Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 19
Evaluation of Potential Therapeutic Targets for Bacterial Infectious Skin Diseases: A Proteome-Wide Mendelian Randomization Study
Authors Yan Z, Shi Z, Wang Y, Yin H
Received 25 October 2025
Accepted for publication 20 February 2026
Published 2 March 2026 Volume 2026:19 576669
DOI https://doi.org/10.2147/CCID.S576669
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Anne-Claire Fougerousse
Zhangren Yan,1 Ziyang Shi,2 Ye Wang,2 Hongwei Yin1
1Department of Traditional Chinese Medicine Surgery, Hospital of Jiangxi University of Chinese Medicine, Nanchang, Jiangxi, People’s Republic of China; 2Graduate School, Jiangxi University of Chinese Medicine, Nanchang, Jiangxi, People’s Republic of China
Correspondence: Hongwei Yin, Email [email protected]
Background: Bacterial infectious skin diseases are common dermatological conditions caused by various pathogenic bacteria. In recent years, their incidence has remained high due to factors such as climate change and the overuse of antibiotics, posing persistent challenges for clinical diagnosis and treatment. This study aimed to identify plasma proteins causally associated with cellulitis, erysipelas, cutaneous abscess, furuncle, and carbuncle through Mendelian randomization (MR) analysis.
Methods: We analyzed genetic instruments of 4907 proteins derived from the Icelandic population together with genome-wide association study (GWAS) data from the FinnGen project to investigate circulating plasma proteins involved in bacterial infectious skin diseases. To validate causal associations, we employed Steiger filtering, reverse MR analysis, phenome-wide association studies (pheWAS), Bayesian colocalization, summary data–based Mendelian randomization (SMR) and (Heterogeneity in Dependent Instruments) HEIDI test. Furthermore, we conducted protein–protein interaction (PPI) network analysis and drug target evaluation to assess therapeutic potential.
Results: We identified five key proteins associated with bacterial infectious skin diseases. Specifically, FN1 was associated with erysipelas; ULK3 and CSK were associated with cellulitis; and ARHGAP25 and ENTPD6 were associated with cutaneous abscess, furuncle, and carbuncle.
Conclusion: Through a systematic proteome-wide Mendelian randomization analysis with multiple layers of validation, this study prioritised a set of host plasma proteins associated with different subtypes of bacterial infectious skin diseases, providing a reference for biomarker exploration, further elucidation of disease-related molecular mechanisms, and subsequent research into non-antibiotic intervention strategies.
Keywords: proteome, mendelian randomization, bacterial infectious skin diseases, erysipelas, cellulitis, cutaneous abscess
Introduction
The skin, the largest organ of the human body, serves as a primary physiological barrier and plays a central role in maintaining water homeostasis, immune defense, and multiple essential physiological functions. Among skin disorders, bacterial skin infections are particularly prevalent, encompassing a wide clinical spectrum ranging from superficial localized infections to deep necrotizing soft-tissue involvement. Owing to differences in pathogen composition, infection depth, and tissue involvement, common clinical subtypes exhibit distinct pathological and phenotypic characteristics. Erysipelas primarily affects the superficial dermis and lymphatic network, typically presenting as sharply demarcated erythematous lesions; cellulitis involves the deeper dermis and subcutaneous tissues, characterized by diffuse inflammation with poorly defined margins; whereas skin abscesses, furuncles, and carbuncles are typically confined to hair follicles and adjacent tissues and are defined by localized purulent inflammation.
Current clinical management relies predominantly on antibiotics; however, the growing prevalence of antimicrobial resistance and the risk of adverse drug reactions—particularly in immunocompromised individuals—pose substantial therapeutic challenges. In response to these challenges, modulation of host immunity has emerged as a promising therapeutic strategy. Unlike conventional antimicrobials that directly target pathogens, immunomodulatory approaches aim to enhance endogenous host defense mechanisms, thereby reducing reliance on traditional antibiotics. Accumulating evidence indicates that bacterial infections trigger inflammatory cascades characterized by the release of cytokines and chemokines, leading to systemic alterations in circulating plasma proteins. It is worth noting that, although plasma proteins circulate systemically, substantial differences in pathogen composition, infection depth, and activation of local host immune pathways across infection subtypes can give rise to disease-specific immune signatures, even among clinically overlapping conditions.1,2
Nevertheless, proteomic investigations in the context of bacterial skin infections remain limited. Proteome-wide Mendelian randomization (MR) offers a powerful causal inference framework that leverages genetic variants as instrumental variables to systematically identify potential biomarkers while minimizing confounding and reverse causation.3,4
Accordingly, we integrated plasma proteomic data with genome-wide association study (GWAS) data on bacterial skin infections to identify genetically prioritized candidate plasma protein targets through a multi-step MR framework. To strengthen causal inference, we further applied reverse MR, colocalization analyses, phenome-wide association studies (PheWAS), Summary-data–based Mendelian Randomization (SMR) and Heterogeneity in Dependent Instruments (HEIDI) test to minimize pleiotropy and bias. Finally, by constructing protein–protein interaction (PPI) networks and assessing druggability. Given the discrepancy between lifelong genetically determined protein exposure and the acute course of infection, the associations identified in this study are framed as genetically prioritized candidates, narrowing the scope of future functional investigations and provides important theoretical insights for the development of novel immunomodulatory therapies. The process is illustrated in Figure 1.
