Back to Journals » International Journal of Nanomedicine » Volume 17
Exosomes and Exosomal Cargos: A Promising World for Ventricular Remodeling Following Myocardial Infarction
Authors Fang J, Zhang Y, Chen D, Zheng Y, Jiang J ![]()
Received 5 June 2022
Accepted for publication 21 September 2022
Published 4 October 2022 Volume 2022:17 Pages 4699—4719
DOI https://doi.org/10.2147/IJN.S377479
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Jiacheng Fang, Yuxuan Zhang, Delong Chen, Yiyue Zheng, Jun Jiang
Department of Cardiology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, 310009, People’s Republic of China
Correspondence: Jun Jiang, Department of Cardiology, The Second Affiliated Hospital, Zhejiang University School of Medicine, No. 88 Jiefang Road, Hangzhou, Zhejiang, 310009, People’s Republic of China, Tel/Fax +86 135 8870 6891, Email [email protected]
Abstract: Exosomes are a pluripotent group of extracellular nanovesicles secreted by all cells that mediate intercellular communications. The effective information within exosomes is primarily reflected in exosomal cargos, including proteins, lipids, DNAs, and non-coding RNAs (ncRNAs), the most intensively studied molecules. Cardiac resident cells (cardiomyocytes, fibroblasts, and endothelial cells) and foreign cells (infiltrated immune cells, cardiac progenitor cells, cardiosphere-derived cells, and mesenchymal stem cells) are involved in the progress of ventricular remodeling (VR) following myocardial infarction (MI) via transferring exosomes into target cells. Here, we summarize the pathological mechanisms of VR following MI, including cardiac myocyte hypertrophy, cardiac fibrosis, inflammation, pyroptosis, apoptosis, autophagy, angiogenesis, and metabolic disorders, and the roles of exosomal cargos in these processes, with a focus on proteins and ncRNAs. Continued research in this field reveals a novel diagnostic and therapeutic strategy for VR.
Keywords: exosomes, ventricular remodeling, extracellular vesicles, heart failure, microRNAs, myocardial infarction
Introduction
Myocardial infarction (MI), a cardiovascular disease (CVD) with a high incidence, occurs when the coronary artery is totally or partially occluded, with the consequent loss of functional cardiomyocytes (CMs). In 2013, 1 of every 7 deaths (370,213 deaths) in the USA died of coronary heart disease.1 Although pharmacological, catheter-based, and surgical interventions have improved the outcomes of acute myocardial infarction (AMI) patients significantly, cardiac damage and pathological change persist. Ventricular remodeling (VR) in response to MI involves a comprehensive series of pathological mechanisms, such as cardiac myocyte hypertrophy, fibrosis, inflammation, angiogenesis, pyroptosis, apoptosis, autophagy, and metabolic disorder, ultimately evolving into advanced heart failure (HF), the final stage of many cardiovascular disorders. Various approaches, such as pharmacological interventions and stem cell therapies, have been shown to attenuate VR after MI.
Exosomes, a subgroup of extracellular vesicles (EVs), are a novel mechanism of intercellular communication. Released from intracellular multivesicular bodies (MVBs) via an exocytic pathway, exosomes transfer abundant biological cargos into recipient cells and regulate physiological activities and pathological responses. The effective molecules within exosomes mainly include proteins, lipids, DNAs, and non-coding RNAs (ncRNAs), the most intensively studied molecules. A growing number of studies have confirmed the critical role of exosomes in pathological VR. Recently, stem cell therapies, such as mesenchymal stem cells (MSCs) and induced pluripotent stem cells (iPSCs), have also shown an anti-remodeling effect through paracrine effects of exosomes.2,3 Here, we have reviewed the recent studies about the mechanisms of ventricular remodeling, and the potential value of exosomes from various origins in VR following MI.
Mechanisms of Ventricular Remodeling Following Myocardial Infarction
Cardiac Myocyte Hypertrophy
Cardiac myocyte hypertrophy is a progressive compensatory response to offset the reduction of cardiac output after MI, as human adult CMs are terminally differentiated and unable to proliferate. It is acknowledged that the Akt/mTOR signaling pathway plays an important role in physiological hypertrophy.4,5 Recent studies have revealed that TRPC6/Calcineurin/NFAT and cWnt/β-catenin signaling pathways are associated with pathological hypertrophy.6–8 Jia et al reported that Bcl2-associated athanogene 3 (BAG3) promotes physiological hypertrophy by activating the Akt/mTOR pathway while attenuating pathological hypertrophy by inhibiting the TRPC6/CaN/NFAT pathway.9 Although a beneficial adaptive response initially, myocardial hypertrophy transforms into a detrimental change and progresses into HF eventually under persistent pathological stimuli. Coronary angiogenesis fails to keep pace with cardiac myocyte growth may help explain this transition.10 Furthermore, the renin–angiotensin system (RAS) is known to play a central role in VR, especially in myocardial hypertrophy. Targeting all levels of the RAS, regulatory miRNAs are closely associated with the progression or attenuation of myocardial hypertrophy. Specifically, miR-21, miR-132, miR-155, miR-208, and miR-212 are involved in promoting hypertrophy, while miR-1, miR-21-3p, miR-30a, miR-101, miR-129-3p, and miR-133a are associated with preventing hypertrophy.11
Cardiac Fibrosis
Cardiac fibrosis is commonly characterized by excessive deposition and abnormal composition of extracellular matrix (ECM) components in the interstitial space. It is an essential response to prevent cardiac rupture after MI.12 However, persistent pathological stimuli may contribute to myocardial stiffness, diastolic and systolic dysfunction, conduction abnormality, and even sudden cardiac death (SCD), or HF ultimately. A recent study has shown that cardiac fibrosis is strongly associated with SCD and promises to serve as a predictor for SCD in coronary artery disease (CAD) patients.13 ECM consists of collagens, proteoglycans, glycoproteins, hyaluronic acids, and elastic fibers, which are mainly synthesized and secreted by cardiac fibroblasts (CFs). Keeping quiescent under physiological conditions, CFs migrate to the infarcted area and differentiate into myofibroblasts after MI. Although the exact origins of these activated CFs (myofibroblasts) remain under debate, recent cellular lineage-tracing analyses indicated that the myofibroblasts mainly arise from epicardium-derived resident fibroblasts.14,15 Myofibroblasts are pivotal for ECM production and scar formation in the healing phase after MI. Of note, it expresses smooth muscle α-actin (α-SMA) to support the infarcted area. Eventually, CFs return to the resting stage or stay in the matured scar with a specialized phenotype called matrifibrocyte after scar maturation.16,17
Cardiac fibrosis is generally classified into three subtypes from the histopathological point of view, namely replacement fibrosis, interstitial fibrosis, and perivascular fibrosis. Replacement fibrosis, as described above, refers to a beneficial reparative process that the infarcted myocardium is replaced by a fibrotic scar after MI. Interstitial fibrosis and perivascular fibrosis, stimulated by persistent maladaptive factors, are characterized by the diffuse ECM deposition and fibrosis formation in the myocardial interstitium or around the cardiac capillary.18,19
Inflammation
Inflammation, which is necessary for the clearance of necrotic CMs, reconstruction of ECM, and neovascularization, plays a central role in the progress of VR.20 Excessive activation of the inflammatory response may contribute to an irreversible pathological cardiac change in structure and function. Main players involved in VR include tumor necrosis factor-α (TNF-α), interleukin-1 β (IL-1β), IL-6, IL-8, IL-10, IL-18, and nuclear factor-κB (NF-κB).20,21 Under the chemotaxis of these proinflammatory molecules, inflammatory cells infiltrate into the infarcted zone, and subsequently, initiate the inflammatory cascade, paving the way for the following remodeling phase. Of them, the role of macrophages in VR has been most intensively studied. Differentiated from monocytes, macrophages can be roughly categorized into M1 and M2 phenotypes depending on their functions. M1 macrophages exhibit proinflammatory activity, producing inflammatory factors such as TNF-α, IL-1β, and IL-6, while M2 macrophages exhibit reparative activity, contributing to CF activation, ECM reconstruction, and angiogenesis.22–24 Recently, it is reported that Lgr4, a typical member of the G protein coupled receptor (GPCR) family, contributes to the proinflammatory phenotype transformation of macrophages by upregulating AP-1 transcriptional activity through the PKA/CREB signaling pathway, serving as a novel therapeutic target for post-infarct VR.25
The nod-like receptor protein 3 (NLRP3) inflammasome, a multiprotein signaling complex, has also been identified as a critical component of cardiac inflammation. Activated by a wide range of stimuli such as Ca2+, reactive oxygen species (ROS), and ncRNAs, the NLRP3 inflammasome mediates the maturation of pro-inflammatory cytokines such as IL-1β and IL-18 by interacting with caspase-1.26,27 Recent studies have revealed that inhibition of the NLRP3 inflammasome may help ameliorate pathological VR. For example, it was found that MCC950, a selective NLRP3 inflammasome inhibitor, is capable of reversing pathological VR by inhibiting the TGF-β/Smad4 pathway and the mitogen-activated protein kinase (MAPK) pathway.28,29 Similarly, triptolide and α-bisabolol are also reported to attenuate VR by targeting the NLRP3 inflammasome signaling pathway.30,31
Microvascular Dysfunction and Angiogenesis
The heart is a highly vascularized organ with a dense and efficient microvascular network. Microcirculation is perceived as vital for supplying oxygen and other nutrients to the heart, while the functional and structural abnormalities of microcirculation are well studied to be associated with HF.32,33 In Akt-1 activated experimental models, physiological cardiac hypertrophy is studied associated with enhanced angiogenesis, which is mainly induced by vascular endothelial growth factor (VEGF) and angiopoietin-2, while pathological myocardial remodeling companies with impaired coronary angiogenesis.10 In an autopsy study, immunohistochemistry and microscopy confirmed the significant reduction of microvascular density in patients with HF with a preserved ejection fraction (HFpEF).34 These studies confirmed that the imbalance between angiogenesis and cardiac repair may contribute to pathological VR and HF, while enhancing cardiac angiogenesis may serve as an ideal therapeutic strategy.35 Vascular endothelial cells (ECs), the most abundant non-cardiomyocytes in the heart, are getting more prominence due to their role in maintaining homeostasis and guiding organ repair.36 Expressed mainly in the endothelium of the coronary artery and capillary, endothelial Bmx tyrosine kinase plays an essential role in cardiac hypertrophy and remodeling by involving in the Ang II-induced signal transducer and activator of transcription 3 (STAT3) signaling pathway. Also, the silence of Bmx helps to abolish the increase in IL-6 and IL-8, which has been implicated in pathological VR.37
