Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
Evaluation of a new tablet formulation of deferasirox to reduce chronic iron overload after long-term blood transfusions
Authors Chalmers A, Shammo J
Received 16 October 2015
Accepted for publication 4 January 2016
Published 15 February 2016 Volume 2016:12 Pages 201—208
DOI https://doi.org/10.2147/TCRM.S82449
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Anna W Chalmers, Jamile M Shammo
Department of Internal Medicine, Division of Hematology/Oncology, Rush University Medical Center, Chicago, IL, USA
Abstract: Transfusion-dependent anemia is a common feature in a wide array of hematological disorders, including thalassemia, sickle cell disease, aplastic anemia, myelofibrosis, and myelodysplastic syndromes. In the absence of a physiological mechanism to excrete excess iron, chronic transfusions ultimately cause iron overload. Without correction, iron overload can lead to end-organ damage, resulting in cardiac, hepatic, and endocrine dysfunction/failure. Iron chelating agents are utilized to reduce iron overload, as they form a complex with iron, leading to its clearance. Iron chelation has been proven to decrease organ dysfunction and improve survival in certain transfusion-dependent anemias, such as β-thalassemia. Several chelating agents have been approved by the United States Food and Drug Administration for the treatment of iron overload, including deferoxamine, deferiprone, and deferasirox. A variety of factors have to be considered when choosing an iron chelator, including dosing schedule, route of administration, tolerability, and side effect profile. Deferasirox is an orally administered iron chelator with proven efficacy and safety in multiple hematological disorders. There are two formulations of deferasirox, a tablet for suspension, and a new tablet form. This paper is intended to provide an overview of iron overload, with a focus on deferasirox, and its recently approved formulation Jadenu® for the reduction of transfusional iron overload in hematological disorders.
Keywords: iron chelation therapy, transfusional iron overload, deferasirox
Introduction to iron overload
Iron homeostasis is a complex system that balances both the absorption of intestinal iron and release of stored iron, with the body’s iron requirements. There are several molecules, such as hepcidin, ferritin, and ferroportin that provide tight regulation of this process, and contribute to iron homeostasis. Under normal circumstances, almost all absorbed iron is rapidly bound to transferrin (TF), an abundant, high-affinity iron-binding protein. Iron occupies approximately 30% of TF iron-binding sites. In heavily transfused patients, non-TF bound iron begins to accumulate. Labile plasma iron (LPI), a directly chelatable form of non-TF bound iron, is readily taken up by cells, leading to expansion of the cellular iron pool and generation of reactive oxygen species, resulting in cellular dysfunction and death.1,2
As there is no regulated mechanism for the excretion of excess iron, patients who require frequent blood transfusions inexorably develop chronic iron overload. Each unit of transfused red blood cells introduces 200–250 mg of elemental iron, with iron overload occurring after approximately ten to 20 transfusions.3 Transfusional iron overload occurs in patients with a wide array of hematological disorders including thalassemia, sickle cell disease, aplastic anemia, myelofibrosis, and myelodysplastic syndromes (MDS). In the United States, an estimated 25,000 patients are transfusion dependent due to sickle cell disease, MDS, refractory anemias, and thalassemia.4 While phlebotomy represents a reasonable chelation method in non-anemic patients with iron overload, it is not feasible in transfusion-dependent patients; in which case, iron chelators represent the treatment of choice for the management of iron overload. It should be noted that ineffective erythropoiesis, noted in patients with non-transfusion-dependent thalassemia (NTDT) syndromes, can also result in clinically relevant iron overload, despite lack of regular blood transfusions primarily due to increased gastrointestinal (GI) absorption.5
Toxicity from iron overload most commonly damages the liver, heart, and endocrine system and can adversely affect survival. For example, in thalassemia without iron chelation, death usually occurs from cardiac failure or arrhythmia at an early age. The median age of death before the widespread use of chelating agents in thalassemia was 17, with observational studies showing improvement in survival rates since the introduction of chelating agents.6,7 Review of several decades of registry data for β-thalassemia patients in the United Kingdom indicates that survival past the age of 24 is attributable to the use of chelating agents.7 For other transfusion-dependent anemias, such as sickle-cell anemia and MDS, long-term efficacy and survival data of chelation therapy is lacking.8
Standard treatment options for iron overload
Iron chelation therapy is intended to reduce iron overload by forming a complex with iron, thereby promoting its excretion. Before initiating treatment with a chelating agent, the degree of iron overload must be quantified. Serum ferritin and TF saturation have traditionally been utilized to diagnose iron overload, determine the need to initiate chelation therapy, and assess its efficacy.