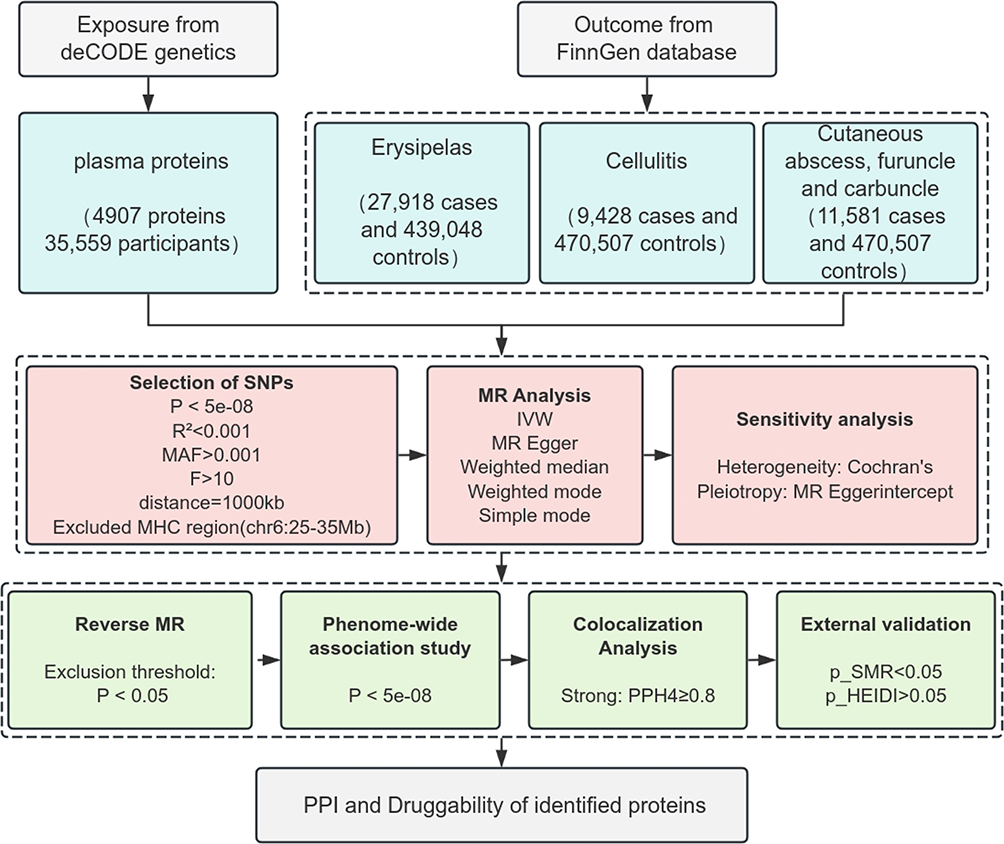 |
Figure 1 Study design flowchart. Note: Bold text indicates key analytical steps in the workflow. |
Materials and Methods
Data Sources and Selection of Instrumental Variables
The exposure data were derived from a large-scale plasma protein quantitative trait loci (pQTL) study comprising 35,559 Icelandic individuals and profiling 4907 plasma proteins.5 To accurately capture the biological effects of cis-regulatory elements and ensure the reliability of causal inference, pQTLs were selected according to the following criteria: (1) only cis-pQTLs located within ±1 Mb of the transcription start site of the target gene were included, ensuring instrument variables exerted cis effects; (2) single nucleotide polymorphisms (SNPs) significantly associated with protein expression at the genome-wide level (P < 5 × 10−8) were retained to enhance instrument reliability; (3) SNPs with minor allele frequency (MAF) < 0.001 and located within the major histocompatibility complex (MHC) region on chromosome 6 (25–35 Mb), which is characterized by extreme pleiotropy and complex linkage disequilibrium (LD) structure, were excluded to minimize potential confounding; (4) only SNPs with F-statistics >10 were retained to remove weak instruments; and (5) independent variants were preserved using LD clumping with an r2 threshold of <0.001.6,7 Instrument characteristics for proteins included in the MR analysis are provided in Supplementary Table 1.
To further reduce potential population stratification bias, the study was restricted to individuals of European ancestry. Moreover, to prevent sample overlap between exposure and outcome datasets, overlapping cohorts were excluded. Outcome data were obtained from the FinnGen R12 dataset, encompassing three bacterial skin infection phenotypes: erysipelas (27,918 cases and 439,048 controls), cellulitis (9428 cases and 470,507 controls), and skin abscess, furuncle, and carbuncle (11,581 cases and 470,507 controls). The outcome data used in this study are provided in Supplementary Table 2. Since this study utilized publicly available data from open databases, ethical approval was not required.
MR Analysis
MR analyses were conducted using R software (version 4.2.2) with the packages “TwoSampleMR”, “gridExtra”, and “Tidyverse”. When multiple SNPs served as instrumental variables, the inverse-variance weighted (IVW) method was applied to estimate causal effects;8 for single-SNP instruments, the Wald ratio method was used.9 To strengthen the robustness of the findings, complementary methods including MR-Egger regression, weighted median, simple mode, and weighted mode were also performed. Heterogeneity was assessed using Cochran’s Q statistic, while horizontal pleiotropy was evaluated with the MR-Egger intercept test.10 To control for the risk of multiple comparisons, false discovery rate (FDR) correction was applied, with a significance threshold set at P < 0.05.11 The results of the Mendelian randomization analysis are presented in Supplementary Table 3. Statistical power for the MR analyses was calculated using the mRnd tool12 and the detailed results are presented in Supplementary Table 4.