Cardiac Cell Death
Pyroptosis
Pyroptosis, a novel form of programmed cell death (PCD), is closely associated with the activation of the NLRP3 inflammasome and caspases. Also known as inflammatory necrosis, pyroptosis is characterized by membrane pore formation, cellular swelling, plasma membrane rupture, the release of proinflammatory cytokines IL-1β and IL-18, and subsequent adverse inflammatory response.38,39 Mechanistically, pyroptosis is mainly driven by the caspase-1 mediated canonical pathway and the caspase-4/5/11 mediated noncanonical pathway. The NLRP3 inflammasome, which consists of a sensor protein pattern recognition receptor (PRR), an adaptor protein apoptosis-associated speck-like protein (ASC), and an effector protein pro-caspase-1, serves as the core of pyroptosis.26,40 Taking the canonical pathway as an example, PRRs recognize intracellular and extracellular signals from damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), thus promoting the combination of pyrin domain with ASC and the recruitment and activation of pro-caspase-1. Activated caspase-1 cleave gasdermin D (GASMD) into N-terminal fragment (GASMDNT), and pro-IL-1β as well as pro-IL-18 into their mature forms. GASMDNT inserts into the plasma membrane and forms stable gasdermin pores, which mediate the release of IL-1β and IL-18 and subsequent cell lysis.41,42 Recently, studies have demonstrated that pyroptosis is involved in the pathogenesis of cardiovascular diseases, including hypertension, diabetic cardiomyopathy, and cardiac fibrosis.43–45 Thus, inhibition of pyroptosis-related molecules, including NLRP3, caspases and GASMD, has developed into a potential therapeutic strategy for myocardial injury and adverse VR.46–48
Autophagy
Autophagy is a lysosomal-associated physiological process involved in degrading and recycling cellular components, such as damaged organelles, excessive cytoplasm, and invading pathogens. Participating in cellular quality control, autophagy is known to play a critical role in the maintenance of cellular homeostasis.49,50 However, the role of autophagy in VR remains controversial.51 Autophagy serves as an adaptive response to limit infarct size and attenuate VR under mild ischemia.52,53 Yang et al suggested that autophagy exerts a protective effect on CMs after hypoxia, and downregulation of exosomal miR-30a derived from hypoxic CMs augments the effect.54 Interestingly, trehalose, a natural disaccharide, is reported to reduce cardiac remodeling efficiently through autophagy activation,55 whereas there is also evidence that excessive activation of autophagy may aggravate myocardial injury and worsen cardiac function.56
Apoptosis
Apoptosis, the most common form of PCD, plays a crucial role in physiological activities and the maintenance of homeostasis. Classically, apoptosis is characterized by cell shrinkage, chromatin condensation, apoptotic body formation, and phagocytosis by macrophages.57 There are three typical apoptotic molecular pathways: the TNF-α induced extrinsic pathway, the mitochondrial-initiated intrinsic pathway, and the newly reported endoplasmic reticulum stress pathway. The caspase cascade serves as the final executioner in three signaling pathways.58 Baldi et al first reported the persistent apoptotic CM loss in the later phases of AMI and the positive correlation between CM apoptotic rate and subsequent left ventricular remodeling in the infarcted area.59 Moreover, it was revealed that upregulation of the proapoptotic gene Nix, a central member of the Bcl-2 family, stimulates the progress of adverse cardiac remodeling.60 Recently, cytotoxic CD8+ T lymphocytes were found to play a proapoptotic role in adverse post-ischemic cardiac remodeling by being recruited and activated in the ischemic cardiac tissue and releasing Granzyme B.61 Intriguingly, apoptosis and autophagy interplay with each other through several common molecules and signaling pathways, such as p53, TNF-α, Beclin 1-Bcl-2/Bcl-xL complex, and mTOR pathway.58,62
Metabolic Disorders
It is indispensable for the human heart, the most metabolically active organ, to have an efficient and stable energy supply system. Mitochondria are considered the primary organelles for the generation of adenosine triphosphate (ATP). Increasing evidence has shown that mitochondrial dysfunction is implicated in pathological VR and HF.63 Specifically, mitochondrial dysfunction is reflected in multiple levels of ATP generation, including shifted metabolic substrate utilization, impaired mitochondrial oxidative phosphorylation (OXPHOS) system activity, increased reactive oxygen species (ROC) generation, and aberrant mitochondrial dynamics.64 Studies have confirmed that carbohydrates such as glucose and ketone act as substitutes for fatty acids to serve as the primary energy substrates in the failing heart.65,66 Recently, a study by Carley et al showed that short-chain fatty acids present an unexplored energy source in the failing heart, with a higher oxidation efficiency compared with ketone.67 Moreover, accumulating shreds of evidence have indicated that impaired mitochondrial function is involved in pathological cardiac hypertrophy.68 An example is that dual-specificity tyrosine-regulated kinase 1B (DYRK1B), which directly binds and activates STAT3, derives cardiac hypertrophy and HF by suppressing mitochondrial bioenergetics.69 Furthermore, Li et al confirmed that miR-27b-3p, which was elevated in transverse aortic constriction (TAC) and Ang II-induced cardiac hypertrophy mouse models, inhibited the OXPHOS activity to accelerate cardiac hypertrophy.70 The pathological mechanisms of ventricular remodeling are depicted in Figure 1.
 |
Figure 1 The pathological mechanisms of ventricular remodeling in response to myocardial infarction. |
Biology of Exosomes
The Biogenesis and Uptake of Exosomes
Almost all cells are empowered to release exosomes as part of their physiological functions and pathological responses. Generally, exosome biogenesis starts with the formation of early endosomes, derived from the endocytosis of extracellular substances. Subsequently, early endosomes grow into late endosomes via interaction with organelles, such as the Golgi complex and endoplasmic reticulum, and other intracellular vesicles. Continuous invagination of the late endosomal membrane contributes to the formation of the multivesicular bodies (MVBs), the effective vehicles of exosomes. With the cooperation of the Rab family of GTPase proteins, MVBs are transported towards and fuse with the plasma membrane, eventually, releasing exosomes into the extracellular microenvironment.71 In addition, there is also evidence that exosomes bud directly from the plasma membrane.72 Membrane integral proteins and adhesion molecules, including tetraspanins, lectins, integrins, and other peripheral surface receptors, mediate the specific binding of exosomes to recipient cells. Classically, exosomes are internalized into target cells through membrane fusion, endocytosis, and specific ligand–receptor interaction.73
The Characteristics and Functions of Exosomes
Exosomes are a subgroup of single-membrane extracellular nanovesicles with a diameter ranging from 40 to 160 nm.73 Composed of various bioactive substances, like proteins, lipids, metabolites, DNAs, and ncRNAs, exosomes participate in regulating the physiological and pathological responses of recipient cells.72 MicroRNAs, a subgroup of ncRNAs with a 21 to 23 nucleotides length, silence the target genes via binding the 3’-untranslational regions (3’-UTRs) or the coding sequences (CDSs) of target mRNA.74,75 Furthermore, exosome secretion and compositions are closely associated with cellular origins and exposed conditions, reflecting specific pathological states and disease progression. For example, serum levels of miR-1 and miR-133a are significantly upregulated in AMI patients, which are enriched in exosomes likely.76 Moreover, Matsumoto et al found that p53-responsive exosomal miR-34a, miR-194, and miR-192 were increased in the serum of progressive HF patients after AMI, serving as reliable prognostic predictors.77 The biomarkers of exosomes are the integral membrane tetraspanin proteins (CD81, CD63, and CD9), ALG-2-interacting protein X (Alix), and heat shock proteins (HSPs), which are ubiquitous to all exosomes. In the past decades, numerous studies have demonstrated the role of exosomes in VR, and the effects of specific exosomal substances on cardiac myocyte hypertrophy, fibrosis, inflammation, angiogenesis, pyroptosis, autophagy, and apoptosis are summarized in Tables 1 and 2.78
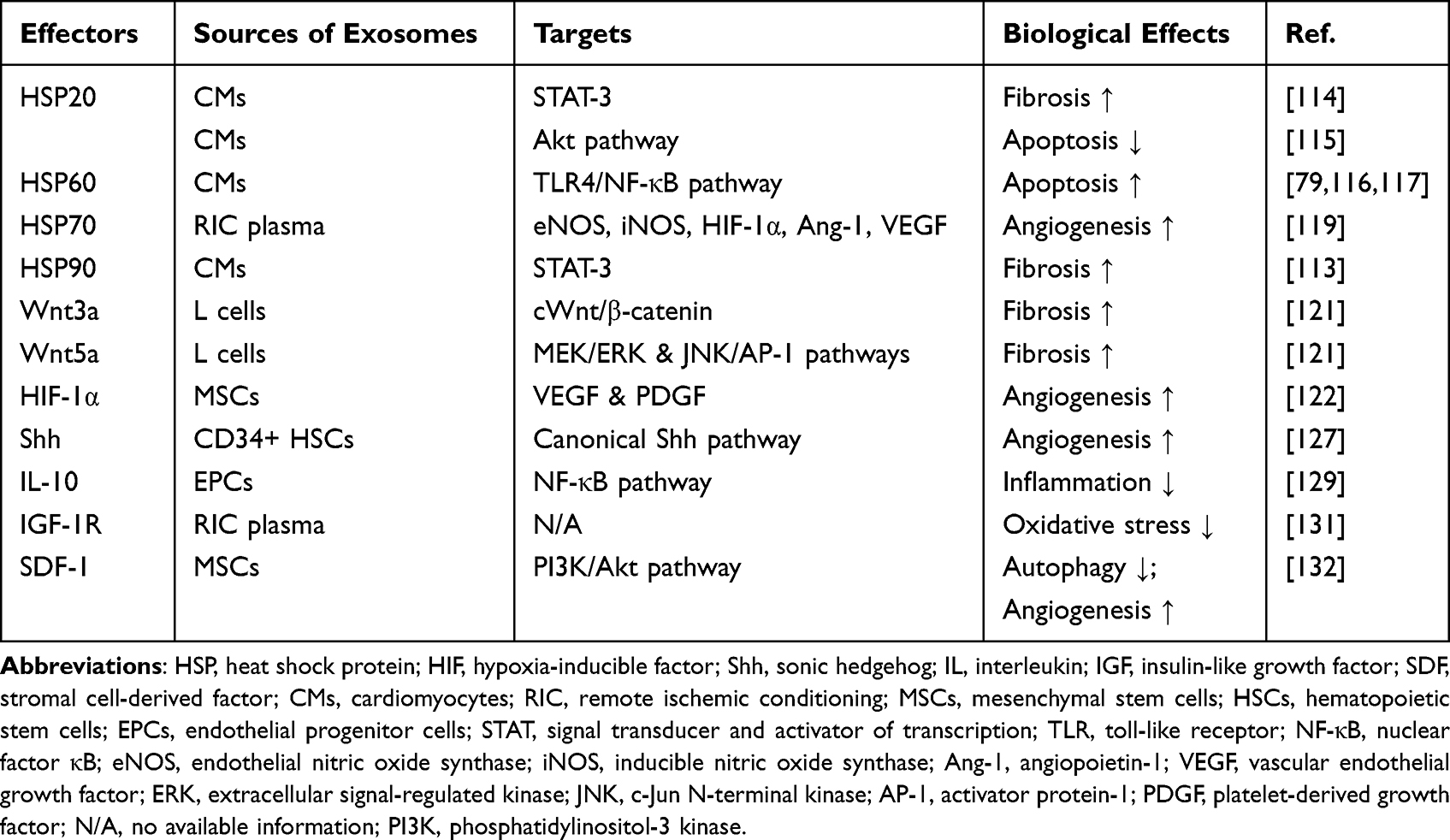 |
Table 1 Exosomal Proteins in Ventricular Remodeling Following Myocardial Infarction |
 |  |  |
Table 2 Exosomal Non-Coding RNAs in Ventricular Remodeling Following Myocardial Infarction |
Exosomes from Different Cellular Origins and Their Modulation Role in Ventricular Remodeling Following Myocardial Infarction
The heart is commonly composed of a wide population of cells with distinct functions, such as cardiomyocytes and non-cardiomyocytes (including fibroblasts, endothelial cells, and resident macrophages). Cardiomyocytes, the primary functional cells in the heart, specialize in myocardial contraction, while fibroblasts are responsible for the synthesis of ECM and tissue repair, endothelial cells constitute the abundant capillary network in the heart, and macrophages are predominantly involved in inflammation and immune response after MI. It is now well recognized that communication and interaction between these cells through exosomes contributes to the development of adverse VR following MI. Additionally, emerging evidence has suggested that infiltrated immune cells and stem/progenitor cells (including cardiosphere-derived cells, cardiac progenitor cells, and mesenchymal stem cells) exhibit a potent potential to release exosomes regulating VR.