Serum ferritin measurement represents an easy and widely available method to assess iron overload as well as a screening tool. However, there are limitations of utilizing ferritin as a sole measure as it can be elevated in the absence of iron overload. This can occur in autoimmune disorders, inflammatory states, malignancy, chronic renal insufficiency, metabolic syndromes, and hepatopathies.9 Additionally, ferritin does not always correlate with other, more accurate measures of iron burden, such as liver iron concentration (LIC).10
Beyond serum laboratory tests, LIC has been shown to accurately assess total body iron stores.11 LIC can be determined directly with a liver biopsy. However, noninvasive measurement with magnetic resonance imaging (MRI) has been shown to have a high sensitivity and specificity for measuring LIC.12 Therefore, MRI frequently supplants the use of liver biopsies.
Due to the risk of cardiac toxicity in iron overload, monitoring cardiac iron stores is important. Similar to liver monitoring, cardiac T2* MRI is validated as a true measure of cardiac iron.13 Cardiac T2* is a relaxation parameter, with <20 ms indicating iron overload and predicting a higher risk for the development of heart failure and arrhythmias.14 It is important to note that cardiac T2* and liver MRI do not always correlate.15 Beyond the initial determination of iron overload, liver and cardiac MRI should be used serially to determine the efficacy of chelation therapy, though there is no universally accepted interval for repeat monitoring.
The indication and timing of initiation of iron chelation therapy for transfusion-dependent anemia vary according to disease states. In some instances, triggers for initiation of iron chelation therapy in transfusion-dependent anemia patients are mostly derived from consensus guidelines as well as expert opinion (Table 1).
 | Table 1 Indications for chelation by diseasea |
There are three iron chelators currently available for use in the United States, deferoxamine, deferiprone, and deferasirox, each with a different route of administration, dosing regimen, and side effect profile (Table 2). In the United States deferasirox is the most frequently utilized iron chelator due to ease of oral administration.4
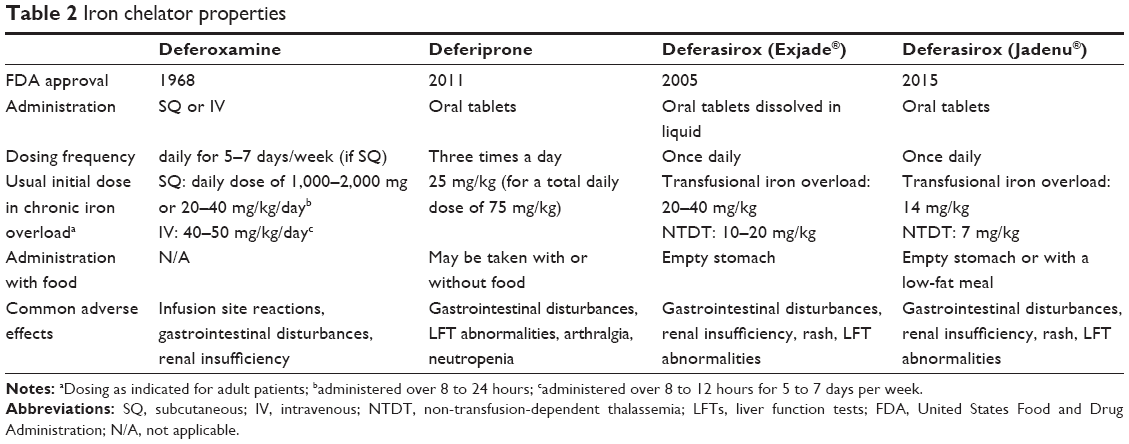 | Table 2 Iron chelator properties |
Deferoxamine became the first iron chelator to be approved by the United States Food and Drug Administration (FDA) in 1968 for use in iron overload, with subcutaneous, muscular or intravenous routes of administration. Due to low oral bioavailability and short half-life of less than 1 hour, it is not suitable for oral administration and is usually given over 8 to 12 hours a day, 5 to 7 days a week.16 Common adverse effects are discomfort at the injection site and GI disturbances. Visual and auditory toxicity have been reported, but can be minimized by avoiding higher doses and over-chelation.17 Increases in serum creatinine (possibly dose-related), acute renal failure and renal tubular disorders, associated with the administration of deferoxamine, have been reported in post-marketing surveillance.18 All patients should be monitored with periodic audiology and ophthalmology exams as well as kidney and liver function tests.