To minimize the possibility of reverse causation, reverse MR analysis and MR Steiger tests were performed for proteins identified as positive in forward MR. The results are presented in Supplementary Tables 5 and 6, respectively. Proteins showing evidence of reverse causality (Steiger P > 0.05 or significant in reverse MR analysis) were excluded from further analyses.
PheWAS Analysis
To further validate the independence assumption and assess potential confounding, proteins identified in MR analyses were subjected to pheWAS using the PheWAS Portal. Genome-wide significance was defined as P < 5×10−8.
Bayesian Colocalization Analysis
To determine whether genetic variants influence both exposure and outcome through regulation of the same causal locus, Bayesian colocalization analysis was performed using the “coloc” package in R. Prior probabilities were set as follows p1 = 1×10−5 (association with disease), p2 = 1×10−5 (association with protein levels), and p12 = 1×10−5 (association with both traits). The method evaluates five mutually exclusive hypotheses H0 (no association with either trait), H1 (association with disease only), H2 (association with protein levels only), H3 (association with both traits via distinct causal variants), and H4 (both traits share a common causal variant). Posterior probabilities (PP) were calculated for each hypothesis, with PPH4 ≥ 0.8 considered strong evidence of colocalization, in line with thresholds adopted in previous studies to reduce false positives.13 The colocalization results are presented in Supplementary Table 7.
External Validation
To validate the causal associations between protein expression and disease, we performed SMR test14 using eQTL data from the eQTLGen Consortium and SMR software (v.1.3.1). The HEIDI test was used to evaluate whether observed associations were driven by linkage with multiple genetic variants.15 A causal relationship was inferred if the SMR test yielded P<0.05 and the HEIDI test P > 0.05. The external validation results are presented in Supplementary Table 8.
Key Protein Selection
Proteins simultaneously meeting the following criteria were defined as key proteins and subjected to follow-up analyses:
(1) statistical significance in forward MR analysis after multiple testing correction;
(2) no evidence of reverse causality based on reverse MR analysis and Steiger tests;
(3) genome-wide significant associations in pheWAS analyses;
(4) strong evidence of colocalization, defined as a PPH4 ≥ 0.8;
(5) significant results in SMR analysis (SMR P < 0.05) with no evidence of HEIDI test (HEIDI P > 0.05).
Gene Network Analysis
Gene network construction was performed using GeneMANIA (http://www.genemania.org/), which integrates multiple biological data sources, including gene co-expression, genetic interactions, signaling pathways, and protein domain similarity. This tool predicts functionally related genes or proteins and generates visualized interaction networks, providing insights into potential molecular mechanisms and biological roles of candidate proteins.16,17
Druggability Analysis
