese patients with pre-diabetes or type 2 diabetes, with biopsy-proven NASH stage 2–3 fibrosis. Primary outcomes are the histologic resolution of NASH, as well as fibrosis improvement without worsening of NASH. The trial also seeks evidence of improved clinical outcomes such as either reduced progression to cirrhosis, development of MELD > 15, hospitalizations related to hepatic decompensating events, as well as the need for liver transplant and mortality.
Anti-Inflammatory/Anti-Fibrotic Agents
SSAO, better known as VAP-1 (Vascular Adhesion Protein-1), is an enzyme that promotes inflammation in the liver with subsequent fibrosis, and is increased in patients with chronic liver disease.64 The SSAO-inhibitor BI 1467335 is being studied in a phase 2 trial (NCT03166735), measuring reduction in the levels of the biomarker and amine oxidase copper-containing 3 as a primary outcome, and secondary outcomes that include a reduction of ALT, AST, alkaline phosphatase, and gamma-glutamyl transferase (GGT).
Hepastem are liver-derived mesenchymal stem cells, which are proposed to decrease inflammation, deactivate HSCs and thereby reduce fibrosis.65 NCT03963921 is an open-label, multi-center study, focused on safety and tolerability in patient with fibrosis or cirrhosis on histology.
Natural Killer (NK) cells are generally protective against infection and inflammation; however, in patients with liver fibrosis they may be dysfunctional, thereby worsening liver injury and inflammation. An NK cell antagonist, GRI0621, is being tested in a double-blind, randomized, placebo-controlled Phase 2 study (NCT02949375), using ALT as a marker of response to treatment in patient with NASH, viral hepatitis, and alcoholic steatohepatitis.
Emricasan, a caspase Inhibitor that is intended to reduce cell death in liver injury due to apoptosis, is currently in a phase 2 trial in patients with biopsy-proven NASH fibrosis and either diabetes mellitus or metabolic syndrome, using the NASH CRN Histologic Scoring System to evaluate improvement in fibrosis in patients receiving the treatment (NCT02686762).
JKB-121 is an antagonist to the Toll-like receptor 4 (TLR-4), important for the activation of inflammatory cytokine signaling and liver injury. TLR4 antagonism inhibits this inflammatory cascade, HSC activation, and the formation of collagen. JKB-121 is currently in a phase 2 trial (NCT02442687), enrolling patients with biopsy-proven NASH, and using MRI-PDFF as a marker for response to treatment.
Cenicriviroc antagonizes the chemokine receptors CCR 2 and CCR5 antagonist, which activate HSCs, and promotes inflammation and angiogenesis.66 It is currently being evaluated in a phase 3 clinical trial, AURORA (NCT03028740). The primary endpoint is a decrease in fibrosis on liver biopsy of at least one stage using the NAFLD Activity Score. A second part of the study includes longitudinal outcomes that assess the progression of cirrhosis on histology, clinical outcomes related to liver disease, as well as all-cause mortality.
The ASK1 (apoptosis signal-regulating kinase 1) inhibitor selonsertib, was expected to block inflammation and fibrosis through necrosis and apoptosis,67 and was studied in 2 phase 3 trials, STELLAR-3 and STELLAR-4 (NCT03053063 and NCT03053050), in patients with NASH stage 3 fibrosis and cirrhosis, respectively. Unfortunately, both trials were completely negative and the agent is no longer being evaluated, although ASK1 may remain a potential target if better antagonists can be developed.
Conclusions
The recognition that NASH is a growing cause of advanced liver disease has precipitated an accelerating effort to develop new therapies, as the disease threatens to precipitate a growing population of patients of advanced liver disease worldwide. As our understanding into the pathogenesis of fibrosis deepens, new targets are rapidly emerging. Although some drugs have failed to show benefit, there is important momentum towards success in the coming years. Coupled with improved noninvasive assessment of fibrosis, the emergence of drugs to treat NASH fibrosis will likely accelerate. Although most investigational drugs are currently in phase 2 trials, many new agents, alone or in combination, promise to transform the treatment landscape for hepatic fibrosis and some success in treating NASH fibrosis will hopefully arrive with the approval of new agent(s) within the next 3–4 years.