Though originally synthesized in 1984, deferiprone is an oral agent that was only approved by the FDA in 2011. This is in part due to initial concerns regarding its efficacy.19,20 Nonetheless, a review of 17 clinical trials indicates that deferiprone results in a significant reduction of iron stores in transfusion-dependent patients.21 The same review noted that adverse events are more common in deferiprone compared to deferoxamine.21 The most common side effects include elevation in hepatic enzymes, GI discomfort, and arthralgia. Agranulocytosis is an uncommon side effect, occurring in 1%–3% of patients, with the potential risk of sepsis.22,23 For most patients, agranulocytosis resolves after stopping the medication.24 All patients should be monitored with complete blood counts, liver function tests, and serum zinc levels.
Deferasirox is an oral iron chelator with a long half-life that allows for once-daily dosing. Two deferasirox formulations are available, the original tablet for oral suspension (Exjade®; Novartis International AG, Basel, Switzerland) and the newly approved film-coated tablet (Jadenu®). The accelerated approval of the new formulation in 2015 was based on results of trials conducted to evaluate the safety and efficacy of the original deferasirox formulation (Exjade®).
Exjade® (deferasirox) was initially approved by the FDA in 2005, for the treatment of chronic iron overload due to blood transfusions in patients 2 years of age and older. Patients must have evidence of iron overload defined as transfusion of ≥100 mL/kg of packed red blood cells and serum ferritin consistently >1,000 μg/L. The approval of deferasirox has recently been expanded to include the management of chronic iron overload, defined as an LIC of 5 mg Fe/g dry weight (dw), and serum ferritin >300 μg/L, in patients with NTDT syndromes in patients 10 years of age and older.
The following represents a summary of the clinical trials conducted in various patient populations to evaluate the safety and efficacy of deferasirox original formulation, Exjade®, in the management of patients with iron overload either due to transfusions or in NTDT.
The pivotal non-inferiority trial that led to the FDA approval of deferasirox was a Phase III randomized study comparing deferasirox to deferoxamine over 1 year, in patients 2 years and older with transfusion-dependent β-thalassemia, and evidence of iron overload as defined by an LIC of >2 mg Fe/g dw. LIC was determined by either liver biopsy, or a special non-invasive technique, termed superconducting quantum interference device.25 Five-hundred and eighty-six patients were randomized at 1:1 ratio to either deferasirox or deferoxamine, with most completing 1 year of therapy in the study. This trial did not meet its primary efficacy end-point of maintenance or reduction of LIC when compared to deferoxamine, which was attributed to a relative under-dosing of deferasirox. Nonetheless, non-inferiority was demonstrated in a subgroup of patients receiving higher doses of either agent (deferasirox 20 or 30 mg/kg and deferoxamine doses of >35 mg/kg) due to higher LIC at baseline. Pooled data from clinical trials indicated that the response to deferasirox was dose-dependent and efficacy was influenced by rate of transfusional iron intake.26–28 Future clinical trials used periodic dose adjustments to account for differences in transfusion requirements of patients, as it varies widely in patients with sickle cell disease and MDS.29,30
The EPIC study was the largest ever investigation conducted for an iron chelator, enrolling 1,744 patients to evaluate whether fixed starting doses of deferasirox, based on transfusional iron intake, with dose titration guided by serum ferritin trends and safety markers, provided clinically acceptable chelation in patients (aged ≥2 years) with transfusional hemosiderosis from various types of anemia. Though the largest subset of patients had thalassemia, the trial also included patients with MDS, sickle cell disease, aplastic anemia, and other rare anemias. The recommended initial dose was 20 mg/kg/day for patients receiving 2–4 packed red blood cell units/month and 10 or 30 mg/kg/day was recommended for patients receiving less or more frequent transfusions, respectively. Dose adjustments were based on 3-month serum