Back to Journals » Therapeutics and Clinical Risk Management » Volume 16
Evaluating the Safety, Efficacy, and Therapeutic Potential of Momelotinib in the Treatment of Intermediate/High-Risk Myelofibrosis: Evidence to Date
Authors Bassiony S, Harrison CN, McLornan DP
Received 22 May 2020
Accepted for publication 14 August 2020
Published 25 September 2020 Volume 2020:16 Pages 889—901
DOI https://doi.org/10.2147/TCRM.S258704
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Sarah Bassiony, Claire N Harrison, Donal P McLornan
Department of Haematology, Guy’s and St Thomas’ NHS Foundation Trust, Great Maze Pond, London, SE1 9RT, UK
Correspondence: Donal P McLornan
Department of Haematology, Guy’s and St Thomas’ NHS Foundation Trust, Guy’s Tower, Great Maze Pond, London, SE1 9RT, UK
Tel +44 20 7188 2742
Email [email protected]
Abstract: Myelofibrosis is a heterogeneous disorder with regard to both molecular pathogenesis and clinical phenotype, ranging from an initial fairly indolent condition in some through to an aggressive and debilitating scenario with profound constitutional symptoms, cytopenia frequently requiring transfusional support, and massive splenomegaly. Many advances have been made within the therapeutic arena, and an increasing array of novel agents are now available for disease management. Within this review, we focus on the current and predicted role of the JAK inhibitor momelotinib (Sierra Oncology) in myelofibrosis, with an emphasis on clinical trial evaluation, drug efficacyand safety, and discuss the suggested place in the therapeutic paradigm of myelofibrosis in 2020 and beyond.
Keywords: myelofibrosis, JAK inhibitors, momelotinib
Introduction
Myelofibrosis (MF) is a Philadelphia chromosome–negative myeloproliferative neoplasm (MPN) with an estimated combined annual incidence of 0.5 per 100,000 population.1 Clinicopathological features can be heterogeneous, ranging from the presence of constitutional symptoms (e.g. night sweats, weight loss and loss of appetite), variable degrees of splenomegaly and cytopenia, with significant anemia being a well-documented adverse prognostic feature, and an inherent risk of development of acute leukemia over the disease course.2–4 Many advances in understanding disease pathogenesis followed the description of the somatic JAK2V617F mutation in MPN by four groups in 2005 and the subsequent delineation of the role of CALR mutations in MF and other MPNs in 2012.5–9 Multiple therapeutic agent targeting, either directly or indirectly, disease-associated deregulated JAK–STAT signaling have entered the clinical arena.2 At the time of writing, two JAK inhibitors — ruxolitinib (Rux; Incyte Pharmaceuticals, Wilmington, DE) and fedratinib (Inrebic; Celgene) — have US Food and Drug Administration (FDA) approval for use in MF (either first or second line), whereas in the UK Rux remains the only licensed agent to date.10 Over the last decade, following the two large pivotal phase III trials COMFORT-1 and −2, Rux has certainly revolutionized the treatment of MF for many patients globally, yet there remains a growing need for effective treatments for patients who are intolerant to this agent or display resistant/refractory disease.11–13 In particular, one of the biggest challenges in managing patients with MF is adequately addressing the significant burden associated with anemia.3 Evidence suggests that anemia may be present in 40% of patients at the time of diagnosis and that up to one in four may be red cell transfusion–dependent, highlighting a significant unmet need.14 In this article, we provide a comprehensive review of currently available preclinical and clinical trial data on momelotinib (Mmb; Sierra Oncology) in MF, with emphasis on drug efficacy and safety and suggested place in the therapeutic paradigm.
Clinical Features of Myelofibrosis and Prognostic Scores for Therapeutic Stratification
As stated, MF is a heterogeneous disease and can present either de novo, termed primary MF, or following antecedent essential thrombocythemia (ET) or polycythemia vera (PV), termed post-ET MF (PET-MF) and post-PV MF (PPV-MF), respectively. The clinical phenotype can vary markedly, with some patients displaying no symptoms through to those with debilitating constitutional symptoms, problematic splenomegaly, cytopenia and risks of both thrombosis and hemorrhage. Therapeutic decisions are frequently made upon considering both clinical features and assigned prognostic group. Commonly used prognostic scores within the clinical setting include the International Prognostic Scoring System (IPSS) for PMF, applied at the time of diagnosis and based upon five factors influencing survival: age >65 years, presence of anemia with hemoglobin <100 g/L, white blood–cell count >25×109/L, presence of constitutional symptoms, and peripheral blood blasts ≥1%.15 This permits delineation of four distinct groups: low risk (0 risk factors, median survival 135 months), intermediate risk 1 (one risk factor, median survival 95 months), intermediate risk 2 (two risk factors, median survival 48 months), and high risk (three or more risk factors, median survival of 27 months). Subsequent refinements have occurred with the Dynamic IPSS (DIPSS; allows prognostication at any time in the disease course of PMF and gives more adverse prognostic weighting to the presence of anemia) and DIPSS-plus prognostic scoring, which includes three additional adverse prognostic factors: presence of thrombocytopenia (platelets <100×109/L), red cell–transfusion dependence and adverse karyotype.16,17 Forward-thinking scores have also been developed based on the presence of conventional risk factors and extended molecular screening and/or karyotypic abnormalities, such as the mutation-enhanced IPSS (MIPSS70) version 2.0 and the genetically inspired prognostic score (GIPSS).18,19 With regard to PET-MF and PPV-MF, the myelofibrosis secondary to PV and ET-prognostic model (MYSEC-PM) permits classification into four groups with markedly different survival estimates.20 Summaries of these scores are highlighted in Table 1. Lastly, individualized scores have also been developed based upon clinical factors and comprehensive genomic characterization that can more accurately predict personalized outcomes.21
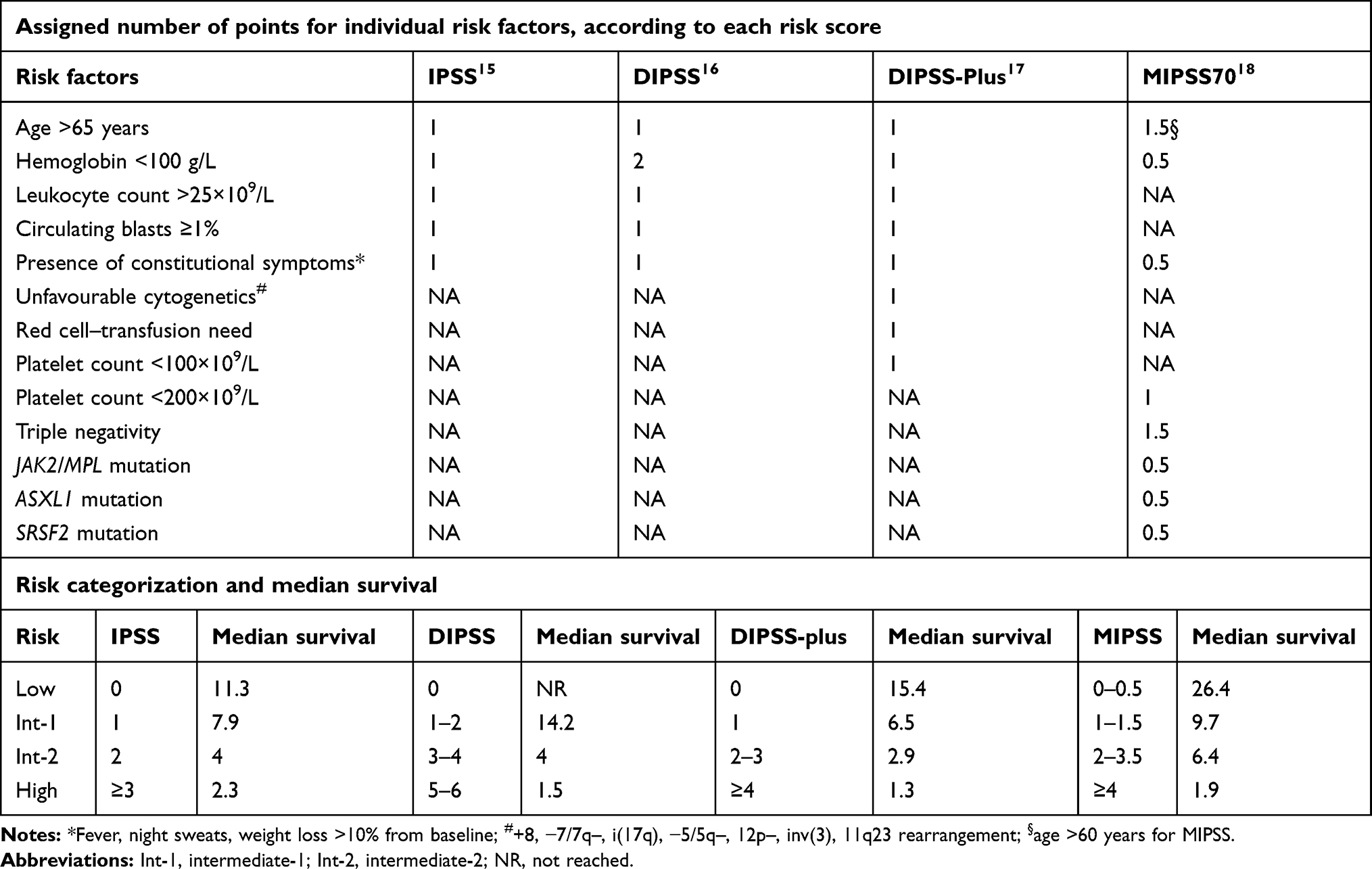 |
Table 1 Summary of prognostic scores for therapeutic stratification in MF |
Anemia in Myelofibrosis: Extent of the Problem
Evidence suggests that approximately 40% of MF patients are anemic at diagnosis and almost 25% red cell transfusion–dependent.14 As the disease progresses, anemia can become more prominent, either through a direct consequence of the progression or indeed the therapies used. The etiology of the anemia is frequently multifactorial, resulting from impaired erythropoiesis because of a hostile and fibrotic marrow microenvironment, increased red blood cell (RBC) turnover, and sequestration due to splenomegaly and a proinflammatory milieu that disrupts normal erythropoiesis.3 Moreover, in MF there is upregulated expression of hepcidin, which abrogates normal iron metabolism and utilization.22 Recent studies have additionally suggested an association between the presence of U2AF-spliceosome mutations and MF-related anemia.4 Management strategies for anemia are varied and dependent on disease stage, degree of anemia, patient age, performance status, baseline endogenous erythropoietin levels, and available therapies. Frequently, those with significant anemia require RBC transfusions on a regular basis, with the inherent risk of alloimmunization, transfusion-related complications, and iron overload. Transfusion dependence has been shown to be an independent adverse prognostic factor, as already discussed.17 Erythropoiesis-stimulating agents have a role in some patients, and appear more effective in those with MF-associated anemia who are RBC transfusion–independent and have low serum erythropoietin (<125 U/L) levels, albeit responses are often heterogeneous and unpredictable.3,23,24
Erythropoiesis-stimulating agents have also been combined with Rux, both in the COMFORT-2 trial and in a non-trial setting.25 Immunomodulatory agents, such as lenalidomide and thalidomide, have been tried with limited success, additionally compromised by toxicities or myelosuppression, and low-dose pomalidomide, albeit initially of great interest in this area, failed to show significant benefit over placebo in a large phase III study in MF with RBC-transfusion dependence.26–28 Danazol is an androgenic steroid effective in approximately 30% of MF patients with anemia. Response rates, however, are slow (median time to response is on average 5 months), less common in the presence of RBC dependency, and appropriate monitoring for potential toxicity is mandated.29 Novel agents in clinical trials, such as the oral BET inhibitor CPI-0610 (Constellation Pharmaceuticals), alone or in combination with Rux, has demonstrated hemoglobin responses alongside spleen/symptom responses, and the erythroid maturation agent luspatercept (Celgene) has demonstrated clinically significant anemia responses in a proportion of MF patients in an ongoing phase II study. Final results from these trials are awaited with interest.30–32 As will be discussed, Mmb has a specific role in the field of MF management in that it can address the three main hallmarks of the disease: splenomegaly, symptom burden, and potentially MF-associated anemia.
