Back to Journals » International Journal of Nanomedicine » Volume 13
Exosome-mediated delivery of functionally active miRNA-142-3p inhibitor reduces tumorigenicity of breast cancer in vitro and in vivo
Authors Naseri Z, Oskuee RK ![]() , Jaafari MR
, Jaafari MR ![]() , Forouzandeh Moghadam M
, Forouzandeh Moghadam M ![]()
Received 2 August 2018
Accepted for publication 3 October 2018
Published 20 November 2018 Volume 2018:13 Pages 7727—7747
DOI https://doi.org/10.2147/IJN.S182384
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Zahra Naseri,1 Reza Kazemi Oskuee,2 Mahmoud Reza Jaafari,3,4 Mehdi Forouzandeh Moghadam5
1Department of Medical Biotechnology, School of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran; 2Neurogenic Inflammation Research Center, Mashhad University of Medical Sciences, Mashhad, Iran; 3Biotechnology Research Center, Pharmaceutical Technology Institute, Mashhad University of Medical Sciences, Mashhad, Iran; 4Department of Pharmaceutical Nanotechnology, School of Pharmacy, Mashhad University of Medical Sciences, Mashhad, Iran; 5Department of Medical Biotechnology, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran
Background: Exosomes, widely recognized natural nanovesicles, represent one of the recently discovered modes of intercellular communication due to their ability to transmit crucial cellular information that can be engineered to have robust delivery and targeting capacity. MiR-142-3p, one of the upregulated microRNAs (miRNAs) in many types of breast cancer, activates the canonical Wnt signaling pathway and transactivates the miR-150 expression, and results in the hyperproliferation of cancer cells in vitro and mammary glands in vivo.
Materials and methods: In this study, we exploited the exosomes isolated from bone marrow-derived mesenchymal stem cells (MSCs-Exo) to deliver LNA (locked nucleic acid)-modified anti-miR-142-3p oligonucleotides to suppress the expression level of miR-142-3p and miR-150 in 4T1 and TUBO breast cancer cell lines.
Results: The in vitro results showed that the MSCs-Exo can efficiently deliver anti-miR-142-3p to reduce the miR-142-3p and miR-150 levels and increase the transcription of the regulatory target genes, APC and P2X7R. We also evaluated in vivo distribution of the MSCs-Exo in tumor-bearing mice. The in vivo result indicated that MSCs-Exo can penetrate the tumor site and are suitable nanovehicles to deliver the inhibitory oligonucleotides into the tumor tissues to downregulate the expression levels of miR-142-3p and miR-150.
Conclusion: We showed that MSCs-derived exosomes could be used as a feasible nanovehicle to deliver drug molecules like LNA-anti-miR-142-3p in both in vitro and in vivo studies.
Keywords: MSCs-derived exosomes, tumor tropism, Wnt/β-catenin signaling pathway, breast cancer, LNA-antimiR-142-3p
Introduction
Exosomes are naïve nanovesicles with an approximate diameter of 30–150 nm, which have attracted considerable attention from the past decade due to their unique characteristics such as immune compatibility, low toxicity, and nano-scale size, and are also relatively homogenous and stable.1,2 Exosomes are generated by almost all cell types through inward budding of the inner endosomal membrane followed by plasma membrane fusion and can be extracted from various biological fluids. Several proteins such as tetraspanins (CD63, CD9, CD81), heat shock proteins (Hsc70), lysosomal proteins (Lamp2b), and fusion proteins (flotillin, annexin) are identified on the surface of exosomes.3 Exosomes exert their physiological roles through the exchange of biomolecules such as proteins, lipids, mRNAs, and microRNAs (miRNAs) between different cells. Recent studies have indicated that exosomes contribute to homeostasis and cell survival by ridding cells of unpleasant substances and also play important roles in different pathophysiological conditions.4 Since exosomes have the potential to act as nanocarrier to deliver drugs and nucleic acid-based molecules, they have attracted much attention in nanomedicine applications. Exosomes have different receptors or membranous proteins, depending on the cell of origin, and accordingly have different tropism in the body.5 In fact, we hypothesize that different exosomes with cell-specific surface proteins may have distinct routes and circulation pattern all over the body. The use of exosomes as nanovehicles in the field of cancer therapy requires the recognition of the tumor tropism of the extracted exosomes.
Mesenchymal stem cells (MSCs) have been used in the treatment of various diseases due to their unique characteristics including homing to injured tissues; multilineage differentiation; tissue repair promotion; and anti-inflammatory, immunosuppression, and neuroprotection effects.6 MSCs have the potential to produce more exosomes in comparison to other cell sources. It was determined that MSCs exert their biological functions through paracrine effectors and exosome release.7 Exosomes derived from MSCs have been tested in various studies, and their therapeutic effects have been shown to be similar to the MSCs of origin, for example, the MSC exosomes have the capacity to promote tissue repair and mediate regeneration.8 Moreover, it was indicated that MSCs-derived exosomes, as opposed to dendritic cells-derived exosomes, are poorly immunogenic. Accordingly, it can be predicted that MSCs-derived exosomes, in addition to injured tissues, exhibit tumor tropism similar to MSCs.9 To the best of our knowledge, tumor tropism of the exosomes derived from the bone marrow-derived MSCs has not been investigated so far. So, in this work, we have tried to examine the biodistribution of the MSC exosomes in vivo in tumor-bearing mouse models to find whether the exosomes can be used as nanovehicles to deliver miRNA inhibitor molecules to the tumor. The imaging results showed that MSCs-derived exosomes have the ability to penetrate the tumor site based on the enhanced permeability and retention (EPR) effect. As a result, the MSCs-derived exosomes, due to the strong tumor tropism, are suitable nanocarriers for transferring the desired biomolecules to the tumor site.
Although significant improvements have been made in the field of nucleic acid-based therapies and numerous carriers have been used to transport molecules such as siRNA and miRNA inhibitors, studies on finding suitable carriers for these kinds of molecules are still ongoing.10 The main problem in the miRNA inhibitors transfer is their instability and negative charge, and hence, even in the presence of a suitable transfection reagent, cannot effectively penetrate the hydrophobic cell membranes.11 In recent years, exosomes have been introduced as a new alternative to the transmission of these kinds of therapeutic molecules.12 Here, we investigated whether MSCs-derived exosomes can act as a carrier to deliver LNA (locked nucleic acid)-modified miR-142-3p inhibitor to 4T1 and TUBO breast cancer cells to decrease cancer cell proliferation in vitro and in vivo. Our results demonstrated that it can be useful to exploit the MSCs-derived exosomes to efficiently deliver LNA-anti-miR-142-3p, which significantly reduced miR-142-3p and miR-150 levels and enhanced transcriptional levels of the corresponding target genes in both in vitro and in vivo studies.
Materials and methods
Materials
DMEM, α-minimum essential medium (α-MEM), FBS, PBS, trypsin, and penicillin-streptomycin (pen/strep) were all obtained from Thermo Fisher Scientific (Waltham, MA, USA). ExoQuick™-TC reagent was purchased from System Biosciences (Mountain View, CA, USA). Exosome antigens and antibodies were obtained from Cell Guidance Systems Inc. (Babraham, Cambridge, UK). DAPI dye and PKH67 Green Fluorescent Cell Linker Kit were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). MiR-142-3p miRCURY LNA miRNA Power Inhibitor (catalog no. YI04100271), miRCURY LNA miRNA Power Inhibitor Negative Control (catalog no. YI00199006), Universal cDNA Synthesis Kit II, and ExiLENT SYBR® Green Master Mix Kit were purchased from Qiagen (Hilden, Germany). CD29, CD44, CD90, CD105, CD34, and CD45 antibodies were obtained from eBioscience (San Diego, CA, USA). Annexin V-Fluos apoptosis detection kit was purchased from Hoffman-La Roche Ltd. (Basel, Switzerland).
Cell lines
TUBO cell line, a cloned cell line that overexpressed the rHER2/neu protein, was established from a lobular carcinoma that had spontaneously arisen in a female BALB/c mouse transgenic for transforming rat neu oncogene. This cell line was kindly provided by Dr Pier-Luigi Lollini (Department of Clinical and Biological Sciences, University of Turin, Orbassano, Italy) and was cultured in DMEM supplemented with 20% FBS, 100 units/mL penicillin, and 100 units/mL streptomycin. All experiments involving in vitro culture of TUBO cell line were carried out under the approval of the Institutional Ethical Committee and Research Advisory Committee of Mashhad University of Medical Sciences based on the specific National Ethical Guidelines for Biomedical Research issued by the Research and Technology Deputy of Ministry of Health and Medical Education (MOHME) of Iran (Education Office, dated May 13, 2015; proposal code 931289). 4T1 cell line was purchased from American Type Culture Collection center and cultured in DMEM supplemented with 10% FBS, 100 units/mL penicillin, and 100 units/mL streptomycin at 37°C in humidified air with 5% CO2.
Mice
Four- to eight-week-old female BALB/c mice were purchased from the Pasteur Institute (Tehran, Iran). All animal experiments and procedures were approved by the Institutional Ethical Committee and Research Advisory Committee of Mashhad University of Medical Sciences (Education Office, dated May 13, 2015; proposal code 931289), based on the Specific National Ethical Guidelines for Biomedical Research issued by the Research and Technology Deputy of Ministry of Health and Medical Education (MOHME) of Iran that was issued in 2005. The mice were kept in cages in a ventilated room with unlimited access to food and water.
Isolation and culture of mesenchymal stem cells
Primary MSCs were isolated from mouse bone marrow obtained from the femurs and tibias of 4–8-week-old BALB/c mice. The bones were aseptically removed, scrubbed carefully to remove the residual soft tissues, and transferred to a sterile culture dish with complete α-MEM medium on ice. The bone cavities were flushed with the medium until the bones appeared pale. The cells were then washed with PBS and suspended in α-MEM medium with 10% FBS. Bone marrow cells (1×107/mL) were placed in 75 cm2 cell culture flasks and incubated at 37°C with 5% CO2. After 3 days, spindle-shaped cells appeared and nonadherent cells were removed, and fresh culture medium was added. MSCs reached 70%–90% confluence within 4–6 days and were subcultured at a split ratio of 1:3.
Phenotypic characterization and differentiation studies of the isolated MSCs
Bone marrow-derived MSCs at passage three were phenotyped using CD29, CD44, CD90, CD105, CD34, and CD45 by a FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA, USA). In order to evaluate the differentiation abilities of MSCs, the cells (2×103/cm2) were seeded into six-chamber slides. Osteogenic differentiation medium (α-MEM containing dexamethasone 1×10−8 M, β-glycerophosphate 10 mM, ascorbic 0.3 mM, and 10% FBS) and adipogenic induction medium (α-MEM containing 1 μM dexamethasone, 10 μg/mL recombinant insulin, 0.5 mM 3-isobutyl-1-methylxanthine, and 10% FBS) were added to the chamber slides. Osteocyte formation was evaluated 3 weeks later using Alizarin red as an indicator of calcium accumulation. Induction of adipocytes was assessed using Oil Red O stain as an indicator of intracellular lipid accumulation.