ferritin trends and continuous assessment of safety markers, including changes in serum creatinine, urinary protein:urinary creatinine ratio, liver function tests, development of skin rash, auditory and ocular disturbances, cytopenias, and hypersensitivity reactions. This trial reached its primary end-point of overall reduction of serum ferritin after 52 weeks of therapy compared with baseline, (−264 ng/mL; P<0.0001). These data confirmed response to chelation therapy across various anemias, and feasibility of dose titration as guided by trends in serum ferritin and safety markers as mentioned. The most common (>5%) adverse events in that trial were GI disturbances (28%) and skin rash (10%).30
Within the EPIC trial, a smaller subset of patients (N: 114) with normal left ventricular ejection fraction and cardiac iron overload, defined as myocardial T2* of <20 ms and >5 ms, were prospectively followed while on deferasirox.31 Myocardial T2 was significantly improved (from 12 ms to 17.1 ms), corresponding to a decrease in cardiac iron concentration.32
The CORDELIA study was a prospective, randomized comparison of deferasirox (target dose 40 mg/kg per day) vs subcutaneous deferoxamine (50–60 mg/kg per day for 5–7 days/week) for the removal of myocardial iron in 197 β-thalassemia major patients who had myocardial siderosis (T2* 6–20 ms) without signs of cardiac dysfunction. The study demonstrated that deferasirox was non-inferior to deferoxamine in reducing cardiac iron as assessed by T2*.33
A reported subgroup analysis from the prospective, 1-year, multicenter ESCALATOR study, on the effect of deferasirox on LPI levels in patients with β-thalassemia, demonstrated that once-daily dosing with deferasirox ≥20 mg/kg/day resulted in sustained reduction in LPI levels in heavily iron-overloaded patients, suggesting 24-hour protection from LPI. It was hypothesized that deferasirox may therefore reduce unregulated tissue iron loading and prevent further end-organ damage.34 Overall, deferasirox has been extensively studied and found to improve hepatic pathology and decrease cardiac iron.32,33,35
The FDA approval of deferasirox was recently expanded to include patients with NTDT based on the results of the THALASSA trial. The study was a 1-year, randomized, double-blind, placebo-controlled study conducted to assess the efficacy and safety of deferasirox in iron-overloaded NTDT patients. The primary efficacy variable was the absolute change in LIC from baseline to week 52. A total of 166 patients were randomized in a 2:1:2:1 ratio to starting doses of 5 or 10 mg/kg/day of deferasirox or placebo. Dose adjustment recommendations were also provided based on continuous safety assessments. If serum ferritin was <100 μg/L, or LIC was <3 mg Fe/g dw at any visit, treatment was to be suspended until LIC increased to ≥5 mg Fe/g dw and serum ferritin to >300 μg/L. At 1 year, the LIC and serum ferritin decreased significantly compared to placebo (P<0.001). In patients receiving placebo, both LIC and serum ferritin increased from baseline. The most common drug-related adverse events were nausea (n=11; 6.6%), rash (n=8; 4.8%), and diarrhea (n=6; 3.6%).36
Pharmacology and pharmacokinetics of the new tablet formulation of deferasirox
Deferasirox is a tridentate iron chelator with high affinity and high selectivity for ferric iron. Deferasirox binds ferric iron in a 2:1 (deferasirox:iron) ratio. It is well absorbed from the GI tract. Once absorbed and bound to iron, it is eliminated predominantly by the hepatobiliary system and excreted via the fecal route, with a small percentage excreted in the urine.37,38
Deferasirox as a tablet for oral suspension (Exjade®) is taken once daily but must be mixed with water, orange juice, or apple juice until a fine suspension is obtained. Any residual medication has to be resuspended in a small volume of liquid and taken. This leads to a lengthy mixing process and the theoretical risk of patients not completely taking the intended dose. Additionally, one third of patients find deferasirox as a tablet for oral suspension unpalatable.39 The new tablet formulation (Jadenu®) was developed in an attempt to overcome these tolerability issues, and is the only once-daily oral iron chelator that can be swallowed whole. It was approved by the FDA on March 31, 2015.