Pre- and Early Clinical Characterization of Momelotinib
Mmb, previously known as CYT387 (N-(cyanomethyl)-4-[2-[[4-(4-morpholinyl)phenyl]amino]-4-pyrimidinyl]-benzamide), is an aminopyrimidine-derivative ATP-competitive small-molecule inhibitor of JAK1 and JAK2, withIC50 values of 11 and 18 nM, respectively. The IC50 for JAK3 is approximately nine times that. Early preclinical characterization more than a decade ago showed that Mmb inhibited in vitro growth of Ba/F3-JAK2V617F, human erythroleukemia (HEL) cells and Ba/F3-MPLW515L cells and induced dose-dependent decreases in both STAT3 and STAT5 phosphorylation.33 Moreover, Mmb effectively inhibited growth of erythroid colonies containing the JAK2V617F mutation in vitro derived from patients with PV. Tyner et al demonstrated efficacy in a murine model of MPN (irradiated BALB/c mice were transplanted with marrow transduced with a JAK2V617F retrovirus) showing normalization of white blood–cell counts, reductions in splenomegaly, and extramedullary hematopoiesis coupled with modulation of proinflammatory cytokines.34 Focusing on erythroid responses, Asshoff et al investigated the erythroid-specific effects of Mmb in a group A streptococcal peptidoglycan-polysaccharide fragment –induced rat model of chronic anemia and found both an increase in RBC numbers and normalization of hemoglobin values.35 Mmb mediates these effects through inhibition of the BMPR kinase ACVR1, a member of the TGFβ superfamily of receptors, with resultant decreased hepcidin production from the liver and increased iron availability, and hence stimulates erythropoiesis. Two phase I open-label adaptive studies investigated the pharmacokinetics of a single dose of Mmb 200 mg administered orally in those with impaired hepatic or renal function compared to matched healthy controls.36 There was no clinically significant difference in plasma exposure of Mmb and its main active metabolite between healthy controls and those with either moderate hepatic impairment or moderate/severe renal impairment. However, for severe hepatic impairment, Mmb plasma exposure was increased compared to healthy control subjects, and consideration of dose/avoidance would be necessitated.
Clinical Trial Data of Momelotinib in Myelofibrosis: What Do We Know and Where Are We Going?
The first trial investigating the utility of Mmb in MF was an open label nonrandomized phase I/II study performed in two parts: part 1 was a dose-escalation study (n=60 patients) to confirm the therapeutic dose, pharmacokinetic behavior and efficacy of Mmb with a second phase enrolling a further 106 patients across multiple centers in the dose confirmation phase of the trial.37 All patients had intermediate 2 or high-risk MF, with some intermediate1–risk patients included with symptomatic splenomegaly or those who had failed other available therapies. A total of 52 (87%) patients completed phase I of the trial, receiving at least nine cycles of Mmb. The median age was 65 (range 34–85) years and median hemoglobin level 94 (range 71–144) g/L. In the initial report, only six patients had discontinued the trial due to adverse events (AEs) within this period, with only one felt to be related to the trial medication and two patients for lack of response. Five patients died during the trial, none of the causes being related to the trial medication. Within the early stages of the dose escalation, it was found that both the 150 mg (n=18) and 300 mg (n=21) doses were “biologically effective” and hence brought forward for the remaining patients enrolled to the study. A total of 52 patients were assessable for spleen response: 25 patients (48%) achieved a spleen response as per International Working Group–Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) criteria, with no differences between the two dose groups (300 mg versus 150 mg daily). There were demonstrable improvements in anemia in those evaluable (n=41), with many transfusion-dependent patients becoming transfusion-independent on IWG-MRT criteria. Median duration of transfusion independence at data cutoff was 9.6 months, with ongoing responses. Of note, there was no significant difference in transfusion responses between the 150 mg and 300 mg doses. Dose-linear Cmax and area under the curve were observed for both the 150- and 300 mg/day doses. Dizziness (mostly grade 1) was seen in approximately a quarter of patients within the first hour of initiating Mmb, on some occasions associated with postural hypotension, but symptoms commonly resolved spontaneously within a few hours. Subsequent management strategies included “holding” antihypertensive therapies the day prior to commencing dosing and encouraging good oral hydration. No correlation existed between this symptom and dose administered. Gastrointestinal AEs were mild, frequently ranging from grade 1 −2, and often self-limiting (incidence of nausea 18%, diarrhea 13%), requiring no dose reductions. In terms of hematologic AEs, thrombocytopenia was the most commonly reported, with 32% grade 3/4, particularly for those with platelet count <100×109/L, while treatment-related anemia and neutropenia were not common, affecting two and three patients, respectively, at grade 3/4 severity. Non hematologic grade 3 or 4 AEs included increased aspartate aminotransferase (3%) and alanine aminotransferase (3%) levels, headache (3%), and hyperlipasemia (5%). Of particular note, 16 patients (27%) reported neurological symptoms, with 13 having new-onset peripheral neuropathy and three worsening of pre-existing neuropathy. Median time to development of reportable peripheral neuropathy was 141 days. Interestingly, half the affected patients had complete resolution of neuropathy despite continuing Mmb therapy, although 38% with neuropathy required dose reduction or treatment discontinuation. Subsequent updated analyses looked at patients both in the core study and enrolled in the extension phase with a data cutoff in February 2015.38 Overall, 124 (75%) patients completed the core study and 120 enrolled in the extension study. Median age was 67 years and approximately two-thirds had PMF. During the course of the core/extension phase, of 111 anemia response–evaluable patients, 75% of transfusion-dependent patients achieved transfusion independence and 28.2% with hemoglobin <100 g/L achieved a hemoglobin response, indicating the beneficial response for those with MF-associated anemia.
Tefferi et al subsequently reported long term (7-year) follow-up of 100 patients (64 with primary MF, 22 PPV-MF, and 14 PET-MF; median age 66 years)) who had been treated with Mmb during 2009–2010 in the aforementioned phase I/II study.39 Distribution of the cohort across DIPSS-plus risk stratification was 63% high risk, 36% intermediate 2, and 1% intermediate 1 risk. As of July 2017, treatment had been discontinued in 91% of patients after a median treatment duration of 1.4 (range 0.02–6.2) years. Clinical improvements occurred in 57% of those treated, with 44% gaining an anemia response and a similar proportion a spleen response, which were more likely to occur in the absence of ASXL1 mutations and with circulating blasts <2%. Most frequent reasons for Mmb discontinuation were suboptimal response and progressive disease. Seven patients discontinued due to peripheral neuropathy, and overall grade 1/2 peripheral neuropathy was reported in 47% of the cohort. Of note, a comparative analysis of the Mmb-treated cohort with a similar risk-matched MF cohort did not reveal significant differences in overall survival. A summary of clinical trial outcomes and safety issues is given in Tables 2 and 3.
