Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
Evaluation of lung parenchyma, blood vessels, and peripheral blood lymphocytes as a potential source of acute phase reactants in patients with COPD
Authors Arellano-Orden E ![]() , Calero C, López-Ramírez C, Sánchez-López V, López-Villalobos JL, Abad Arranz M, Blanco-Orozco A
, Calero C, López-Ramírez C, Sánchez-López V, López-Villalobos JL, Abad Arranz M, Blanco-Orozco A ![]() , Otero-Candelera R
, Otero-Candelera R ![]() , López-Campos JL
, López-Campos JL ![]()
Received 24 September 2018
Accepted for publication 18 March 2019
Published 20 June 2019 Volume 2019:14 Pages 1323—1332
DOI https://doi.org/10.2147/COPD.S188567
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Richard Russell
Elena Arellano-Orden,1,2 Carmen Calero,1,2 Cecilia López-Ramírez,1,2 Verónica Sánchez-López,1,2 José Luis López-Villalobos,1,2 María Abad Arranz,1,2 Ana Blanco-Orozco,1 Remedios Otero-Candelera,1,2 José Luis López-Campos1,2
1Unidad Médico-Quirúrgica de Enfermedades Respiratorias, Instituto de Biomedicina de Sevilla (IBiS), Hospital Universitario Virgen del Rocío, Universidad de Sevilla, Seville, Spain; 2CIBER de Enfermedades Respiratorias (CIBERES), Instituto de Salud Carlos III, Madrid, Spain
Background: Previous studies have shown that the arterial wall is a potential source of inflammatory markers in COPD. Here, we sought to compare the expression of acute phase reactants (APRs) in COPD patients and controls both at the local (pulmonary arteries and lung parenchyma) and systemic (peripheral blood leukocytes and plasma) compartments.
Methods: Consecutive patients undergoing elective surgery for suspected primary lung cancer were eligible for the study. Patients were categorized either as COPD or control group based on the spirometry results. Pulmonary arteries and lung parenchyma sections, peripheral blood leukocytes, and plasma samples were obtained from all participants. Gene expression levels of C-reactive protein (CRP) and serum amyloid A (SAA1, SAA2, and SAA4) were evaluated in tissue samples and peripheral blood leukocytes by reverse transciption-PCR. Plasma CRP and SAA protein levels were measured by enzyme-linked immunosorbent assays. Proteins were evaluated in paraffin-embedded lung tissues by immunohistochemistry.
Results: A total of 40 patients with COPD and 62 controls were enrolled. We did not find significant differences in the gene expression between COPD and control group. Both CRP and SAA were overexpressed in the lung parenchyma compared with pulmonary arteries and peripheral blood leukocytes. The expression of SAA was significantly higher in the lung parenchyma than in the pulmonary artery (2-fold higher for SAA1 and SAA4, P=0.015 and P<0.001, respectively; 8-fold higher for SAA2, P<0.001) and peripheral blood leukocytes (16-fold higher for SAA1, 439-fold higher for SAA2, and 5-fold higher for SAA4; P<0.001). No correlation between plasma levels of inflammatory markers and their expression in the lung and peripheral blood leukocytes was observed.
Conclusions: The expression of SAA in lung parenchyma is higher than in pulmonary artery and peripheral blood leukocytes. Notably, no associations were noted between lung expression of APRs and their circulating plasma levels, making the leakage of inflammatory proteins from the lung to the bloodstream unlikely. Based on these results, other potential sources of systemic inflammation in COPD (eg, the liver) need further scrutiny.
Keywords: COPD, inflammation, C-reactive protein, serum amyloid A, arterial wall, parenchyma
Introduction
Although the presence of systemic inflammation has been extensively documented in patients with COPD, its exact sources are still unclear.1 Moreover, the question as to whether the systemic inflammatory response may influence the clinical manifestations and the expression of different COPD phenotypes remains open.2
C-reactive protein (CRP) and serum amyloid A (SAA) are among the most common acute phase reactants (APRs) found to be increased in COPD,3,4 even though their biological sources have not been yet identified.5 It has been hypothesized that the excess production of local inflammatory mediators in the pulmonary compartment may spill over into the systemic circulation.6 However, this possibility is contradicted by the lack of correlation between local and systemic expression of inflammatory biomarkers in COPD.7 Moreover, markers of inflammation in the induced sputum did not show a significant association with their plasma levels.6
Previous investigations conducted in normal tissues as well as animal and in vitro studies demonstrated that lung tissues are able to synthesize and secrete APRs.3,8,9 We have previously shown that bronchi and the lung parenchyma are able to synthesize CRP and SAA in COPD as compared with smokers who did not develop the disease (control group),10 with potential distinct roles for fibroblasts, epithelial cells, and macrophages.11 Notably, our immunohistochemical observations revealed an increased expression of APRs in the arterial wall of pulmonary vessels. Therefore, it is possible that blood vessels or cells may be responsible for these observations. The role of pulmonary arteries and peripheral blood leukocytes as a potential source of systemic inflammatory markers in COPD has not yet been evaluated. The potential involvement of the arterial wall and/or peripheral blood leukocytes in COPD inflammation would also explain the well-known association of this condition with adverse cardiovascular events.12
We therefore designed the current case-control study to compare the expression of APRs in COPD patients and controls both at the local (pulmonary arteries and lung parenchyma) and systemic (peripheral blood leukocytes and plasma) compartments.
