Back to Journals » International Medical Case Reports Journal » Volume 15
Exceptional Clinical Response to Surgery in Somalian Child Affected by Hyper Secretive Adrenal Cortical Carcinoma
Authors Osman AA ![]() , Omar Abdi A, Mohamud Abdullahi I
, Omar Abdi A, Mohamud Abdullahi I ![]() , Tahtabasi M, Kaya V
, Tahtabasi M, Kaya V ![]()
Received 9 April 2022
Accepted for publication 23 June 2022
Published 4 July 2022 Volume 2022:15 Pages 343—348
DOI https://doi.org/10.2147/IMCRJ.S370022
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ronald Prineas
Ahmed Adam Osman,1 Ahmed Omar Abdi,2 Ismail Mohamud Abdullahi,3 Mehmet Tahtabasi,4 Veysel Kaya5
1Department of Radiology, University of Health Sciences-Somalia Turkey Recep Tayyip Erdogan Education and Research Hospital, Mogadishu, Somalia; 2Department of General Surgery, Shaafi Hospital, Mogadishu, Somalia; 3Department of Pathology, University of Health Sciences-Somalia Turkey Recep Tayyip Erdogan Education and Research Hospital, Mogadishu, Somalia; 4Department of Radiology, University of Health Sciences-Mehmet Akif Inan Education and Research Hospital, Sanliurfa, Turkey; 5Department of Radiology, Harran University-Faculty of Medicine, Sanliurfa, Turkey
Correspondence: Ahmed Adam Osman, Department of Radiology, University of Health Sciences- Somalia Turkey Recep Tayyip Erdogan Education and Research Hospital, Mogadishu, Somalia, Tel +252615570262, Email [email protected]
Abstract: Adrenocortical cancers in childhood are very rare tumors. They are categorized as functional (hormone-secreting) or silent and as either benign or malignant. They have a bimodal distribution. Although in most adults they are non-functional, in the pediatric age group they may present as hormonal active or as an active tumor presenting with either virilizing forms or Cushing’s syndrome or both sometimes. In children, due to the rapid development of symptoms, they come to attention early. However, if not diagnosed and treated early, they can develop into serious medical conditions. We present here a 6-year-old girl complaining of voice changes (deepening), extremely overweight, excessive hair growth over her body, and clitoromegaly for one year. Abdominal ultrasound and computed tomography revealed a well-defined adrenal mass with a slightly heterogeneous appearance and heterogeneous-contrast enhancement containing some necrotic areas. The patient was discharged one week after unilateral right adrenalectomy in good condition, and oral medications were given along with high-dose corticosteroid medications, which were reduced gradually. All the symptoms disappeared 6 months after the operation.
Keywords: childhood adrenocortical carcinoma, Cushing’s syndrome, virilizing tumors’ pediatric cancers
Introduction
Adrenocortical carcinoma (ACC) is an orphan malignancy with an annual incidence between 0.7 and 2 cases per million population. ACC is more frequent in women (55–60%) with a peak incidence in the fourth and fifth decades of life.1 Adrenocortical tumors (ACTs) are uncommon in children, accounting for only 0.2% of all neoplasms and 6% of pediatric adrenal tumors.2 Vast majority (91.8%) of pediatric ACC occurs among Caucasians and Asians are affected only in 5.9% of cases.3 Although most adult ACCs are non-functional, in the pediatric age group tumors are more likely to be functioning.4
Virilization is the most common abnormality and Cushing syndrome is the second most prevalent ACT presentation in children, with moon facies and hyperaldosteronism are less frequent.5 In this paper, we report a rare case of a 6-year-old girl with an adrenal tumor who came to Mogadishu Somali Turkey Training and Research Hospital pediatric outpatient department, which was diagnosed clinically, radiologically, and treated surgically at Shaafi hospital.
Case Presentation
We report here a 6-year-old girl came to the outpatient department of Mogadishu Somali Turkey Hospital complaining of voice changes (deepening), extensive overweight, excessive hair growth over the body and clitoromegaly for one year. Initially, her mother was noticed that she had weight gain and her clitoris was more protruded than the normal until become elongated and enlarged associated with labial enlargement. Within one year, throughout the body, hair grew, especially chin and pubis, and bad smelling odor and revealed male characterization.