Potential therapeutic agents targeting the identified proteins were explored using the Drug Signature Database (DSigDB), which integrates multiple data resources on drug–gene expression signatures. DSigDB contains information on approved drugs, clinical trial candidates, and experimental compounds, including drug identifiers, mechanisms of action, and development.18 The list of the druggability analysis are provided in Supplementary Table 9.
Results
Mendelian Randomization Analysis
A total of 1746 cis-pQTLs from the deCODE study were included in the MR analysis. The inverse-variance weighted (IVW) method or the Wald ratio was used as the primary analytical approach, with odds ratios (ORs) and 95% confidence intervals (CIs) estimated to assess risk associations. After FDR correction and power calculation verification, several significant protein–disease relationships were identified. The results are as shown in Figures 2 and 3.
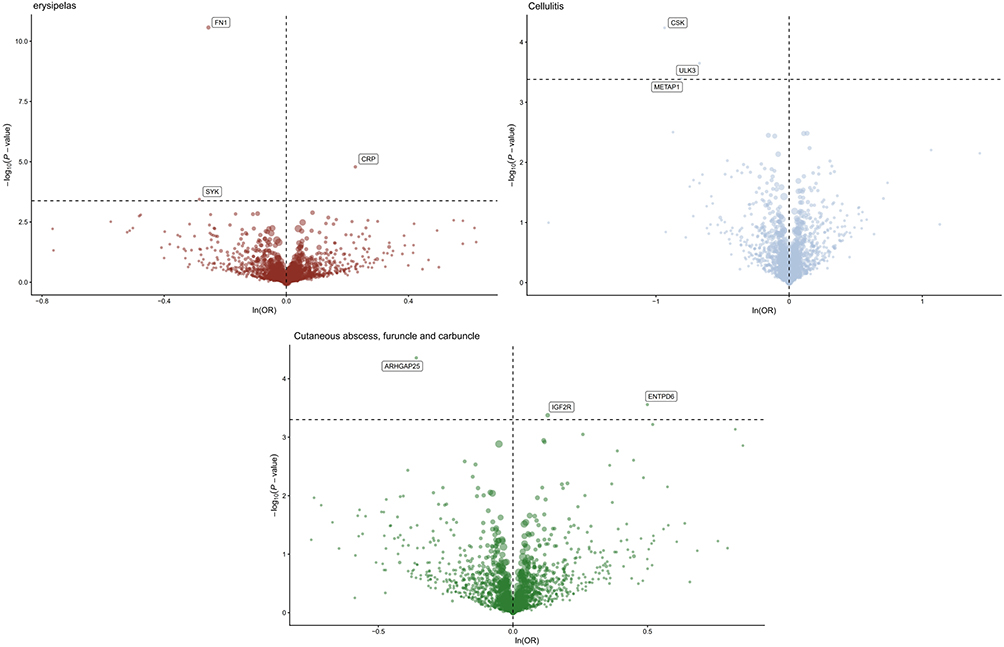 |
Figure 2 Proteome-wide MR results. |
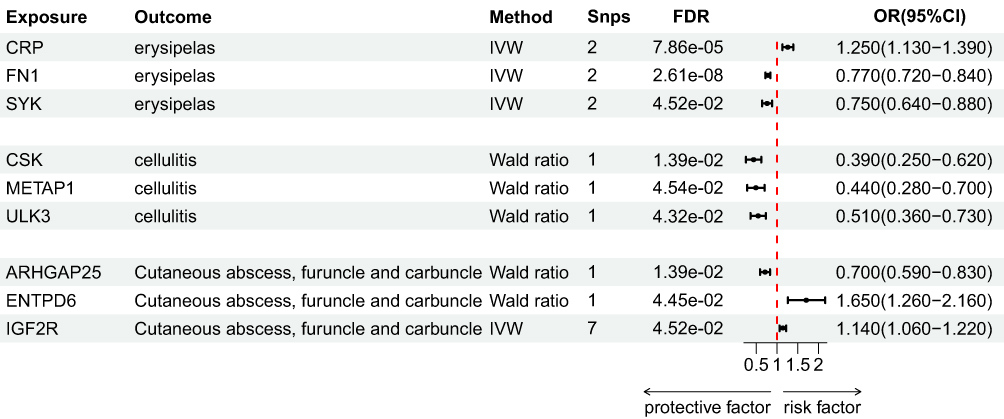 |
Figure 3 Forest plot of MR analysis. Note: Statistical significance was defined as FDR < 0.05. |
For erysipelas, CRP showed a positive association (OR = 1.25, 95% CI [1.13–1.39]), whereas FN1 (OR = 0.77, 95% CI [0.72–0.84]) and SYK (OR = 0.75, 95% CI [0.64–0.88]) demonstrated negative associations.
For cellulitis, negative associations were observed with CSK (OR = 0.39, 95% CI [0.25–0.62]), METAP1 (OR = 0.44, 95% CI [0.28–0.70]), and ULK3 (OR = 0.51, 95% CI [0.36–0.73]).
For cutaneous abscess, carbuncle, and furuncle, positive associations were detected for IGF2R (OR = 1.14, 95% CI [1.06–1.22]) and ENTPD6 (OR = 1.65, 95% CI [1.26–2.16]), while a negative association was observed for ARHGAP25 (OR = 0.70, 95% CI [0.59–0.83]).
Sensitivity analyses showed no evidence of horizontal pleiotropy or heterogeneity, as indicated by non-significant P values (P > 0.05) in MR-Egger regression and Cochran’s Q tests.
Reverse Causality Analysis
Reverse MR analysis and MR Steiger tests were performed to evaluate the possibility of reverse causality in the MR findings. Among the proteins identified in forward MR, only CRP showed a significant association in reverse MR (P < 0.05), suggesting that its relationship with erysipelas may be driven by reverse causation. Therefore, CRP was excluded from subsequent analyses to avoid erroneous causal inference.
PheWAS Analysis Results
PheWAS analysis revealed that, with the exception of ENTPD6, none of the other candidate drug targets showed significant associations with alternative traits at the genetic level. This finding indicates a relatively low risk of potential side effects or pleiotropy for these targets, thereby strengthening the robustness of our results. In contrast, ENTPD6 was found to be associated with cardiometabolic traits, suggesting that drugs acting on this protein may exert effects on cardiometabolic health.
Colocalization Analysis
To enhance the robustness of our findings, Bayesian colocalization analysis was conducted with a threshold of PPH≥0.8. Strong colocalization evidence was observed between proteins and their respective diseases FN1 with erysipelas (PPH4 = 0.84); CSK (PPH4 = 0.91) and ULK3 (PPH4 = 0.87) with cellulitis; and ARHGAP25 (PPH4 = 0.94) and ENTPD6 (PPH4 = 0.91) with cutaneous abscess, carbuncle, and furuncle. These proteins, validated through colocalization, were defined as key causal proteins in this study. The results are as shown in Figure 4.