Methods
Study participants
Consecutive patients with suspected primary lung cancer who were scheduled for elective pneumectomy or lobectomy were eligible. Patients were approached for the study the day before scheduled surgery. Exclusion criteria were as follows: 1) age <40 years, 2) a history of acute respiratory infections in the previous 2months, 3) a previously diagnosed neoplasm, 4) treatment with radiotherapy and/or chemotherapy, 5) presence of chronic inflammatory diseases, and 6) autoimmune diseases. The amount of time spent between the opening of the cutaneous layer and the extraction of the anatomical sample was measured in all participants. Cases with an elapsed time >3 hrs were excluded because of the potential surgery-related increase in inflammatory markers. Patients had their medical records checked to ensure that they recently underwent a lung function test. Participants with a /FVC ratio <0.7 post-bronchodilator were considered as having COPD. The patients were classified according to the stage of the disease following the instructions of the GOLD. All of the remaining was considered controls. The study participants completed a standardized questionnaire focusing on their medical history, tobacco smoking habits, and current use of drugs. The Charlson-age comorbidity index is a measure of comorbidity that was developed as a prospectively applicable method for classifying comorbid conditions which might alter the risk of morality for use in longitudinal studies.13 The TNM staging of the primary lesion14 and the surgical approach were also recorded. A lung specimen was submitted for microbiological examination before surgical resection to evaluate microbial colonization. Microbiological samples obtained with bronchoscopy were also examined. After surgery, a section of the lung parenchyma and the largest available section of the resected pulmonary artery were collected for further study. Samples that were most distally located from the primary lesion were identified and immediately processed. The remaining anatomical sample was submitted for pathological diagnosis of the primary lesion. Peripheral venous blood samples were collected before surgery in all participants. This study was conducted in accordance with the Declaration of Helsinki.
Gene expression levels in tissues and blood
Expression of inflammatory markers in tissues and peripheral blood leukocytes was analyzed using reverse transcription quantitative PCR(RT-qPCR), a method consisting of three steps (RNA extraction, reverse transcription to cDNA, and gene amplification). RNA was isolated from fresh tissues with a TriSure kit (Bioline, London, UK). Samples subsequently underwent DNase treatment using RNase-free DNase (QIAGEN GmbH, Hilden, Germany) and cDNA was synthesized using an iScript™ cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). Each 25 µL amplification reaction mixture comprised 5 µL cDNA (10 ng/µL), 12.5 µL SYBR® Green PCR Master Mix (Stratagene, La Jolla, CA, USA) and 7.5 µL primers/H2O. RT-qPCR was performed on an Applied Biosystems® 7900 Real-Time PCR System (Life Technologies, Grand Island, NY, USA) under the following cycling conditions: 95 ºC for 15 s, 60ºC for 30 s, and 72ºC for 40 s, 40 cycles. 18S ribosomal RNA (rRNA) expression was used as an internal control for normalization of target gene expression. The list of primers used to amplify the genes of interest is provided in Table 1. Three tightly linked SAA genes have been identified in humans (SAA1, SAA2, and SAA4). Their synthesis is triggered by proinflammatory cytokines released upon activation of monocytes/macrophages.15
 | Table 1 Primers used for reverse transcription quantitative PCR |
Measurements of plasma inflammatory markers
Blood samples (5 mL) were collected by venipuncture in K3-EDTA tubes (Vacuette, Bio-One GmbH, Greiner, Austria). Samples were immediately centrifuged, and plasma aliquots were stored at −80°C until immediately before analysis. Plasma CRP (R&D System, Minneapolis, MN, USA) and SAA (Anogen, Mississauga, Ontario, Canada) levels were measured using commercially available enzyme immunoassay kits according to the manufacturers’ protocol.