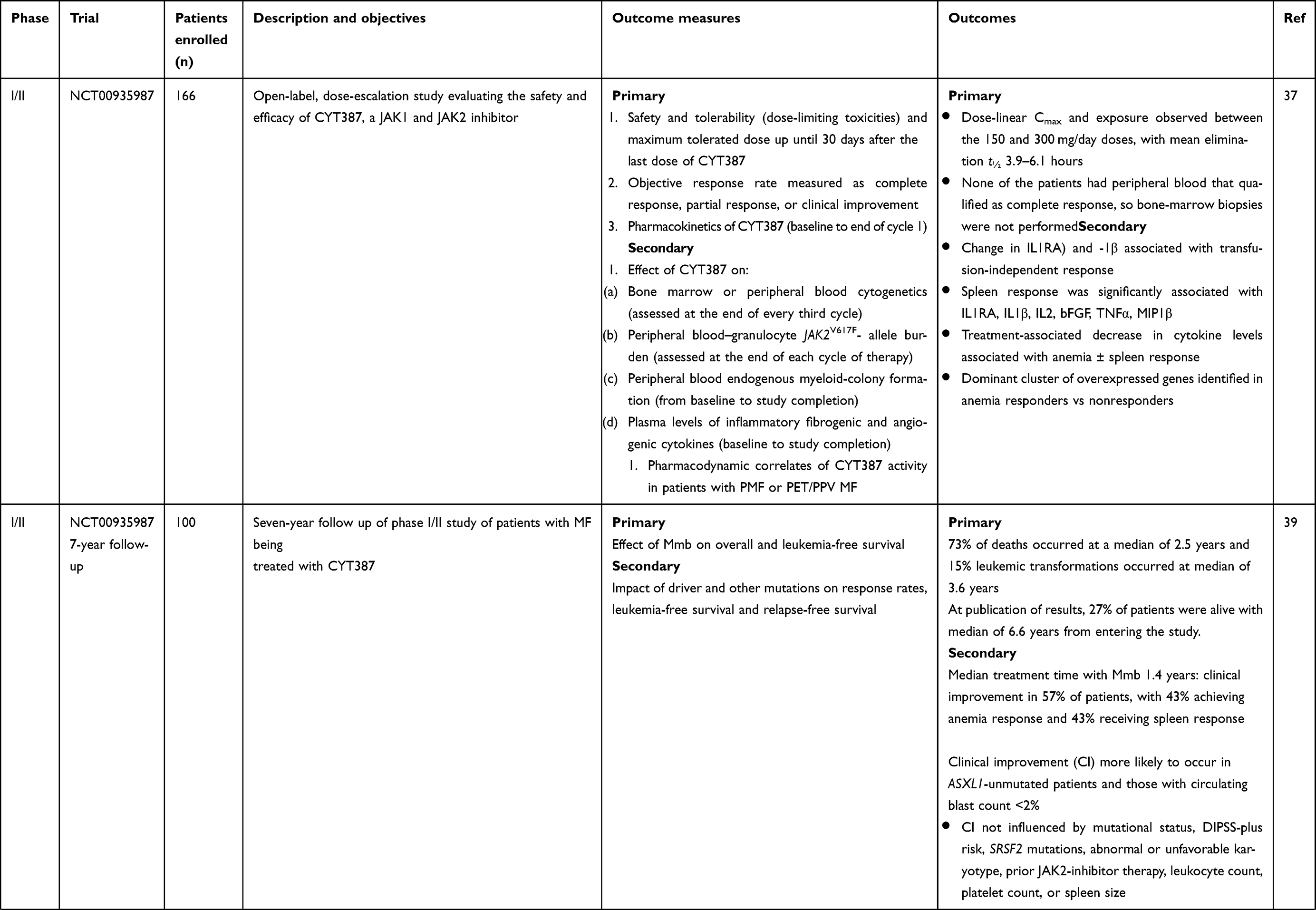 | 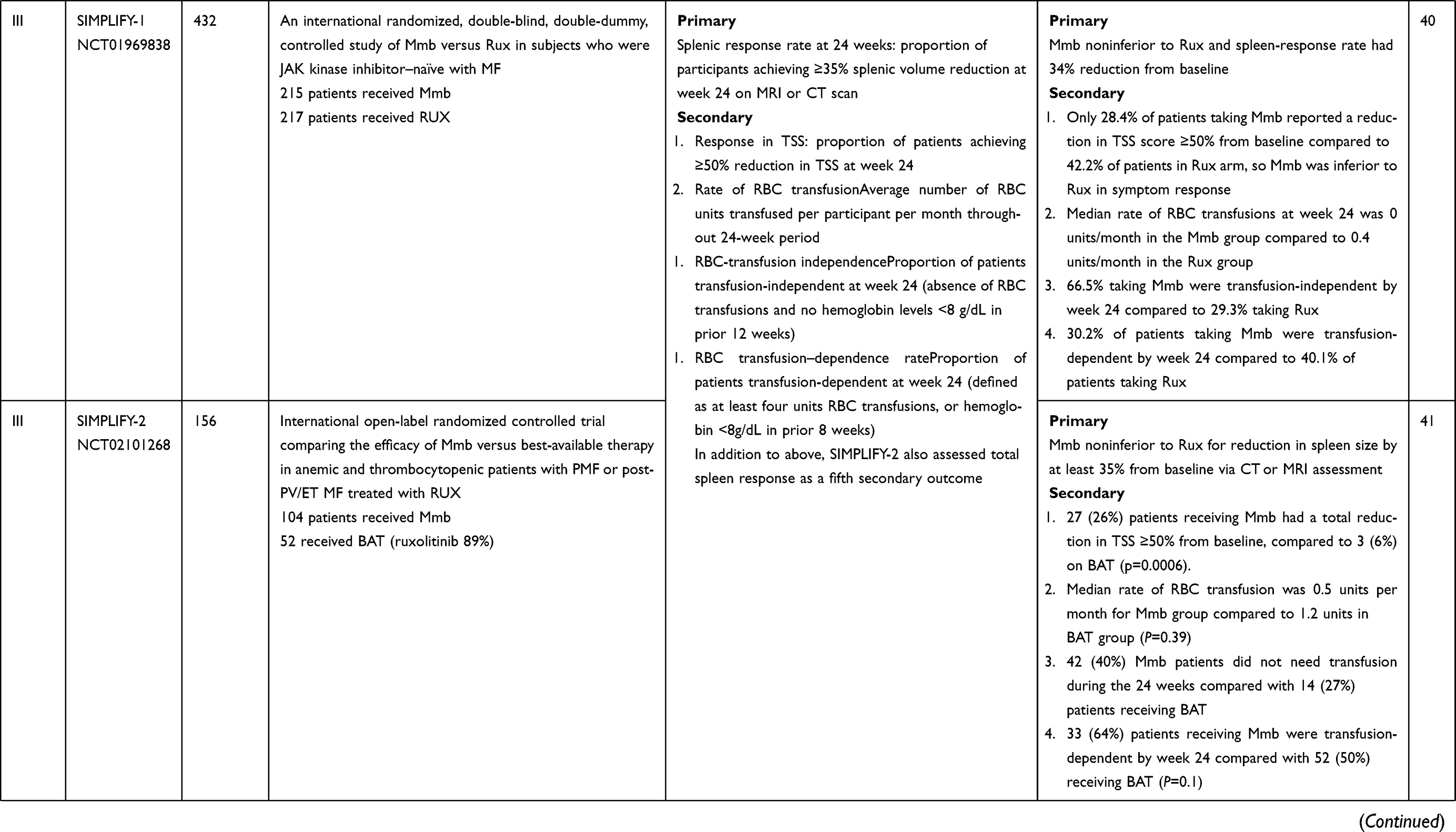 | 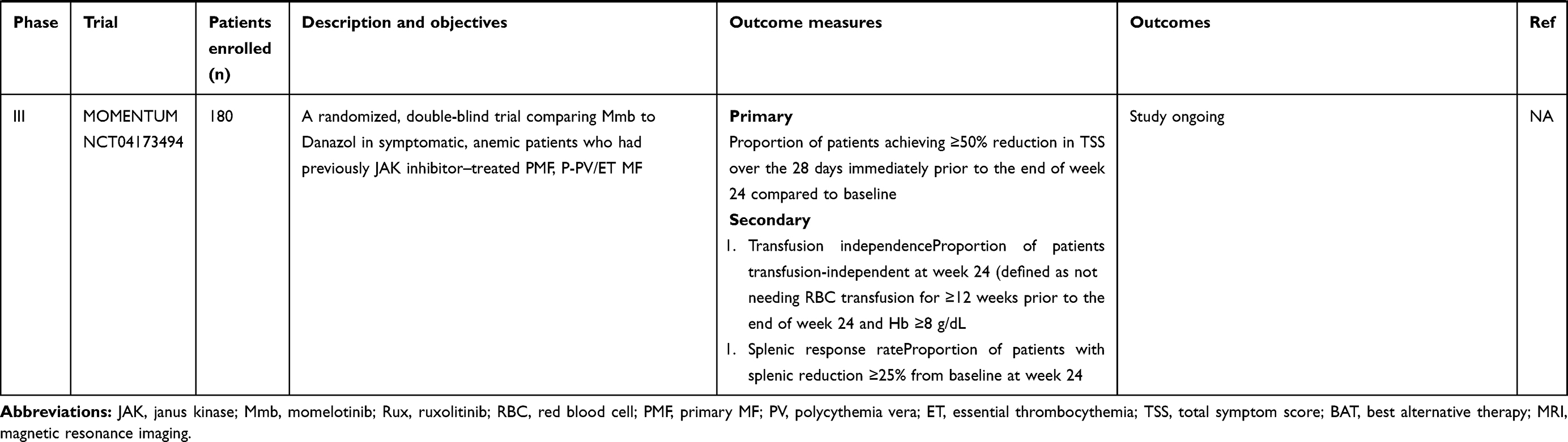 |
Table 2 Summary of clinical trials using momelotinib in management of MF |
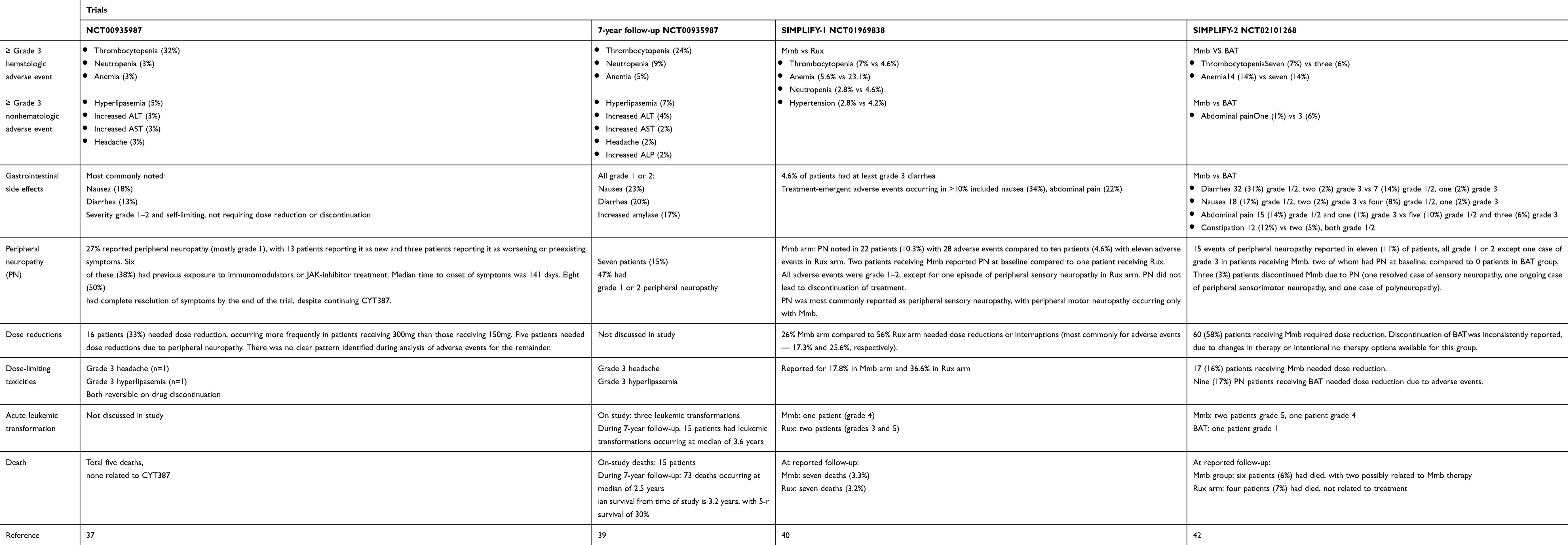 |
Table 3 Summary of momelotinib safety from clinical trials |
SIMPLIFY-1
SIMPLIFY-1 (S1) was a randomized double-blind phase III trial that compared Mmb to Rux in JAK inhibitor–naïve patients with intermediate 2, high-risk, or symptomatic intermediate-risk 1 disease.40 There was 1:1 allocation to either treatment arm, with 214 patients receiving Mmb 200 mg once daily and 216 receiving Rux 20 mg twice a day (dosing could be modified according to license). Patients in the Rux cohort had the opportunity to switch to open-label Mmb therapy at 24 weeks. The primary end point was spleen-volume reduction (SVR) ≥35% from baseline at 24 weeks, and four secondary end points were analyzed: total symptom score (TSS) response rate, RBC transfusion–independence rate (proportion of patients transfusion-independent at week 24), RBC transfusion–dependence rate, and rate of RBC transfusion (average RBC units per subject-month during treatment). Treatment discontinuations were 18.6% (Mmb) and 7.4% (Rux). At week 24, 184 Mmb and 204 Rux patients had spleen volume assessment: 26.5% of the Mmb group and 29% of the Rux group (noninferior; p=0.011) achieved this end point, and thus with regard to SVR Mmb was noninferior to Rux. In contrast, with regard to TSS response at week 24, fewer patients in the Mmb arm (28.4%) than the Rux arm (44.2%) achieved a reduction in TSS of ≥50% from baseline. Initial analyses demonstrated the potential benefit of Mmb for anemia: at week 24, 