Deferasirox tablets (Jadenu®) contain the same active ingredient as deferasirox tablets for oral suspension (Exjade®). In a single-dose healthy volunteer study37 (ICL670F2102) the new formulation was shown to have comparable PK to the dispersible tablet formulation. However, peak serum concentrations (Cmax) were approximately 30% higher. The new formulation is also 36% more bioavailable than the original formulation, Exjade®.37 Therefore, when converting a patient from deferasirox tablets for oral suspension (Exjade®) to deferasirox tablets (Jadenu®), the dosage should be decreased by 30%. For instance, a patient who is receiving Exjade at a dose of 30 mg/kg/day should be given Jadenu at 21 mg/kg/day (Table 3).
 | Table 3 Dosage conversion to the new formulation of deferasirox |
In addition, a food effect study37 (ICL670F2103) indicated that the new deferasirox formulation can be taken with a light meal.37 Since the absorption of Jadenu is increased when taken with a high fat meal, with an 18% increase in the area under the curve,37 patients should be informed of this property, and educated as to food choices when initiating therapy.
Treatment with deferasirox tablets (Jadenu®) should be initiated at 14 mg/kg/day and titrated up by 3.5–7 mg/kg/day.37 Dose adjustment should be made every 3 to 6 months based on iron stores assessments, such as serum ferritin or LIC.37 The maximum dosage is 28 mg/kg/day. It is available as 90, 180, and 360 mg tablets. It is recommend to hold chelation therapy in patients with transfusional iron overload of ferritin <500 μg/L. In patients with NTDT, chelation therapy should be held if LIC is <3 mg Fe/g dw or if ferritin is <300 μg/L.37
Efficacy, safety, and tolerability of the new formulation
To date, the new formulation of deferasirox has only been evaluated in pharmacokinetic studies in healthy volunteers. No clinical data are available yet in patients taking this formulation; however, a Phase 2 randomized, open-label, multicenter, two-arm study comparing the two formulations of deferasirox in patients with transfusion-dependent thalassemia or low risk MDS has met its accrual goal. The study’s primary end-point is safety, with several secondary end-points including comparative assessment of pharmacokinetics, GI side effects, palatability, and compliance. Until the results of this trial are available, the side effect profile of deferasirox tablets (Jadenu®) is considered similar to deferasirox tablets for oral suspension (Exjade®), since it contains the same active ingredient.
The most common side effect with deferasirox tablets for oral suspension is GI discomfort, with 10%–33% of patients experiencing abdominal pain, diarrhea, nausea, and/or vomiting.1,40 Though most patients are able to tolerate this side effect, 7% of patients cite GI side effects as a reason for stopping deferasirox.1 Treatment algorithms developed for the management of GI side effects recommend a combination of pre-prandial evening dosing, starting at a low dose and titrating up as tolerated, and loperamide use as needed for diarrhea.41 As deferasirox tablets (Jadenu®) lack lactose and sodium sulfate, both found in the original formulation and possibly implicated in GI side effects, it is hypothesized it will have improved tolerability.41 Confirmation of this hypothesis is pending the results of the prospective trial comparing the two formulations of deferasirox.
GI hemorrhage has also been reported in patients with advanced hematological malignancies and thrombocytopenia with the original formulation of deferasirox tablets for oral suspension. Though the exact risk is unknown with the new formulation, the same black box warning applies. Therefore, patients should be monitored for symptoms and signs of GI bleeding. Deferasirox is contraindicated in patients with platelet count <50 th/μL.
As deferasirox can cause renal toxicity and proteinuria, creatinine should be measured twice prior to the initiation of therapy and monthly thereafter (Table 4). Patients at an increased risk of complications for renal impairment, such as elderly patients or those taking other nephrotoxic medications, are at risk of acute renal failure with deferasirox and should be monitored with weekly serum creatinine.37 For patients with a creatinine clearance of 40–60 mL/min, the starting dose is reduced by 50%. Its use is not advised in patients with creatinine clearance <40 mL/min. If a patient develops an elevation in creatinine during treatment >33% off baseline that is sustained for 1 week, the dose should be decreased by 7 mg/kg/day for deferasirox tablets.37 All patients should have the medication held if creatinine clearance is <40 mL/min.
 | Table 4 Efficacy and toxicity monitoring for deferasirox tablets (Jadenu®) |
Liver function should be measured at baseline, then every 2 weeks, then at least monthly thereafter (Table 4). In general, the elderly are at higher risk of complications with all formulations of deferasirox and they should be monitored more frequently for toxicity.37
The dose adjustment in liver disease depends on the degree of liver dysfunction. While Child-Pugh score A requires no dose reduction, moderate (Child-Pugh B) hepatic impairment requires a 50% dose reduction.37 It should not be used in Child-Pugh C.