Immunohistochemistry
Tissue blocks from the subpleural parenchyma (paying attention to avoid areas involved by the tumor) were fixed in 10% formalin and embedded in paraffin. Five-μm-thick sections were prepared for immunohistochemical analysis. Mouse monoclonal antibodies against SAA1 (Novus Biologicals, Cambridge, UK) and CRP (Abcam, Cambridge UK) were used. Antigen retrieval was achieved by microwave heat treatment in citrate buffer (Dako, Glostrup, Denmark) at 98°C for 15 mins. The bound antibody was developed with diaminobenzidine using a Dako Envision staining kit (K4065) according to manufacturer’s recommendations. Stained sections were observed under light microscope by two independent observers. All immunohistochemical studies included appropriate standard quality controls. As a negative control, immunohistochemistry was performed using secondary antibodies without the primary antibody.
Statistical analysis
Means and SDs for continuous variables and frequencies and percentages for categorical variables were estimated. Relative gene expression values were evaluated with the 2−∆∆Ct method16 to compare COPD and control group, and according to the anatomical location. The Student’s t-test was used to test for differences in continuous variables between COPD patients and controls. Categorical data were compared with the χ2 test. The associations between tissue gene expression and plasma levels of inflammatory biomarkers were analyzed with the Spearman’s rank correlation coefficient. Analyses were adjusted for gender and pack-years smoked. All testing hypotheses were performed at a 5% level of significance (two-tailed), using the statistical software IBM SPSS Statistics version 20.0 (IBM Corporation, Somers, NY, USA).
Results
Patients and procedures
Forty patients with COPD were included in the study. The main characteristics of the study participants are summarized in Table 2. The surgical procedures consisted of pneumectomies (17.6%), lobectomies (78.4%), and atypical resections (4%). Operations were performed in the right and left hemi-thorax in 54.9% and 45.1% of the patients, respectively. Squamous cell (epidermoid) carcinoma (42.2%) and adenocarcinomas (31.4%) were the most common histological types. The results of pathological examination revealed no malignancies in 7.8% of the study patients.
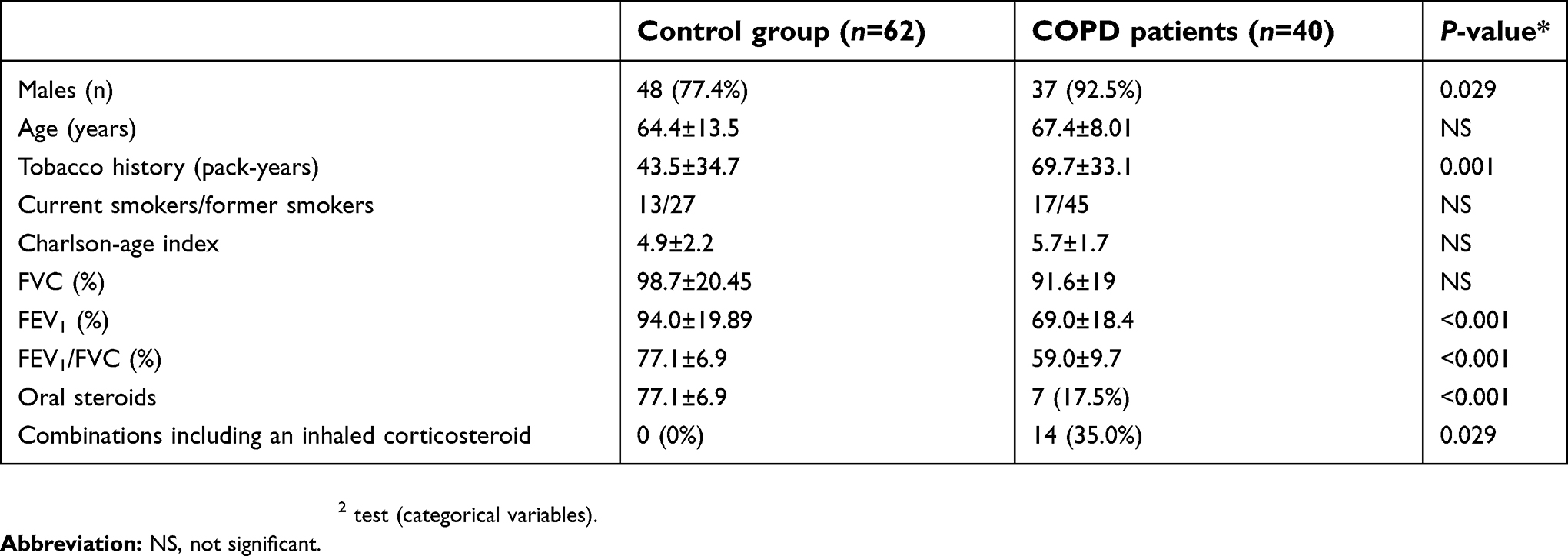 | Table 2 General characteristics of the study participants |
Eleven patients were in Stage I, 24 in Stage II and 5 in Stage III.