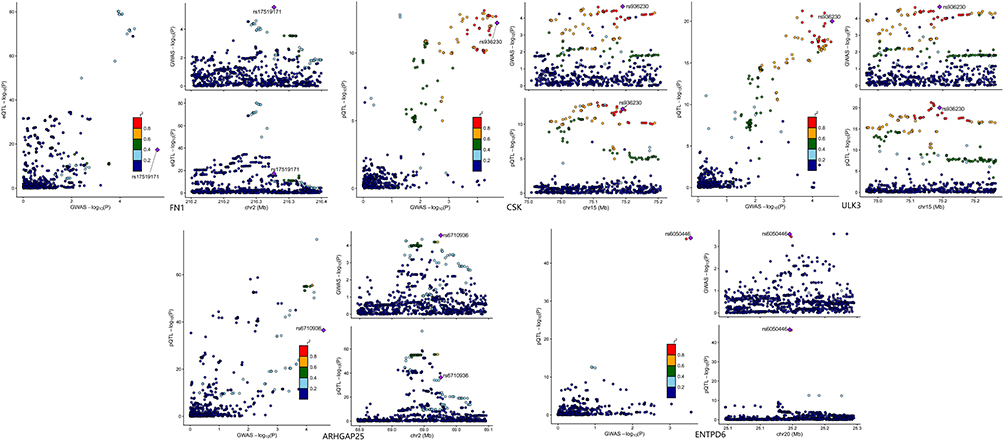 |
Figure 4 Proteins with colocalization results of PPH4≥0.8. |
SMR Analysis Results
Using eQTL data corresponding to the identified drug-target proteins, we performed SMR analyses together with HEIDI tests. Seven proteins demonstrated significant associations in SMR analyses FN1 and SYK for erysipelas; CSK, METAP1, and ULK3 for cellulitis; and ENTPD6 and ARHGAP25 for cutaneous abscess, carbuncle, and furuncle. These results confirmed that the causal associations between these proteins and the respective diseases were independent of linkage disequilibrium, thereby further supporting the robustness of our findings.
Results of Key Protein Selection
Table 1 summarizes the proteins prioritized based on forward MR, reverse MR, pheWAS, colocalization, HEIDI test and SMR test. Among them, five proteins met all prioritization criteria and were defined as key proteins: FN1, ULK3, CSK, ARHGAP25, and ENTPD6.
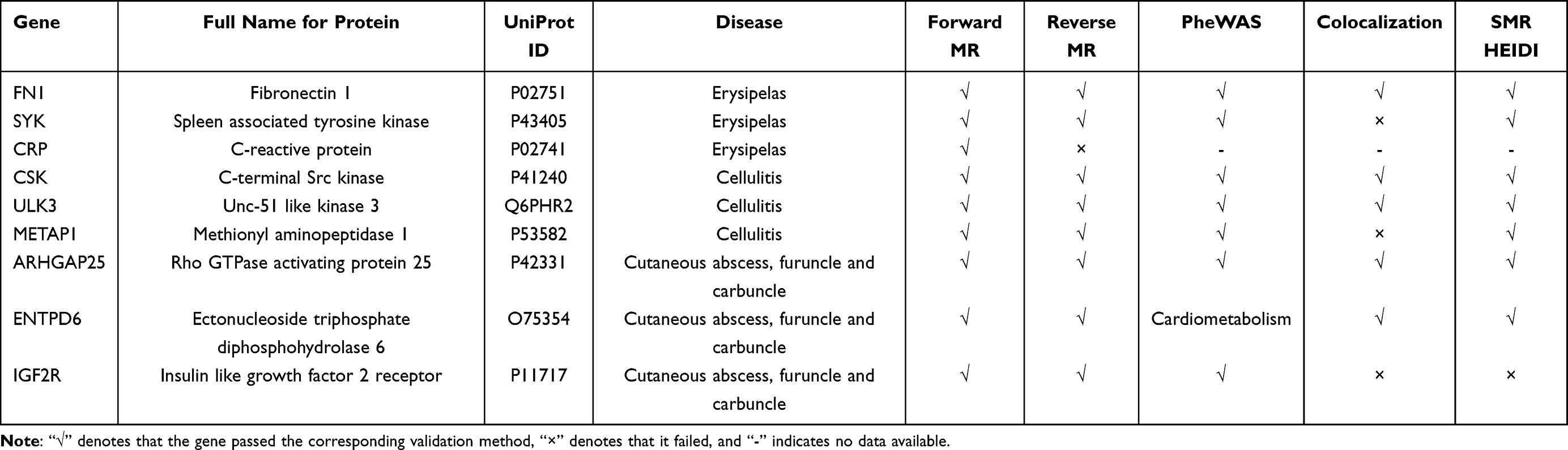 |
Table 1 Screening of Risk Proteins Validated Through Multiple Approaches |
PPI Network and Drug Identification
The PPI network constructed using GeneMANIA highlighted interactions between the identified proteins and their hub genes, providing insights into their potential functional roles in the pathogenesis of bacterial infectious skin diseases. Our findings revealed distinct immunological and metabolic characteristics across disease subtypes erysipelas was primarily associated with humoral immune responses, characterized by B cell activation, antibody production, complement activation, and phagocytosis; cellulitis was more closely linked to cellular immunity, with enrichment in T cell activation and regulation, leukocyte adhesion, and tyrosine kinase signaling; while cutaneous abscesses exhibited unique metabolic features, involving diverse cellular metabolic processes and biosynthetic pathways. The results are as shown in Figure 5.
 |
Figure 5 PPI network of plasma proteins from MR analysis. |
Through the DSigDB, we identified several potential therapeutic agents targeting these proteins. For FN1, candidate drugs included sitaxentan, formoterol, and mercaptopurine. For CSK and ULK3, multiple drug candidates were identified, such as axitinib, dabrafenib, and vandetanib. Notably, only ciclopirox was found to be associated with ARHGAP25, whereas no candidate drugs were identified for ENTPD6. The results are as shown in Figure 6.
 |
Figure 6 Drug enrichment analysis of key proteins. |
Discussion
In recent years, the rapid global escalation of antibiotic resistance has created an urgent need for therapeutic strategies that operate independently of conventional antimicrobial mechanisms.19 Unlike bacterial resistance mechanisms such as efflux pumps or target modifications, host-directed immune modulation harnesses the body’s intrinsic defense systems to combat infection.20 As central mediators of host immune responses, plasma proteins exhibit dynamic circulating levels that not only reflect the host’s defensive state but also provide critical insights for assessing immune responses, monitoring disease progression, and informing clinical management.