Preparation of conditioned medium and exosomes isolation
The MSCs at passage three or higher were used for preparing conditioned medium (CM). The exosomes were isolated from CM according to the International Society of Extracellular Vesicles recommendations. In brief, 150 mL of CM was collected from 150 mm plates (n=10), each containing 13 million MSCs after 48 hours of culture in CM containing exo-depleted FBS. Cell debris was removed by centrifuging at 300× g for 5 minutes, then 2,000× g for 20 minutes, and finally 16,000× g for 1 hour. The cleared CM was passed through a 0.22 μm filter and concentrated using 100 kDa molecular weight Amicon Ultra-15 Centrifugal Filter (Merck Millipore, Billerica, MA, USA). The filtered supernatants were incubated with the appropriate volume of Exoquick-TC precipitation solution according to the manufacturer’s instructions (System Biosciences) for 16 hours at 4°C, and then centrifuged for 30 minutes at 1,500× g to pellet exosomes. Some studies have indicated that there are no significant differences in exosome population isolated by the Exoquick protocol compared with ultracentrifugation methods.13,14
Characterization of isolated exosomes
Morphological assessment of the exosomes was performed using transmission electron microscopy (TEM, Philips CM30 electron microscope, Eindhoven, Netherlands) at 80 kV. Briefly, the exosome preparation was fixed for 1 hour in 4% paraformaldehyde and washed once with PBS. Then, the pellets were fixed in 2.5% glutaraldehyde, loaded on formvar-/carbon-coated electron microscopy EM grids. The grids were blocked with 5% BSA for 10 minutes. The blocked grids were incubated with anti-CD63 antibody overnight at 4°C, washed six times in 0.1% BSA, and then incubated with the recommended dilution of a 10 nm-gold-coupled secondary antibody (Abcam, Cambridge, UK) for 1 hour at room temperature. The grids were then postfixed in 1% glutaraldehyde and contrasted successively in 2% methylcellulose/0.4% uranyl acetate (pH 4.0). Size distribution of purified exosomes was evaluated using dynamic light scattering (DLS). Briefly, about 20 μL of exosome sample was diluted in 1 mL PBS and shaken at 4°C for 20 minutes prior to DLS measurement. DLS measurements were conducted at 25°C using Nano Zetasizer (Malvern Instruments Ltd., Malvern, UK). To identify the exosomal marker using Western blot, exosome proteins or whole cells were lysed in reducing sample buffer and boiled for 10 minutes at 95°C. Proteins were resolved on a 10% SDS-PAGE, transferred to nitrocellulose membranes, blocked in 5% non-fat powdered milk in PBS-T (0.5% Tween-20) and incubated separately with CD81, CD63, and calnexin-specific primary antibodies at the supplier recommended dilutions overnight at 4°C. After subsequent washing, the membranes were further incubated with horseradish peroxidase-coupled secondary antibodies. Protein bands were detected using enhanced chemiluminescence reagent (Amersham ECL Select GE healthcare life sciences, USA).
Cellular uptake of PKH67-labeled exosomes
MSCs-derived exosomes were fluorescently labeled using PKH67 dye, which is a green fluorescent dye that labels the lipid membranes. In brief, 100 μg of exosomes was resuspended in 100 μL of diluent C and then mixed with 4 μL of PKH67 dye diluted in 100 μL of diluent C and then incubated for 20 minutes at room temperature; 1 mL of PBS containing 1% BSA was added to stop the labeling reaction and labeled exosomes were reisolated by Exoquick precipitation solution. 4T1 and TUBO cells were cultured in 24-well plate in complete DMEM and when a confluency of 60%–70% was reached, 5 μg of PKH67-labeled exosomes was added to each well and cells were incubated for 24 hours at 37°C with 5% CO2. After incubation, the cells were washed with PBS and fixed in 4% paraformaldehyde for 20 minutes at room temperature. About 0.2 μg/mL of DAPI was added to nuclear staining and then cellular uptake of PKH67-labeled exosomes was visualized using confocal laser scanning microscopy (Leica TCS SPE; Leica Microsystems, Wetzlar, Germany).
Loading the exosomes with LNA-anti-miR-142-3p by electroporation
In order to load the exosomes with LNA-anti-miR-142-3p and miRNA inhibitor negative control, electroporation method with the validated conditions was used (Figure S1).15 For this purpose, the pellet of exosomes was suspended in pre-chilled EDTA (1 mM) and trehalose (25 mM) containing hypo-osmolar electroporation buffer (Eppendorf Multiporator, Hamburg, Germany). MiRNA inhibitor and scrambled control molecules at a final concentration of 150 pmol were added to 1 μg/μL of the exosomes sample and the mixture was transferred into a cold 0.4 cm electroporation cuvette. Electroporation was performed at 0.200 kV and 100 μF with three pulses (all containers and buffers were RNase free). The sample was then incubated at room temperature for 30 minutes and subsequently treated with one unit of RNase H to eliminate free unincorporated anti-miR molecules, and the loaded exosomes were reisolated using the Exoquick protocol.
Determination of LNA-anti-miR-142-3p encapsulated in MSCs-Exo
In order to estimate the amount of LNA-anti-miR-142-3p and LNA-anti-miR negative control oligonucleotides in MSCs-derived exosomes, the sample preparations were centrifuged twice at 100,000× g for 1 hour to precipitate the exosomes loaded with the anti-miRNA oligonucleotides. The supernatant was carefully collected and the pellet (with loaded exosomes) was lysed by adding 5% Triton X-100 and subsequently subjected to sonication. The anti-miRNA concentration was determined by measurement of absorbance at 260 nm (Abs260). The percentage of the loaded anti-miRNA oligonucleotides was calculated as follows:
|

RNA isolation, mRNA and miRNA analysis
Expression levels of miRNA-142 and miRNA-150 were quantified using the ExiLENT SYBR Green Master Mix Kit, and the expression levels of putative miRNA-142 and miRNA-150 target genes, APC and P2X7R, respectively, were measured using the Roche Master Mix containing SYBR green on a Rotor-Gene RG-3000 real-time PCR machine (Corbett Research, Cambridge, UK). MiRNA quantitations were normalized to an endogenous control U6 snRNA, and expression levels of target genes were normalized to the β-actin gene. The miR-specific LNA-enhanced primers for miR-150 and miR-142-3p, and primers for U6 snRNA were obtained from Exiqon. Following primers were used for amplification of the target genes and β-actin gene: APC forward primer: TGTCCCTCCGTTCTTATGGAA, APC reverse primer: TCTTGGAAATGAACCCATAGGAA; P2X7R forward primer: ATCGGCTCAACCCTCTCCTAC, P2X7R reverse primer: CTGGAGTAAGTGTCGATG AGGAAG; β-actin forward primer: TCCCTGGAGAAGAGCTACG, β-actin reverse primer: GTAGTTTCGTGGATGCCACA. MiRNA and gene quantification levels were calculated using the 2−ΔΔCt method.
Cell viability assay
Cell viability was determined by using the MTT assay. Each of the 4T1 and TUBO cell lines was seeded at a density of 104 cells/well in 96-well plate before treatment. After overnight plating, cells were treated with unloaded MSCs-Exo (exosomes isolated from bone marrow-derived mesenchymal stem cells), MSCs-Exo loaded with LNA-anti-miR-142-3p, or MSCs-Exo loaded with LNA-miRNA inhibitor negative control for 48 hours. The same amount of exosomes (equal to 5 μg) was added to each treated group.
Quantification of apoptosis by flow cytometry
Cell apoptosis was measured by an Annexin V-Fluos apoptosis detection kit according to the manufacturer’s protocol. In brief, 4T1 and TUBO cell lines in each treated group, unloaded MSCs-Exo, MSCs-Exo loaded with LNA-anti-miR-142-3p, or MSCs-Exo loaded with LNA-miRNA inhibitor negative control, were harvested and washed twice with PBS. Each pellet was resuspended in 400 μL PBS at a concentration of 1×106 cells/mL and then 100 μL of incubation buffer containing 2 μL of annexin V and 2 μL of propidium iodide was added to the cells of each experimental group. Treated cells, as well as control cells, were analyzed using a BD flow cytometer within 1 hour.
In vivo anti-tumor analyses
Female BALB/c mice were inoculated subcutaneously in the flank region with 4T1 or TUBO tumor cells (1×106 cells in 100 μL PBS) in two separate experimental groups. Eight days after 4T1 cells and 12 days after TUBO cells inoculation, when the tumors were palpable, 4T1 tumor-bearing mice or TUBO tumor-bearing mice were randomly divided into four different treatment groups. Treatment groups (n=5/group) included: PBS, unloaded MSCs-Exo, MSCs-Exo loaded with LNA-anti-miR negative control, and MSCs-Exo loaded with LNA-anti-miR-142-3p. Mice were injected intravenously with 30 μg of unloaded MSCs-Exo, 30 μg of MSCs-Exo loaded with LNA-anti-miR-142-3p, 30 μg of MSCs-Exo loaded with LNA-anti-miR negative control, or with PBS as a control every 48 hours for 36 or 40 days. The rate of tumor growth in terms of tumor volume was inspected every 4 days by measurement of tumor diameters with a vernier caliper. Tumor volume (TV) was calculated using the formula: TV (mm3) = (height×length×width)×0.5 cm3. Survival of mice was monitored for 36 days in 4T1 tumor-bearing mice and for 40 days in TUBO tumor-bearing mice. A Kaplan–Meier method was used to evaluate the probability of mice survival, and the log-rank test was used to compare the fraction of surviving mice between groups (α=0.05). The mice at the end of the treatments were sacrificed by cervical dislocation, the tumors were removed, and the weight of each tumor tissue was measured.
In vivo visualization of intravenously injected exosomes
To assess the biodistribution of MSCs-derived exosomes, female 6–8-week-old BALB/c mice were injected with 106 TUBO or 4T1 cells in the flank area. The mice bearing TUBO tumor were monitored daily. When the volume of tumors reached 400 mm3, 30 μg of PKH67-labeled exosomes in a volume of 200 μL PBS containing 5% glucose was injected intravenously into tumor-bearing mice and a mouse without tumor as a control. A tumor-bearing mouse was also injected with 200 μL (PBS +5% glucose) as background control. The tumor-bearing mice were anesthetized with 2.5% isoflurane, and images were acquired after 3, 12, 24, and 48 hours post-exosome administration using small animal imaging system (in vivo imaging system [IVIS]; Kodak, Rochester, NY, USA). The fluorescent signal of each tissue sample was quantified in region of interest (ROI) draw freehand. The relative mean fluorescence intensity of each ROI was obtained by subtracting the mean fluorescence intensity of the corresponding ROI on the blank mouse from the measured mean fluorescence intensity and was plotted as a function of time.
Tissue dissection and fluorescent microscopy
At the end of the experiments, the animals were euthanized by cervical dislocation. In addition to tumor tissue, the vital tissues including lung, liver, kidney, and spleen were dissected, cryopreserved in 5%, 10%, and 14% sucrose, frozen at −80°C in optimal cutting temperature (OCT) medium, and cryosectioned using Leica CM1800 cryostat (Leica Microsystems). Frozen tissue sections (8 μm thick) were fixed in acetone and their nuclei were stained with DAPI. Fluorescently labeled exosomes visualized in dissected tissues using Cytation-3 Cell Imaging Multi-Mode Reader (Bio-Tek Instruments, Winooski, VT, USA). Frozen sections were also stained with H&E to verify the structure of the tissues.
Statistical analysis
Based on data distribution, Mann–Whitney nonparametric test or one-way ANOVA was used in this study to make comparisons between different groups, and the error bars are shown as the average ± standard error of the mean (SEM). Statistical significance was set as P<0.05.
Results
Characterization of bone marrow derived-mesenchymal stem cells and MSCs-Exo
Murine bone marrow-derived MSCs had a defined spindle-shaped fibroblastic morphology (Figure 1A). Differentiation of the MSCs into osteoblasts and adipocytes by culturing in appropriate culture medium confirmed their multipotent nature (Figure 1A). Analysis of phenotypic markers showed that isolated MSCs were positive for CD29, CD44, CD90, and CD105 and negative for CD34 and CD45 (Figure 1B). TEM analysis verified the disc-shape of MSCs-Exo and showed that the exosomes had an average size between 30 and 150 nm (Figure 2A). Expressions of some important exosomal markers such as CD81 and CD63, and not expressions of calnexin and endoplasmic reticulum marker, were observed using Western blot (Figure 2B). The average size of the MSCs-derived exosomes was found to be 103 nm according to Zetasizer results (Figure 2C). These results indicated that MSCs and their secreted exosomes were successfully isolated and that they met the required verification criteria according to previously defined studies.43–45 We measured the amount of isolated exosomes from MSCs based on their protein content using the Bradford assay. According to the results, about 200 μg of exosomes were obtained from 13×107 mesenchymal stem cells (150 mL supernatant).