On examination, her height was 1.0 m, her weight was 60 kg, and her blood pressure was 190/150 mm Hg (refractory hypertension). She had cushingoid facies, signs of virilization in the form of pustular acne, hirsutism (axillary and facial hair in Figure 1A), and clitoromegaly. She also had an unpleasant body odor. On palpation, she had a palpable mass on the right side of her abdomen. In laboratory investigations, elevation of Dehydroepiandrosterone sulfate (DHEA-SO4) 419.03 µg/d (normal range 24.4–209.7 µg/d) indicating production of excess hormones related to adrenal tumors. Other hematological and biochemical parameters were normal.
 |
Figure 1 (A) In pre-operative photos seems overweight with excessive hair growth all over her body (hirsutism) and pustular acne on her face. (B) 6-month post-op photos show a dramatically reduced body weight, appearing similar to her age group, and a total absence of body hair. |
As usual in pediatric cases, in the beginning, an abdominal ultrasound (US) was performed, showing a well-defined adrenal mass with a slightly heterogeneous mix of hyper- and hypoechoic consistency with smooth margins (Figure 2). For further investigation and lesion characterization, computed tomography (CT) of the abdomen has done, revealing that the right adrenal fossa well-defined a 6 cm sized mass lesion with heterogeneous contrast enhancement containing hypodense necrotic areas and slightly displaces right kidney inferiorly (Figure 3). To bring her blood pressure back into normal range, the child was admitted to the intensive care unit two days before the operation.
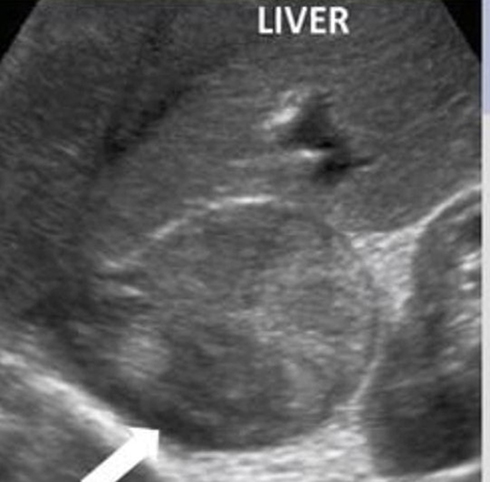 |
Figure 2 Abdominal ultrasound shows well-defined adrenal mass appearing slightly heterogeneous mixed hyper and hypoechoic consistent with smooth margins (white arrow). |
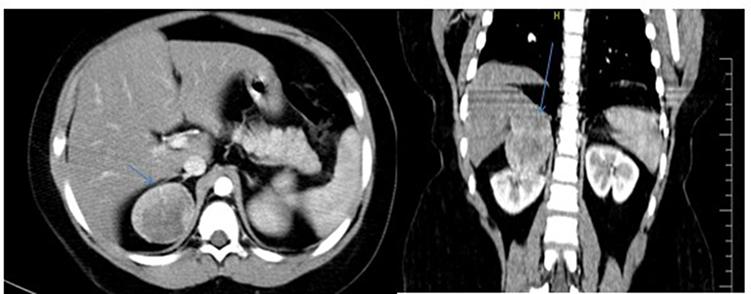 |
Figure 3 Axial and coronal contrast-enhanced abdominal computed tomography showed that 6 cm right adrenal fossa well-defined heterogeneously enhanced mass lesion containing hypodense necrotic areas and slightly displaces right kidney inferiorly (blue arrow). |
A diagnosis of an adrenocortical tumor was made. Correction of general condition was done. Details about the disease, plan of management, and risks were explained to the parents. After written consent and pre-anesthetic checkup, during the surgery, a Makuuchi incision was made to the patient’s abdomen. Next, the solid mass was excised by means of unilateral right adrenalectomy (Figure 4). Then, abdominal layers were repaired using a vicryl 1. And the mass was sent for histopathological study. Histopathological diagnosis was made using the Li-Weiss-Bisceglia criteria. In Table 1, the histological features of the current case are indicated. Microscopic examination revealed >75% oncocytic cells, diffuse architecture (>33% of tumor) and high nuclear grade. In addition, Ki-67 proliferation index was calculated as 15–20%. According to these findings, the case was diagnosed with oncocytic ACC (Figure 4). The patient was admitted, and appropriate post-operative care was given. And discharged after one week with good condition and oral medications were given. Corticosteroid drugs were started with a high dose and the dose was gradually reduced. And all the symptoms disappeared 6 months after the operation (Figure 1B).