Similar to deferoxamine, neurosensory deafness and hypoacusis are reported. All patients should be monitored with periodic audiology and ophthalmology exams.
There are several drug interactions with deferasirox tablets (Jadenu®). Bile acid sequestrates and uridine 5′-diphospho-glucuronosyltransferase inducers, such as phenytoin or ritonavir, have the potential to lower the concentration of deferasirox. Aluminum-containing antacid preparations should not be administered with deferasirox tablets (Jadenu®). Though their concomitant use has not been formally studied, the rationale for avoiding this medication is the theoretical risk of aluminum binding to the chelator in the GI tract. Drugs metabolized by CYP3A4 may be made less effective by deferasirox. Additionally, due to the known risk of GI hemorrhage, medications that can cause ulcers or those with a hemorrhagic potential should be avoided.37
Patient focused perspectives
Due to the chronic nature of chelation therapy and the adverse consequences of iron overload, patient adherence to therapy is an important issue. Patients taking deferasirox tablets for oral suspension (Exjade®) reported superior satisfaction scores compared to those reported by patients taking deferoxamine.42 Patient satisfaction rates for deferasirox tablets for oral suspension (Exjade®) were reported as high as 90%.43 Nonetheless, palatability studies show 37% of patients disliked deferasirox tablets for oral suspension (Exjade®).39 Self-reported adherence to the medication was reported at 67%–85%.44 Patients prefer to take deferasirox with food.39 As the earlier discussed pharmacokinetic data allow the new formulation to be taken with a light, low-fat meal, it is hoped this will improve patient satisfaction, and thereby adherence. The previously mentioned Phase 2 clinical trial is prospectively comparing the two formulations of deferasirox and will compare efficacy, and further address the question of patient satisfaction, palatability, and adherence.
Conclusion
Once the need for iron chelators is established in patients with transfusional iron overload, the ideal agent should be determined by the practitioner and patient. Deferasirox is a frequent choice due to ease of once-daily oral administration, but adherence may be hampered by palatability of the tablet for oral suspension. The new formulation of deferasirox tablets has similar pharmacological parameters, but hopes to address the prior issue of palatability. An additional advantage is that it can be taken with food. Ongoing clinical trials will address whether this new formulation improves patient satisfaction and compliance, without trade-offs in adverse events or safety.
Disclosure
Dr Shammo would like to report that she has received research funding, consultancy fees and honoraria from Novartis. The authors report no other conflicts of interest in this work.
References
Gattermann N, Rachmilewitz EA. Iron overload in MDS-pathophysiology, diagnosis, and complications. Ann Hematol. 2011;90(1):1–10. | ||
Cabantchik ZI, Breuer W, Zanninelli G, Cianciulli P. LPI-labile plasma iron in iron overload. Best Pract Res Clin Haematol. 2005;18(2):277–287. | ||
Andrews N. Disorders of iron metabolism. N Engl J Med. 1999;341(26):1986–1995. | ||
Brittenham GM. Iron-chelating therapy for transfusional iron overload. N Engl J Med. 2011;364(2):146–156. | ||
Musallam KM, Cappellini MD, Wood JC, et al. Elevated liver iron concentration is a marker of increased morbidity in patients with β thalassemia intermedia. Haematologica. 2011;96(11):1605–1612. | ||
Borgna-Pignatti C, Rugolotto S, De Stefano P, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. 2004;89(10):1187–1193. | ||
Modell B, Khan M, Darlison M. Survival in beta-thalassaemia major in the UK: data from the UK Thalassaemia Register. Lancet. 2000;10;355(9220):2051–2052. | ||