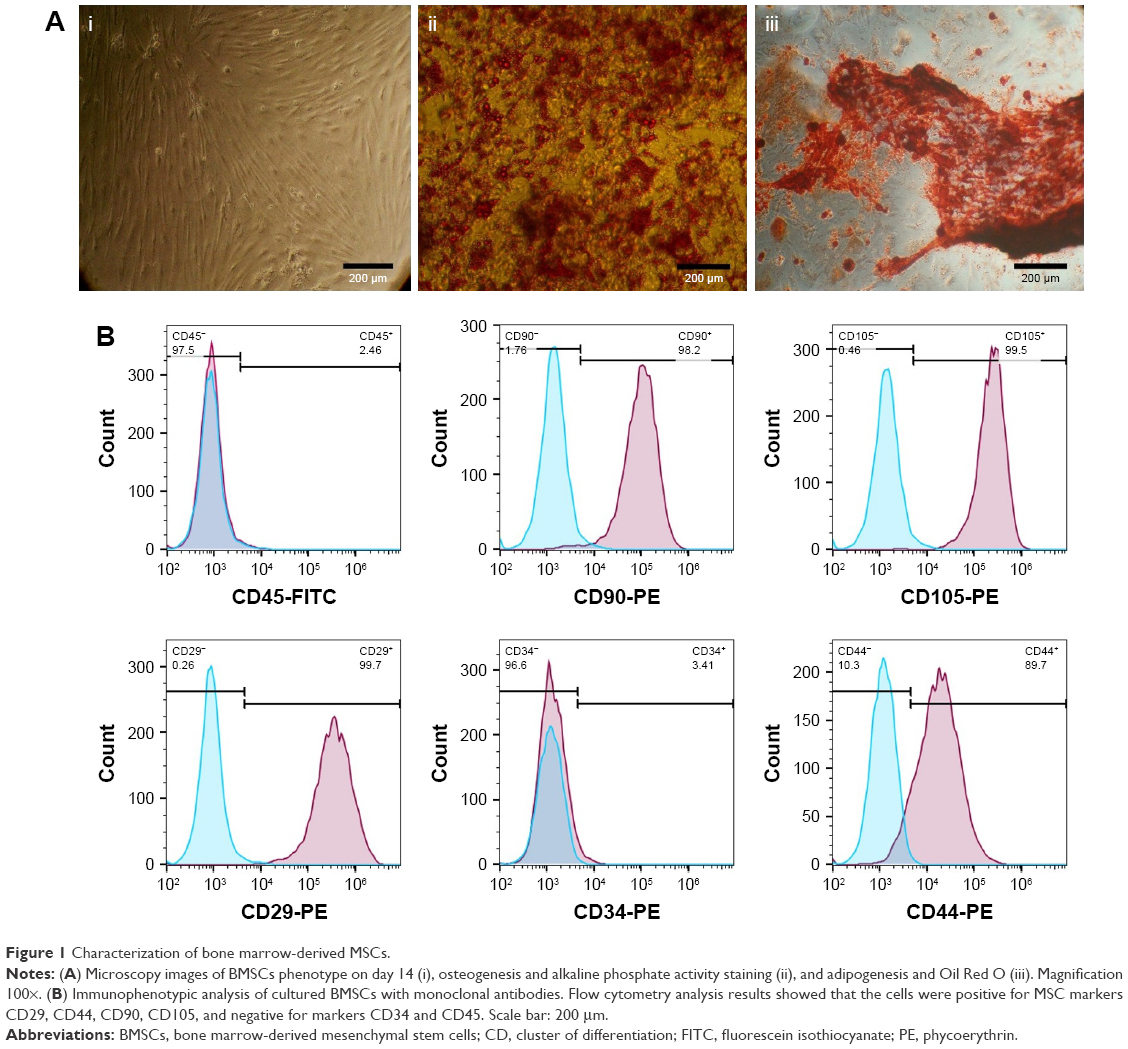 | Figure 1 Characterization of bone marrow-derived MSCs. |
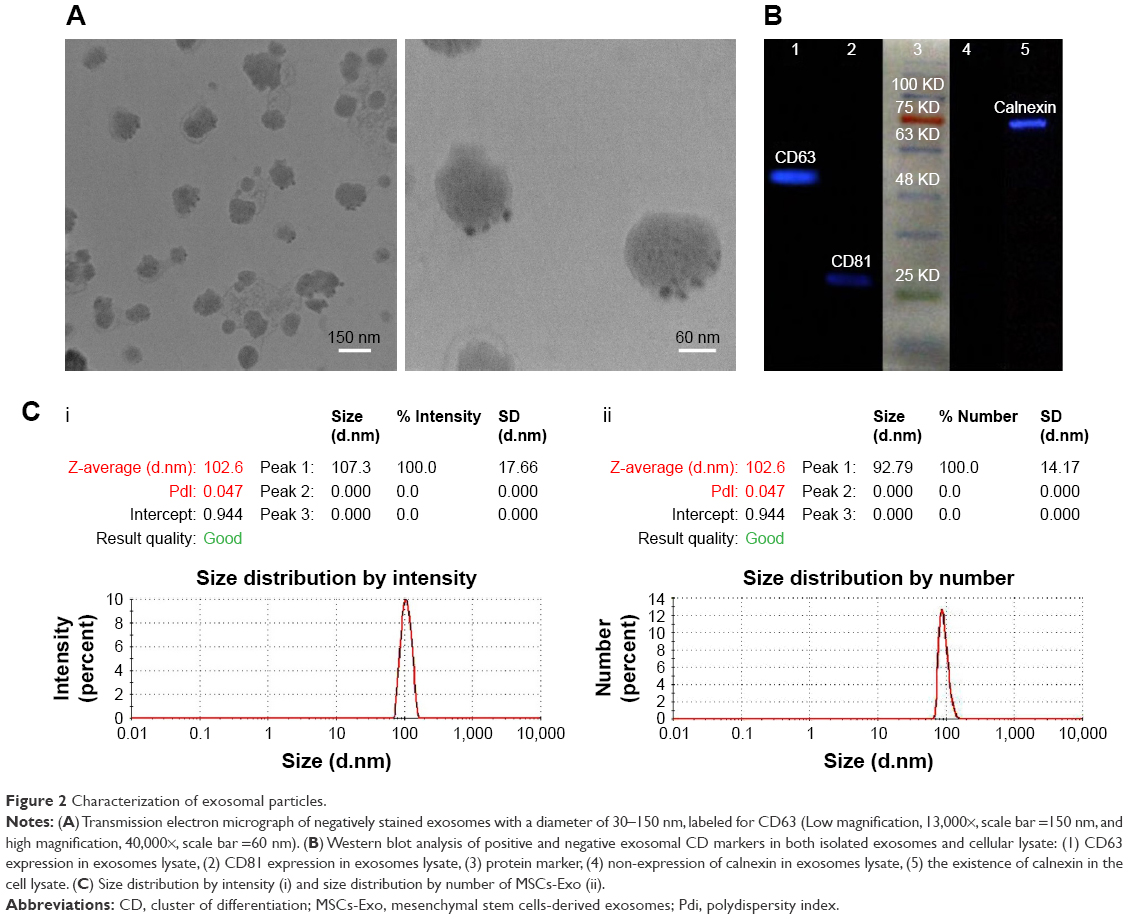 | Figure 2 Characterization of exosomal particles. |
Efficient delivery of LNA-miRNA-142-3p via MSCs-Exo after incorporating into 4T1 and TUBO cells
One of the unique properties of exosomes compared to other carriers is incorporation into target cells simply through the co-culture method, and they can penetrate the cells through pathways such as endocytosis and membrane fusion. Confocal laser scanning microscopy showed the penetration of fluorescent PKH67-labeled MSCs-Exo into 4T1 and TUBO cells after 24 hours of co-culturing (Figure 3). Furthermore, our results indicated efficient loading of the MSCs-Exo with LNA-anti-miR-142-3p (about 57%) and LNA-anti-miR negative control (about 55%) oligonucleotides. Evidence of MSCs-derived exosome entry into the cells by fluorescent microscopy demonstrated that the MSCs-Exo could be used to transfer LNA-based anti-miRNA oligonucleotides to exert their inhibitory function. Therefore, we used the MSCs-Exo to transfer the LNA-anti-miR-142-3p to inhibit miR-142 and investigate the functional effects on miR-150 expression and their respective target genes, APC and P2X7R receptor. We assessed the functional effects of the MSCs-Exo mediated delivery of the anti-miR-142-3p on 4T1 and TUBO cells in four groups, including untreated, unloaded MSCs-Exo, MSCs-Exo loaded with anti-miR-142-3p, and MSCs-Exo loaded with LNA-anti-miR negative control. In order to inhibit miR-142 using MSCs-Exo, MSC-derived exosomes should be free of the miRNA-142 molecules. So, we first extracted the total RNA of the exosomes and demonstrated by quantitative real-time PCR (qRT-PCR) that MSCs-derived exosomes did not contain miR-142 and miR-150 (Figure S2). MSCs-Exo showed efficient functional delivery of anti-miR-142-3p, which is indicated by a statistically significant decrease in miR-142-3p levels and subsequent statistically significant increase in APC mRNA levels in the cells treated with MSCs-Exo loaded with anti-miR-142-3p compared to other three groups of the treated cells (P<0.05) (Figure 4A). Additionally, exosome-mediated miR-142-3p inhibition resulted in a significant decrease of miR-150 levels and a significant increase of P2X7R receptor mRNA levels compared to the cells treated with unloaded MSCs-Exo or MSCs-Exo loaded with anti-miR negative control (P<0.05) (Figure 4B).