Abbreviations
HCV, hepatitis C virus; HBV, hepatitis B virus; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; PBC, primary biliary cholangitis; PSC, primary sclerosing cholangitis; HCC, hepatocellular carcinoma; HSCs, hepatic stellate cells; ECM, extracellular matrix; FXR, farnesoid X receptor; PPAR, peroxisome proliferator-activated receptors; FGF-21, fibroblast growth factor 21; PDGF, platelet-derived growth factor; VEGF, vascular endothelial growth factor; NK, natural killer; DM, diabetes mellitus; ALT, alanine transaminase; AST, aspartate aminotransferase; LDL-C, low-density lipoprotein cholesterol; GGT, gamma-glutamyl transferase; OCA, obeticholic acid; MR, magnetic resonance; MRI-PDFF, magnetic resonance imaging derived proton density fat fraction; TE, transient elastography.
Disclosure
Scott L. Friedman reports consulting for 89 Bio, Amgen, Axcella Health, Blade Therapeutics, Bristol Myers Squibb, Can-Fite Biopharma, ChemomAb, Escient Pharmaceuticals, Forbion, Foresite laboratories, Galmed, Gordian Biotechnology, Glycotest, Glympse Bio, Hepgene, Insitro, Morphic Therapeutics, North Sea Therapeutics, Novartis, Ono Pharmaceuticals, Pfizer Pharmaceuticals, Scholar Rock, Surrozen. He owns stock options from Blade Therapeutics, Escient, Galectin Galmed, Genfit, Glympse, Hepgene, Lifemax, Metacrine, Morphic Therapeutics, Nimbus, North Sea, Therapeutics, Scholar Rock. He is also involved in research activities with commercial entities for Morphic Therapeutics, Novo Nordisk, Abalone Bio (SBIR Grant), and Galmed. Douglas Dieterich reports consulting with Gilead, Arbutus, and Abbvie. The authors report no other conflicts of interest in this work.
References
1. Younossi Z, Tacke F, Arrese M, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2019;69(6):2672–2682. doi:10.1002/hep.30251
2. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214(2):199–210. doi:10.1002/path.2277
3. Roehlen N, Crouchet E, Baumert TF. Liver fibrosis: mechanistic concepts and therapeutic perspectives. Cells. 2020;9(4):875. doi:10.3390/cells9040875
4. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397–411. doi:10.1038/nrgastro.2017.38
5. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2020.
6. Lemoinne S, Friedman SL. New and emerging anti-fibrotic therapeutics entering or already in clinical trials in chronic liver diseases. Curr Opin Pharmacol. 2019;49:60–70. doi:10.1016/j.coph.2019.09.006
7. Mohagheghi S, Geramizadeh B, Nikeghbalian S, et al. Intricate role of yes-associated protein1 in human liver cirrhosis: TGF-beta1 still is a giant player. IUBMB Life. 2019;71(10):1453–1464. doi:10.1002/iub.2052
8. Dewidar B, Meyer C, Dooley S, Meindl-Beinker AN. TGF-beta in hepatic stellate cell activation and liver fibrogenesis-updated 2019. Cells. 2019;8(11):1419. doi:10.3390/cells8111419
9. Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, Brenner DA. The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J Hepatol. 1999;30(1):77–87. doi:10.1016/S0168-8278(99)80010-5
10. Arthur MJ, Fibrogenesis II. Metalloproteinases and their inhibitors in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2000;279(2):G245–249. doi:10.1152/ajpgi.2000.279.2.G245
11. Campbell JS, Hughes SD, Gilbertson DG, et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2005;102(9):3389–3394. doi:10.1073/pnas.0409722102
12. Yang L, Kwon J, Popov Y, et al. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenterology. 2014;146(5):1339–1350e1331. doi:10.1053/j.gastro.2014.01.061
13. Noureddin M, Anstee QM, Loomba R. Review article: emerging anti-fibrotic therapies in the treatment of non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2016;43(11):1109–1123. doi:10.1111/apt.13620
14. Duran A, Hernandez ED, Reina-Campos M, et al. p62/SQSTM1 by binding to vitamin d receptor inhibits hepatic stellate cell activity, fibrosis, and liver cancer. Cancer Cell. 2016;30(4):595–609. doi:10.1016/j.ccell.2016.09.004
15. An P, Wei LL, Zhao S, et al. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat Commun. 2020;11(1):2362. doi:10.1038/s41467-020-16092-0
16. Ioannou GN. The role of cholesterol in the pathogenesis of NASH. Trends Endocrinol Metab. 2016;27(2):84–95. doi:10.1016/j.tem.2015.11.008
17. Davison BA, Harrison SA, Cotter G, et al. Suboptimal reliability of liver biopsy evaluation has implications for randomized clinical trials. J Hepatol. 2020.