Cardiomyocytes
Cardiomyocytes (CMs) are considered the most critical functional cells in the human heart and the main origin of cardiac exosomes. Gupta et al first reported that highly differentiated CMs are capable of releasing exosomes under physiological and pathological conditions, and HSP60, which is released from CMs through exosomes, induces myocardial apoptosis via the activation of TLR-4.79 It is also studied that exosomes secreted by CMs promote cardiac angiogenesis under ischemic conditions, and exosome-derived miR-222 and miR-143 play a major role in this process.80 Exosomes from CMs regulate the activity of CFs and ECs through internalization by target cells. A recent study indicated that miR-217-containing exosomes secreted by CMs promote CFs proliferation and cardiac fibrosis.81
Cardiac Fibroblasts
Numerically, cardiac fibroblasts (CFs) constitute ~65–70% of cardiac cells in the human heart.82 Bang et al studied for the first time that miRNA-containing exosomes are involved in the interaction between CMs and CFs. CF-derived exosomes transport miR-21-3p into CMs under stress conditions. Directly targeting and silencing sorbin and SH3 domain-containing protein 2 (SORBS2) and PDZ and LIM domain 5 (PDLIM5), miR-21-3p participate in the development of cardiac hypertrophy.83 Recently, it has been found that TNF-β-activated myofibroblasts produce more miR-200a-3p, transferring it to ECs via exosomes. Enriched miR-200a-3p impairs the angiogenic potential of ECs by inhibiting the expression of placental growth factor (PIGF), which is of great significance in endothelial biology and function.84 Interestingly, TGF-β1 expressed by cancer cell-derived exosomes is responsible for the activation of fibroblasts, which means fibroblasts also serve as the recipient cells of exosomes.85
Endothelial Cells
Endothelial cells (ECs), which are pivotal to microvascular integrity and angiogenesis, serve as a vital source of cardiac exosomes in physiological and pathological conditions. In a murine model of MI, the releasing of exosomes and microvesicles, originating mainly from ECs and CMs, is significantly increased and participates in regulating local inflammatory responses.86 Besides, it has been studied that the protein and RNA composition of EC-derived exosomes varied under hypoxic conditions, involving proteins like fibronectin and genes like BCL2.87 The significance of exosomal miRNAs for endothelial function has also been widely studied. By enhancing the activity of proangiogenic factors, such as VEGF and fibroblast growth factor (FGF), and inhibiting the expression of Spred-1, an inhibitor of angiogenesis, endothelial-specific miR-126 enhances the effect of the downstream Ras/MAPK signaling pathway and promotes microvessel formation.88 Furthermore, van Balkom et al indicated that miR-214-containing exosomes secreted from ECs stimulate endothelial migration and tube formation by suppressing the cell cycle arrest of recipient ECs.89
Immune Cells
Resident and infiltrating immune cells respond quickly after MI and persist throughout cardiac remodeling. There is rising evidence that immune cell-derived exosomes play a central role in this process.90 In the infarcted myocardium, activated macrophages inhibit proliferation of fibroblasts by downregulating Son of Sevenless 1 via exosomal miR-155, enhancing inflammation by downregulating Suppressor of Cytokine Signaling 1.91 Exosomal miR-155 from M1 macrophages has also been reported to impair the angiogenic ability of endothelial cells after MI.92 Meanwhile, M1 macrophage-derived exosomes aggregate fibrosis and exacerbate cardiac dysfunction by silencing tissue inhibitors of metalloproteinase 3 (TIM3) via exosomal miR-21-5p.93 Additionally, emerging studies indicate that long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs), like MALAT1 and circUbe3a, are also involved in ventricular remodeling via macrophage exosomes.94,95 Dendritic cells (DCs), pivotal antigen-presenting cells, also participate in regulating the myocardial microenvironment after MI via recruiting and activating other immune cells, especially T cells. Zhang et al found that DC-derived exosomes (DEXs) contribute to the activation of regulatory T cells (Tregs) and the transition of macrophages towards the anti-inflammatory phenotype, and alginate hydrogel loaded with DEXs prolongs the effect.96 Furthermore, DEXs directly activate CD4+ T cells through Th1 pathway and upregulate the inflammatory cytokines IFN-γ, TNF and IL-2, and injection of DEXs helps ameliorate cardiac dysfunction in mice post-MI.97 Activated CD4+ T cells also aggravate cardiac fibrosis via exosomal miR-142-3p-WNT signaling.98
Cardiac Progenitor Cells and Cardiosphere-Derived Cells
Cardiac progenitor cells (CPCs), a pluripotent group of stem cells, are capable of differentiating into major cardiac cells such as CMs, ECs, and smooth muscle cells after MI. Stem cell implantation or intracoronary infusion therapy has been considered an effective approach to attenuate cardiac remodeling after MI.99,100 In 2008, Rota et al found that activation or implantation of CPCs in the infarct zone contributes to replacing myocardial scar, expanding working myocardium, and enhancing cardiac function eventually.101 Deeper studies indicate that exosomes and exosomal miRNAs serve as the powerful effectors behind the beneficial effect of CPC implantation.102 Data from Barile et al indicated that injection of CPC-derived EVs into the infarct border zone inhibited cardiomyocyte apoptosis, stimulated angiogenesis, and improved cardiac function 7 days after MI. According to miRNA transcriptional profiles, anti-apoptotic miR-210 and miR-132 and proangiogenic miR-146a-3p are enriched in these extracellular vesicles.103 Targeting genes GDF-11 and ROCK-2, miR-373 enriched in EV-CPC has also been reported to suppress fibrosis after MI.104
Cardiosphere-derived cells (CDCs), which tend to aggregate into spheres spontaneously in suspension culture, are a special group of CPCs and also have been proven to attenuate cardiac remodeling after MI through exosomes.99,105 In the CADUCEUS (Cardiosphere-Derived Cells for Heart Regeneration After MI) trial, Malliaras et al demonstrated that intracoronary administration of autologous exosomes from CDCs post-MI contributes to decreased scar mass, increased viable myocardial mass, and improved regional cardiac function.106 The beneficial effect was attributed to the phenotypic conversion of fibroblasts and macrophages in subsequent studies. Priming with exosomes from CDCs in vitro, fibroblasts downregulate the expression of TGF-β and increase the secretion of stromal cell-derived factor 1 (SDF-1) and VEGF. Hence, intramyocardial injection of exosome-primed fibroblasts conduces to enhance cardiac pump function, increase vessel density and reduce scar mass.107 Besides, CDC exosome-primed macrophages were also studied to participate in cardioprotection in rats with AMI. Exosomal miR-181b inhibits the expression of PKCδ, which mediates the polarization of macrophages, and contributes to the cardioprotective effect.108
Mesenchymal Stem Cells
Mesenchymal stem cells (MSCs) have been extensively investigated due to their capacity to improve cardiac function following MI. In a 52-week follow-up study, intramyocardial administration of autologous MSCs was found to have a therapeutic effect on left ventricular remodeling, both on LV end-diastolic volume (LVEDV) and end-systolic volume (LVESV), in patients with advanced HF. At 52 weeks, LVEDV and LVESV decreased by a mean of 17.0 mL and 12.8 mL more in treated patients than controls (P = 0.006 and P = 0.017, respectively).109 Poor cell survival rates of in situ implantation hamper the beneficial effect of MSCs, while paracrine action of MSC-derived exosomes has been proved to attenuate adverse VR effectively.2 Recently, Yao et al designed and tested an MSC-derived exosome spray (EXOS) in a rat model of AMI and further in a pig model. Via a minimally invasive pathway, EXOS effectively reduced the infarct size, preserved viable myocardium, and enhanced angiomyogenesis in the post-MI hearts.110 Intramyocardial delivery of exosomes exerts similar therapeutic effects in other studies.111 It has been proved that MSC-derived EVs deliver miR-302d-3p to attenuate cardiac remodeling and repress inflammation by inhibiting the Bcl-6/MD-2/NF-κB axis, a critical proinflammatory signal pathway activated under hypoxia conditions.112
Exosomal Proteins in Ventricular Remodeling Following Myocardial Infarction
Heat Shock Proteins
It has been studied that heat shock proteins (HSPs), the most abundant chaperonins in cells, play a pivotal role in cellular stress resistance as a defense mechanism after exposure to pathological conditions such as MI. HSP90, expressed in almost all eukaryotic cells, has been identified as the critical modulator of cardiac fibrosis. Increasing the synthesis and secretion of exosomal IL-6 in CMs, HSP90 transmigrates to fibroblasts together with IL-6 via exosomes during hypertrophy. Enriched IL-6 promotes collagen synthesis in CFs via biphasic activation of STAT-3 subsequently.113 Recently, phosphorylated HSP20 has also been proved to exhibit the pro-fibrotic effect through the same regulatory pathway.114 Besides, HSP20 also negatively affects the process of myocardial apoptosis following MI. Activating the PI3K/Akt signaling pathway and inhibiting the expression of TNF-α and IL-1β, HSP20-overexpressed exosomes relieve apoptosis and maintain cardiac function.115 HSP60 is tightly bound to the exosomal membrane in physiological conditions and keeps stable in the exosome, while extracellular HSP60 induces cardiac apoptosis through binding to TLR-4 and activating NF-κB.116,117 Intriguingly, HSP27 has also been visually confirmed to be located at the exosomal membrane via immunogold labeling and transmission electron microscopy (TEM), and HSP27-laden exosomes exert an anti-inflammatory effect via promoting the secretion of IL-10, the anti-inflammatory cytokines.118 Remote ischemic conditioning (RIC) serves as a potential therapeutic strategy to relieve cardiac remodeling after MI via temporary ligation of the distal arteries. Exosomes isolated from the plasma of RIC animal models markedly promote the expression of vasoactive substances through HSP70, including endothelial nitric oxide synthase (eNOS), inducible nitric oxide synthase (iNOS), hypoxia-inducible factor-1α (HIF-1α), angiopoietin-1 (Ang-1), and VEGF, and improve cardiac remodeling and angiogenesis subsequently.119
Wnt
Wnt signaling pathways are critical for the fibrotic process after MI, and exosomes have been identified as the novel transporters for Wnt proteins.120 In the canonical Wnt/β-catenin pathway, Wnt proteins protect β-catenin from degradation by inhibiting the activity of glycogen synthase kinase 3β (GSK3β) and induce β-catenin to translocate into the nucleus, where β-catenin activates T-cell factor (TCF)/lymphoid enhancer factor (LEF) transcription factors as well as regulates the expression of target genes. Działo et al revealed that Wnt 3a-rich exosomes contribute to the activation of CFs by activating the Wnt/β-catenin pathway, while Wnt 5a-rich exosomes activate the noncanonical MEK/ERK and JNK/AP-1 pathways and increase the expression of profibrotic IL-6.121
Hypoxia Inducible Factor-1α
Hypoxia inducible factor-1 (HIF-1) has been recognized as a master transcriptional regulator of cellular adaptation under hypoxia conditions. Notably, HIF-1α-overexpressed exosomes secreted by MSCs exhibit a strong proangiogenic effect on the infarct heart by upregulating proangiogenic factors, including vascular VEGF and platelet-derived growth factor (PDGF).122 Further, miR-221-3p has been found to be involved in the proangiogenic and antiapoptotic effects of HIF-1α-engineered EVs.123 TNF-α is generally accepted as the fundamental factor related to cardiac inflammation and remodeling. Initiated by HIF-1α under hypoxia conditions, TNF-α exhibits the autocrine effects and contributes to cardiac remodeling via the exosomal pathway.124
Sonic Hedgehog
Sonic hedgehog (Shh) exhibits a pleiotropic effect on cardiac remodeling by promoting angiogenesis and decreasing cardiomyocyte apoptosis.125 Previous studies have demonstrated that Shh signaling is required for the maintenance of the coronary vasculature and Shh-containing microparticles enhance the vasculogenic capacity of EPCs and increase coronary artery density by stimulating the production of NO.126,127 Further, CD34+ hematopoietic stem cell has shown a pro-angiogenic effect via Shh-containing exosomes as well.128
Other Proteins
IL-10 has been identified as an anti-inflammatory cytokine, which significantly downregulated under systemic inflammation conditions following MI. Yue et al found that integrin-linked kinase (ILK) is highly overexpressed in exosomes derived from EPCs in IL-10 knockout mice, which mimics the post-MI IL-10 deficient condition. Phosphorylating p65 subunit of NF-κB, ILK in EPC-derived exosomes is involved in the activation of the NF-κB pathway and subsequent inflammatory response.129 Insulin-like growth factor 1 (IGF-1) plays a pivotal role in the progress and prevention of pathological cardiac remodeling by regulating cardiac metabolism, apoptosis, autophagy, growth, and aging.130 Yamaguchi et al found that IGF-1R is recruited and enriched in the exosomes after repeated RIC treatment, which attenuates cardiac remodeling and oxidative stress by transporting IGF-1R-enriched exosomes into remote non-infarcted myocardium.131 By activating PI3K/Akt pathway, SDF-1 has been revealed to inhibit autophagy of ischemic CMs via MSC exosomes, as well as promote regeneration of ECs.132 The roles of exosomal proteins are summarized in Table 1.