Expression of acute phase reactants in the lung parenchyma
CRP and SAA gene expression in the lung parenchyma was higher in COPD patients than in controls (Figure 1A); however, the expression did not differ significantly between the two groups (P>0.05). In the lung parenchyma, we found significant positive correlations between the expression of CRP with SAA1 and SAA4; SAA1 with SAA2 and SAA4; and SAA2 with SAA4 (Table 3). Significant differences in terms of SAA4 expression were observed between controls and GOLD III COPD (P=0.034), as well as between GOLD II and GOLD III COPD (P=0.044). SAA4 expression in the lung parenchyma increased in parallel with the severity of the disease (Figure 1B).
 | Table 3 Correlations between expressions of acute phase reactant |
 | Figure 1 Gene expression in COPD patients and control group. Gene expression of acute phase reactants in resistant smokers (controls) and COPD patients in lung parenchyma (panel A). SAA4 expression profile in the lung parenchyma according to COPD stage (Panel B). I, GOLD I COPD; II, GOLD II COPD; III, GOLD III COPD. No statistically significant differences were identified (P>0.05). Abbreviations: CNT, resistant smokers (controls); SAA, serum amyloid A. |
Expression of acute phase reactants in the pulmonary artery
CRP expression in the pulmonary artery was higher in COPD patients than in controls (Figure 2A). Moreover, SAA2 expression was 100% higher in COPD patients than in controls (P=0.028; Figure 2A). In the pulmonary artery, we found significant positive correlations between the expression of CRP and SAA1, 2 and 4, as well as between SAA1 and SAA2 (Table 3). Significant differences in terms of SAA2 expression were observed between resistant smokers and GOLD I COPD patients (P=0.010), as well as between GOLD I and GOLD II COPD (P=0.012; Figure 2B).
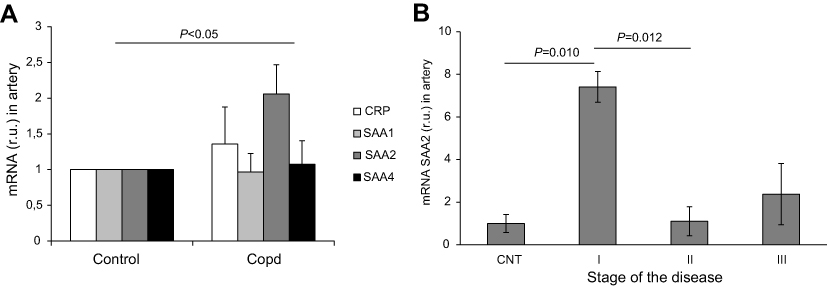 | Figure 2 Gene expression in COPD patients and control group. Gene expression of acute phase reactants in resistant smokers (controls) and COPD patients in pulmonary artery (panel A). SAA2 expression profile in the pulmonary artery according to COPD stage (B). I, GOLD I COPD; II, GOLD II COPD; III, GOLD III COPD. No statistically significant differences were identified (P>0.05). Abbreviations: CNT, resistant smokers (controls); SAA, serum amyloid A. |
Expression of acute phase reactants in peripheral blood leukocytes
Expression of CRP, SAA1, and SAA4 in peripheral blood leukocytes was found to differ significantly between COPD patients and controls. In line with the results obtained in the pulmonary artery, SAA2 expression was twofold higher in COPD patients than in controls (Figure 3A).
 | Figure 3 Gene expression of acute phase reactants in resistant smokers (controls) and COPD patients in blood cells (A). SAA1 (Panel B) and SAA4 (Panel C) expression in blood cells according to COPD stage. I, GOLD I COPD; II, GOLD II COPD; III, GOLD III COPD. No statistically significant differences were identified (P>0.05). Abbreviations: CNT, resistant smokers (controls); SAA, serum amyloid A. |
There was a significant positive correlation between the expression of CRP and SAA1 and SAA4; and SAA1 and SAA4 (Table 3). We found significant differences between GOLD I COPD and controls in terms of SAA1 (P=0.037) and SAA4 (P=0.018) expression (Figure 3B and C). In addition, significant differences in terms of SAA4 expression were observed between GOLD I and GOLD II COPD (P=0.030).