18. Vali Y, Lee J, Boursier J, et al. Enhanced liver fibrosis test for the non-invasive diagnosis of fibrosis in patients with NAFLD: a systematic review and meta-analysis. J Hepatol. 2020;73(2):252–262. doi:10.1016/j.jhep.2020.03.036
19. Boursier J, Guillaume M, Leroy V, et al. New sequential combinations of noninvasive fibrosis tests provide an accurate diagnosis of advanced fibrosis in NAFLD. J Hepatol. 2019;71(2):389–396. doi:10.1016/j.jhep.2019.04.020
20. Sterling RK, Lissen E, Clumeck N. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology. 2006;43(6):1317–1325. doi:10.1002/hep.21178
21. Boursier J, Vergniol J, Guillet A, et al. Diagnostic accuracy and prognostic significance of blood fibrosis tests and liver stiffness measurement by FibroScan in non-alcoholic fatty liver disease. J Hepatol. 2016;65(3):570–578. doi:10.1016/j.jhep.2016.04.023
22. de Ledinghen V, Vergniol J. Transient elastography for the diagnosis of liver fibrosis. Expert Rev Med Devices. 2010;7(6):811–823. doi:10.1586/erd.10.46
23. de Ledinghen V, Vergniol J, Foucher J, El-Hajbi F, Merrouche W, Rigalleau V. Feasibility of liver transient elastography with FibroScan using a new probe for obese patients. Liver Int. 2010;30(7):1043–1048. doi:10.1111/j.1478-3231.2010.02258.x
24. Lee SJ, Kim SU. Noninvasive monitoring of hepatic steatosis: controlled attenuation parameter and magnetic resonance imaging-proton density fat fraction in patients with nonalcoholic fatty liver disease. Expert Rev Gastroenterol Hepatol. 2019;13(6):523–530. doi:10.1080/17474124.2019.1608820
25. Smith AD, Porter KK, Elkassem AA, Sanyal R, Lockhart ME. Current imaging techniques for noninvasive staging of hepatic fibrosis. AJR Am J Roentgenol. 2019;1–13.
26. Jayakumar S, Middleton MS, Lawitz EJ, et al. Longitudinal correlations between MRE, MRI-PDFF, and liver histology in patients with non-alcoholic steatohepatitis: analysis of data from a Phase II trial of selonsertib. J Hepatol. 2019;70(1):133–141. doi:10.1016/j.jhep.2018.09.024
27. Han MAT, Altayar O, Hamdeh S, et al. Rates of and factors associated with placebo response in trials of pharmacotherapies for nonalcoholic steatohepatitis: systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2019;17(4):616–629e626. doi:10.1016/j.cgh.2018.06.011
28. Lin SC, Heba E, Bettencourt R, et al. Assessment of treatment response in non-alcoholic steatohepatitis using advanced magnetic resonance imaging. Aliment Pharmacol Ther. 2017;45(6):844–854. doi:10.1111/apt.13951
29. Roeb E. Matrix metalloproteinases and liver fibrosis (translational aspects). Matrix Biol. 2018;68–69:463–473. doi:10.1016/j.matbio.2017.12.012
30. Roeb E, Geier A. Nonalcoholic steatohepatitis (NASH) – current treatment recommendations and future developments. Z Gastroenterol. 2019;57(4):508–517. doi:10.1055/a-0784-8827
31. Rizzo G, Renga B, Mencarelli A, Pellicciari R, Fiorucci S. Role of FXR in regulating bile acid homeostasis and relevance for human diseases. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5(3):289–303. doi:10.2174/1568008054863781