66.5% of patients in the Mmb cohort were transfusion-independent compared to 49.3% of those in the Rux cohort (nominal P<0.001), and the median rate of RBC transfusion was 0 units/month in the Mmb group compared with 0.4 units/month in the Rux group (nominal P<0.001). Although Mmb did not meet the TSS end point with regard to Rux noninferiority, the proportion of patients who achieved the collective end points of SVR ≥35%, TSS ≥50%, and transfusion independence was slightly higher in the Mmb (10.2%) than the Rux (7.8%) group.
Updated analyses presented at the American Society of Hematology meeting in 2019 investigated further novel anemia endpoints, recognising the clinical importance of hemoglobin responses and transfusion independence within this cohort of MF patients. For Mmb and Rux, the proportion of patients receiving ≤4 RBC units was 83% and 62% (p<0.0001), respectively, and analyses revealed that Mmb increased the odds of having zero units transfused by a factor of 9.3 versus Rux (P<0.0001).41 Relative transfusion events between groups were analysed by Kaplan–Meir estimates of time to first, third, and fifth RBC units. An immediate and sustained Mmb effect compared to Rux was identified with regard to time to first transfusion (log-rank P<0.0001). First-dose effects were reported in both groups, with 14 patients (6.5%) in the Mmb group and two (0.9%) in the Rux group experiencing these. Symptoms included hypotension, dizziness, flushing, nausea, or headache, either in isolation or combination. All episodes were grade 1 or 2, excluding one grade 3 event reported in a patient receiving Mmb that required dose reduction. Only 25% of patients receiving Mmb had AEs of grade ≥3 compared to 43.5% of