Comparison of acute phase reactants expression in different tissues
We then performed a comparison of APRs expression in the lung parenchyma, pulmonary artery, and peripheral blood leukocytes (Figure 4). The expression of SAA was significantly higher in the lung parenchyma than in the pulmonary artery (2-fold higher for SAA1 and SAA4, P=0.015 and P<0.001, respectively; eightfold higher for SAA2, P<0.001) and peripheral blood leukocytes (16-fold higher for SAA1, 439-fold higher for SAA2, and fivefold higher for SAA4; P<0.001). Compared with peripheral blood leukocytes, the expression of SAA was significantly higher in the pulmonary artery (eightfold higher for SAA1, P<0.001; 50-fold higher for SAA2, P<0.001; and 2.3-fold higher for SAA4, P=0.019). However, no differences in CRP expression were observed in the lung parenchyma, pulmonary artery, and peripheral blood leukocytes. All findings were maintained after adjusting for gender and pack-years smoked. We failed to identify significant differences in the APRs expression according to the use of treatment.
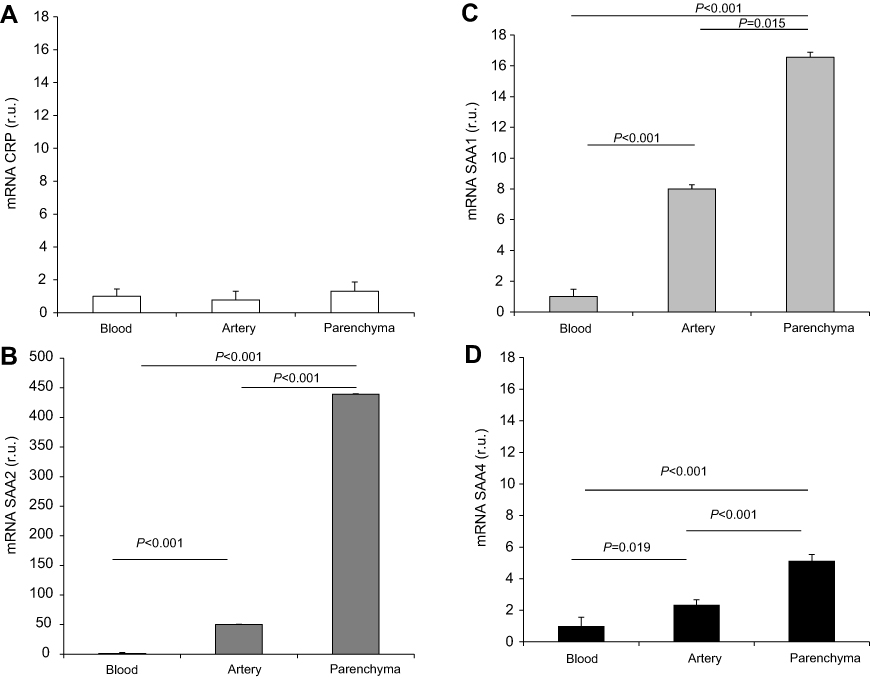 | Figure 4 Comparison of acute phase reactants gene expression levels in different tissues (blood cells, pulmonary artery, and lung parenchyma) of COPD patients. Panel (A): CRP expression; panel (B): SAA2 expression; panel (C): SAA1 expression and panel (D): SAA4 expression. Abbreviations: CRP, C-reactive protein; SAA, serum amyloid A. |
Correlations between acute phase reactants expression in different tissues
Significant positive correlations were identified in the different tissues (Table 4). These findings were maintained after adjusting for gender and pack-years smoked. We failed to identify significant differences in the APRs expression according to the use of treatment.