Given the heterogeneity in tissue involvement and pathogenic features among subtypes of bacterial skin infections, the identification of subtype-specific plasma proteins is critical for precisely delineating their pathogenic mechanisms. Accordingly, we employed a genome-wide proteomic MR framework to genetically prioritize plasma proteins with potential causal associations with these infections. Following rigorous multi-step filtering to exclude pleiotropic effects and reverse causality, we identified FN1 as genetically prioritized in erysipelas, CSK and ULK3 in cellulitis, and ENTPD6 and ARHGAP25 in skin abscesses. These proteins were thus designated as the key candidates in our study. In the subsequent discussion, we focus on the functional and clinical implications of these key proteins.
FN1 is a ubiquitously expressed extracellular matrix (ECM) protein that regulates fundamental biological processes, including cell adhesion, migration, proliferation, and survival, and has been implicated in diverse pathological contexts such as cancer, inflammation, and infection.21 Through Mendelian randomization analysis, we identified FN1 as a genetically prioritized signal in erysipelas, potentially reflecting its established roles in immune responses. Previous studies have demonstrated that FN1 mediates host–pathogen interactions by binding bacterial fibronectin-binding proteins and integrin receptors on phagocytic cells, thereby facilitating pathogen recognition and internalization and enhancing phagocytic efficiency.22 Moreover, FN1 has been reported to participate in T cell–mediated immune regulation23 and to play a critical role in cutaneous wound healing and tissue repair,24 suggesting a potential function in coordinating local immune responses with tissue homeostasis.
The Src kinase family, a group of non-receptor tyrosine kinases, plays a central role in regulating inflammation, cell proliferation, motility, and adhesion. CSK, a principal negative regulator of Src family kinases (SFKs), phosphorylates the C-terminal tyrosine residues of SFKs to maintain them in an inactive state, thereby precisely modulating neutrophil-mediated inflammatory responses.25 Skin-specific studies have further demonstrated that epidermal deletion of CSK leads to constitutive Src activation, resulting in epidermal hyperplasia, skin thickening, and chronic inflammation, highlighting a critical role for CSK in maintaining cutaneous homeostasis and restraining SFK-driven inflammatory signaling.26
ULK3, a member of the small kinase subfamily, is known to participate in diverse cellular processes, including autophagy, cell division, and Sonic Hedgehog (Shh) signaling.27 In epidermal keratinocytes, ULK3 predominantly localizes to the nucleus, where it regulates transcriptional programs to influence keratinocyte self-renewal and differentiation balance. Additionally, ULK3 has been reported to interact with epigenetic regulators PRMT1 and PRMT5, modulating gene expression and chromatin state to maintain epithelial homeostasis.28 Although current evidence largely derives from keratinocyte carcinoma models and in vitro studies, these findings suggest that ULK3 may play a pivotal role in coordinating host epithelial responses to external stimuli, including microbial invasion, potentially via autophagy-related mechanisms.
ARHGAP25 is a leukocyte-specific Rac GTPase-activating protein that functions as a negative regulator of neutrophil phagocytosis. It is highly expressed in peripheral blood leukocytes and granulocyte cell lines, and its silencing enhances phagocytic activity and associated superoxide production, whereas overexpression suppresses these processes in a GAP-dependent manner.29 Mechanistically, ARHGAP25 likely modulates phagosome formation by regulating Rac-dependent F-actin remodeling and acts upstream of NADPH oxidase complex assembly within Fcγ receptor and complement receptor signaling pathways. In vivo, ARHGAP25 expression is upregulated in leukocytes in murine models of contact hypersensitivity, and its deficiency attenuates immune cell infiltration into inflamed skin and alters the pro-inflammatory cytokine milieu, implicating ARHGAP25 as a critical modulator of leukocyte recruitment during cutaneous inflammation.30
ENTPD6, also known as CD39L2, is an extracellular nucleotidase predominantly expressed in the heart, where it plays a critical role in regulating cardiac microcirculation, inhibiting platelet aggregation, and preventing thrombosis.31 Although our study identified a statistically significant association between genetically predicted circulating ENTPD6 levels and susceptibility to skin infections, it is important to consider potential off-target cardiovascular effects when contemplating therapeutic interventions. Strategies such as dose optimization or local targeting may be required to mitigate cardiac risk, underscoring the necessity of thorough safety evaluation before clinical translation of ENTPD6 as a potential therapeutic target.
Our PPI network analysis further refines the mechanistic understanding of subtype-specific disease processes. FN1, associated with erysipelas, is integral to networks governing humoral immune regulation, whereas the cellulitis-associated proteins CSK and ULK3 preferentially converge on pathways linked to T-cell activation and broader cellular immune responses. This integrative mapping—from systemic genetic associations to localized immune network interactions—highlights potentially divergent immunopathological architectures underlying distinct clinical subtypes.
Collectively, our findings highlight specific plasma proteins that regulate local inflammation and infection in bacterial skin diseases. These proteins likely influence wound infection and healing through mechanisms such as immune cell recruitment and complement activation. Although we discussed only validated targets, other proteins identified in this study may also play important roles and merit future investigation.