Exosomal microRNAs in Ventricular Remodeling Following Myocardial Infarction
MiRNAs in Cardiac Myocyte Hypertrophy
Cardiac myocyte hypertrophy serves as an adaptive response to hemodynamic stress after MI, as well as a critical component of VR. It is clear that miRNAs, like miR-1 and miR-133, play an essential regulatory role in the development of cardiac hypertrophy.133,134 In a mouse model of Ang II–induced cardiac hypertrophy, CF-derived exosomal miR-21 exerts a pro-hypertrophy effect via targeting SORBS2 and PDLIM5, the Z-line-associated proteins.83 Similarly, a recent study showed that miR-27a is upregulated in infarcted hearts, and promotes cardiomyocyte hypertrophy through targeting PDLIM5, subsequently, contributing to hypertrophic gene expression.135 TAC-induced mouse model is commonly used for studying cardiac hypertrophy and HF. It has been found that the expression of miR-217 is upregulated both in TAC mouse models and HF patients. Specifically, targeting phosphatase and tension homolog deleted on chromosome 10 (PTEN), exosomal miR-217 derived from CMs participates in cardiac hypertrophy as well as fibroblast proliferation.81 A recent study showed that reprogramming fibroblasts into iPSCs attenuate cardiac hypertrophy by decreasing exosomal miR-22, which is known to be upregulated in cardiac hypertrophy and remodeling.136,137
MiRNAs in Cardiac Fibrosis
Cardiac fibrosis after MI is primarily mediated by activated CFs, namely myofibroblasts. Studies have demonstrated that exosomes derived from hypoxic CMs contribute to CF differentiation into myofibroblasts. MiR-92a and miR-208a, transferred into CFs via exosomes, target Smad7 and Dyrk2, respectively, and promote the phenotypic conversion of CFs, evidenced by the increased expression of α-smooth muscle actin (α-SMA) and collagen I.138,139 A cardiac fibrosis model was established in C57BL/6 male mice using the left anterior descending (LAD) coronary artery ligation method and the study discovered miR-212-5p, enriched in MSC-EVs, exerts an anti-fibrotic effect by inhibiting hypoxia-mediated activation of CFs via targeting the TGF-β1/Smad pathway.140 Similarly, exosomal miR-30d promotes the proliferation of CFs by targeting integrin α-5 (itga5) directly and inhibiting the TGF-β1/Smad pathway, the key profibrotic pathway.141 Moreover, Ramanujam et al indicated that macrophage miR-21 is necessary to maintain the proinflammatory phenotype of M1 macrophages and activate fibroblasts into myofibroblasts, while further study demonstrated miR-21-5p derived from M1 exosomes induces cardiac fibrosis via targeting tissue inhibitor of metalloproteinase 3 (TIMP3).93,142 Sun et al showed that endothelial progenitor cell-derived exosomes enhance CF proliferation and angiogenesis in the SD rat model of MI through the delivery of miR-218-5p and miR-363-3p. Targeting the p53/JMY signaling pathway, miR-218-5p and miR-363-3p inhibitor or mimic exert a significantly decreased or increased profibrotic effect both in vitro and in vivo, respectively.143 In addition, mi-320a in serum exosomes promotes the CF proliferation via regulating the PI3K/Akt/mTOR pathway, as well as serves as a potential diagnostic biomarker of CHF.144 Simulating the mechanical stress condition after MI via TAC in vivo and stretching silicon dishes in vitro respectively, Yuan et al confirmed that miR-378 secreted from CMs inhibits excessive cardiac fibrosis by regulating the p38 MAPK pathways through a paracrine mechanism.145 Intriguingly, it has been studied that sacubitril/valsartan, the novel component of HF treatment, exerts an effect of attenuating cardiac fibrosis by downregulating the expression of miR-181a in the payload of circulating exosomes. In a rodent model of chronic MI, miR-181a antagomir markedly reduced the fibrosis burden versus NC (11.86 ± 3.54 versus 42.74 ± 10.31%, P < 0.05), while miR-181a mimic + sacubitril/valsartan treatment group reduced the region of collagen-rich tissue compared with miR-181a mimic treatment group.146
MiRNAs in Cardiac Angiogenesis
Cardiac ECs are considered the primary functional cells mediating angiogenesis following MI and the significant recipient cells of exosomes and miRNAs. A recent study reported that MI patients showed an increased level of miR-193a-5p in circulating exosomes, protecting ECs from oxidative stress damage to promote angiogenesis.147 Moreover, downregulated exosomal miR-143 in coronary serum of MI patients exerts a pro-angiogenic effect via activating IGF-1R signaling and promoting NO production.148 ECs are commonly regulated by surrounding cardiac cells via intercellular communication. Reportedly, the intercellular transfer of miR-19a-3p and miR-200c-3p from CMs to cardiac ECs impairs endothelial cell proliferation and angiogenesis.149,150 Moreover, activated CFs (myofibroblasts) impair the angiogenic potential of ECs via exosomal miR-200a-3p.84 Recently, Liao et al demonstrated that telocytes, a novel population of cardiac interstitial cells, inhibit EC apoptosis via silencing of exosomal miR-21-5p-targeted cell death inducing p53 target 1 (Cdip1) gene and, subsequently, downregulating caspase-3 to facilitate cardiac angiogenesis after MI.151 EC proliferation, migration, and tube formation have been commonly used to assess angiogenesis capacity. Ribeiro-Rodrigues et al confirmed that H9C2 and rat fetal CM-derived exosomes promote EC migration, tube formation, and sprouting via miR-222 and miR-143 under ischemia, and intramyocardial administration of ischemic exosomes facilitates neovascularization following MI.80 In addition, infiltrated inflammatory cells in the MI microenvironment are also involved in angiogenesis modulation. Liu et al suggested that M1 macrophage-derived exosomes suppress the angiogenic capacity of ECs by targeting five novel genes simultaneously, including Rac family small GTPase 1 (RAC1), RAC1-activated kinase 2 (RAK2), Sirt1, AMPKα2, and eNOS.92 Differently, infiltrated dendritic cells exert a pro-angiogenic effect characterized by enhanced tube formation along with increased VEGF expression.152 Furthermore, a growing number of studies have shown that progenitor and stem cell-derived exosomal miRNAs, such as miR-31, miR-132, miR-210, miR-322, and miR-1246, present pro-angiogenic activity in cardiac ECs.153–157 EPC-derived exosomal miR-1246 and miR-1290 reportedly induce the phenotypic shift from FBs to ECs, thus enhancing angiogenesis after MI.158 Besides, Zhu et al confirmed that macrophage migration inhibitory factor (MIF) enhances the pro-angiogenic effect of MSCs through upregulating exosomal miR-133a-3p.159 Interestingly, Gollmann-Tepeköylü et al found that mechanical stimulation of ischemic myocardium via shock wave therapy stimulates miR-19a-3p containing exosomes released from ECs, so as to enhance EC proliferation and tube formation through downregulating thrombospondin 1, a potent inhibitor of angiogenesis.160
MiRNAs in Cardiac Inflammation and Pyroptosis
The TLR4/NF-κB pathway has been considered the canonical inflammatory pathway as well as the target of exosomal miRNAs. In a mouse model of LAD ligation, miR-302d-3p deriving from MSCs targets BCL6 directly and then inhibits downstream NF-κB, decreasing TNF-α and IL-6 expression, so as to alleviate inflammatory response after MI.112 Similarly, miR-93-5p decreases inflammatory factor expression to suppress inflammation after MI by inhibiting infarction-induced TLR4 expression as well.161 Examining the proteomic alterations in peri-infarct areas, Kore et al found that inflammatory markers (NLRP3 and LOX-1) and cytokines of pyroptosis (caspase 1, caspase 3, and GASMD) were dramatically reduced after MSC exosome treatment.162 Recently, exosomal miR-182-5p was found to reduce cell inflammation and pyroptosis by downregulating the expression of GASMD.163 Moreover, exosomal miRNAs were also implicated in regulating macrophage activation and polarization. Transferred into macrophages from hypertrophic CMs, miR-155 promotes macrophage activation and secretion of inflammatory cytokines IL-6 and IL-8 via the MAPK pathway.164 In addition, exosomal miR-155 deriving from activated macrophages suppresses the proliferation of CFs and upregulates inflammatory cytokine expression, such as IL-1β, IL-6, TNF-α, and C-C motif chemokine ligand 2 (CCL2), so as to increase the incidence of cardiac rupture after MI.91
MiRNAs in Cardiac Apoptosis and Autophagy
Apoptosis has been considered an integral part of VR, as well as the critical target of miRNAs. A previous study suggested that the direct transfer of exosomal miR-328-3p from infarcted CMs to neighboring surviving CMs accelerates apoptosis via the caspase signaling, the canonical apoptosis regulating signaling pathway.165 Conversely, miRNAs such as miR-30e and miR-132 regulate caspase pathways negatively to ameliorate apoptosis.166,167 Moreover, miRNAs have been implicated in apoptosis pathogenesis by acting on classical pro-apoptotic genes such as B-cell lymphoma-associated X (Bax), the Fas ligand (FasL), PTEN, and p53. Studies have indicated that exosomal miR-150-5p and miR-338 derived from MSCs attenuate CM apoptosis in the MI model via reducing Bax expression.168,169 Peng et al confirmed that exosomal miR-25-3p from MSCs protects CMs against oxygen-glucose deprivation (OGD)-induced apoptosis by inhibiting the pro-apoptotic proteins PTEN and FasL, as well as attenuates inflammation via downregulating the enhancer of zest homolog 2 (EZH2)/suppressor of cytokine signaling 3 (SOCS3) axis.170 Recently, it was found that miR-221-3p showed a higher expression in young MSC exosomes than in aged MSC exosomes and exerted a greater anti-apoptotic effect through the PTEN/Akt pathway.171 Moreover, miR-21 reportedly inhibits CM apoptosis by downregulating proapoptotic genes PTEN and the programmed cell death 4 (PDCD4).172,173 Stem cell therapy, including CPCs and MSCs, has also been reported to restore cardiac function by inhibiting apoptosis and enhancing cell survival through the exosomal miR-21/PDCD4 pathway.174,175 Furthermore, Zhu et al confirmed that hypoxia-elicited exosomes (Hypo-Exo) derived from MSCs ameliorate CM apoptosis via miR-125b-5p/p53 axis, and subsequently, intravenous administration of ischemic myocardium-targeted peptide-conjugated Hypo-Exo markedly decreases the apoptotic area compared with the PBS and Scr-Exo control groups (6.26% ± 1.17% vs 20.50% ± 1.63% and 12.27% ± 1.52%, P < 0.01 and P < 0.05, respectively).176 Also, apoptosis-inducing factor 3 (AIFM3) and autophagy-related 12 (Atg12) were also confirmed to be suppressed by exosomal miRNAs to attenuate post-infarction cardiac apoptosis.177,178 In the models of ischemic HF, Li et al showed that overexpression of miR-30 (transgenic, lentivirus, or agomiR-mediated) ameliorates adverse cardiac remodeling, while miR-30 loss (locked nucleic acid or knockout) exaggerates adverse cardiac remodeling, which is mediated by MAPK pathway-associated apoptosis.141 Additionally, miR-210 from CPCs is also involved in improving cardiac function after MI through apoptosis inhibition.103