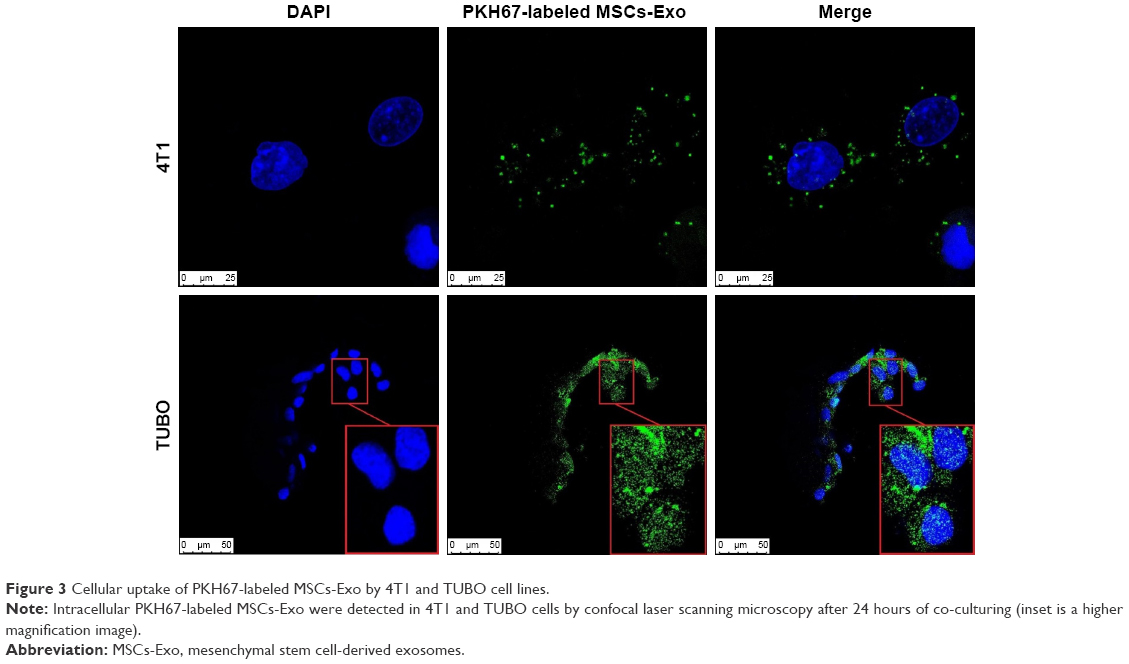 | Figure 3 Cellular uptake of PKH67-labeled MSCs-Exo by 4T1 and TUBO cell lines. |
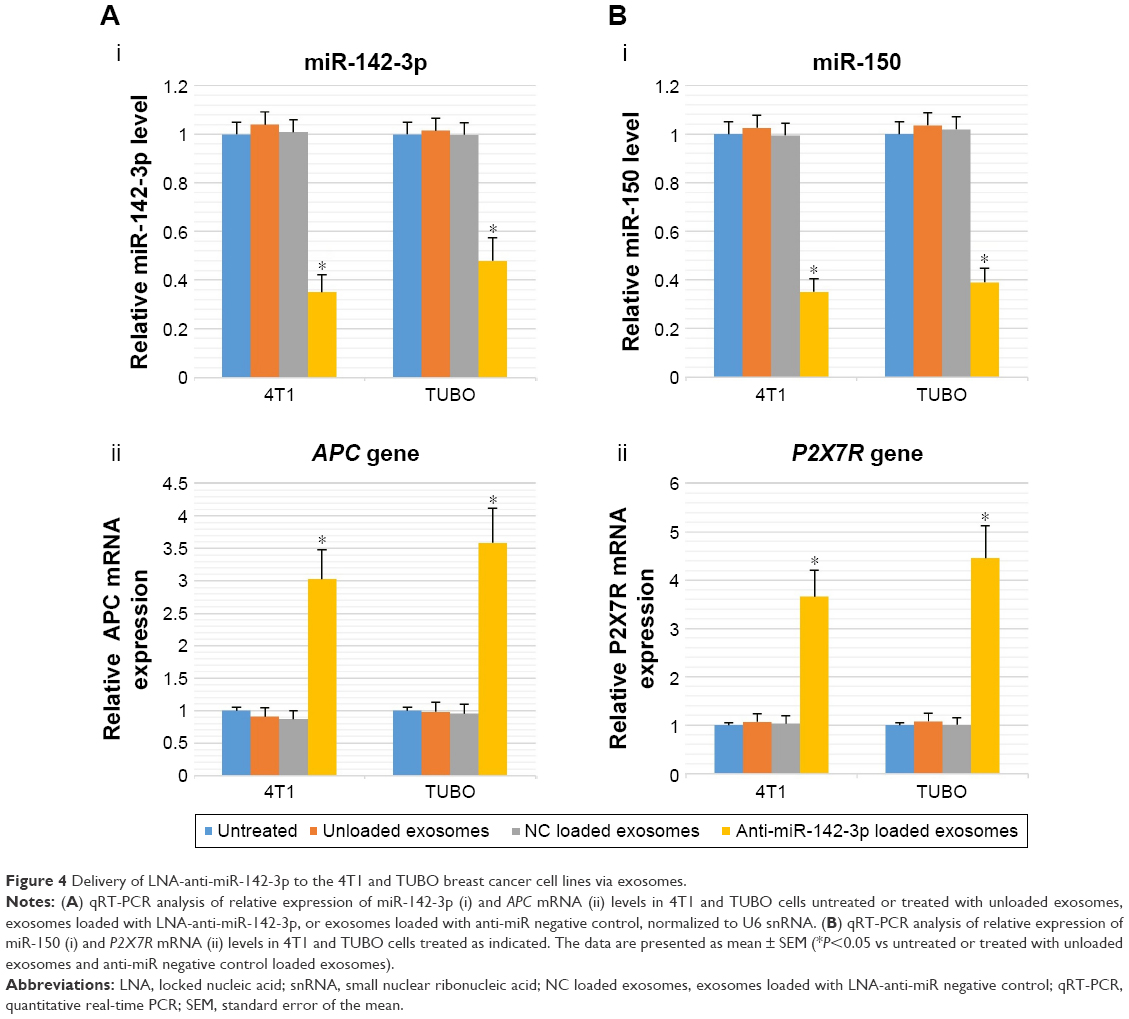 | Figure 4 Delivery of LNA-anti-miR-142-3p to the 4T1 and TUBO breast cancer cell lines via exosomes. |
MSCs-derived exosomes had minimal cytotoxicity in 4T1 and TUBO cell lines
Confocal laser scanning microscopy (CLSM) observations indicated that MSCs-derived exosomes have potential to be taken up by 4T1 and TUBO cells to deliver LNA-anti-miR-142-3p molecules; hence, cytotoxicity effect of the unloaded MSCs-Exo, LNA-anti-miR-142-3p loaded MSCs-Exo, and LNA-anti-miR negative control on 4T1 and TUBO cells were then investigated by MTT assay after 48 hours. Cell viability and proliferation of 4T1 and TUBO cells treated with MSCs-derived exosomes were moderately increased compared to the untreated cells. On the other hand, cell viability of 4T1 and TUBO cells treated with LNA-anti-miR-142-3p loaded MSCs-Exo was slightly reduced compared to MSCs-Exo and LNA-anti-miR negative control, probably due to the transfer of LNA-anti-miR-142-3p to the mentioned cells, as shown in Figure 5.
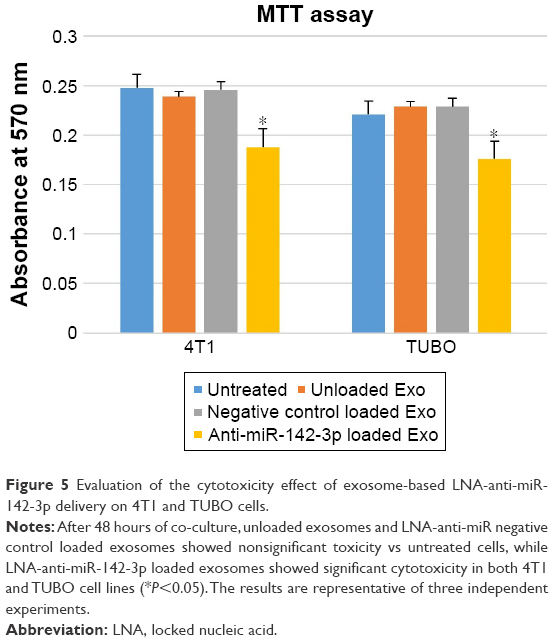 | Figure 5 Evaluation of the cytotoxicity effect of exosome-based LNA-anti-miR-142-3p delivery on 4T1 and TUBO cells. |
Delivery of LNA-anti-miR-142-3p molecules using MSCs-derived exosomes induces apoptosis in 4T1 and TUBO breast cancer cell lines
To further investigate the functional role of LNA-anti-miR-142-3p in 4T1 and TUBO cells, the effect of LNA-anti-miR-142-3p delivery via MSCs-derived exosomes on apoptosis of 4T1 and TUBO cells after 48 hours of treatment with the unloaded MSCs-Exo, LNA-anti-miR-142-3p loaded MSCs-Exo, and LNA-anti-miR negative control loaded MSCs-Exo was assessed. To quantify the apoptosis levels of 4T1 and TUBO cells, annexin V/PI staining was performed according to the method previously mentioned in the “Materials and methods” section. As presented in Figure 6, MSCs-derived exosomes had no significant effects on apoptosis of 4T1 or TUBO cells compared to the untreated cells. But apoptosis percentage of 4T1 or TUBO cells was significantly increased following treatment with LNA-anti-miR-142-3p loaded MSCs-Exo compared to unloaded MSCs-Exo or anti-miR negative control loaded MSCs-Exo (P<0.05).
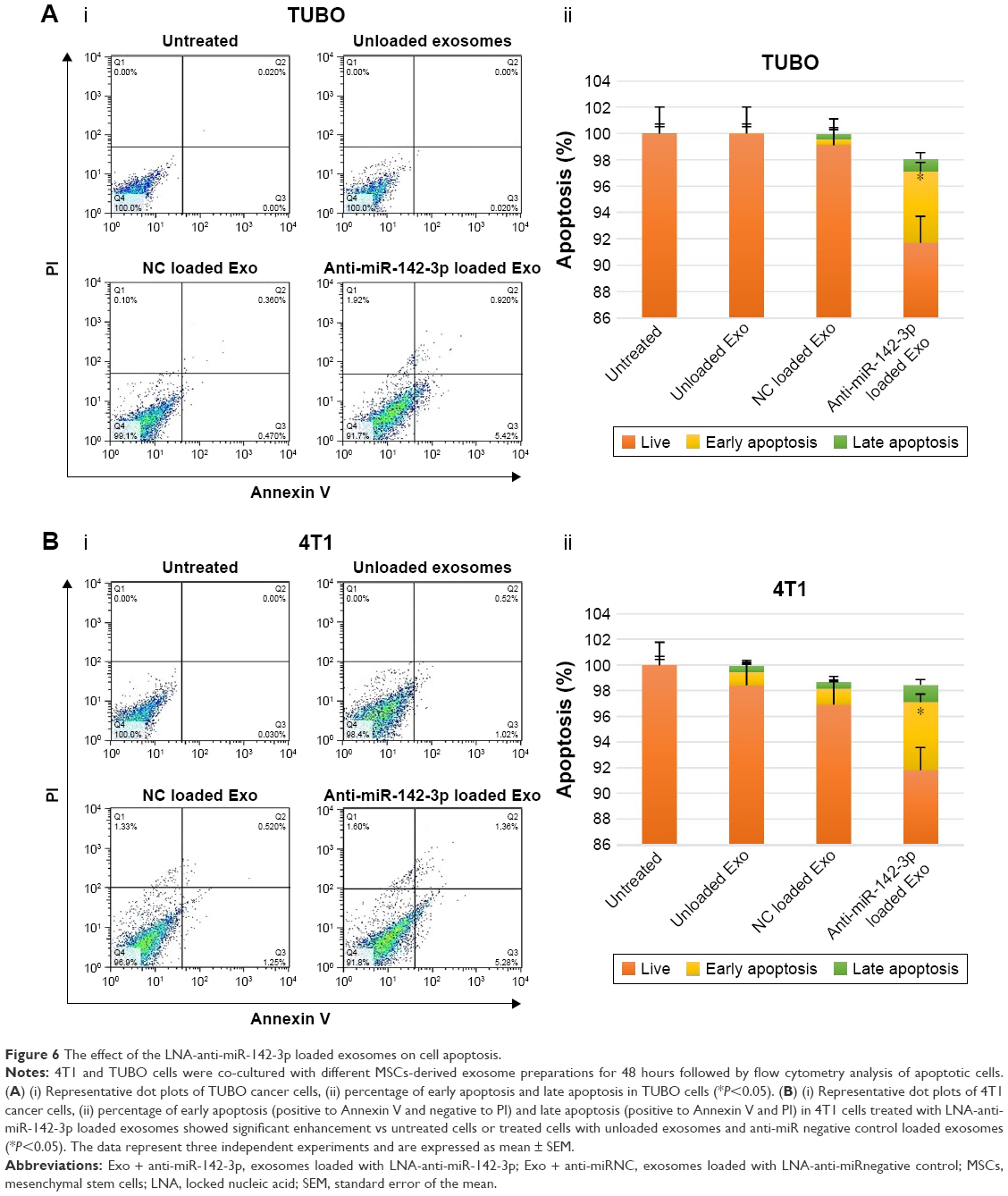 | Figure 6 The effect of the LNA-anti-miR-142-3p loaded exosomes on cell apoptosis. |
In vivo visualization of fluorescently labeled MSCs-Exo in tumor-bearing mice
Next, we assessed biodistribution and tumor penetration of MSCs-Exo in mice bearing TUBO tumor. To observe the existence of PKH67-labeled exosomes in the tumors as well as other tissues, 30 μg of MSCs-Exo was injected intravenously into BALB/c tumor-bearing mice in a volume of 200 μL PBS containing 5% glucose. Fluorescence intensity of PKH67-labeled exosomes from the injected mice was monitored from 3 hours to 48 hours postinjection. PBS was injected into one tumor-bearing mouse as a control and one mouse without tumor was used as a background subtraction control. As shown in Figure 7, fluorescent signal from PKH67-labeled exosomes can be readily observed in different tissues of the body as well as at the tumor site in tumor-bearing mouse compared to mouse injected with PBS 3 hours after injection. Despite a gradual decrease in the fluorescent signal from labeled exosomes all over the body 12 and 24 hours postinjection, it remained visible in tumor tissue. At 48 hours after injection, most of the labeled exosomes were cleared from the main tissues of the body, but few were still visible in the tumor site.