32. Venetsanaki V, Karabouta Z, Polyzos SA. Farnesoid X nuclear receptor agonists for the treatment of nonalcoholic steatohepatitis. Eur J Pharmacol. 2019;863:172661. doi:10.1016/j.ejphar.2019.172661
33. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–965. doi:10.1016/S0140-6736(14)61933-4
34. Younossi ZM, Ratziu V, Loomba R, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2019;394(10215):2184–2196. doi:10.1016/S0140-6736(19)33041-7
35. Eaton JE, Gores GJ. Long-term outcomes with obeticholic acid in primary biliary cholangitis: reassuring, but still an itch we need to scratch. Lancet Gastroenterol Hepatol. 2019;4(6):417–418. doi:10.1016/S2468-1253(19)30084-6
36. Pedrosa M, Seyedkazemi S, Francque S, et al. A randomized, double-blind, multicenter, phase 2b study to evaluate the safety and efficacy of a combination of tropifexor and cenicriviroc in patients with nonalcoholic steatohepatitis and liver fibrosis: study design of the TANDEM trial. Contemp Clin Trials. 2020;88:105889. doi:10.1016/j.cct.2019.105889
37. Loomba R, Noureddin M, Kowdley KV, et al. Combination therapies including cilofexor and firsocostat for bridging fibrosis and cirrhosis due to NASH. Hepatology. 2020;73:S116–S117. doi:10.1016/S0168-8278(20)30753-4
38. Polyzos SA, Kang ES, Boutari C, Rhee EJ, Mantzoros CS. Current and emerging pharmacological options for the treatment of nonalcoholic steatohepatitis. Metabolism. 2020;111S:154203. doi:10.1016/j.metabol.2020.154203
39. Harrison SA, Bashir MR, Guy CD, et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2019;394(10213):2012–2024. doi:10.1016/S0140-6736(19)32517-6
40. Zhou J, Waskowicz LR, Lim A, et al. A liver-specific thyromimetic, VK2809, decreases hepatosteatosis in glycogen storage disease type Ia. Thyroid. 2019;29(8):1158–1167. doi:10.1089/thy.2019.0007
41. Mondal S, Mugesh G. Novel thyroid hormone analogues, enzyme inhibitors and mimetics, and their action. Mol Cell Endocrinol. 2017;458:91–104. doi:10.1016/j.mce.2017.04.006
42. Yilmaz Y, Eren F, Yonal O, et al. Increased serum FGF21 levels in patients with nonalcoholic fatty liver disease. Eur J Clin Invest. 2010;40(10):887–892. doi:10.1111/j.1365-2362.2010.02338.x
43. Gong Q, Hu Z, Zhang F, et al. Fibroblast growth factor 21 improves hepatic insulin sensitivity by inhibiting mammalian target of rapamycin complex 1 in mice. Hepatology. 2016;64(2):425–438. doi:10.1002/hep.28523
44. Degirolamo C, Sabba C, Moschetta A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat Rev Drug Discov. 2016;15(1):51–69.
45. Sanyal A, Charles ED, Neuschwander-Tetri BA, et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet. 2019;392(10165):2705–2717. doi:10.1016/S0140-6736(18)31785-9
46. Harrison SA, Neff G, Guy CD, et al. Efficacy and safety of aldafermin, an engineered FGF19 analog, in a randomized, double-blind, placebo-controlled trial of patients with nonalcoholic steatohepatitis. Gastroenterology. 2020.