those receiving Rux, albeit 13.1% of the Mmb cohort had AEs requiring drug discontinuation compared to 5.6% in the Rux cohort. There was no statistical significance detected between the cohort arms with regard to leukemic transformation or death. Peripheral neuropathy was reported in both groups, with 28 events in the Mmb arm and eleven in the Rux arm, all graded as grade 1 or 2, with the exception of one grade 3 sensory neuropathy reported in a patient receiving Rux. There was no indicated need for discontinuation as a result of peripheral neuropathy in any cases at the time of study report.
SIMPLIFY-2
SIMPLIFY-2 was a multinational phase III open-label trial comparing Mmb with best alternative therapy (BAT) in MF patients who had previously received at least 28 days of Rux therapy and had either required red blood–cell transfusions while receiving Rux (defined as ≥4 units of RBCs or hemoglobin <8 g/dL in the 8 weeks prior to randomization) or a dose reduction to <20 mg twice daily with at least one of grade 3 anemia, thrombocytopenia, hemorrhage at least grade 3, an absence of grade 2 or greater peripheral neuropathy, and a spleen that was at least 5 cm palpable below the costal margin.42 Those with previous splenectomy, had undergone splenic irradiation, or who had a concomitant uncontrolled concurrent illness were ineligible. Stratification prior to randomization included evaluation of transfusion requirements and degree of symptom burden assessed by the TSS. A 2:1 randomization (Mmb:BAT) was applied to 24 weeks therapy of either 200 mg once daily Mmb or BAT (which could include multiple agents including RUX), and after this period all patients were able to receive extended access to Mmb.