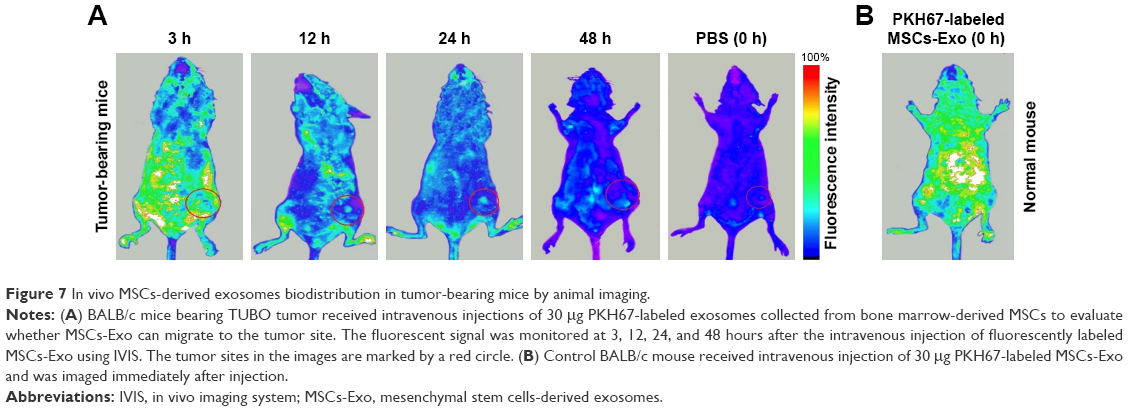 | Figure 7 In vivo MSCs-derived exosomes biodistribution in tumor-bearing mice by animal imaging. |
Ex vivo analysis of dissected organs
All mice including control mice were sacrificed at 0, 3, 12, 24, and 48 hours after intravenous (iv) injection of labeled exosomes or PBS, and fluorescent signal was observed from freshly dissected tissues immediately using IVIS. The fluorescent signal from PKH67-labeled exosomes was readily observed in the tumor tissue as well as in the most tissues of the body including lung, liver, spleen, and kidneys (Figure 8A and B). After 12 hours postinjection of the labeled exosomes, the fluorescence intensity gradually decreased in the lung and liver tissues and increased in the spleen and kidneys, but the fluorescent signal was still visible in the tumor tissue. After 24 hours, the signal intensity of the injected exosomes was still detectable in tumor tissue but decreased in spleen and kidneys. However, at 48 hours post-iv injection, the fluorescent signal almost declined to baseline in almost all tissues and was similar to the signal in tissues from the mouse injected with PBS, but was visible to some extent in the tumor tissue in the tumor-bearing mouse. According to the quantitative fluorescence intensity results, the relative signal of the fluorescently labeled exosomes in the tumor site was significantly higher compared to the fluorescent signal of the tumor region of the control mouse injected with PBS (Figure 8C). The presence of PKH67-labeled MSCs-derived exosomes within tumor tissue section as well as in other tissue sections was confirmed by imaging using Cytation-3 multi-mode reader system with an appropriate wavelength. Fluorescent signal of labeled exosomes in the tumor section derived from tumor-bearing mice was obviously visible after 3 hours of injection (Figure 9A). Accordingly, the time of reinjection of the MSCs-derived exosomes loaded with LNA-anti-miR-142-3p was set as 48 hours. H&E staining was performed to evaluate the histomorphology of the different tissues after injection. As shown in Figure 9B, no histological changes were observed in the lung, liver, spleen, kidneys, and tumor tissues in tumor-bearing mice injected with MSCs-derived exosomes compared to normal mice without injection.
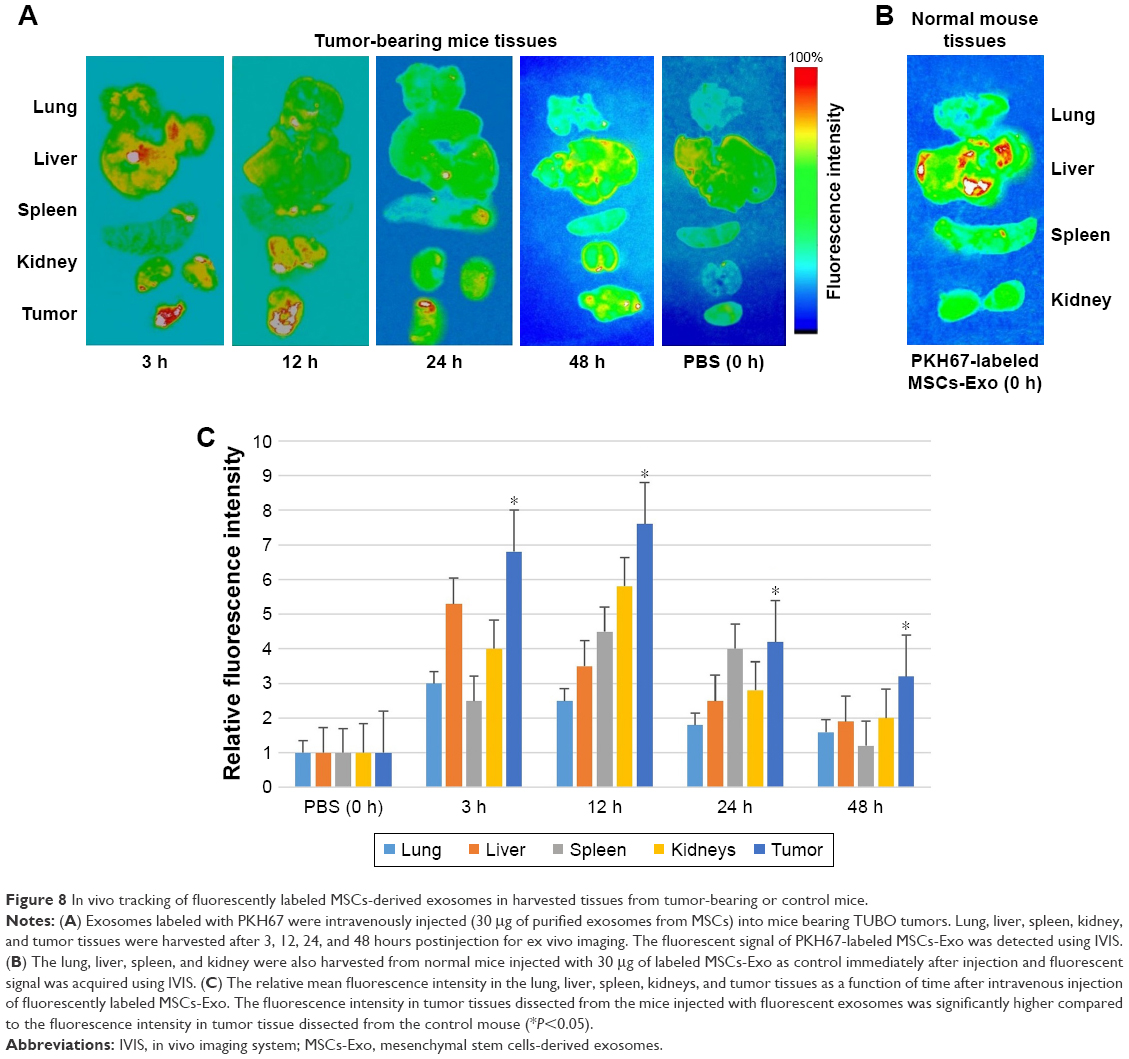 | Figure 8 In vivo tracking of fluorescently labeled MSCs-derived exosomes in harvested tissues from tumor-bearing or control mice. |
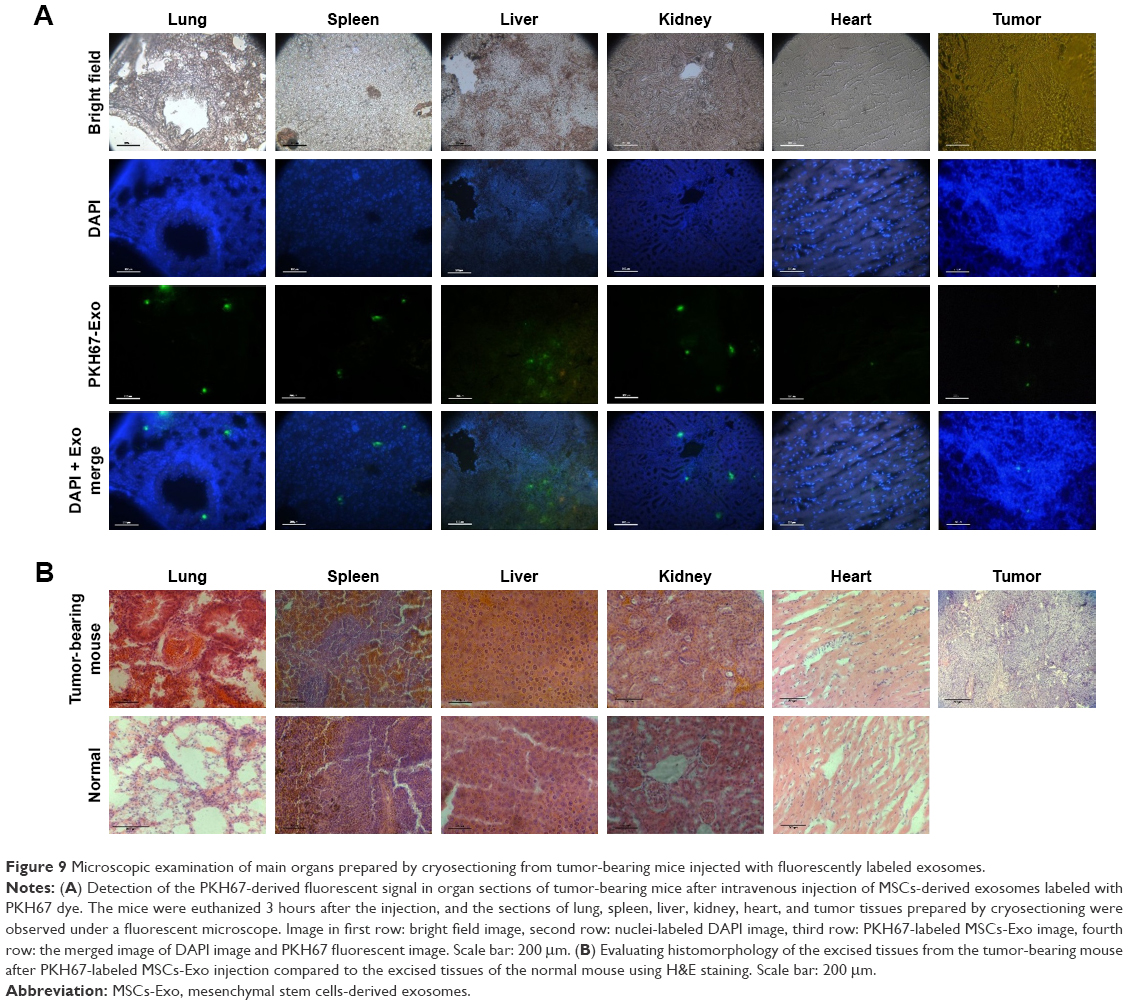 | Figure 9 Microscopic examination of main organs prepared by cryosectioning from tumor-bearing mice injected with fluorescently labeled exosomes. |
In vivo anti-tumor efficiency of the exosomes
Anti-tumor effects of the LNA-anti-miR-142-3p delivery via MSC exosomes were evaluated in both murine model of 4T1 and TUBO breast cancer. Tumor growth analysis in 4T1 tumor group (Figure 10A) revealed that while unloaded exosomes and exosomes loaded with LNA-anti-miR negative control had an identical impact on tumor growth rate, exosomes loaded with LNA-anti-miR-142-3p induced a significant decrease in tumor volume compared to the PBS-treated mice. Similar results were observed in TUBO tumor group. Tumor growth curve analysis (Figure 10B) indicated that injection of exosomes loaded with LNA-anti-miR-142-3p led to a significant reduction in tumor growth rate compared to the three other control groups (the mice treated with unloaded exosomes, the mice treated with exosomes loaded with LNA-anti-miR negative control, and the PBS-treated mice). Survival analyses showed that the 4T1 tumor-bearing mice treated with MSCs-Exo loaded with LNA-anti-miR-142-3p had a significantly longer survival (mean survival [ms], 37.0 days) than PBS-treated mice (ms, 21.2 days) or mice treated with control preparations, including both unloaded MSCs-Exo (ms, 18.0 days) and LNA-anti-miR negative control loaded MSCs-Exo (ms, 21.8 days) (Figure 10A). Mean survival time of the TUBO tumor-bearing mice treated with MSCs-Exo loaded with LNA-anti-miR-142-3p was 29.3 days, which was significantly higher than the survival rate of PBS-treated mice (ms, 17.0 days) or mice treated with unloaded MSCs-Exo (ms, 18.5 days) and LNA-anti-miR negative control loaded MSCs-Exo (ms, 19.8 days) (P<0.05) (Figure 10B). When the tumor volume exceeded 800 mm3, at the end of day 36 for 4T1 tumors and at the end of day 40 for TUBO tumors, the mice were sacrificed by cervical dislocation and tumor tissues were removed (Figure S3) and weighed. While the actual tumor weights in mice treated with unloaded exosomes and the mice treated with LNA-anti-miR negative control were similar to the control mice injected with PBS, actual tumor weight was significantly reduced in mice injected with LNA-anti-miR-142-3p encapsulating exosomes compared to the control groups in both 4T1 and TUBO tumor models (Figure 10).