47. Baruch A, Wong C, Chinn LW, et al. Antibody-mediated activation of the FGFR1/Klothobeta complex corrects metabolic dysfunction and alters food preference in obese humans. Proc Natl Acad Sci U S A. 2020;117(46):28992–29000. doi:10.1073/pnas.2012073117
48. Zarei M, Aguilar-Recarte D, Palomer X, Vazquez-Carrera M. Revealing the role of peroxisome proliferator-activated receptor beta/delta in nonalcoholic fatty liver disease. Metabolism. 2021;114:154342. doi:10.1016/j.metabol.2020.154342
49. Fougerat A, Montagner A, Loiseau N, Guillou H, Wahli W. Peroxisome proliferator-activated receptors and their novel ligands as candidates for the treatment of non-alcoholic fatty liver disease. Cells. 2020;9(7):1638. doi:10.3390/cells9071638
50. Choudhary NS, Kumar N, Duseja A. Peroxisome proliferator-activated receptors and their agonists in nonalcoholic fatty liver disease. J Clin Exp Hepatol. 2019;9(6):731–739. doi:10.1016/j.jceh.2019.06.004
51. Kumar DP, Caffrey R, Marioneaux J, et al. The PPAR alpha/gamma agonist saroglitazar improves insulin resistance and steatohepatitis in a diet induced animal model of nonalcoholic fatty liver disease. Sci Rep. 2020;10(1):9330. doi:10.1038/s41598-020-66458-z
52. Boeckmans J, Natale A, Rombaut M, et al. Anti-NASH drug development hitches a lift on PPAR agonism. Cells. 2019;9(1):37. doi:10.3390/cells9010037
53. Jones D, Boudes PF, Swain MG, et al. Seladelpar (MBX-8025), a selective PPAR-delta agonist, in patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid: a double-blind, randomised, placebo-controlled, phase 2, proof-of-concept study. Lancet Gastroenterol Hepatol. 2017;2(10):716–726. doi:10.1016/S2468-1253(17)30246-7
54. Haczeyni F, Wang H, Barn V, et al. The selective peroxisome proliferator-activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol Commun. 2017;1(7):663–674. doi:10.1002/hep4.1072
55. Ratziu V, Harrison SA, Francque S, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and -delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. 2016;150(5):1147–1159e1145. doi:10.1053/j.gastro.2016.01.038
56. Newsome PN, Buchholtz K, Cusi K, et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2020. doi:10.1056/NEJMoa2028395
57. Liang Z, Li T, Jiang S, et al. AMPK: a novel target for treating hepatic fibrosis. Oncotarget. 2017;8(37):62780–62792. doi:10.18632/oncotarget.19376
58. Colca JR, McDonald WG, McCommis KS, Finck BN. Treating fatty liver disease by modulating mitochondrial pyruvate metabolism. Hepatol Commun. 2017;1(3):193–197. doi:10.1002/hep4.1036
59. van den Hoek AM, Pieterman EJ, van der Hoorn JW, et al. Icosabutate exerts beneficial effects upon insulin sensitivity, hepatic inflammation, lipotoxicity, and fibrosis in mice. Hepatol Commun. 2020;4(2):193–207. doi:10.1002/hep4.1453
60. Stokman G, van den Hoek AM, Denker Thorbekk D, et al. Dual targeting of hepatic fibrosis and atherogenesis by icosabutate, an engineered eicosapentaenoic acid derivative. Liver Int. 2020;40(11):2860–2876. doi:10.1111/liv.14643
61. Iruarrizaga-Lejarreta M, Varela-Rey M, Fernandez-Ramos D, et al. Role of aramchol in steatohepatitis and fibrosis in mice. Hepatol Commun. 2017;1(9):911–927. doi:10.1002/hep4.1107
62. Ratziu V, de Guevara L, Safadi R, et al. One-year results of the global phase 2b randomized placebo controlled ARREST trial of aramchol, a stearoyl CoA desaturase modulator in NASH patients. Hepatology. 2018;68:LB–5.
63. Safadi R, Konikoff FM, Mahamid M, et al. The fatty acid-bile acid conjugate aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2014;12(12):2085–2091e2081. doi:10.1016/j.cgh.2014.04.038
64. Weston CJ, Shepherd EL, Claridge LC, et al. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J Clin Invest. 2015;125(2):501–520. doi:10.1172/JCI73722
65. Tricot T, De Boeck J, Verfaillie C. Alternative cell sources for liver parenchyma repopulation: where do we stand? Cells. 2020;9(3):566. doi:10.3390/cells9030566
66. Lefere S, Devisscher L, Tacke F. Targeting CCR2/5 in the treatment of nonalcoholic steatohepatitis (NASH) and fibrosis: opportunities and challenges. Expert Opin Investig Drugs. 2020;29(2):89–92. doi:10.1080/13543784.2020.1718106
67. Younossi ZM, Stepanova M, Lawitz E, et al. Improvement of hepatic fibrosis and patient-reported outcomes in non-alcoholic steatohepatitis treated with selonsertib. Liver Int. 2018;38(10):1849–1859. doi:10.1111/liv.13706
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.