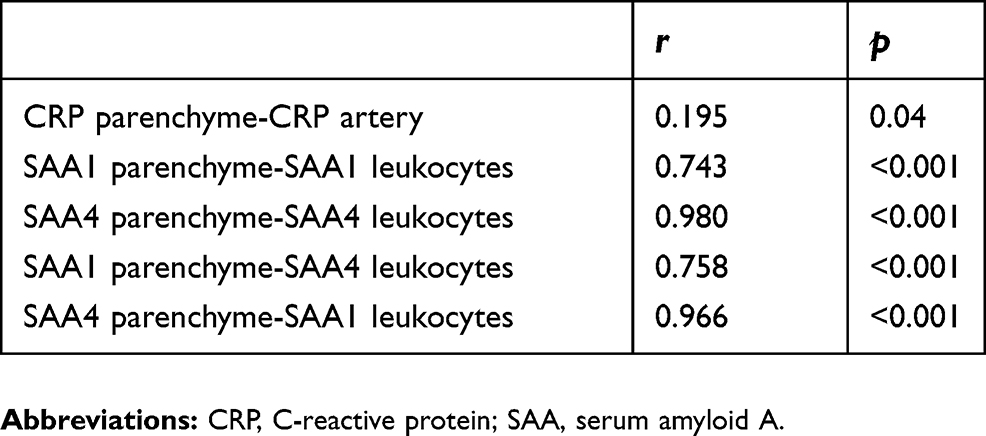 | Table 4 Correlations between different tissues |
Associations between acute phase reactants gene expression and their plasma concentrations
Plasma CRP showed a trend toward higher levels in COPD patients (4.4±1.73 mg/mL) than in control group (3.8±2.04 mg/mL, P>0.05). SAA concentrations in plasma were significantly higher in COPD patients (5.6±1.3 mg/mL) than in controls (4.6±21 mg/mL, P=0.031). A significant positive association between plasma CRP and SAA levels was evident (P=0.617, P<0.001). However, there was no significant correlation between plasma levels of APRs and their gene expression in the lung parenchyma, pulmonary artery, and peripheral blood leukocytes. SAA levels increased in parallel with the severity of COPD (Figure 5A) and CRP levels increased with the stage of the disease however these differences were not significant (Figure 5B).
 | Figure 5 Plasma concentrations of serum amyloid A (SAA) (panel A) and CRP (panel B) in CNTs and patients with different stages of COPD. Abbreviations: CRP, C-reactive protein; CNT, resistant smokers (controls). |
Immunohistochemistry
Immunohistochemistry revealed an increased expression of both CRP and SAA in the lung tissue of COPD patients as compared to controls. As shown in Figure 6, staining was chiefly captured by the small vessel wall, most markedly for SAA.
 | Figure 6 Immunostaining of C-reactive protein (CRP) and serum amyloid A (SAA) in lung biopsies of patients. Pulmonary parenchyma stained with CRP of a COPD (panel A) and control (panel B). Pulmonary parenchyma stained with SAA of COPD patient (panel C) and control patients (panel D). Scale bar represents 100 μm in each case. |
Discussion
In the current study, we performed a comparison of APRs expression in the lung parenchyma, pulmonary artery, and peripheral blood leukocytes of COPD patients. We also measured plasma CRP and SAA concentrations with the goal of establishing whether significant associations exist with their tissue gene expression. Our results indicate that the expression of ARPs is higher in the lung compared than in peripheral blood. Notably, we also observed that both SAA and CRP expression is more pronounced in the lung parenchyma than in the pulmonary artery. However, no association was found between the expression of inflammatory molecules in the lung and their circulating plasma levels.
Previous studies have consistently shown that COPD inflammation is not limited to the airways and lung,17 but it can also be found at the systemic level.18 Although increased circulating levels of CRP, IL-6, IL-8, and tumor necrosis factor-α have been reported in COPD patients,19,20 the exact sources of proinflammatory molecules in this clinical entity remain unclear. The most widely accepted model explaining systemic inflammation in COPD postulates that the excess production of local inflammatory mediators in the pulmonary compartment may spill over into the systemic circulation.6 CRP is a well-known biomarker of both airflow obstruction21 and systemic inflammation,22 but less data are available on SAA as an inflammatory biomarker in COPD. Although our current findings generally indicate a more marked inflammatory reaction at the local rather than the systemic level, it seems that the lack of correlation with serum levels do not support the spillover hypothesis.
The compartmentalization of CRP and SAA gene expression is a key factor that needs to be considered when analyzing inflammatory biomarkers in COPD. Although the disease is characterized by marked inflammation of the airways, lung parenchyma, and pulmonary vasculature,23 the anatomical involvement may show a significant interindividual variability that ultimately results in different disease phenotypes.24,25 Aortic endothelial cells may secrete CRP26 and SAA has been shown to be expressed in the endothelium.27 However, no studies to date have focused on the potential role of the pulmonary vascular bed as a source of systemic inflammation in COPD. Our results indicate that SAA expression in COPD patients is higher in the lung parenchyma than in the pulmonary artery. In a previous immunohistochemical investigation, we have shown that SAA and CRP in COPD are both expressed in the pulmonary vessel wall.10 Here, we expanded our previous observations by assessing their gene expression levels both in the lung and blood compartments. The higher expression levels of SAA in the lung parenchyma could be ascribed to small vessels (capillaries), which showed the most marked staining on immunohistochemistry. Interestingly, APRs were under-expressed in PBL from COPD as compared to controls. Although the differences are not significant, our interpretation is that probably the blood is a bad candidate as a potential contributor of systemic inflammation in COPD.