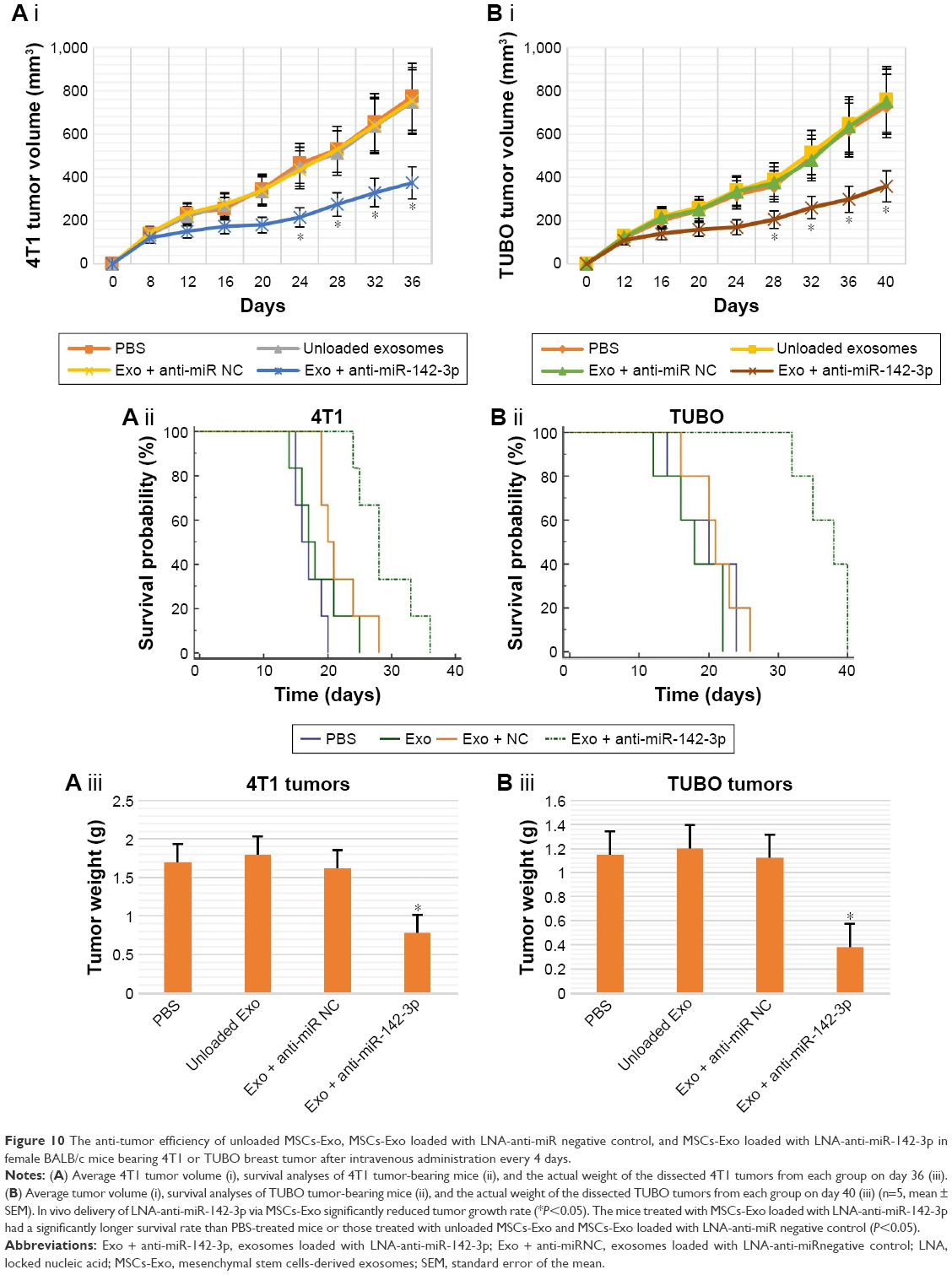 | Figure 10 The anti-tumor efficiency of unloaded MSCs-Exo, MSCs-Exo loaded with LNA-anti-miR negative control, and MSCs-Exo loaded with LNA-anti-miR-142-3p in female BALB/c mice bearing 4T1 or TUBO breast tumor after intravenous administration every 4 days. |
Mir-142-3p inhibition in tumor tissue after the systemic injection of LNA-miR-142-3p inhibitor loaded MSCs-Exo
To evaluate the efficiency of the MSCs-derived exosomes in the transfer of LNA-anti-miR-142-3p in vivo, all mice in each group were sacrificed and 4T1 or TUBO tumor tissues were removed for RNA extraction and qRT-PCR analyses. We observed a significant decrease in miR-142-3p level in 4T1 and TUBO tumor-bearing mice injected with LNA-anti-miR-142-3p loaded exosomes in comparison to three other groups as indicated. Next, to evaluate the functional effect of anti-miR-142-3p on target miRNA and target genes, we measured the expression level of miR-150, APC, and P2X7R genes by real-time PCR. As expected, reduction of miR-142-3p led to a significant reduction of the miR-150 level (P<0.05) and a significant increase of target genes APC and P2X7R (P<0.05) (Figure 11). These results confirmed that MSC exosomes can be harnessed as an efficient carrier to deliver anti-miR molecules like LNA-anti-miR-142-3p into tumor tissue.
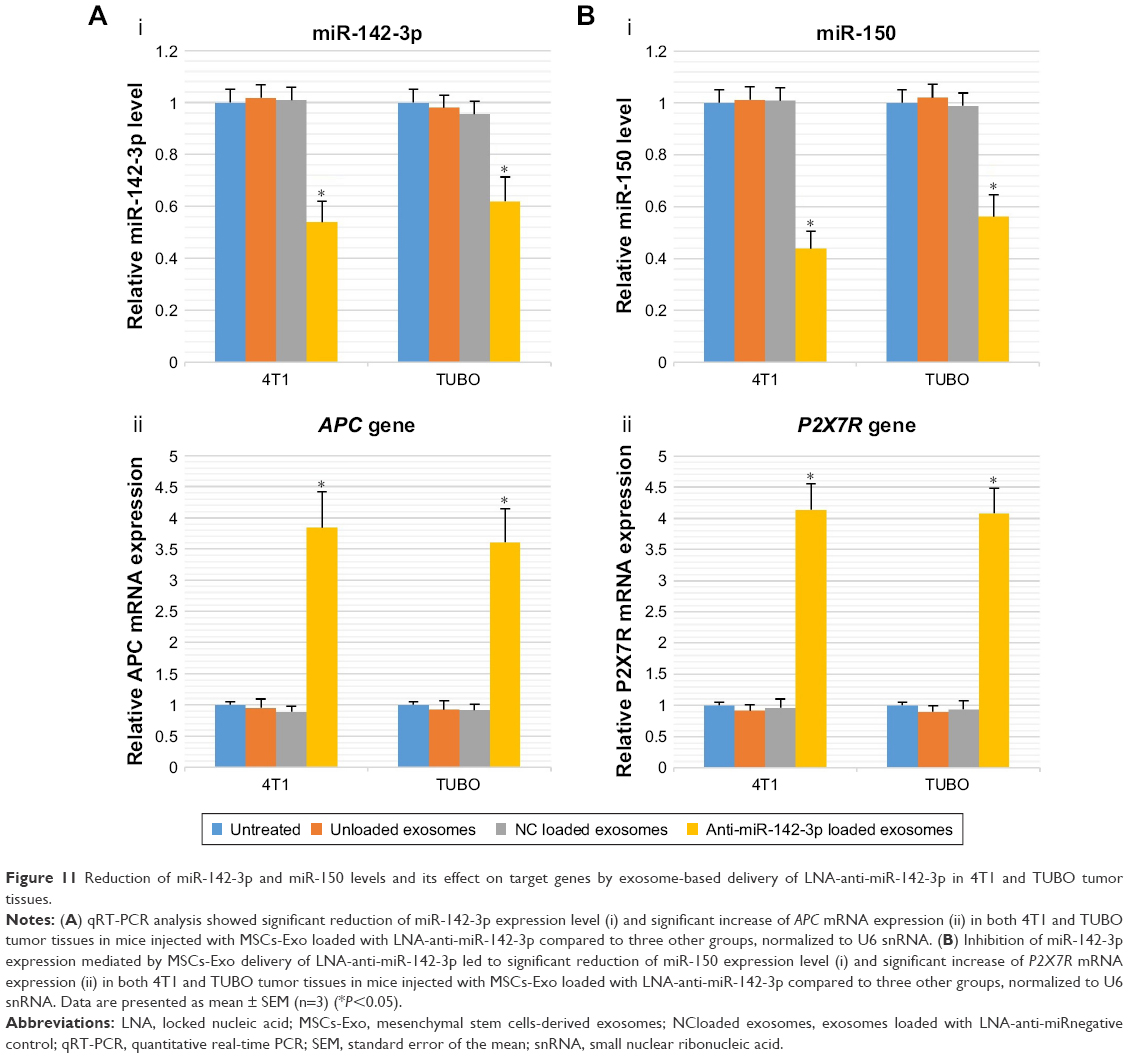 | Figure 11 Reduction of miR-142-3p and miR-150 levels and its effect on target genes by exosome-based delivery of LNA-anti-miR-142-3p in 4T1 and TUBO tumor tissues. |
Discussion
Scientists in recent years have highlighted the role of exosomes in the field of cancer diagnosis and treatment.16 Exosomes are known to be naïve nanovesicles that play an important role in intercellular communications and the transmission of messenger molecules such as miRNAs and fragments of DNA molecules in the body. These observations have led scientists to use exosomes as nanocarriers to deliver siRNAs, miRNAs, and even chemotherapy drugs such as doxorubicin. For the first time, Valadi et al in 2007 found that exosomes contain mRNAs and miRNAs which can be delivered to recipient cell and can be functional in the new location.17 Exosomes carry specific molecules, depending on the cell of origin, and deliver their contents to the recipient cells through the pathways such as endocytosis and membrane fusion.18 Since then, exosomes isolated from different sources have been used to transfer nucleic acid-based therapeutics in the treatment and diagnosis of cancer or many other types of diseases.15,19–24 Various types of cells have been used as sources of producing exosomes, including B cells, HEK-293 and melanoma cell lines, epithelial cells, immature dendritic cells, and MSCs.25–29 In this context, stem cell-derived exosomes have become more prominent due to their more functional properties than other cells in the clinical field. Since MSCs have the potential to produce large quantities of exosomes, these cells can be exploited to produce exosomes on a clinical scale. They also have a large capacity for ex vivo expansion and great ability to differentiate into many cell types.7 MSCs are also known to have immunosuppressive properties and could be used in allogeneic transplantation. There is increasing evidence that the therapeutic efficiency of MSCs is mediated through soluble factors, especially exosomes. MSCs-derived exosomes, unlike exosomes derived from dendritic cells, do not stimulate the immune system. So, MSCs-derived exosomes are likely to be safe and would not elicit an intrinsic adverse effect or immune rejection response when used as drug delivery vehicles.9 Although some studies have so far examined the exosomal tropism in the body, the mechanisms of clearance and biodistribution of MSCs-derived exosomes have not yet been fully disclosed.29–32 It was observed that MSCs-derived exosomes accumulated in the kidneys of mice, resulting in acute kidney injury. However, the accumulation of MSC exosomes was mainly observed in the liver and spleen.33 Considering that tumor tropism of exosomes plays an important role in cancer treatment, we first investigated the tumor tropism and biodistribution of the MSC exosomes in tumor-bearing mice. We found that MSCs-derived exosomes have the ability to penetrate the tumor site after iv injection of the exosomes. In addition, the MSC exosomes were observed to accumulate in the spleen and liver, and to a lesser extent in the lung 3 hours after iv injection of the exosomes, while their concentration gradually decreased in the liver and spleen, and increased in the lung and kidneys 12 hours postinjection. After 48 hours, they were almost cleared from lung, liver, spleen, and kidneys, but were still detectable in the tumor site. This observation confirmed that MSC exosomes mainly excrete by spleen and liver and secondarily by lung and kidneys.