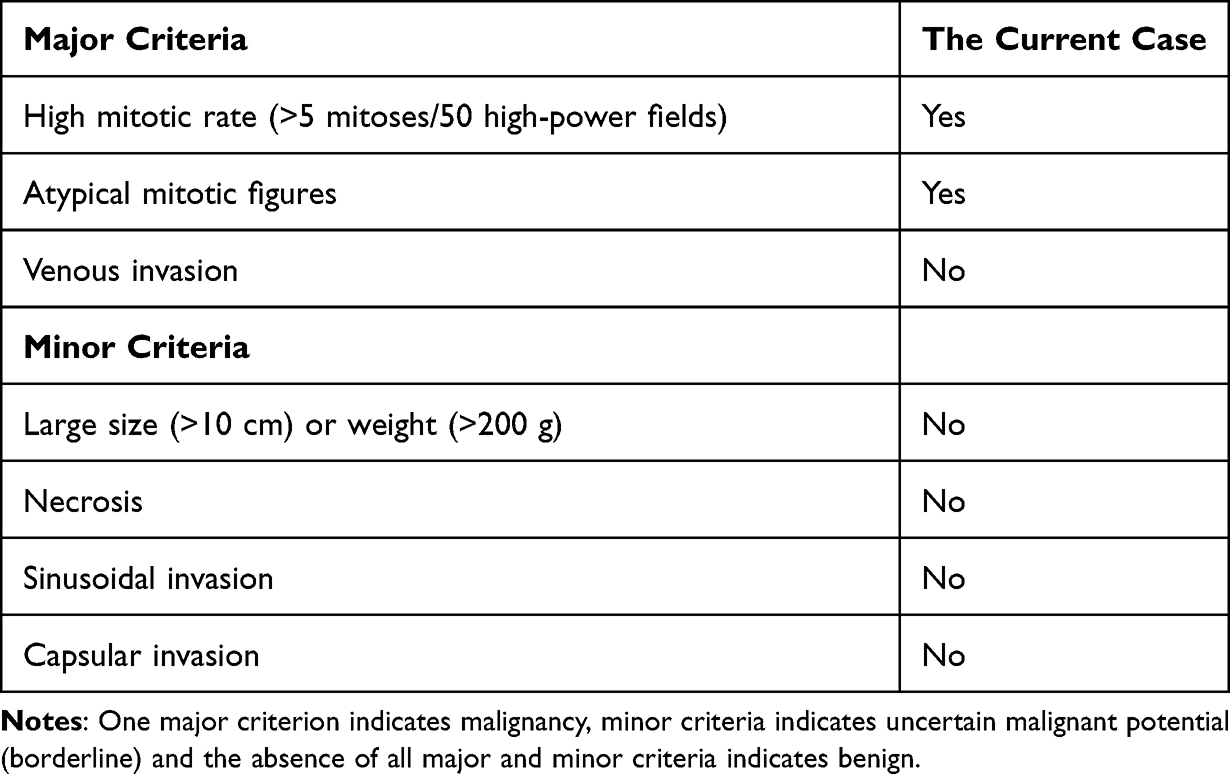 |
Table 1 Li- Weiss-Bisceglia Diagnostic Algorithm for Oncocytic ACC |
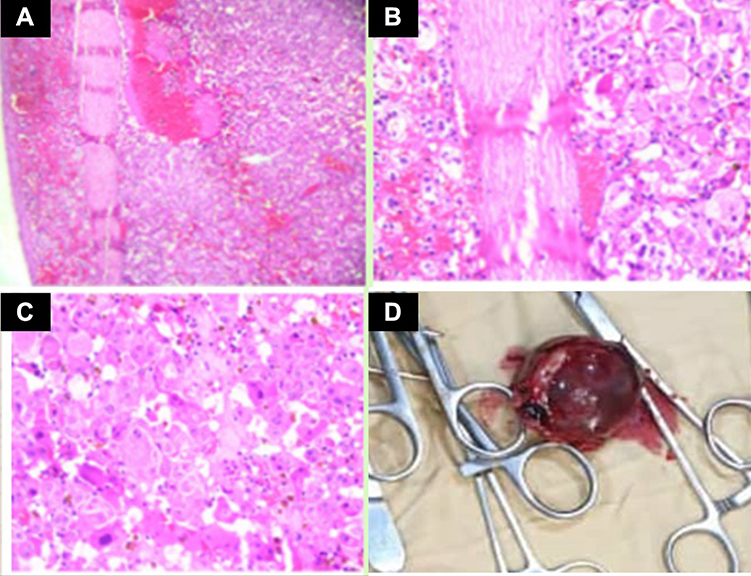 |
Figure 4 Histopathological examination shows (A) encapsulated tumor composed of variably sized nests, large sheets, and trabeculae, areas of necrosis, hemorrhage, and degeneration areas. (B) Low grade: ≤20 mitoses/50 high power fields morphologic subtypes, Oncocytic (>75% oncocytic cells). (C) Large cells with granular clear to eosinophilic cytoplasm, pleomorphic Intranuclear inclusions, mitoses, and atypical mitoses. (D) Gross surgical specimen of a removed adrenal mass. |
Discussion
Adrenal adenomas or ACCs are tumors that arise from the adrenal cortex, whereas neuroblastomas and pheochromocytomas arise from the adrenal medulla (PCCs). They are traditionally divided into functional and nonfunctional tumors. Neuroblastomas are nonfunctional tumors, whereas adenomas, ACCs, and PCCs are functional tumors. Adrenocortical tumors are uncommon in children, accounting for only 0.2% of all neoplasms.2 The ACTs come in a variety of forms, including virilizing forms, presentations with Cushing’s syndrome, or both. Children become aware early due to the quick development of symptoms; nevertheless, if not diagnosed and treated early, it can progress to serious medical conditions. New diagnostic imaging methods have emerged in the recent decade. There is also a lot of research going on as these tumors can be unresectable or have a high recurrence rate.
According to the International Pediatric Adrenocortical Tumor Registry (IPATR), 90% of childhood ACCs are functional, and their clinical signs and biological behavior are different from adult ACCs. The most common clinical presentation is virilization (84.3%).6 The most common symptoms in ACC cases are acne, pubic hair, and clitoris enlargement.7 Cushing’s syndrome is the second most common clinical presentation with moon facies, centripetal fat distribution, striae over the skin, and a plethora in one-third of patients.8 Similar to these data, our case had a functional and hormone-producing mass. Moreover, another study reported that 51% of 16 pediatric ACCs were functional.9 However, recent studies have shown that the incidence of functional tumors is slightly higher. The reason for these differences may be related to the size of the hormonal evaluation performed in different centers. The virilizing tumor of pediatric ACCs is explained by their origin in the persistent fetal region of the adrenal cortex. This is also related to the tendency to produce DHEA-S.10