Of note, in the original protocol there was no designated washout period from the previous therapy. The primary end point was SVR ≥35% from baseline at 24 weeks. A total of 156 patients were recruited (Mmb 104 and BAT 52), and in the BAT arm 46 patients (89%) received Rux, with 14 of these receiving Rux in combination with another therapy (e.g. hydroxycarbamide), this being many more individuals on Rux in the BAT that had been anticipated. A total of 73 patients in the Mmb cohort and 40 in the BAT cohort completed the 24-week therapy phase. Median Mmb exposure was 23.9 weeks. At the 24-week analysis, Mmb was not shown to be superior to BAT for achieving SVR ≥35% from baseline: only seven (Mmb) and three (BAT) patients achieved this end point. Of note, all responders in the BAT group were receiving Rux. Most common grade 3/4 AEs were anemia and thrombocytopenia. Other AEs in the Mmb cohort included peripheral neuropathy, occurring in 11%, with one patient having grade 3 peripheral neuropathy. For reductions in TSS, 26% of patients in the Mmb cohort had >50% reduction in TSS compared to only 6% in the BAT cohort. In each group, >50% of patients were transfusion-dependent, and importantly, at week 24 more patients in the Mmb arm were transfusion-independent than the BAT arm (43% versus 21%, P=0.0012). Therefore, in conclusion, although the primary end points were not reached, Mmb displayed efficacy in amelioration of symptoms, improved anemia responses, and less red blood–cell transfusion need. Longer-term efficacy data are currently being analyzed.
Ongoing Studies: MOMENTUM
MOMENTUM is a multinational phase III trial aiming to confirm the efficacy of Mmb in comparison to danazol in addressing splenomegaly, symptoms, and MF-associated anemia in MF patients who have previously been treated with JAK inhibitors. Patients will be randomized 2:1 between Mmb and danazol, and after 24 weeks of therapy, patients enrolled on the danazol arm will be permitted to cross over to Mmb. The primary end point is TSS reduction at week 24, and secondary end points include 24-week splenic response rates, patient-reported measures of fatigue and physical function, transfusion-independence rates and other anemia end points, such as transfusion-dependence responses, cumulative transfusion burden, and duration of TSS responses at week 48. Planned recruitment is 180 patients globally, with top line data expected in late 2021, and results are eagerly awaited.
Conclusion
Mmb is a potent JAK1, JAK2, and ACVR1 inhibitor, an attribute that potentially places it in a novel position with competing JAK-inhibitor compounds. Clinical trials to date have suggested that Mmb can not only address MF-associated splenomegaly and symptoms but also has a clear and consistent signature of anemia responses via profound inhibition of the hepcidin–ACVR1 axis. However, despite these findings, the two large phase III trials SIMPLIFY-1 and SIMPLIFY-2 just failed to meet their primary clinical end points, and hence the drug remains unlicensed at present. The FDA, however, granted fast-track designation to Mmb in June 2019, recognizing the potential benefits of this agent. It is important to note that in SIMPLIFY-1, Mmb was associated with clinically significant anemia and transfusion-independence responses and higher response rates for the collective end point of SVR, TSS reduction, and anemia response than Rux. Moreover, the end points may have been somewhat limited by the 24-week duration, and hence extended exposure may demonstrate differential efficacy. Hemoglobin responses were additionally evident in those initially treated with Rux who subsequently switched to Mmb. With regard to overall tolerability, side-effect profile and safety of Mmb, clinical trial data suggest clinicians and patients alike need to be aware of the risk of first-dose hypotension, with some patients additionally noting headache, dizziness, or flushing following dosing. Low-grade gastrointestinal side effects, such as nausea and diarrhea, may be apparent. Mmb-related transaminitis may occur, and serum amylase and lipase levels can rise in the absence of clinical pancreatitis. Clinicians should additionally be aware of the potential risk of low-grade peripheral neuropathy — frequently grade 1 — which is predominantly sensory in nature.
It appears to date that unlike Rux for some patients, Mmb-dose intensification and exposure is not as limited by myelosuppression, potentially allowing sustained dose intensification for a longer period across the MF continuum, but this requires confirmation and is currently undergoing evaluation. There remains an unmet need for patients who fail initially to respond to Mmb, those who have an initial response but subsequently become intolerant or resistant, and those who have to stop the drug due to to AEs. Mitigating these issues will only evolve as clinical experience grows. For many patients with MF-associated anemia for whom current treatment options may be limited, a role for Mmb is clear. Dependent on results from the MOMENTUM study, potential may arise in future to use Mmb not only as a sole agent but also in combination with other agents, such as antifibrotics, checkpoint inhibitors and BET inhibitors, to name but a few potential strategies.
Acknowledgments
The authors would like to acknowledge the MPN team at Guy’s and St Thomas’ NHS Foundation Trust, our patients, and collaborating teams globally.
Disclosure
CNH declares speaker fees and honoraria from and advisory boards for Novartis, Celgene, CTI Biopharma, Constellation Pharmaceuticals, Sierra Oncology, and IOP Pharma. DPM reports speaker fees from and advisory boards for Novartis, Celgene, and Jazz Pharmaceuticals. The authors report no other conflicts of interest in this work.
References
1. Titmarsh GJ, Duncombe AS, McMullin MF, et al. How common are myeloproliferative neoplasms? A systematic review and meta-analysis: how Common are Myeloproliferative Neoplasms? A Systematic Review and Meta-Analysis. Am J Hematol. 2014;89(6):581–587. doi:10.1002/ajh.23690
2. Harrison CN, McLornan DP. Current treatment algorithm for the management of patients with myelofibrosis, JAK inhibitors, and beyond. Hematology Am Soc Hematol Educ Program. 2017;2017(1):489–497. doi:10.1182/asheducation-2017.1.489
3. Naymagon L, Mascarenhas J. Myelofibrosis-Related Anemia: current and Emerging Therapeutic Strategies. HemaSphere. 2017;1(1):e1. doi:10.1097/HS9.0000000000000001
4. Tefferi A. Anemia in myelofibrosis—prevalence, the U2AF1 connection, new treatments. Blood Cancer J. 2017;7(12):648. doi:10.1038/s41408-017-0032-9
5. James C, Ugo V, Le Couédic J-P, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi:10.1038/nature03546
6. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi:10.1016/j.ccr.2005.03.023
7. Kralovics R, Passamonti F, Buser AS, et al. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N Engl J Med. 2005;352(17):1779–1790. doi:10.1056/NEJMoa051113
8. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. doi:10.1016/S0140-6736(05)71142-9
9. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N Engl J Med. 2013;369(25):2379–2390. doi:10.1056/NEJMoa1311347
10. Celgene Corporation Press release. Aug. 16, 2019 – Summit, N.J. – Celgene Corporation (NASDAQ: CELG) Today Announced the U.S. Food and Drug Administration (FDA) Has Approved INREBIC® (Fedratinib) for the Treatment of Adult Patients with Intermediate-2 or High-Risk Primary or Secondary (Post-Polycythemia Vera or Post-Essential Thrombocythemia) Myelofibrosis; 2019.