The basal level of autophagy is critical for cardioprotection, while excessively promotes cell death and ventricular remodeling. Exosomes and exosomal miRNAs are also involved in cardiac autophagy regulation. Autophagosome markers, the microtubule-associated protein 1 light chain 3-II (LC3-II) and the autophagy receptor P62, are commonly used to monitor autophagy. Xiao et al demonstrated that MSC transplantation exerts the cardioprotective effect through inhibition of infarction-induced autophagy, and molecules responsible for the effect, exosomal miR-125b, targeting the p53/Bcl2-interacting protein 3 (Bnip3) pathway.179 Moreover, miR-301 enriched exosomes also contribute to suppressing cardiac autophagy and reducing infarction area in MI rats.180 A recent study showed that iPSC-derived CM-derived exosomes (iCM-Ex) increase autophagosome production and autophagy flux via mTOR and the Bcl-2 family signaling, the known regulators of autophagy.3 Also, inhibition of exosomal miR-30 reportedly contributes to maintaining the protective autophagic response in hypoxic CMs by abolishing repression to target gene Beclin-1 and Atg12.54
Exosomal Long Noncoding RNAs in Ventricular Remodeling Following Myocardial Infarction
MALAT1
Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is considered a proangiogenic lncRNA regulating the proliferation of ECs in vitro and vascularization in vivo.181 One study from Wu et al found that MALAT1 derived from CPC-EVs partially contributes to improving cardiomyocyte survival and promoting angiogenesis through targeting miR-497 under hypoxia.182 Interestingly, Shyu et al confirmed that hyperbaric oxygen (HBO) therapy promotes angiogenesis following MI via exosomal MALAT1. Specifically sponging miR-92a, MALAT1 upregulates the expression of krüppel-like factor 2 (KLF2) and CD31 to enhance neovascularization in the rat model of AMI.183 Further, it has been shown that MALAT1 exerts an inhibitory effect on angiogenesis by competitively binding to miR-25-3p, increasing the expression of CDC42, subsequently, activating the MEK/ERK pathway.94
Other lncRNAs
LncRNA Neat1 and AK139128 were found to be highly expressed in hypoxic cardiomyocyte-derived exosomes. Regulated by HIF2A under hypoxia, Neat1 is indispensable for fibroblast survival and functions, such as migration capacity and fibrotic gene expression, while AK139128 stimulates fibroblast apoptosis and inhibits proliferation, migration, and invasion in the rat model of MI.184,185 MSCs serve as an important source of lncRNA-containing exosomes. Recently, lncRNA KLF3-AS1 derived from MSCs was found to ameliorate cardiomyocyte inflammatory response and pyroptosis in MI rats via reacting with miR-138-5p to upregulate Sirt1.186 Moreover, lncRNA urothelial carcinoma associated 1 (UCA1) increases the level of antiapoptotic protein BCL2 to exert a cardioprotective effect via the miR-873-5p/XIAP axis.187 Interestingly, Huang et al found that atorvastatin enhances the cardioprotective effects of MSCs. In the atorvastatin-pretreated MI model, exosomal lncRNA H19 activates the proangiogenic factor VEGF to promote angiogenesis in the peri-infarct region, as well as downregulates IL-6 and TNF-α to inhibit apoptosis, via upregulating the expression of miR-675.188
Exosomal Circular RNAs in Ventricular Remodeling Following Myocardial Infarction
Circular RNAs (circRNAs), a special type of ncRNAs, are recently studied expressed in exosomes and participate in the pathogenesis of cardiac remodeling. Wang et al reported that exosomal circHIPK3 derived from hypoxic CMs effectively promotes angiogenesis in the peri-infarct area, as well as reduces the infarct area. Mechanistically, circHIPK3 sponges miR-29 to upregulate proangiogenic factor VEGFA in cardiac ECs and subsequently enhance the proliferation, migration, and tube formation of cardiac ECs.189 Further, it is also reported that circUbe3a exacerbates myocardial fibrosis after MI by promoting the phenotypic transformation of CFs via targeting miR-138-5p.95 The roles of exosomal ncRNAs are summarized in Table 2.
Conclusion
Myocardial infarction is a progressively prevalent health problem worldwide, and ventricular remodeling following MI is still a troublesome challenge. It has been widely researched that VR is a multiphasic maladaptive response to various stimuli, including MI, which involves a complex series of adverse mechanisms. In addition to cardiac myocyte hypertrophy, fibrosis, angiogenesis and metabolic disorders, other novel pathologic changes have recently been appreciated, including inflammatory response, immune cell activation and programmed cell death. A host of cell types, including cardiomyocytes (promoting cardiac hypertrophy), fibroblasts (promoting fibrosis), endothelial cells (promoting microvascular dysfunction), immune cells (promoting inflammation and immune response), and stem/progenitor cells (promoting proliferation and regeneration), are involved in orchestrating this complicated pathological process, and it is now clear that exosomes secreted by these cells are pivotal in the modulation of VR.
Current studies on exosomes and exosomal molecules have provided novel strategies for optimized VR diagnosis and treatment. Derived from different cells, exosomes are heterogeneous in size and contents, reflecting the physiological and pathological states. For example, exosomes enriched with p53-responsive miR-34a, miR-192 and miR-194 contribute to the prediction of HF after MI.77 Similarly, exosomal molecules, especially miRNAs, show promise to be biomarkers for the diagnosis of VR.190 Transferring biological cargos into recipient cells, exosomes regulate signaling pathways associated with modulation of VR and thus promote cardiac function. Nevertheless, exosomes fail to induce cardiomyocyte proliferation in the infarcted heart.191 Moreover, the transportation properties, biocompatibility, and low immunogenicity characteristics of exosomes make it possible to become drug delivery vesicles and clinical tools.192 For example, Vandergriff A et al conjugated exosomes with cardiac homing peptide to effectively target exosomes to the infarcted heart, promoting cellular proliferation and angiogenesis.193 Furthermore, stem cell therapy, such as MSCs and induced pluripotent stem cells, has also shown an anti-remodeling effect through paracrine effects of exosomes.
However, there are still several problems remaining to be solved. Owing to technical limitations, there is still a lack of standard isolation and purification procedures to distinguish exosomes from microvesicles and apoptotic bodies. In addition, the rapid clearance of exosomes from bodies issues a challenge to persistent beneficial effects. Moreover, their susceptibility to change makes it difficult to isolate precisely. Nonetheless, we expect to see tremendous advances in the exosome field and breakthroughs in exosomal applications for ventricular remodeling.
Acknowledgments
The authors thank members of the laboratory for helpful discussions and critiques.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Mozaffarian D, Benjamin EJ, Go AS, et al. Executive summary: Heart disease and stroke statistics--2016 update: a report from the American Heart Association. Circulation. 2016;133(4):447–454. doi:10.1161/CIR.0000000000000366
2. Wang X, Tang Y, Liu Z, et al. The application potential and advance of mesenchymal stem cell-derived exosomes in myocardial infarction. Stem Cells Int. 2021;2021:5579904.
3. Santoso MR, Ikeda G, Tada Y, et al. Exosomes from induced pluripotent stem cell-derived cardiomyocytes promote autophagy for myocardial repair. J Am Heart Assoc. 2020;9(6):e014345.
4. DeBosch B, Treskov I, Lupu TS, et al. Akt1 is required for physiological cardiac growth. Circulation. 2006;113(17):2097–2104.
5. McMullen JR, Shioi T, Zhang L, et al. Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci U S A. 2003;100(21):12355–12360.
6. Bogdanova E, Beresneva O, Galkina O, et al. Myocardial hypertrophy and fibrosis are associated with cardiomyocyte beta-catenin and TRPC6/Calcineurin/NFAT signaling in spontaneously hypertensive rats with 5/6 Nephrectomy. Int J Mol Sci. 2021;22(9):34.
7. Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14(1):38–48.
8. Nakayama H, Wilkin BJ, Bodi I, Molkentin JD. Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J. 2006;20(10):1660–1670.
9. Jia P, Wu N, Yang H, Guo Y, Guo X, Sun Y. Different roles of BAG3 in cardiac physiological hypertrophy and pathological remodeling. Transl Res. 2021;233:47–61.
10. Shiojima I, Sato K, Izumiya Y, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115(8):2108–2118. doi:10.1172/JCI24682
11. Adamcova M, Kawano I, Simko F. The impact of microRNAs in renin-angiotensin-system-induced cardiac remodelling. Int J Mol Sci. 2021;22(9):22.
12. Gao X-M, White DA, Dart AM, Du X-J. Post-infarct cardiac rupture: recent insights on pathogenesis and therapeutic interventions. Pharmacol Ther. 2012;134(2):156–179.
13. Zegard A, Okafor O, de Bono J, et al. Myocardial fibrosis as a predictor of sudden death in patients with coronary artery disease. J Am Coll Cardiol. 2021;77(1):29–41.
14. Kanisicak O, Khalil H, Ivey MJ, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260.
15. Ruiz-Villalba A, Simón AM, Pogontke C, et al. Interacting resident epicardium-derived fibroblasts and recruited bone marrow cells form myocardial infarction scar. J Am Coll Cardiol. 2015;65(19):2057–2066.
16. Fu X, Khalil H, Kanisicak O, et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest. 2018;128(5):2127–2143.
17. Tallquist MD. Cardiac fibroblast diversity. Annu Rev Physiol. 2020;82:63–78.
18. de Boer RA, De Keulenaer G, Bauersachs J, et al. Towards better definition, quantification and treatment of fibrosis in heart failure. A scientific roadmap by the Committee of translational research of the Heart Failure Association (HFA) of the European Society of Cardiology. Eur J Heart Fail. 2019;21(3):272–285.
19. Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res. 2021;117(6):1450–1488.
20. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11(5):255–265.
21. Dutka M, Bobiński R, Ulman-Włodarz I, et al. Various aspects of inflammation in heart failure. Heart Fail Rev. 2020;25(3):537–548.