The strengths of this study are manifold. First, stringent thresholds were applied in the selection of instrumental variables to minimize bias from weak instruments, linkage disequilibrium, and false-positive associations. Second, multiple complementary approaches—including reverse Mendelian randomization, phenome-wide association analysis, Bayesian colocalization, and SMR-HEIDI testing—were employed to cross-validate findings, thereby establishing convergent lines of evidence and substantially enhancing the robustness of causal inference. Third, protein–protein interaction network analysis revealed putative biological pathways, offering novel insights into disease mechanisms. Finally, druggability assessment identified several candidate molecules, highlighting these proteins as potential therapeutic targets for further investigation.
Nevertheless, several limitations of this study warrant consideration. First, the GWAS datasets employed were predominantly derived from European populations, with limited representation of other ancestries, potentially restricting the generalizability of our findings to populations with distinct genetic architectures and epidemiological profiles of infection. Second, although multiple statistical approaches were implemented to mitigate confounding, Mendelian randomization analyses cannot fully exclude bias from unknown or unmeasured genetic confounders, and the high pleiotropy observed in some candidate proteins may further complicate causal inference. Third, our analyses focused primarily on circulating plasma proteins, encompassing both secreted and leakage-derived species. Given that protein expression may differ between blood and skin tissue, circulating protein levels may not fully capture tissue-specific activity, introducing a degree of uncertainty. Finally, as our study infers lifelong protein exposure from genetic instruments, these estimates may not directly reflect dynamic fluctuations occurring during acute infection. Consequently, interpretation of these results should be approached with caution, and validation through experimental or clinical studies is warranted.
Conclusion
In this study, we employed a proteome-wide Mendelian randomization approach to identify a set of plasma proteins whose genetically predicted levels are associated with bacterial skin infections. These candidate proteins were validated through complementary analyses, including reverse Mendelian randomization, phenome-wide association studies, Bayesian colocalization, and SMR-HEIDI testing, thereby highlighting potential targets for future functional investigations. It is important to note that our analyses were based on genetically predicted plasma protein levels in European populations, representing lifelong exposure, which may not fully capture dynamic changes in local skin environments during acute infection. Consequently, these findings should be interpreted as exploratory and warrant further validation through experimental and clinical studies to elucidate their biological mechanisms and potential clinical relevance.
Abbreviations
MR, Mendelian Randomization; GWAS, Genome-Wide Association Study; pheWAS, Phenome-Wide Association Study; SMR: Summary data–based Mendelian Randomization; PPI, Protein–Protein Interaction; pQTL, Protein Quantitative Trait Locus; SNP, Single Nucleotide Polymorphism; LD, Linkage Disequilibrium; MAF, Minor Allele Frequency; IVW, Inverse-Variance Weighted; FDR, False Discovery Rate; HEIDI, Heterogeneity in Dependent Instruments; PP, Posterior Probability; ECM, Extracellular Matrix; SFK, Src Family Kinase; RacGAP, Rac GTPase-Activating Protein; DSigDB, Drug Signature Database; eQTL, Expression Quantitative Trait Locus.
Data Sharing Statement
The cis-pQTL GWAS summary statistics are publicly available from the deCODE genetics research database (https://www.decode.com/summarydata/).GWAS data were obtained from the FinnGen Biobank (https://www.finngen.fi/en).
Ethical Declarations
The design and implementation of this study followed the relevant regulations and ethical guidelines of China. According to Article 32 of the “Ethical Review Measures for Human Life Sciences and Medical Research” jointly issued by relevant Chinese departments on February 18, 2023, the following two types of studies can be exempted from ethical review: (1): Non-intrusive observational studies conducted in public places, and those that do not involve the collection of private behaviors and information that cannot identify individual identities; (2): Studies using legally available database data or information collected through anonymous methods, and those that cannot be traced back to specific individuals and will not pose any risks to the subjects. After assessment, this study falls under the circumstances stipulated in the second item of the above-mentioned provisions. Therefore, this study is exempt from the review and approval of the institutional ethics committee.