Lucania G, Vitrano A, Filosa A, Maggio A. Chelation treatment in sickle-cell-anaemia: much ado about nothing? Br J Haematol. 2011;154(5):545–555. | ||
Knovich MA, Storey JA, Coffman LG, Torti SV, Torti FM. Ferritin for the clinician. Blood Rev. 2009;23(3):95–104. | ||
Harmatz P, Butensky E, Quirolo K, et al. Severity of iron overload in patients with sickle cell disease receiving chronic red blood cell transfusion therapy. Blood. 2000;96(1):76–79. | ||
Angelucci E, Brittenham GM, McLaren CE, et al. Hepatic iron concentration and total body iron stores in thalassemia major. N Engl J Med. 2000;343(5):327–331. | ||
Voskaridou E, Douskou M, Terpos E, et al. Magnetic resonance imaging in the evaluation of iron overload in patients with beta thalassaemia and sickle cell disease. Br J Haematol. 2004;126(5):736–742. | ||
Carpenter JP, He T, Kirk P, et al. On T2* magnetic resonance and cardiac iron. Circulation. 2011;123(14):1519–1528. | ||
Kirk P, Roughton M, Porter JB, et al. Cardiac T2* magnetic resonance for prediction of cardiac complications in thalassemia major. Circulation. 2009;120(20):1961–1968. | ||
Anderson LJ, Westwood MA, Holden S, et al. Myocardial iron clearance during reversal of siderotic cardiomyopathy with intravenous desferrioxamine: a prospective study using T2* cardiovascular magnetic resonance. Br J Haematol. 2004;127(3):348–355. | ||
Oliveri N, Brittenham GM. Iron-chelating therapy and the treatment of thalassemia. Blood. 1997;89(3):739–761. | ||
Porter JB, Jaswon MS, Huehns ER, East CA, Hazell JW. Desferrioxamine ototoxicity: evaluation of risk factors in thalassaemic patients and guidelines for safe dosage. Br J Haematol. 1989;73(3):403–409. | ||
Bhandari S, Galanello R. Renal aspects of thalassaemia a changing paradigm. Eur J Haematol. 2012;89(3):187–197. | ||
Olivieri NF, Brittenham GM, McLaren CE, et al. Long-term safety and effectiveness of iron-chelation therapy with deferiprone for thalassemia major. N Engl J Med. 1998;339(7):417–423. | ||
Hoffbrand AV, AL-Refaie F, Davis B, et al. Long-term trial of deferiprone in 51 transfusion-dependent iron overloaded patients. Blood. 1998;91(1):295–300. | ||
Fisher SA, Brunskill SJ, Doree C, et al. Oral deferiprone for iron chelation in people with thalassaemia. Cochrane Database Syst Rev. 2013;8:CD004839. | ||
Piga A, Roggero S, Salussolia I, Massano D, Serra M, Longo F. Deferiprone. Ann N Y Acad Sci. 2010;1202:75–78. | ||
Henter JI, Karlén J. Fatal agranulocytosis after deferiprone therapy in a child with Diamond-Blackfan anemia. Blood. 2007;109(12):5157–5159. | ||
Cohen AR, Galanello R, Piga A, Dipalma A, Vullo C, Tricta F. Safety profile of the oral iron chelator deferiprone: a multicentre study. Br J Haematol. 2000;108(2):305–312. | ||
Cappellini MD, Cohen A, Piga A, et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood. 2006;107(9):3455–3462. | ||
Cappellini MD. Exjade® (deferasirox, ICL670) in the treatment of chronic iron overload associated with blood transfusion. Ther Clin Risk Manag. 2007;3(2):291–299. | ||
Cohen AR, Glimm E, Porter JB. Effect of transfusional iron intake on response to chelation therapy in beta-thalassemia major. Blood. 2008;111(2):583–587. | ||
Meerpohl JJ, Antes G, Rücker G, et al. Deferasirox for managing iron overload in people with thalassaemia. Cochrane Database Syst Rev. 2012;2:CD007476. | ||
Vichinsky E, Onyekwere O, Porter J, et al. A randomised comparison of deferasirox versus deferoxamine for the treatment of transfusional iron overload in sickle cell disease. Br J Haematol. 2007;136(3):501–508. | ||
Cappellini MD, Porter J, El-Beshlawy A, et al. Tailoring iron chelation by iron intake and serum ferritin: the prospective EPIC study of deferasirox in 1,744 patients with transfusion-dependent anemias. Haematologica. 2010;95(4):557–566. | ||
Pennell DJ, Porter JB, Cappellini MD, et al. Efficacy of deferasirox in reducing and preventing cardiac iron overload in beta-thalassemia. Blood. 2010;115(12):2364–2371. | ||