22. Kologrivova I, Shtatolkina M, Suslova T, Ryabov V. Cells of the immune system in cardiac remodeling: main players in resolution of inflammation and repair after myocardial infarction. Front Immunol. 2021;12:664457.
23. Ma Y, Mouton AJ, Lindsey ML. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl Res. 2018;191:15–28.
24. Peet C, Ivetic A, Bromage DI, Shah AM. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc Res. 2020;116(6):1101–1112.
25. Huang C-K, Dai D, Xie H, et al. Lgr4 governs a pro-inflammatory program in macrophages to antagonize post-infarction cardiac repair. Circ Res. 2020;127(8):953–973.
26. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157(5):1013–1022.
27. Yan Z, Qi Z, Yang X, et al. The NLRP3 inflammasome: multiple activation pathways and its role in primary cells during ventricular remodeling. J Cell Physiol. 2021;236(8):5547–5563.
28. Li X, Yang W, Ma W, et al. F-FDG PET imaging-monitored anti-inflammatory therapy for acute myocardial infarction: exploring the role of MCC950 in murine model. J Nucl Cardiol. 2021;28(5):2346–2357.
29. Zhao M, Zhang J, Xu Y, et al. Selective inhibition of NLRP3 inflammasome reverses pressure overload-induced pathological cardiac remodeling by attenuating hypertrophy, fibrosis, and inflammation. Int Immunopharmacol. 2021;99:108046.
30. Nagoor Meeran MF, Azimullah S, Laham F, et al. α-Bisabolol protects against β-adrenergic agonist-induced myocardial infarction in rats by attenuating inflammation, lysosomal dysfunction, NLRP3 inflammasome activation and modulating autophagic flux. Food Funct. 2020;11(1):965–976.
31. Pan X-C, Liu Y, Cen -Y-Y, et al. Dual role of triptolide in interrupting the NLRP3 inflammasome pathway to attenuate cardiac fibrosis. Int J Mol Sci. 2019;20(2):11.
32. Camici PG, Tschöpe C, Di Carli MF, Rimoldi O, Van Linthout S. Coronary microvascular dysfunction in hypertrophy and heart failure. Cardiovasc Res. 2020;116(4):806–816.
33. Oka T, Akazawa H, Naito AT, Komuro I. Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ Res. 2014;114(3):565–571.
34. Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131(6):550–559.
35. Dittrich GM, Froese N, Wang X, et al. Fibroblast GATA-4 and GATA-6 promote myocardial adaptation to pressure overload by enhancing cardiac angiogenesis. Basic Res Cardiol. 2021;116(1):26.
36. Rafii S, Butler JM, Ding B-S. Angiocrine functions of organ-specific endothelial cells. Nature. 2016;529(7586):316–325.
37. Holopainen T, Räsänen M, Anisimov A, et al. Endothelial Bmx tyrosine kinase activity is essential for myocardial hypertrophy and remodeling. Proc Natl Acad Sci U S A. 2015;112(42):13063–13068.
38. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):23.
39. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–254.
40. Anand PK. Lipids, inflammasomes, metabolism, and disease. Immunol Rev. 2020;297(1):108–122.
41. Julien O, Wells JA. Caspases and their substrates. Cell Death Differ. 2017;24(8):1380–1389.
42. Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 2017;27(9):673–684.
43. De Miguel C, Pelegrín P, Baroja-Mazo A, Cuevas S. Emerging role of the inflammasome and pyroptosis in hypertension. Int J Mol Sci. 2021;22(3):32.
44. Jia C, Chen H, Zhang J, et al. Role of pyroptosis in cardiovascular diseases. Int Immunopharmacol. 2019;67:311–318.
45. Song Z, Gong Q, Pyroptosis: GJ. Mechanisms and links with fibrosis. Cells. 2021;10(12):34.
46. Aluganti Narasimhulu C, Singla DK. Amelioration of diabetes-induced inflammation mediated pyroptosis, sarcopenia, and adverse muscle remodelling by bone morphogenetic protein-7. J Cachexia Sarcopenia Muscle. 2021;12(2):403–420.
47. Shen J, Fan Z, Sun G, Qi G. Sacubitril/valsartan (LCZ696) reduces myocardial injury following myocardial infarction by inhibiting NLRP3‑induced pyroptosis via the TAK1/JNK signaling pathway. Mol Med Rep. 2021;24(3):1.
48. Zhang L, Jiang Y-H, Fan C, et al. MCC950 attenuates doxorubicin-induced myocardial injury in vivo and in vitro by inhibiting NLRP3-mediated pyroptosis. Biomed Pharmacother. 2021;143:112133.
49. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42.
50. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176(1–2):11–42.
51. Chaanine AH. Autophagy and myocardial remodeling: is it autophagy or autophagic machinery and signaling pathways regulating it? J Am Coll Cardiol. 2018;71(18):2011–2014.
52. Matsui Y, Takagi H, Qu X, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100(6):914–922.
53. Buss SJ, Riffel JH, Katus HA, Hardt SE. Augmentation of autophagy by mTOR-inhibition in myocardial infarction: when size matters. Autophagy. 2010;6(2):304–306.
54. Yang Y, Li Y, Chen X, Cheng X, Liao Y, Yu X. Exosomal transfer of miR-30a between cardiomyocytes regulates autophagy after hypoxia. J Mol Med (Berl). 2016;94(6):711–724.
55. Sciarretta S, Yee D, Nagarajan N, et al. Trehalose-induced activation of autophagy improves cardiac remodeling after myocardial infarction. J Am Coll Cardiol. 2018;71(18):1999–2010.
56. Liu C-Y, Zhang Y-H, Li R-B, et al. LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nat Commun. 2018;9(1):29.
57. Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. 2019;99(4):1765–1817.
58. Dong Y, Chen H, Gao J, Liu Y, Li J, Wang J. Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J Mol Cell Cardiol. 2019;136:27–41.
59. Baldi A, Abbate A, Bussani R, et al. Apoptosis and post-infarction left ventricular remodeling. J Mol Cell Cardiol. 2002;34(2):165–174.
60. Diwan A, Wansapura J, Syed FM, Matkovich SJ, Lorenz JN, Dorn GW. Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation. Circulation. 2008;117(3):396–404.
61. Santos-Zas I, Lemarié J, Zlatanova I, et al. Cytotoxic CD8 T cells promote granzyme B-dependent adverse post-ischemic cardiac remodeling. Nat Commun. 2021;12(1):1483.
62. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–752.
63. Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol. 2018;15(8):457–470.
64. Wu C, Zhang Z, Zhang W, Liu X. Mitochondrial dysfunction and mitochondrial therapies in heart failure. Pharmacol Res. 2022;175:106038.
65. Aubert G, Martin OJ, Horton JL, et al. The failing heart relies on ketone bodies as a fuel. Circulation. 2016;133(8):698–705.
66. Badolia R, Ramadurai DKA, Abel ED, et al. The role of nonglycolytic glucose metabolism in myocardial recovery upon mechanical unloading and circulatory support in chronic heart failure. Circulation. 2020;142(3):259–274.
67. Carley AN, Maurya SK, Fasano M, et al. Short-Chain fatty acids outpace ketone oxidation in the failing heart. Circulation. 2021;143(18):1797–1808.
68. Yang D, Liu H-Q, Liu F-Y, et al. Mitochondria in pathological cardiac hypertrophy research and therapy. Front Cardiovasc Med. 2021;8:822969.
69. Zhuang L, Jia K, Chen C, et al. DYRK1B-STAT3 drives cardiac hypertrophy and heart failure by impairing mitochondrial bioenergetics. Circulation. 2022;145(11):829–846.
70. Li G, Shao Y, Guo HC, et al. MicroRNA-27b-3p down-regulates FGF1 and aggravates pathological cardiac remodelling. Cardiovasc Res. 2022;118(9):2139–2151.
71. Zhang Y, Liu Y, Liu H, Tang WH. Exosomes: biogenesis, biologic function and clinical potential. Cell Biosci. 2019;9:19.
72. Pegtel DM, Gould SJ. Exosomes. Annu Rev Biochem. 2019;88:487–514.
73. Kalluri R, LeBleu VS. The biology function and biomedical applications of exosomes. Science. 2020;367:6478.
74. Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141(4):1202–1207.
75. Liu G, Zhang R, Xu J, Wu C-I LX. Functional conservation of both CDS- and 3’-UTR-located microRNA binding sites between species. Mol Biol Evol. 2015;32(3):623–628.
76. Kuwabara Y, Ono K, Horie T, et al. Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circ Cardiovasc Genet. 2011;4(4):446–454.
77. Matsumoto S, Sakata Y, Suna S, et al. Circulating p53-responsive microRNAs are predictive indicators of heart failure after acute myocardial infarction. Circ Res. 2013;113(3):322–326.
78. Ibrahim A, Marbán E. Exosomes: fundamental biology and roles in cardiovascular physiology. Annu Rev Physiol. 2016;78:67–83.
79. Gupta S, Knowlton AA. HSP60 trafficking in adult cardiac myocytes: role of the exosomal pathway. Am J Physiol Heart Circ Physiol. 2007;292(6):H3052–H3056.
80. Ribeiro-Rodrigues TM, Laundos TL, Pereira-Carvalho R, et al. Exosomes secreted by cardiomyocytes subjected to ischaemia promote cardiac angiogenesis. Cardiovasc Res. 2017;113(11):1338–1350.
81. Nie X, Fan J, Li H, et al. miR-217 promotes cardiac hypertrophy and dysfunction by targeting PTEN. Mol Ther Nucleic Acids. 2018;12:254–266.
82. Nag AC. Study of non-muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios. 1980;28(109):41–61.
83. Bang C, Batkai S, Dangwal S, et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124(5):2136–2146.
84. Ranjan P, Kumari R, Goswami SK, et al. Myofibroblast-derived exosome induce cardiac endothelial cell dysfunction. Front Cardiovasc Med. 2021;8:676267.
85. Webber J, Steadman R, Mason MD, Tabi Z, Clayton A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010;70(23):9621–9630.
86. Loyer X, Zlatanova I, Devue C, et al. Intra-cardiac release of extracellular vesicles shapes inflammation following myocardial infarction. Circ Res. 2018;123(1):100–106.
87. de Jong OG, Verhaar MC, Chen Y, et al. Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes. J Extracell Vesicles. 2012;1:34.
88. Wang S, Aurora AB, Johnson BA, et al. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15(2):261–271.
89. van Balkom BWM, de Jong OG, Smits M, et al. Endothelial cells require miR-214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood. 2013;121(19):34.
90. Xiong -Y-Y, Gong Z-T, Tang R-J, Yang Y-J. The pivotal roles of exosomes derived from endogenous immune cells and exogenous stem cells in myocardial repair after acute myocardial infarction. Theranostics. 2021;11(3):1046–1058.
91. Wang C, Zhang C, Liu L, et al. Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Mol Ther. 2017;25(1):192–204.
92. Liu S, Chen J, Shi J, et al. M1-like macrophage-derived exosomes suppress angiogenesis and exacerbate cardiac dysfunction in a myocardial infarction microenvironment. Basic Res Cardiol. 2020;115(2):22.
93. Dong J, Zhu W, Wan D. Downregulation of microRNA-21-5p from macrophages-derived exosomes represses ventricular remodeling after myocardial infarction via inhibiting tissue inhibitors of metalloproteinase 3. Int Immunopharmacol. 2021;96:107611.