Percutaneous biopsies are not recommended for the diagnosis of ACC. There is a risk of metastasis in the needle tract and tumor pillage due to capsular breach.10
The role of imaging is very important in the diagnosis of ACC. Although ultrasonography, which is a non-invasive method, is the first choice in pediatric patients, its sensitivity is low. Therefore, cross-sectional imaging with CT and magnetic resonance imaging (MRI) is essential for diagnosis and detection of local and distant tumor spread. Information from CT and MRI is important for guiding surgery and further patient treatment. The typical appearance of ACC on unenhanced CT is a large and heterogeneous suprarenal mass that replaces adjacent structures. It is a large, heterogeneous but well-defined suprarenal mass that displaces adjacent structures. Unenhanced on CT, areas with low attenuation mostly due to necrosis are seen. However, lesions smaller than 6 cm may be homogeneous. After the administration of IV contrast agent, there are peripherally enhanced solid areas and centrally less contrasted areas of necrosis. CT is also very valuable in demonstrating local and distant spread. Preservation of fat around the tumor indicates no local invasion. However, in cases where there is little retroperitoneal fat, it may be impossible to detect organ invasion.11 In our case, an adrenal lesion was detected by the US, and we performed a contrast-enhanced abdominal CT to characterize the mass. On CT, we detected a well-defined suprarenal mass. Typically, we detected greater enhancement peripherally and relatively little enhancement centrally due to central necrosis. Since the size of the mass was not very large, retroperitoneal fatty tissue was preserved, and there was no adjacent organ invasion.
Complete surgical resection is currently the only potentially curative treatment for ACC, and surgery is the cornerstone of treatment. Laparoscopic adrenalectomy, which is a minimally invasive method compared to open surgery, has been used in the treatment of adrenal masses in recent years. In Conzo et al’s series of 126 diseases, only one conversion to open surgery occurred. No post-operative major complications were observed, while minor complications occurred in 8 patients (0.79%).12,13 The contralateral adrenal gland is thought to be suppressed functional ACTs.14 The modified Weiss criteria are a well-established technique for ACC diagnosis.14,15 Age at diagnosis is a significant prognostic factor, with children under the age of two years having a favorable result. Children above the age of two are said to have a survival rate of only 29%,16 However, our case is 6 years old, and she is alive and well 1 year after the operation.