According to the EPR effect, the nanoparticle-based drug can protrude preferentially at leaky tumor vasculatures and remain in the tumor interstitium for an extended time.34 Since exosomes have an ideal particle size of 40–150 nm, they have potential to avoid rapid renal clearance and phagocytosis by mononuclear phagocyte system, and to accumulate in the tumor site passively via EPR effect. As shown in Figures 7 and 8, MSCs-derived exosomes penetrated the tumor site 3 hours postinjection due to their EPR effect, and even after clearance from other tissues, they were still present in the tumor tissue. On the other hand, tumor tropism of MSCs has been indicated in some studies.35 So, MSCs-derived exosomes can also behave like their maternal stem cells and exhibit tumor tropism. Furthermore, the histopathology findings demonstrated the low systematic toxicity of the intravenously injected MSCs-derived exosomes.
MiRNAs are a class of endogenous small non-coding single-stranded RNA molecules that regulate gene expression and affect multiple cellular processes including proliferation and development, differentiation, apoptosis, and angiogenesis. Gain or loss of function of specific miRNA contributes to tumorigenesis and cancer progression.36 Since many of the miRNAs can behave functionally as oncogenes, miR-targeting therapeutics such as 2′-O-methyl antisense and LNA-miRNA inhibitor oligonucleotides is an area of extensive research.37 Due to the unstable nature, and potentially immunogenic and negative charge of anti-miR oligonucleotides, various carriers have been used to transmit the molecules into target cells in many studies.38 Exosomes as natural membranous nanocarriers can transfer bio-macromolecules without immunogenicity and cytotoxicity, in contrast to conventional gene therapy carriers such as viral or non-viral vectors.12 Autologous MSCs-derived exosomes can be isolated from the recipient’s own cell culture and would be a perfect source of personalized drug vehicles without immunogenicity or cytotoxicity.
Wnt/β-catenin signaling is an ancient, highly conserved pathway that regulates cell proliferation, differentiation, adhesion, and migration through the β-catenin-dependent pathway. APC is a pivotal component of the demolition complex that destabilizes β-catenin and suppresses the activity of the Wnt/β-catenin signaling pathway.39 Mutations in the APC gene have been linked to colon cancer, but the APC gene is expressed not only in the intestine but also in other tissues such as mammary tissue. Breast cancer cells rarely have mutations in the APC gene, but recent studies have indicated that some miRNAs that target this gene might promote the growth of these tumor cells.40 A couple of studies have indicated that miR-142-3p expression is upregulated in several breast cancer subtypes. On the other hand, miR-142-3p targets APC to upregulate Wnt/β-catenin signaling pathway and thereby enhances miR-150 expression which is also upregulated in breast cancer cells.41 Both miRNAs have the ability to induce hyperproliferation in mammary tissue. MiR-150 overexpression promotes growth and clonogenicity, and reduces apoptosis in breast cancer cells by targeting the proapoptotic purinergic P2X7R receptor gene.42 Our in vitro experiments indicated that the MSC exosomes can be efficiently loaded with LNA-anti-miR-142-3p and the loaded exosomes can deliver the LNA-anti-miR molecules to the 4T1 and TUBO breast cancer cells, causing inhibition of miRNA-142-3p, and functionally led to a statistically significant increase of APC gene level. On the other hand, miRNA-142-3p inhibition led to a significant decrease of miRNA-150 expression level. Since P2X7R is a putative target of miRNA-150 and its expression is inversely correlated with miRNA-150 level, successful delivery of LNA-anti-miR-142-3p to the 4T1 and TUBO cells led to a significant increase of P2X7R mRNA level. The anti-tumor effects of MSC exosomes containing LNA-anti-miR-142-3p oligonucleotides were evaluated in mice bearing 4T1 or TUBO breast tumors. As the clearance of the MSCs-Exo from major organs was observed by 48 hours postinjection with no observable lasting toxicity, the IV injection of the MSCs-derived exosomes loaded with LNA-anti-miR-142-3p into mice bearing 4T1 or TUBO tumor was repeated every 48 hours. According to in vivo anti-tumor studies, the lowest tumor volume and highest survival time were seen in exosomes loaded with LNA-anti-miR-142-3p group that showed significant anti-tumor efficiency in comparison with unloaded exosomes and exosomes containing LNA-anti-miR negative control in both 4T1 and TUBO breast tumor-bearing mice. Moreover, qRT-PCR results confirmed successful inhibition of miR-142-3p in both 4T1 and TUBO tumor tissues, causing a significant increase in APC target gene of miR-142-3p. It was also shown that miR-142-3p inhibition led to significant inhibition of miR-150 in tumor tissues and significant increase of P2X7R mRNA level. The in vivo studies using tumor-bearing mice generally indicated that MSC exosomes mediated efficient delivery of LNA-anti-miR-142-3p to the tumor tissues after IV injection.
In general, MSCs-derived exosomes can offer safer and more efficient carriers to deliver RNA-based therapeutics compared to other common synthetic vehicles in nanomedicine applications. So, the exosomes have provided new insights into the generation of new nanovehicles of anticancer drugs.
Conclusion
Our results demonstrate that bone marrow-derived MSCs are reliable producers of exosomes and that the MSCs-Exo can be used as suitable nanovectors for RNA-based therapeutics in vitro and in vivo applications. In vitro studies using CLSM and qRT-PCR provided strong evidence of binding and uptake of MSCs-derived exosomes via fusion with lipid bilayer of the breast cancer cells, suggesting that the MSCs-derived exosomes mediate effective functional delivery of LNA-based anti-miR-142-3p oligonucleotides, which leads to the downregulation of miR-142-3p and miR-150, and subsequently results in upregulation of the associated tumor suppressor genes including APC and P2X7R in the recipient breast tumor cells. The biodistribution findings convincingly indicated that MSCs-derived exosomes have the homing ability to the tumor site postinjection, and based on the EPR effect they remain in the tumor tissue even after the elimination from body’s main organs, suggesting that the MSCs-Exo can mediate successful delivery of LNA-based anti-miR-142-3p oligonucleotides to the tumor site as early as 3 hours after iv injection. Notably, in vivo anti-tumor analyses indicated that iv injection of LNA-anti-miR-142-3p loaded MSCs-Exo had a significant inhibitory effect on tumor growth rate. Accordingly, quantitative analyses of RNA expression in the tumor tissues suggested that in vivo transfer of LNA-anti-miR-142-3p loaded MSCs-Exo results in a significant reduction of the miR-142-3p and miR-150 levels in the tumor tissues in mouse models. Our results provide evidence for the concept of using MSCs-derived exosomes as efficient nanovehicles for RNA-based therapeutics in nanomedicine applications, especially in cancer therapy field.
Acknowledgments
This research was funded by Mashhad University of Medical Sciences and Tarbiat Modares University, Tehran (no. 931289), and a grant from Cancer Research Center of Cancer Institute of Iran (Shams Cancer Charity, grant no. 37604-202-01-97). We are grateful to our colleagues and PhD students, for their help and support during this work, in the Department of Medical Biotechnology, Faculty of Medical Sciences, at Tarbiat Modares University, Tehran.