Pennell DJ, Porter JB, Cappellini MD, et al. Deferasirox for up to 3 years leads to continued improvement of myocardial T2* in patients with β-thalassemia major. Haematologica. 2012;97(6):842–848. | ||
Pennell DJ, Porter JB, Piga A, et al. A 1-year randomized controlled trial of deferasirox vs deferoxamine for myocardial iron removal in β-thalassemia major (CORDELIA). Blood. 2014;123(10):1447–1454. | ||
Daar S, Pathare A, Nick H, et al. Reduction in labile plasma iron during treatment with deferasirox, a once-daily oral iron chelator, in heavily iron-overloaded patients with beta-thalassaemia. Eur J Haematol. 2009;82(6):454–457. | ||
Deugnier Y, Turlin B, Ropert M, et al. Improvement in liver pathology of patients with β-thalassemia treated with deferasirox for at least 3 years. Gastroenterology. 2011;141(4):1202–1211. | ||
Taher AT, Porter J, Viprakasit V, et al. Deferasirox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood. 2012;120(5):970–977. | ||
Novartis Pharmaceuticals Corporation. Highlights of prescribing information: JADENU®. New Jersey, US: Novartis; 2015. Available from: http://www.pharma.us.novartis.com/product/pi/pdf/jadenu.pdf. Accessed January 7, 2016. | ||
Waldmeier F, Bruin GJ, Glaenzel U, et al. Pharmacokinetics, metabolism, and disposition of deferasirox in beta-thalassemic patients with transfusion-dependent iron overload who are at pharmacokinetic steady state. Drug Metab Dispos. 2010;38(5):808–816. | ||
Goldberg SL, Giardina PJ, Chirnomas D, Esposito J, Paley C, Vichinsky E. The palatability and tolerability of deferasirox taken with different beverages or foods. Pediatr Blood Cancer. 2013;60(9):1507–1512. | ||
Angelucci E, Santini V, Di Tucci AA, et al. Deferasirox for transfusion-dependent patients with myelodysplastic syndromes: safety, efficacy, and beyond (GIMEMA MDS0306 Trial). Eur J Haematol. 2014;92(6):527–536. | ||
Nolte F, Angelucci E, Breccia M, et al. Updated recommendations on the management of gastrointestinal disturbances during iron chelation therapy with Deferasirox in transfusion dependent patients with myelodysplastic syndrome – Emphasis on optimized dosing schedules and new formulations. Leuk Res. 2015;39(10):1028–1033. | ||
Vichinsky E, Pakbaz Z, Onyekwere O, et al. Patient-reported outcomes of deferasirox (Exjade, ICL670) versus deferoxamine in sickle cell disease patients with transfusional hemosiderosis. Substudy of a randomized open-label phase II trial. Acta Haematol. 2008;119(3):133–141. | ||
Taher A, Al Jefri A, Elalfy MS, et al. Improved treatment satisfaction and convenience with deferasirox in iron-overloaded patients with beta-Thalassemia: Results from the ESCALATOR Trial. Acta Haematol. 2010;123(4):220–225. | ||
Porter J, Bowden DK, Economou M, et al. Health-Related Quality of Life, Treatment Satisfaction, Adherence and Persistence in β-Thalassemia and Myelodysplastic Syndrome Patients with Iron Overload Receiving Deferasirox: Results from the EPIC Clinical Trial. Anemia. 2012;2012:297641. | ||
Bennett JM; MDS Foundation’s Working Group on Transfusional Iron Overload. Consensus statement on iron overload in myelodysplastic syndromes. Am J Hematol. 2008;83(11):858–861. | ||
Gattermann N. Overview of guidelines on iron chelation therapy in patients with myelodysplastic syndromes and transfusional iron overload. Int J Hematol. 2008;88(1):24–29. | ||
Porter J, Garbowski M. Consequences and management of iron overload in sickle cell disease. Hematology Am Soc Hematol Educ Program. 2013;2013:447–456. | ||
Cappellini MD, Cohen A, Eleftheriou A, Piga A, Porter J, Taher A. Guidelines for the Clinical Management of Thalassemia, 2nd Revised edition. Cyprus: Thalassaemia International Federation; 2008. | ||
Vlachos A, Muir E. How I treat Diamond-Blackfan anemia. Blood. 2010;116(19):3715–3723. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.