Conclusion
An interdisciplinary approach is needed in the management of childhood adrenal tumors, which is a challenging task. Adrenal tumor-related hypertension in children is a life-threatening condition. Surgery is the best option, if the diagnosis is made early and the prognosis is good. ACT in children can be diagnosed early due to the rapid development of symptoms. When cancer is discovered later, the prognosis is poor. Management of diagnosis and treatment of ACCs can be done effectively with a multidisciplinary approach under the guidance of currently approved guidelines.
Ethics Committee Approval and Consent for Publication
Written informed consent was obtained from the patient’s parent for publication of this case report and accompanying images. Approval from the Ethics Committee of the Somali Turkey Training and Research Hospital was not required to publish case details. A copy of the written permission is available for review by the Editor of this journal.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jasim S, Habra MA. Management of adrenocortical carcinoma. Curr Oncol Rep. 2019;21:20. doi:10.1007/s11912-019-0773-7
2. Stewart JN, Flageole H, Kavan P. A surgical approach to adrenocortical tumors in children: the mainstay of treatment. J Pediatr Surg. 2004;39:759–763. doi:10.1016/j.jpedsurg.2004.01.029
3. McAteer JP, Huaco JA, Gow KW. Predictors of survival in pediatric adrenocortical carcinoma: a Surveillance, Epidemiology, and End Results (SEER) program study. J Pediatr Surg. 2013;48:1025–1031. doi:10.1016/j.jpedsurg.2013.02.017
4. Zaremba LS, Smoleński WH. Optimal portfolio choice under a liability constraint. Ann Oper Res. 2000;97:131–141. doi:10.1023/A:1018996712442
5. Cagle PT, Hough AJ, Jeffrey TP, et al. Comparison of adrenal cortical tumors in children and adults. Cancer. 1986;57:2235–2237. doi:10.1002/1097-0142(19860601)57:11<2235::AID-CNCR2820571127>3.0.CO;2-O
6. Michalkiewicz E, Sandrini R, Figueiredo B, et al. Clinical and outcome characteristics of children with adrenocortical tumors: a report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol. 2004;22:838–845. doi:10.1200/JCO.2004.08.085
7. Sandrini R, Ribeiro RC, DeLacerda L. Childhood adrenocortical tumors 1. J Clin Endocrinol Metab. 1997;82:2027–2031. doi:10.1210/jcem.82.7.4057
8. Garg M, Jacob M, Kharb S, et al. Childhood adrenocortical carcinoma: case report and review. Indian J Endocrinol Metab. 2012;16:431. doi:10.4103/2230-8210.95699
9. Sabaretnam M, Mishra A, Agarwal G, et al. Adrenocortical carcinoma in children and adults: two decades experience in a single institution. Indian J Cancer. 2016;53(2):317. doi:10.4103/0019-509X.197737
10. Yadav SK, Jha CK, Mishra SK, et al. Pediatric mixed functioning adrenocortical carcinoma: a case report and review of literature. Middle East J Cancer. 2019;10:372–377.
11. Bharwani N, Rockall AG, Sahdev A, et al. Adrenocortical carcinoma: the range of appearances on CT and MRI. Am J Roentgenol. 2011;196:W706–W714. doi:10.2214/AJR.10.5540
12. Conzo G, Gambardella C, Candela G, et al. Single center experience with laparoscopic adrenalectomy on a large clinical series. BMC Surg. 2018;18:2. doi:10.1186/s12893-017-0333-8
13. Conzo G, Pasquali D, Gambardella C, et al. Long-term outcomes of laparoscopic adrenalectomy for Cushing disease. Int J Surg. 2014;12:S107–S111. doi:10.1016/j.ijsu.2014.05.036
14. Lau SK, Weiss LM. The Weiss system for evaluating adrenocortical neoplasms: 25 years later. Hum Pathol. 2009;40:757–768. doi:10.1016/j.humpath.2009.03.010
15. Aubert S, Wacrenier A, Leroy X, et al. Weiss system revisited. Am J Surg Pathol. 2002;26:1612–1619. doi:10.1097/00000478-200212000-00009
16. Ciftci AO, Şenocak ME, Tanyel FC, et al. Adrenocortical tumors in children. J Pediatr Surg. 2001;36:549–554. doi:10.1053/jpsu.2001.22280
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.