Acknowledgments
We thank all participants who consented to participate in this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by funding from the Jiangxi University of Chinese Medicine Science and Technology Innovation Team Development Program (CXTD22009).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Willems E, Gloerich J, Suppers A, et al. Impact of infection on proteome-wide glycosylation revealed by distinct signatures for bacterial and viral pathogens. Iscience. 2023;26(8):107257. doi:10.1016/j.isci.2023.107257
2. Irani Shemirani M, Pernestig AK, Björkman J, et al. Identification of protein biomarkers to differentiate between gram-negative and gram-positive infections in adults suspected of sepsis. BMC Infect Dis. 2025;25:1576. doi:10.1186/s12879-025-11973-5
3. Wu J, Yang Z, Ding J, et al. Proteome-wide mendelian randomization identifies causal plasma proteins in prostate cancer development. Human Genomics. 2025;19(1):17. doi:10.1186/s40246-025-00724-x
4. Yang C, Fagan AM, Perrin RJ, et al. Mendelian randomization and genetic colocalization infer the effects of the multi-tissue proteome on 211 complex disease-related phenotypes. Genome Med. 2022;14(1):140. doi:10.1186/s13073-022-01140-9
5. Ferkingstad E, Sulem P, Atlason BA, et al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet. 2021;53(12):1712–13. doi:10.1038/s41588-021-00978-w
6. Pierce BL, Ahsan H, Vanderweele TJ. Power and instrument strength requirements for mendelian randomization studies using multiple genetic variants. Int J Epidemiol. 2011;40(3):740–752. doi:10.1093/ije/dyq151
7. Burgess S, Thompson SG. CRP CHD Genetics Collaboration. Avoiding bias from weak instruments in mendelian randomization studies. Int J Epidemiol. 2011;40(3):755–764. doi:10.1093/ije/dyr036
8. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658–665. doi:10.1002/gepi.21758
9. Burgess S, Small DS, Thompson SG. A review of instrumental variable estimators for mendelian randomization. Stat Methods Med Res. 2015;26(5):2333–2355. doi:10.1177/0962280215597579
10. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through egger regression. Int J Epidemiol. 2015;44(2):512–525. doi:10.1093/ije/dyv080
11. Chen X, Robinson DG, Storey JD. The functional false discovery rate with applications to genomics. Biostatistics. 2019;22(1):68–81. doi:10.1093/biostatistics/kxz010
12. Brion MJA, Shakhbazov K, Visscher PM. Calculating statistical power in mendelian randomization studies. Int J Epidemiol. 2013;42(5):1497–1501. doi:10.1093/ije/dyt179
13. Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLOS Genet. 2014;10(5):e1004383. doi:10.1371/journal.pgen.1004383
14. Wu Y, Zeng J, Zhang F, et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun. 2018;9(1):918. doi:10.1038/s41467-018-03371-0
15. Zhu Z, Zhang F, Hu H, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–487. doi:10.1038/ng.3538
16. Franz M, Rodriguez H, Lopes C, et al. GeneMANIA update 2018. Nucleic Acids Res. 2018;46(W1):W60–W64. doi:10.1093/nar/gky311
17. Warde-Farley D, Donaldson SL, Comes O, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(suppl_2):W214–W220. doi:10.1093/nar/gkq537
18. Yoo M, Shin J, Kim J, et al. DSigDB: drug signatures database for gene set analysis. Bioinformatics. 2015;31(18):3069–3071. doi:10.1093/bioinformatics/btv313
19. Murray CJL, Ikuta KS, Sharara F; Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet. 2022;399(10325):629–655. doi:10.1016/S0140-6736(21)02724-0
20. Kaufmann SHE, Dorhoi A, Hotchkiss RS, et al. Host-directed therapies for bacterial and viral infections. Nat Rev Drug Discov. 2018;17(1):35–56. doi:10.1038/nrd.2017.162
21. Wang H, Zhang J, Li H, et al. FN1 is a prognostic biomarker and correlated with immune infiltrates in gastric cancers. Front Oncol. 2022;12:918719. doi:10.3389/fonc.2022.918719
22. Pommier CG, Inada S, Fries LF, Takahashi T, Frank MM, Brown EJ. Plasma fibronectin enhances phagocytosis of opsonized particles by human peripheral blood monocytes. J Exp Med. 1983;157(6):1844–1854. doi:10.1084/jem.157.6.1844
23. Fernandes NRJ, Reilly NS, Schrock DC, et al. CD4+ T cell interstitial migration controlled by fibronectin in the inflamed skin. Front Immunol. 2020;11:1501. doi:10.3389/fimmu.2020.01501
24. Muro AF, Chauhan AK, Gajovic S, et al. Regulated splicing of the fibronectin EDA exon is essential for proper skin wound healing and normal lifespan. J Cell Biol. 2003;162(1):149–160. doi:10.1083/jcb.200212079
25. Berman-Booty LD, Eraslan R, Hanumegowda U, et al. Systemic loss of C-terminal src kinase expression elicits spontaneous suppurative inflammation in conditional knockout mice. Vet Pathol. 2018;55(2):331–340. doi:10.1177/0300985817747330
26. Paik SJ, Kim DJ, Jung SK. Preventive effect of pharmaceutical phytochemicals targeting the src family of protein tyrosine kinases and aryl hydrocarbon receptor on environmental stress-induced skin disease. Int J Mol Sci. 2023;24(6):5953. doi:10.3390/ijms24065953
27. Kasak L, Näks M, Eek P, et al. Characterization of protein kinase ULK3 regulation by phosphorylation and inhibition by small molecule SU6668. Biochemistry. 2018;57(37):5456–5465. doi:10.1021/acs.biochem.8b00356
28. Goruppi S, Clocchiatti A, Bottoni G, et al. The ULK3 kinase is a determinant of keratinocyte self-renewal and tumorigenesis targeting the arginine methylome. Nat Commun. 2023;14(1):887. doi:10.1038/s41467-023-36410-6
29. Csépányi-Kömi R, Sirokmány G, Geiszt M, et al. ARHGAP25, a novel rac GTPase-activating protein, regulates phagocytosis in human neutrophilic granulocytes. Blood. 2012;119(2):573–582. doi:10.1182/blood-2010-12-324053
30. Schlam D, Bagshaw RD, Freeman SA, et al. Phosphoinositide 3-kinase enables phagocytosis of large particles by terminating actin assembly through rac/Cdc42 GTPase-activating proteins. Nat Commun. 2015;6(1):8623. doi:10.1038/ncomms9623
31. Yeung G, Mulero JJ, McGowan DW, et al. CD39L2, a Gene Encoding a Human Nucleoside Diphosphatase, Predominantly Expressed in the Heart. Biochemistry. 2000;39(42):12916–12923. doi:10.1021/bi000959z
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.