Disclosure
The authors report no conflicts of interest in this work.
References
van Dommelen SM, Vader P, Lakhal S, et al. Microvesicles and exosomes: opportunities for cell-derived membrane vesicles in drug delivery. J Control Release. 2012;161(2):635–644. | ||
Vlassov AV, Magdaleno S, Setterquist R, Conrad R. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim Biophys Acta. 2012;1820(7):940–948. | ||
Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. 2014;3(1):24641. | ||
Tan A, Rajadas J, Seifalian AM. Exosomes as nano-theranostic delivery platforms for gene therapy. Adv Drug Deliv Rev. 2013;65(3):357–367. | ||
Kooijmans SAA, Schiffelers RM, Zarovni N, Vago R. Modulation of tissue tropism and biological activity of exosomes and other extracellular vesicles: New nanotools for cancer treatment. Pharmacol Res. 2016;111:487–500. | ||
Greco SJ, Rameshwar P. Mesenchymal stem cells in drug/gene delivery: implications for cell therapy. Ther Deliv. 2012;3(8):997–1004. | ||
Yeo RW, Lai RC, Zhang B, et al. Mesenchymal stem cell: an efficient mass producer of exosomes for drug delivery. Adv Drug Deliv Rev. 2013;65(3):336–341. | ||
Yu B, Zhang X, Li X. Exosomes derived from mesenchymal stem cells. Int J Mol Sci. 2014;15(3):4142–4157. | ||
Rani S, Ryan AE, Griffin MD, Ritter T. Mesenchymal stem cell-derived extracellular vesicles: toward cell-free therapeutic applications. Mol Ther. 2015;23(5):812–823. | ||
Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chem Biol. 2012;19(1):60–71. | ||
Aagaard L, Rossi JJ. RNAi therapeutics: principles, prospects and challenges. Adv Drug Deliv Rev. 2007;59(2–3):75–86. | ||
Lakhal S, Wood MJA. Exosome nanotechnology: An emerging paradigm shift in drug delivery. Bioessays. 2011;33(10):737–741. | ||
Umezu T, Ohyashiki K, Kuroda M, Ohyashiki JH. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene. 2013;32(22):2747–2755. | ||
Zhu L, Qu XH, Sun YL, Qian YM, Zhao XH. Novel method for extracting exosomes of hepatocellular carcinoma cells. World J Gastroenterol. 2014;20(21):6651. | ||
Momen-Heravi F, Bala S, Bukong T, Szabo G. Exosome-mediated delivery of functionally active miRNA-155 inhibitor to macrophages. Nanomedicine. 2014;10(7):1517–1527. | ||
Johnsen KB, Gudbergsson JM, Skov MN, Pilgaard L, Moos T, Duroux M. A comprehensive overview of exosomes as drug delivery vehicles – endogenous nanocarriers for targeted cancer therapy. Biochim Biophys Acta. 2014;1846(1):75–87. | ||
Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–659. | ||
Ren J, He W, Zheng L, Duan H. From structures to functions: insights into exosomes as promising drug delivery vehicles. Biomater Sci. 2016;4(6):910–921. | ||
Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29(4):341–345. | ||
Dai S, Wei D, Wu Z, et al. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol Ther. 2008;16(4):782–790. | ||
Lee HK, Finniss S, Cazacu S, et al. Mesenchymal stem cells deliver synthetic microRNA mimics to glioma cells and glioma stem cells and inhibit their cell migration and self-renewal. Oncotarget. 2013;4(2):346. | ||
Lee JK, Park SR, Jung BK, et al. Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS One. 2013;8(12):e84256. | ||
Munoz JL, Bliss SA, Greco SJ, Ramkissoon SH, Ligon KL, Rameshwar P. Delivery of Functional Anti-miR-9 by Mesenchymal Stem Cell-derived Exosomes to Glioblastoma Multiforme Cells Conferred Chemosensitivity. Mol Ther Nucleic Acids. 2013;2:e126. | ||
Ohno S, Takanashi M, Sudo K, et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013;21(1):185–191. | ||
Kim SH, Bianco NR, Shufesky WJ, Morelli AE, Robbins PD. Effective treatment of inflammatory disease models with exosomes derived from dendritic cells genetically modified to express IL-4. J Immunol. 2007;179(4):2242–2249. | ||
Lai RC, Yeo RWY, Lim SK. Mesenchymal stem cell exosomes. Semin Cell Dev Biol. 2015;40:82–88. | ||
Mclellan AD. Exosome release by primary B cells. Crit Rev Immunol. 2009;29(3):203–217. | ||
Sokolova V, Ludwig AK, Hornung S, et al. Characterisation of exosomes derived from human cells by nanoparticle tracking analysis and scanning electron microscopy. Colloids Surf B Biointerfaces. 2011;87(1):146–150. | ||
Takahashi Y, Nishikawa M, Shinotsuka H, et al. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J Biotechnol. 2013;165(2):77–84. | ||
Smyth T, Kullberg M, Malik N, Smith-Jones P, Graner MW, Anchordoquy TJ. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J Control Release. 2015;199:145–155. | ||
Bala S, Csak T, Momen-Heravi F, et al. Biodistribution and function of extracellular miRNA-155 in mice. Sci Rep. 2015;5(1):10721. | ||
Tian T, Wang Y, Wang H, Zhu Z, Xiao Z. Visualizing of the cellular uptake and intracellular trafficking of exosomes by live-cell microscopy. J Cell Biochem. 2010;111(2):488–496. | ||
Grange C, Tapparo M, Bruno S, et al. Biodistribution of mesenchymal stem cell-derived extracellular vesicles in a model of acute kidney injury monitored by optical imaging. Int J Mol Med. 2014;33(5):1055–1063. | ||
Greish K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. In: Grobmyer S, Moudgil B, editors. Cancer Nanotechnology: Methods in Molecular Biology (Methods and Protocols). New York, NY: Springer; 2010:25–37. | ||
Lam PY, Ho IA. Tumor tropism of mesenchymal stem cells. In: Shah K, editor. Stem Cell Therapeutics for Cancer. John Wiley & Sons, Inc; 2013:21–38. | ||
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. | ||
Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S. Inhibition of microRNA function by antimiR oligonucleotides. Silence. 2012;3(1):1. | ||
Zhang Y, Wang Z, Gemeinhart RA. Progress in microRNA delivery. J Control Release. 2013;172(3):962–974. | ||
Zeng YA, Nusse R. Wnt proteins are self-renewal factors for mammary stem cells and promote their long-term expansion in culture. Cell Stem Cell. 2010;6(6):568–577. | ||
Liu Y, Huang T, Zhao X, Cheng L. MicroRNAs modulate the Wnt signaling pathway through targeting its inhibitors. Biochem Biophys Res Commun. 2011;408(2):259–264. | ||
Hu T, Phiwpan K, Guo J, et al. MicroRNA-142-3p negatively regulates canonical Wnt signaling pathway. PLoS One. 2016;11(6):e0158432. | ||
Huang S, Chen Y, Wu W, et al. miR-150 promotes human breast cancer growth and malignant behavior by targeting the pro-apoptotic purinergic P2X7 receptor. PLoS One. 2013;8(12):e80707. | ||
Huang S, Xu L, Sun Y, Wu T, Wang K, Li G. An improved protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. J Orthop Translat. 2015;3(1):26–33. | ||
Mead B, Tomarev S. Bone Marrow-Derived Mesenchymal Stem Cells-Derived Exosomes Promote Survival of Retinal Ganglion Cells Through miRNA-Dependent Mechanisms. Stem cells Transl Med. 2017;6(4):1273–1285. | ||
Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. 2014;3(1):26913. |
Supplementary materials
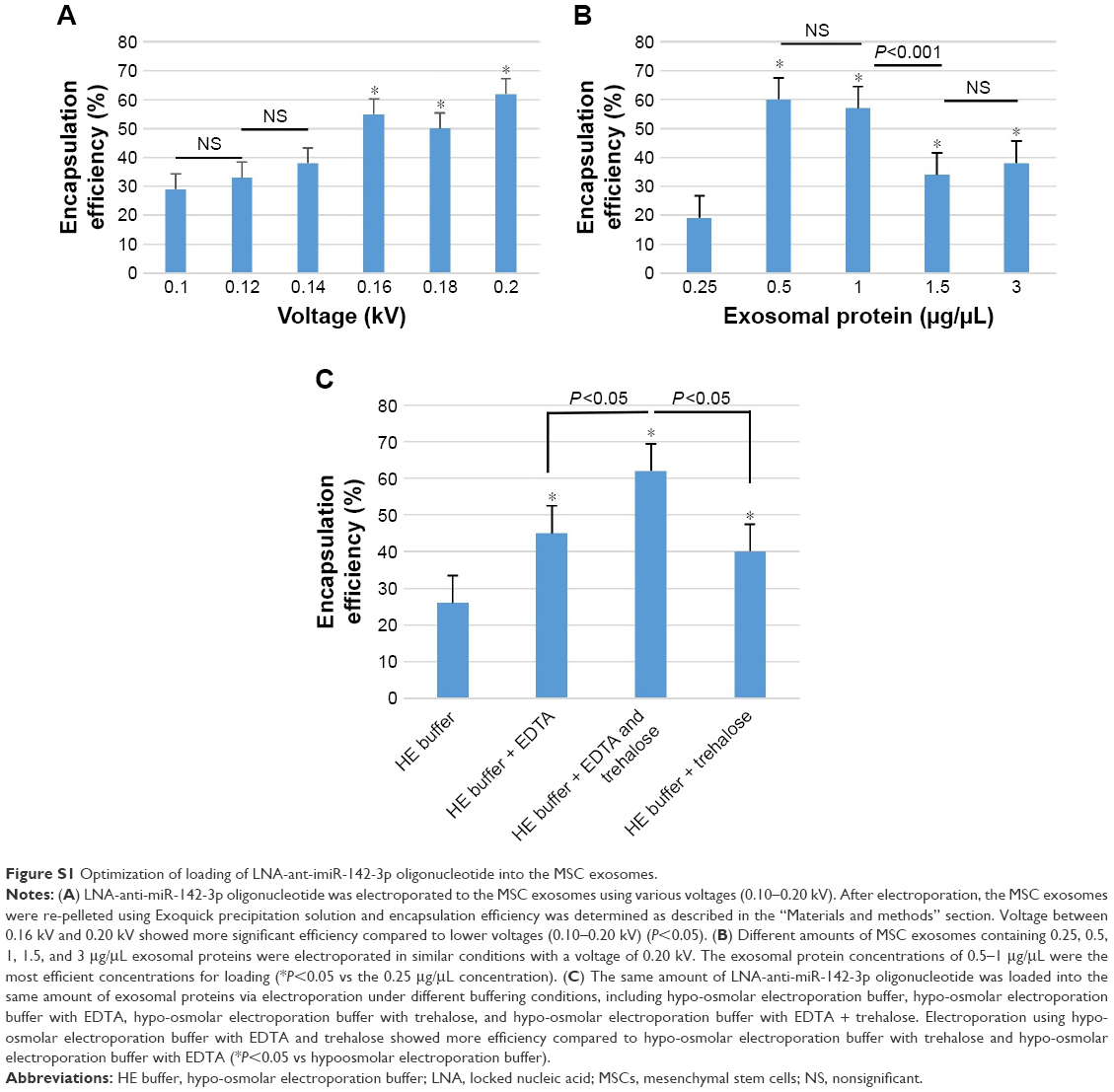 | Figure S1 Optimization of loading of LNA-ant-imiR-142-3p oligonucleotide into the MSC exosomes. |
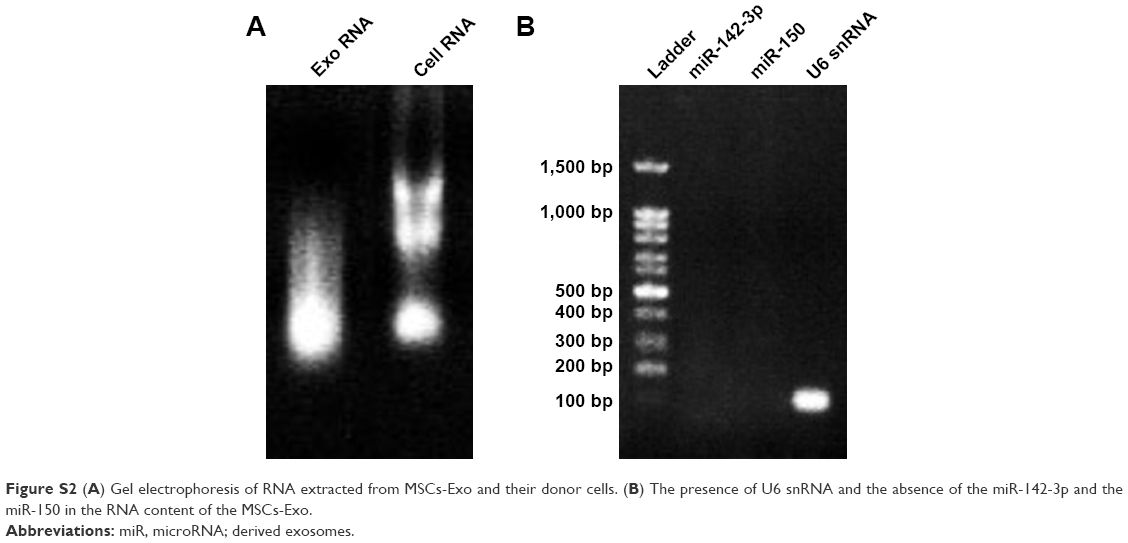 | Figure S2 (A) Gel electrophoresis of RNA extracted from MSCs-Exo and their donor cells. (B) The presence of U6 snRNA and the absence of the miR-142-3p and the miR-150 in the RNA content of the MSCs-Exo. |
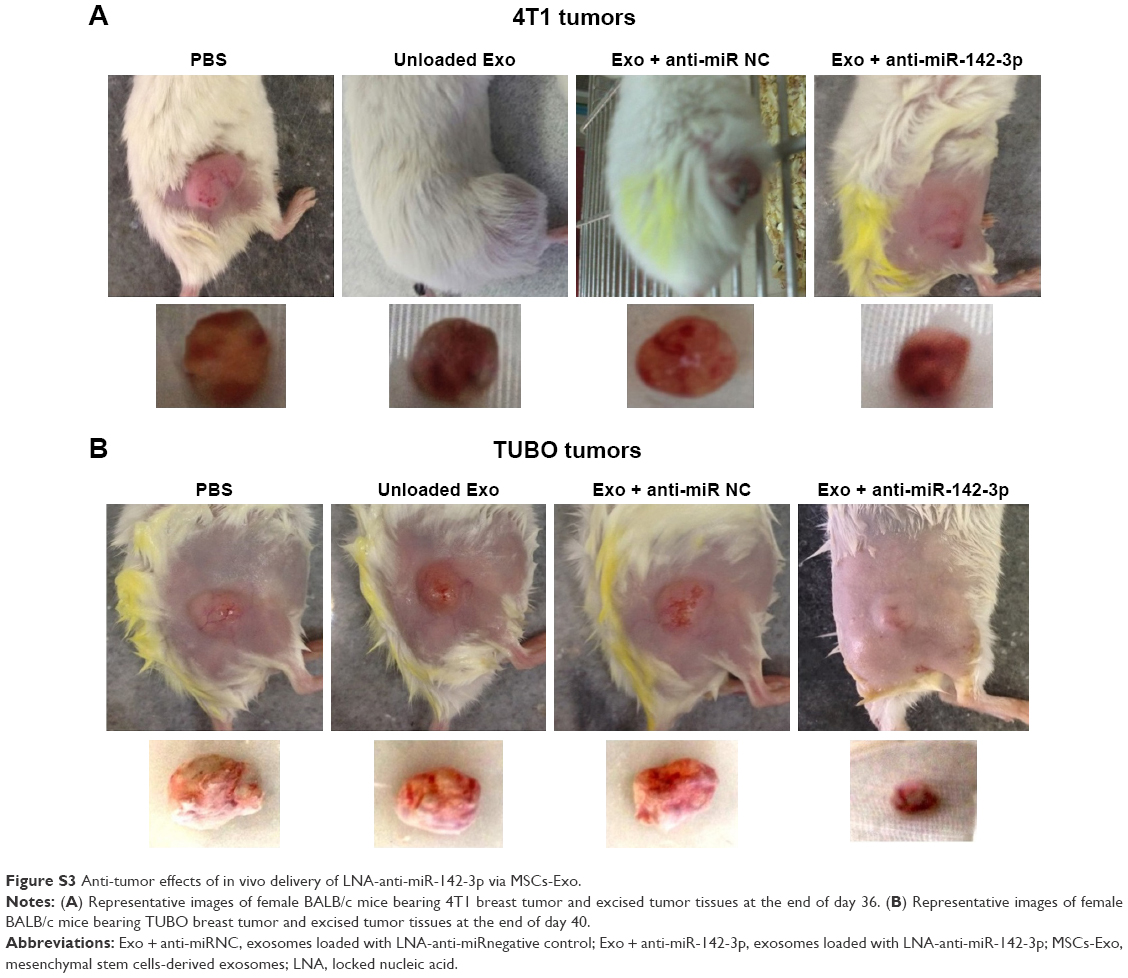 | Figure S3 Anti-tumor effects of in vivo delivery of LNA-anti-miR-142-3p via MSCs-Exo. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.