Some caveats of the current study merit comment. First, although the number of patients may be a limitation, the groups were balanced in the number of patients to make groups comparable in sample size. Second, quantification of mRNA does not provide information on the final production of a functional protein; also the methods used to analyze the gene expression and proteins are to relative quantification, not absolute. Third, numerous APRs (including CRP and SAA) can be synthesized in the liver, an organ which was not specifically investigated in the current report. Ji and coworkers28 have previously studied the inflammatory activity in different compartments (eg, saliva, sputum, bronchoalveolar lavage, and serum/blood) of COPD patients stratified according to their smoking status. The results were adjusted for gender and pack-years smoked because had significant differences in both groups, these not occur for BMI or age. The paper of BMI is especially relevant, since previous reports have informed about this potential association in different clinical conditions.29 In our cohort, however, we were not able to show this relationship. The authors identified several associations between different inflammatory markers both at the local and systemic level. Further studies of the inflammatory responses in various compartments (eg, liver, sputum, or saliva) will be necessary to identify other potential sources of systemic inflammation in COPD.
COPD is a heterogeneous disorder defined by the pulmonary function test. The pathogenesis would be different between subgroups such as the chronic bronchitis and pulmonary emphysema. Nevertheless, we had not differenced by phenotypes, specifically, we had not determinate the grade of emphysema, but all patients had centrilobular emphysema. Other limitation of our study is that the enrolled patients were all under surgery for suspected primary lung cancer and the number of patients in all groups is different, being the number of COPD patients in stage 3 small.
Conclusions
The inflammatory burden of COPD seems to be higher in the lung parenchyma than pulmonary arteries and peripheral blood leukocytes. Notably, no associations were noted between lung expression of APRs and their circulating plasma levels, making the leakage of inflammatory proteins from the lung to the bloodstream unlikely. Based on these results, other potential sources of systemic inflammation in COPD (eg, the liver) need further scrutiny.
Ethics approval and consent to participate
The study protocol was approved by the Institutional Review Board of the Virgen del Rocío Hospital, Seville, Spain (CEI code 2013/023). All patients provided written informed consent before inclusion in the study.
Abbreviations list
CRP, C-reactive protein; SAA, serum amyloid A; APRs, acute phase reactants; RT-qPCR, reverse transcription quantitative PCR; rRNA, 18S ribosomal RNA.
Acknowledgments
The authors thank the HUVR-IBiS Biobank (Andalusian Public Health System Biobank and ISCIII-Red de Biobanco PT13/0010/0056) for the assessment and technical support provided. This study was financially supported by grants from Fundación de Neumología y Cirugía Torácica del Sur (Neumosur) No. 4/2012 and from the Instituto de Salud Carlos III, Ministerio de Economía (PI12/01576).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Garcia-Rio F, Miravitlles M, Soriano JB, et al. Systemic inflammation in chronic obstructive pulmonary disease: a population-based study. Respir Res. 2010;11:63. doi:10.1186/1465-9921-11-63
2. Nussbaumer-Ochsner Y, Rabe KF. Systemic manifestations of COPD. Chest. 2011;139(1):165–173. doi:10.1378/chest.10-1252
3. Urieli-Shoval S, Cohen P, Eisenberg S, Matzner Y. Widespread expression of serum amyloid A in histologically normal human tissues. Predominant localization to the epithelium. J Histochem Cytochem. 1998;46(12):1377–1384. doi:10.1177/002215549804601206
4. Pinto-Plata VM, Mullerova H, Toso JF, et al. C-reactive protein in patients with COPD, control smokers and non-smokers. Thorax. 2006;61(1):23–28. doi:10.1136/thx.2005.042200
5. Sunyer J, Pistelli R, Plana E, et al. Systemic inflammation, genetic susceptibility and lung function. Eur Respir J. 2008;32(1):92–97. doi:10.1183/09031936.00052507
6. Sinden NJ, Stockley RA. Systemic inflammation and comorbidity in COPD: a result of ‘overspill‘ of inflammatory mediators from the lungs? Review of the evidence. Thorax. 2010;65(10):930–936. doi:10.1136/thx.2009.130260
7. Vernooy JH, Kucukaycan M, Jacobs JA, et al. Local and systemic inflammation in patients with chronic obstructive pulmonary disease: soluble tumor necrosis factor receptors are increased in sputum. Am J Respir Crit Care Med. 2002;166(9):1218–1224. doi:10.1164/rccm.2202023
8. Paivaniemi OE, Maasilta PK, Vainikka TL, Alho HS, Karhunen PJ, Salminen US. Local C-reactive protein expression in obliterative lesions and the bronchial wall in posttransplant obliterative bronchiolitis. Mediators Inflamm. 2009;2009:510254. doi:10.1155/2009/510254
9. Ramage L, Proudfoot L, Guy K. Expression of C-reactive protein in human lung epithelial cells and upregulation by cytokines and carbon particles. Inhal Toxicol. 2004;16(9):607–613. doi:10.1080/08958370490464599
10. Lopez-Campos JL, Calero C, Rojano B, et al. C-reactive protein and serum amyloid a overexpression in lung tissues of chronic obstructive pulmonary disease patients: a case-control study. Int J Med Sci. 2013;10(8):938–947. doi:10.7150/ijms.6152
11. Calero C, Arellano E, Lopez-Villalobos JL, Sanchez-Lopez V, Moreno-Mata N, Lopez-Campos JL. Differential expression of C-reactive protein and serum amyloid A in different cell types in the lung tissue of chronic obstructive pulmonary disease patients. BMC Pulm Med. 2014;14:95. doi:10.1186/1471-2466-14-95
12.