94. Chen B, Luo L, Wei X, et al. M1 bone marrow-derived macrophage-derived extracellular vesicles inhibit angiogenesis and myocardial regeneration following myocardial infarction via the MALAT1/MicroRNA-25-3p/CDC42 Axis. Oxid Med Cell Longev. 2021;2021:9959746.
95. Wang Y, Li C, Zhao R, et al. CircUbe3a from M2 macrophage-derived small extracellular vesicles mediates myocardial fibrosis after acute myocardial infarction. Theranostics. 2021;11(13):6315–6333.
96. Zhang Y, Cai Z, Shen Y, et al. Hydrogel-load exosomes derived from dendritic cells improve cardiac function via Treg cells and the polarization of macrophages following myocardial infarction. J Nanobiotechnology. 2021;19(1):271.
97. Liu H, Gao W, Yuan J, et al. Exosomes derived from dendritic cells improve cardiac function via activation of CD4(+) T lymphocytes after myocardial infarction. J Mol Cell Cardiol. 2016;91:123–133.
98. Cai L, Chao G, Li W, et al. Activated CD4 T cells-derived exosomal miR-142-3p boosts post-ischemic ventricular remodeling by activating myofibroblast. Aging. 2020;12(8):7380–7396.
99. Tseliou E, Pollan S, Malliaras K, et al. Allogeneic cardiospheres safely boost cardiac function and attenuate adverse remodeling after myocardial infarction in immunologically mismatched rat strains. J Am Coll Cardiol. 2013;61(10):1108–1119.
100. Bolli R, Chugh AR, D’Amario D, et al. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised Phase 1 trial. Lancet. 2011;378(9806):1847–1857. doi:10.1016/S0140-6736(11)61590-0
101. Rota M, Padin-Iruegas ME, Misao Y, et al. Local activation or implantation of cardiac progenitor cells rescues scarred infarcted myocardium improving cardiac function. Circ Res. 2008;103(1):107–116.
102. Balbi C, Vassalli G. Exosomes: beyond stem cells for cardiac protection and repair. Stem Cells. 2020;38(11):1387–1399.
103. Barile L, Lionetti V, Cervio E, et al. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc Res. 2014;103(4):530–541.
104. Xuan W, Wang L, Xu M, Weintraub NL, Ashraf M. miRNAs in extracellular vesicles from iPS-derived cardiac progenitor cells effectively reduce fibrosis and promote angiogenesis in infarcted heart. Stem Cells Int. 2019;2019:3726392.
105. Gallet R, Dawkins J, Valle J, et al. Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. Eur Heart J. 2017;38(3):201–211.
106. Malliaras K, Makkar RR, Smith RR, et al. Intracoronary cardiosphere-derived cells after myocardial infarction: evidence of therapeutic regeneration in the final 1-year results of the CADUCEUS trial (CArdiosphere-Derived aUtologous stem CElls to reverse ventricUlar dySfunction). J Am Coll Cardiol. 2014;63(2):110–122.
107. Tseliou E, Fouad J, Reich H, et al. Fibroblasts rendered antifibrotic, antiapoptotic, and angiogenic by priming with cardiosphere-derived extracellular membrane vesicles. J Am Coll Cardiol. 2015;66(6):599–611.
108. de Couto G, Gallet R, Cambier L, et al. Exosomal MicroRNA transfer into macrophages mediates cellular postconditioning. Circulation. 2017;136(2):200–214.
109. Teerlink JR, Metra M, Filippatos GS, et al. Benefit of cardiopoietic mesenchymal stem cell therapy on left ventricular remodelling: results from the Congestive Heart Failure Cardiopoietic Regenerative Therapy (CHART-1) study. Eur J Heart Fail. 2017;19(11):1520–1529.
110. Yao J, Huang K, Zhu D, et al. A minimally invasive exosome spray repairs heart after myocardial infarction. ACS Nano. 2021;10:1–2.
111. Huang P, Wang L, Li Q, et al. Combinatorial treatment of acute myocardial infarction using stem cells and their derived exosomes resulted in improved heart performance. Stem Cell Res Ther. 2019;10(1):300.
112. Liu Y, Guan R, Yan J, Zhu Y, Sun S, Qu Y. Mesenchymal stem cell-derived extracellular vesicle-shuttled microRNA-302d-3p represses inflammation and cardiac remodeling following acute myocardial infarction. J Cardiovasc Transl Res. 2022;3:1–8.
113. Datta R, Bansal T, Rana S, et al. Myocyte-derived Hsp90 modulates collagen upregulation via biphasic activation of STAT-3 in fibroblasts during cardiac hypertrophy. Mol Cell Biol. 2017;37:6.
114. Gardner GT, Travers JG, Qian J, et al. Phosphorylation of Hsp20 promotes fibrotic remodeling and heart failure. J Am Coll Cardiol Basic Trans Science. 2019;4(2):188–199.
115. Yu DW, Ge PP, Liu AL, Yu XY, Liu TT. HSP20-mediated cardiomyocyte exosomes improve cardiac function in mice with myocardial infarction by activating Akt signaling pathway. Eur Rev Med Pharmacol Sci. 2019;23(11):4873–4881.
116. Malik ZA, Kott KS, Poe AJ, et al. Cardiac myocyte exosomes: stability, HSP60, and proteomics. Am J Physiol Heart Circ Physiol. 2013;304(7):H954–H965.
117. Kim S-C, Stice JP, Chen L, et al. Extracellular heat shock protein 60, cardiac myocytes, and apoptosis. Circ Res. 2009;105(12):1186–1195.
118. Shi C, Ulke-Lemée A, Deng J, Batulan Z, O’Brien ER. Characterization of heat shock protein 27 in extracellular vesicles: a potential anti-inflammatory therapy. FASEB J. 2019;33(2):1617–1630.
119. Chen Q, Huang M, Wu J, Jiang Q, Zheng X. Exosomes isolated from the plasma of remote ischemic conditioning rats improved cardiac function and angiogenesis after myocardial infarction through targeting Hsp70. Aging. 2020;12(4):3682–3693.
120. Routledge D, Scholpp S. Mechanisms of intercellular Wnt transport. Development. 2019;146(10):4.
121. Działo E, Rudnik M, Koning RI, et al. WNT3a and WNT5a transported by exosomes activate WNT signaling pathways in human cardiac fibroblasts. Int J Mol Sci. 2019;20(6):34.
122. Sun J, Shen H, Shao L, et al. HIF-1α overexpression in mesenchymal stem cell-derived exosomes mediates cardioprotection in myocardial infarction by enhanced angiogenesis. Stem Cell Res Ther. 2020;11(1):373.
123. Wang Q, Zhang L, Sun Z, et al. HIF-1α overexpression in mesenchymal stem cell-derived exosome-encapsulated arginine-glycine-aspartate (RGD) hydrogels boost therapeutic efficacy of cardiac repair after myocardial infarction. Mater Today Bio. 2021;12:100171.
124. Yu X, Deng L, Wang D, et al. Mechanism of TNF-α autocrine effects in hypoxic cardiomyocytes: initiated by hypoxia inducible factor 1α, presented by exosomes. J Mol Cell Cardiol. 2012;53(6):848–857.
125. Zhang Q, Wang L, Wang S, et al. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct Target Ther. 2022;7(1):78.
126. Lavine KJ, Kovacs A, Ornitz DM. Hedgehog signaling is critical for maintenance of the adult coronary vasculature in mice. J Clin Invest. 2008;118(7):2404–2414.
127. Bueno-Betí C, Novella S, Soleti R, et al. Microparticles harbouring Sonic hedgehog morphogen improve the vasculogenesis capacity of endothelial progenitor cells derived from myocardial infarction patients. Cardiovasc Res. 2019;115(2):409–418.
128. Mackie AR, Klyachko E, Thorne T, et al. Sonic hedgehog-modified human CD34+ cells preserve cardiac function after acute myocardial infarction. Circ Res. 2012;111(3):312–321.
129. Yue Y, Wang C, Benedict C, et al. Interleukin-10 deficiency alters endothelial progenitor cell-derived exosome reparative effect on myocardial repair via integrin-linked kinase enrichment. Circ Res. 2020;126(3):315–329.
130. Troncoso R, Ibarra C, Vicencio JM, Jaimovich E, Lavandero S. New insights into IGF-1 signaling in the heart. Trends Endocrinol Metab. 2014;25(3):128–137.
131. Yamaguchi T, Izumi Y, Nakamura Y, et al. Repeated remote ischemic conditioning attenuates left ventricular remodeling via exosome-mediated intercellular communication on chronic heart failure after myocardial infarction. Int J Cardiol. 2015;178:239–246.
132. Gong X-H, Liu H, Wang S-J, Liang S-W, Wang -G-G. Exosomes derived from SDF1-overexpressing mesenchymal stem cells inhibit ischemic myocardial cell apoptosis and promote cardiac endothelial microvascular regeneration in mice with myocardial infarction. J Cell Physiol. 2019;234(8):13878–13893.
133. Sayed D, Hong C, Chen I-Y, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. 2007;100(3):416–424.
134. Carè A, Catalucci D, Felicetti F, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13(5):613–618.
135. Tian C, Hu G, Gao L, Hackfort BT, Zucker IH. Extracellular vesicular MicroRNA-27a* contributes to cardiac hypertrophy in chronic heart failure. J Mol Cell Cardiol. 2020;143:120–131.
136. Kurtzwald-Josefson E, Zeevi-Levin N, Rubchevsky V, et al. Cardiac fibroblast-induced pluripotent stem cell-derived exosomes as a potential therapeutic mean for heart failure. Int J Mol Sci. 2020;21(19):34.
137. Huang Z-P, Chen J, Seok HY, et al. MicroRNA-22 regulates cardiac hypertrophy and remodeling in response to stress. Circ Res. 2013;112(9):1234–1243.
138. Wang X, Morelli MB, Matarese A, Sardu C, Santulli G. Cardiomyocyte-derived exosomal microRNA-92a mediates post-ischemic myofibroblast activation both in vitro and ex vivo. ESC Heart Fail. 2020;7(1):284–288.
139. Yang J, Yu X, Xue F, Li Y, Liu W, Zhang S. Exosomes derived from cardiomyocytes promote cardiac fibrosis via myocyte-fibroblast cross-talk. Am J Transl Res. 2018;10(12):4350–4366.
140. Wu Y, Peng W, Fang M, Wu M, Wu M. MSCs-derived extracellular vesicles carrying miR-212-5p alleviate myocardial infarction-induced cardiac fibrosis via NLRC5/VEGF/TGF-β1/SMAD Axis. J Cardiovasc Transl Res. 2022;15(2):302–316.
141. Li J, Salvador AM, Li G, et al. Mir-30d regulates cardiac remodeling by intracellular and paracrine signaling. Circ Res. 2021;128(1):2.
142. Ramanujam D, Schön AP, Beck C, et al. MicroRNA-21-dependent macrophage-to-fibroblast signaling determines the cardiac response to pressure overload. Circulation. 2021;143(15):1513–1525.
143. Ke X, Yang R, Wu F, et al. Exosomal miR-218-5p/miR-363-3p from endothelial progenitor cells ameliorate myocardial infarction by targeting the p53/JMY signaling pathway. Oxid Med Cell Longev. 2021;2021:5529430.
144. Wang Q-G, Cheng BC-Y, He Y-Z, et al. miR-320a in serum exosomes promotes myocardial fibroblast proliferation via regulating the PIK3CA/Akt/mTOR signaling pathway in HEH2 cells. Exp Ther Med. 2021;22(2):873.
145. Yuan J, Liu H, Gao W, et al. MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress. Theranostics. 2018;8(9):2565–2582.