13. Charlson M, Szatrowski TP, Peterson J, Gold J. Validation of a combined comorbidity index. J Clin Epidemiol. 1994;47(11):1245–1251.
14. Leon-Atance P, Moreno-Mata N, Gonzalez-Aragoneses F, et al. Multicenter analysis of survival and prognostic factors in pathologic stage I non-small-cell lung cancer according to the new 2009 TNM classification. Arch Bronconeumol. 2011;47(9):441–446. doi:10.1016/j.arbres.2011.04.004
15. Malle E, De Beer FC. Human serum amyloid A (SAA) protein: a prominent acute-phase reactant for clinical practice. Eur J Clin Invest. 1996;26(6):427–435.
16. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108.
17. Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. doi:10.1056/NEJMoa032158
18. Gan WQ, Man SF, Senthilselvan A, Sin DD. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax. 2004;59(7):574–580. doi:10.1136/thx.2003.019588
19. Tanni SE, Pelegrino NR, Angeleli AY, Correa C, Godoy I. Smoking status and tumor necrosis factor-alpha mediated systemic inflammation in COPD patients. J Inflamm (Lond). 2010;7:29. doi:10.1186/1476-9255-7-59
20. Agusti A, Edwards LD, Rennard SI, et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One. 2012;7(5):e37483. doi:10.1371/journal.pone.0037483
21. de Torres JP, Cordoba-Lanus E, Lopez-Aguilar C, et al. C-reactive protein levels and clinically important predictive outcomes in stable COPD patients. Eur Respir J. 2006;27(5):902–907. doi:10.1183/09031936.06.00109605
22. Ghobadi H, Fouladi N, Beukaghazadeh K, Ansarin K. Association of high sensitive CRP level and COPD assessment test scores with clinically important predictive outcomes in stable COPD patients. Tanaffos. 2015;14(1):34–41.
23. Wei J, Xiong XF, Lin YH, Zheng BX, Cheng DY. Association between serum interleukin-6 concentrations and chronic obstructive pulmonary disease: a systematic review and meta-analysis. PeerJ. 2015;3:e1199. doi:10.7717/peerj.1199
24. Kim V, Criner GJ. The chronic bronchitis phenotype in chronic obstructive pulmonary disease: features and implications. Curr Opin Pulm Med. 2015;21(2):133–141. doi:10.1097/MCP.0000000000000145
25. Bafadhel M, Umar I, Gupta S, et al. The role of CT scanning in multidimensional phenotyping of COPD. Chest. 2011;140(3):634–642. doi:10.1378/chest.10-3007
26. Venugopal SK, Devaraj S, Jialal I. Macrophage conditioned medium induces the expression of C-reactive protein in human aortic endothelial cells: potential for paracrine/autocrine effects. Am J Pathol. 2005;166(4):1265–1271. doi:10.1016/S0002-9440(10)62345-0
27. Lakota K, Resnik N, Mrak-Poljsak K, Sodin-Semrl S, Veranic P. Colocalization of serum amyloid a with microtubules in human coronary artery endothelial cells. J Biomed Biotechnol. 2011;2011:528276. doi:10.1155/2011/528276
28. Ji J, von Scheele I, Bergstrom J, et al. Compartment differences of inflammatory activity in chronic obstructive pulmonary disease. Respir Res. 2014;15:104. doi:10.1186/s12931-014-0104-3
29. Baynard T, Hilgenkamp TIM, Schroeder EC, Motl RW, Fernhall B. Measures of adiposity differentially correlate with C-reactive protein among persons with multiple sclerosis. Mult Scler Relat Disord. 2018;25:1–4. doi:10.1016/j.msard.2018.07.010
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.