146. Vaskova E, Ikeda G, Tada Y, Wahlquist C, Mercola M, Yang PC. Sacubitril/Valsartan improves cardiac function and decreases myocardial fibrosis via downregulation of exosomal miR-181a in a rodent chronic myocardial infarction model. J Am Heart Assoc. 2020;9(13):e015640.
147. Cao C, Wang B, Tang J, et al. Circulating exosomes repair endothelial cell damage by delivering miR-193a-5p. J Cell Mol Med. 2021;25(4):2176–2189.
148. Geng T, Song Z-Y, Xing J-X, Wang B-X, Dai S-P, Xu Z-S. Exosome derived from coronary serum of patients with myocardial infarction promotes angiogenesis through the miRNA-143/IGF-IR pathway. Int J Nanomedicine. 2020;15:2647–2658.
149. Gou L, Xue C, Tang X, Fang Z. Inhibition of Exo-miR-19a-3p derived from cardiomyocytes promotes angiogenesis and improves heart function in mice with myocardial infarction via targeting HIF-1α. Aging. 2020;12(23):23609–23618.
150. Ottaviani L, Juni RP, de Abreu RC, et al. Intercellular transfer of miR-200c-3p impairs the angiogenic capacity of cardiac endothelial cells. Mol Ther. 2022;30(6):2257–2273.
151. Liao Z, Chen Y, Duan C, et al. Cardiac telocytes inhibit cardiac microvascular endothelial cell apoptosis through exosomal miRNA-21-5p-targeted silencing to improve angiogenesis following myocardial infarction. Theranostics. 2021;11(1):268–291.
152. Liu H, Zhang Y, Yuan J, et al. Dendritic cell‑derived exosomal miR‑494‑3p promotes angiogenesis following myocardial infarction. Int J Mol Med. 2021;47(1):315–325.
153. Zhu D, Wang Y, Thomas M, et al. Exosomes from adipose-derived stem cells alleviate myocardial infarction via microRNA-31/FIH1/HIF-1α pathway. J Mol Cell Cardiol. 2022;162:10–19.
154. Ma T, Chen Y, Chen Y, et al. MicroRNA-132, delivered by mesenchymal stem cell-derived exosomes, promote angiogenesis in myocardial infarction. Stem Cells Int. 2018;2018:3290372.
155. Wang N, Chen C, Yang D, et al. Mesenchymal stem cells-derived extracellular vesicles, via miR-210, improve infarcted cardiac function by promotion of angiogenesis. Biochim Biophys Acta Mol Basis Dis. 2017;1863(8):2085–2092.
156. Youn S-W, Li Y, Kim Y-M, et al. Modification of cardiac progenitor cell-derived exosomes by miR-322 provides protection against myocardial infarction through Nox2-dependent angiogenesis. Antioxidants. 2019;8(1):3.
157. Wang Z, Gao D, Wang S, Lin H, Wang Y, Xu W. Exosomal microRNA-1246 from human umbilical cord mesenchymal stem cells potentiates myocardial angiogenesis in chronic heart failure. Cell Biol Int. 2021;45(11):2211–2225.
158. Huang Y, Chen L, Feng Z, et al. EPC-derived exosomal miR-1246 and miR-1290 regulate phenotypic changes of fibroblasts to endothelial cells to exert protective effects on myocardial infarction by targeting ELF5 and SP1. Front Cell Dev Biol. 2021;9:647763.
159. Zhu W, Sun L, Zhao P, et al. Macrophage migration inhibitory factor facilitates the therapeutic efficacy of mesenchymal stem cells derived exosomes in acute myocardial infarction through upregulating miR-133a-3p. J Nanobiotechnology. 2021;19(1):61.
160. Gollmann-Tepeköylü C, Pölzl L, Graber M, et al. miR-19a-3p containing exosomes improve function of ischaemic myocardium upon shock wave therapy. Cardiovasc Res. 2020;116(6):1226–1236.
161. Liu J, Jiang M, Deng S, et al. miR-93-5p-Containing exosomes treatment attenuates acute myocardial infarction-induced myocardial damage. Mol Ther Nucleic Acids. 2018;11:103–115.
162. Kore RA, Wang X, Ding Z, Griffin RJ, Tackett AJ, Mehta JL. MSC exosome-mediated cardioprotection in ischemic mouse heart comparative proteomics of infarct and peri-infarct areas. Mol Cell Biochem. 2021;476(4):1691–1704.
163. Yue R, Lu S, Luo Y, et al. Mesenchymal stem cell-derived exosomal microRNA-182-5p alleviates myocardial ischemia/reperfusion injury by targeting GSDMD in mice. Cell Death Discov. 2022;8(1):202.
164. Yu H, Qin L, Peng Y, Bai W, Wang Z. Exosomes derived from hypertrophic cardiomyocytes induce inflammation in macrophages miR-155 mediated MAPK pathway. Front Immunol. 2020;11:606045.
165. Huang J, Wang F, Sun X, et al. Myocardial infarction cardiomyocytes-derived exosomal miR-328-3p promote apoptosis via Caspase signaling. Am J Transl Res. 2021;13(4):2365–2378.
166. Pu L, Kong X, Li H, He X. Exosomes released from mesenchymal stem cells overexpressing microRNA-30e ameliorate heart failure in rats with myocardial infarction. Am J Transl Res. 2021;13(5):4007–4025.
167. Zhao Z, Du S, Shen S, Wang L. microRNA-132 inhibits cardiomyocyte apoptosis and myocardial remodeling in myocardial infarction by targeting IL-1β. J Cell Physiol. 2020;235(3):2710–2721.
168. Wu Z, Cheng S, Wang S, Li W, Liu J. BMSCs-derived exosomal microRNA-150-5p attenuates myocardial infarction in mice. Int Immunopharmacol. 2021;93:107389.
169. Fu DL, Jiang H, Li CY, Gao T, Liu MR, Li HW. MicroRNA-338 in MSCs-derived exosomes inhibits cardiomyocyte apoptosis in myocardial infarction. Eur Rev Med Pharmacol Sci. 2020;24(19):10107–10117.
170. Peng Y, Zhao J-L, Peng Z-Y, Xu W-F, Yu G-L. Exosomal miR-25-3p from mesenchymal stem cells alleviates myocardial infarction by targeting pro-apoptotic proteins and EZH2. Cell Death Dis. 2020;11(5):317.
171. Sun L, Zhu W, Zhao P, et al. Down-regulated exosomal MicroRNA-221-3p derived from senescent mesenchymal stem cells impairs heart repair. Front Cell Dev Biol. 2020;8:263.
172. Song Y, Zhang C, Zhang J, et al. Localized injection of miRNA-21-enriched extracellular vesicles effectively restores cardiac function after myocardial infarction. Theranostics. 2019;9(8):2346–2360.
173. Qiao L, Hu S, Liu S, et al. microRNA-21-5p dysregulation in exosomes derived from heart failure patients impairs regenerative potential. J Clin Invest. 2019;129(6):2237–2250.
174. Xiao J, Pan Y, Li XH, et al. Cardiac progenitor cell-derived exosomes prevent cardiomyocytes apoptosis through exosomal miR-21 by targeting PDCD4. Cell Death Dis. 2016;7(6):e2277.
175. Luther KM, Haar L, McGuinness M, et al. Exosomal miR-21a-5p mediates cardioprotection by mesenchymal stem cells. J Mol Cell Cardiol. 2018;119:125–137.
176. Zhu L-P, Tian T, Wang J-Y, et al. Hypoxia-elicited mesenchymal stem cell-derived exosomes facilitates cardiac repair through miR-125b-mediated prevention of cell death in myocardial infarction. Theranostics. 2018;8(22):6163–6177.
177. Cheng H, Chang S, Xu R, et al. Hypoxia-challenged MSC-derived exosomes deliver miR-210 to attenuate post-infarction cardiac apoptosis. Stem Cell Res Ther. 2020;11(1):224.
178. Zhang J, Ma J, Long K, et al. Overexpression of exosomal cardioprotective miRNAs mitigates hypoxia-induced H9c2 Cells apoptosis. Int J Mol Sci. 2017;18(4):5.
179. Xiao C, Wang K, Xu Y, et al. Transplanted mesenchymal stem cells reduce autophagic flux in infarcted hearts via the exosomal transfer of miR-125b. Circ Res. 2018;123(5):564–578.
180. Li Y, Yang R, Guo B, et al. Exosomal miR-301 derived from mesenchymal stem cells protects myocardial infarction by inhibiting myocardial autophagy. Biochem Biophys Res Commun. 2019;514(1):323–328.
181. Michalik KM, You X, Manavski Y, et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114(9):1389–1397.
182. Wu Q, Wang J, Tan WLW, et al. Extracellular vesicles from human embryonic stem cell-derived cardiovascular progenitor cells promote cardiac infarct healing through reducing cardiomyocyte death and promoting angiogenesis. Cell Death Dis. 2020;11(5):354.
183. Shyu K-G, Wang B-W, Fang W-J, Pan C-M, Lin C-M. Hyperbaric oxygen-induced long non-coding RNA MALAT1 exosomes suppress MicroRNA-92a expression in a rat model of acute myocardial infarction. J Cell Mol Med. 2020;24(22):12945–12954.
184. Kenneweg F, Bang C, Xiao K, et al. Long noncoding RNA-enriched vesicles secreted by hypoxic cardiomyocytes drive cardiac fibrosis. Mol Ther Nucleic Acids. 2019;18:363–374.
185. Wang L, Zhang J. Exosomal lncRNA AK139128 derived from hypoxic cardiomyocytes promotes apoptosis and inhibits cell proliferation in cardiac fibroblasts. Int J Nanomedicine. 2020;15:3363–3376.
186. Mao Q, Liang X-L, Zhang C-L, Pang Y-H, Lu Y-X. LncRNA KLF3-AS1 in human mesenchymal stem cell-derived exosomes ameliorates pyroptosis of cardiomyocytes and myocardial infarction through miR-138-5p/Sirt1 axis. Stem Cell Res Ther. 2019;10(1):393.
187. Sun L, Zhu W, Zhao P, et al. Long noncoding RNA UCA1 from hypoxia-conditioned hMSC-derived exosomes: a novel molecular target for cardioprotection through miR-873-5p/XIAP axis. Cell Death Dis. 2020;11(8):696.
188. Huang P, Wang L, Li Q, et al. Atorvastatin enhances the therapeutic efficacy of mesenchymal stem cells-derived exosomes in acute myocardial infarction via up-regulating long non-coding RNA H19. Cardiovasc Res. 2020;116(2):353–367.
189. Wang Y, Zhao R, Shen C, et al. Exosomal CircHIPK3 released from hypoxia-induced cardiomyocytes regulates cardiac angiogenesis after myocardial infarction. Oxid Med Cell Longev. 2020;2020:8418407.
190. Goren Y, Kushnir M, Zafrir B, Tabak S, Lewis BS, Amir O. Serum levels of microRNAs in patients with heart failure. Eur J Heart Fail. 2012;14(2):147–154.
191. Lima Correa B, El Harane N, Desgres M, et al. Extracellular vesicles fail to trigger the generation of new cardiomyocytes in chronically infarcted hearts. Theranostics. 2021;11(20):10114–10124.
192. Liang Y, Duan L, Lu J, Xia J. Engineering exosomes for targeted drug delivery. Theranostics. 2021;11(7):3183–3195.
193. Vandergriff A, Huang K, Shen D, et al. Targeting regenerative exosomes to myocardial infarction using cardiac homing peptide. Theranostics. 2018;8(7):1869–1878.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.