11. Harrison C, Kiladjian -J-J, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. doi:10.1056/NEJMoa1110556
12. Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. doi:10.1056/NEJMoa1110557
13. Palandri F, Elli EM, Polverelli N, et al. Outcome of Patients with Myelofibrosis after Ruxolitinib Failure: role of Disease Status and Treatment Strategies in 214 Patients. Blood. 2018;132(Supplement 1):4277. doi:10.1182/blood-2018-99-116289
14. Tefferi A, Lasho TL, Jimma T, et al. One Thousand Patients With Primary Myelofibrosis: the Mayo Clinic Experience. Mayo Clinic Proc. 2012;87(1):25–33. doi:10.1016/j.mayocp.2011.11.001
15. Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901. doi:10.1182/blood-2008-07-170449
16. Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115(9):1703–1708. doi:10.1182/blood-2009-09-245837
17. Gangat N, Caramazza D, Vaidya R, et al. DIPSS Plus: A Refined Dynamic International Prognostic Scoring System for Primary Myelofibrosis That Incorporates Prognostic Information From Karyotype, Platelet Count, and Transfusion Status. JCO. 2011;29(4):392–397. doi:10.1200/JCO.2010.32.2446
18. Tefferi A, Guglielmelli P, Nicolosi M, et al. GIPSS: genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia. 2018;32(7):1631–1642. doi:10.1038/s41375-018-0107-z
19. Tefferi A, Guglielmelli P, Lasho TL, et al. MIPSS70+ Version 2.0: mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J Clin Oncol. 2018;36(17):1769–1770. doi:10.1200/JCO.2018.78.9867
20. Passamonti F, Giorgino T, Mora B, et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia. 2017;31(12):2726–2731. doi:10.1038/leu.2017.169
21. Grinfeld J, Nangalia J, Baxter EJ, et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N Engl J Med. 2018;379(15):1416–1430. doi:10.1056/NEJMoa1716614
22. Pardanani A, Finke C, Abdelrahman RA, Lasho TL, Tefferi A. Associations and prognostic interactions between circulating levels of hepcidin, ferritin and inflammatory cytokines in primary myelofibrosis. Am J Hematol. 2013;88(4):312–316. doi:10.1002/ajh.23406
23. Cervantes F, Alvarez-Larran A, Hernandez-Boluda J-C, et al. Darbepoetin-alpha for the anaemia of myelofibrosis with myeloid metaplasia. Br J Haematol. 2006;134(2):184–186. doi:10.1111/j.1365-2141.2006.06142.x
24. Tsiara SN, Chaidos A, Bourantas LK, Kapsali HD, Bourantas KL. Recombinant Human Erythropoietin for the Treatment of Anaemia in Patients with Chronic Idiopathic Myelofibrosis. Acta Haematol. 2007;117(3):156–161. doi:10.1159/000097463
25. McMullin MF, Harrison CN, Niederwieser D, et al. The use of erythropoiesis-stimulating agents with ruxolitinib in patients with myelofibrosis in COMFORT-II: an open-label, Phase 3 study assessing efficacy and safety of ruxolitinib versus best available therapy in the treatment of myelofibrosis. Exp Hematol Oncol. 2015;4(1):26. doi:10.1186/s40164-015-0021-2
26. Thapaliya P, Tefferi A, Pardanani A, et al. International working group for myelofibrosis research and treatment response assessment and long-term follow-up of 50 myelofibrosis patients treated with thalidomide-prednisone based regimens. Am J Hematol. 2011;86(1):96–98. doi:10.1002/ajh.21892
27. Quintás-Cardama A, Kantarjian HM, Manshouri T, et al. Lenalidomide Plus Prednisone Results in Durable Clinical, Histopathologic, and Molecular Responses in Patients With Myelofibrosis. JCO. 2009;27(28):4760–4766. doi:10.1200/JCO.2009.22.6548
28. Tefferi A, Al-Ali HK, Barosi G, et al. for the RESUME trialists. A randomized study of pomalidomide vs placebo in persons with myeloproliferative neoplasm-associated myelofibrosis and RBC-transfusion dependence. Leukemia. 2017;31(4):896–902. doi:10.1038/leu.2016.300
29. Cervantes F, Isola IM, Alvarez-Larrán A, Hernández-Boluda J-C, Correa J-G, Pereira A. Danazol therapy for the anemia of myelofibrosis: assessment of efficacy with current criteria of response and long-term results. Ann Hematol. 2015;94(11):1791–1796. doi:10.1007/s00277-015-2435-7
30. Harrison CN, Patriarca A, Mascarenhas J, et al. Preliminary Report of MANIFEST, a Phase 2 Study of CPI-0610, a Bromodomain and Extraterminal Domain Inhibitor (BETi), in Combination with Ruxolitinib, in JAK Inhibitor (JAKi) Treatment Naïve Myelofibrosis Patients. Blood. 2019;134(Supplement_1):4164. doi:10.1182/blood-2019-128211
31. Mascarenhas J, Kremyanskaya M, Hoffman R, et al. MANIFEST, a Phase 2 Study of CPI-0610, a Bromodomain and Extraterminal Domain Inhibitor (BETi), As Monotherapy or ‘Add-on’ to Ruxolitinib, in Patients with Refractory or Intolerant Advanced Myelofibrosis. Blood. 2019;134(Supplement_1):670. doi:10.1182/blood-2019-127119
32. Gerds AT, Vannucchi AM, Passamonti F, et al. A Phase 2 Study of Luspatercept in Patients with Myelofibrosis-Associated Anemia. Blood. 2019;134(Supplement_1):557. doi:10.1182/blood-2019-122546
33. Pardanani A, Lasho T, Smith G, Burns CJ, Fantino E, Tefferi A. CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia. 2009;23(8):1441–1445. doi:10.1038/leu.2009.50
34. Tyner JW, Bumm TG, Deininger J, et al. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood. 2010;115(25):5232–5240. doi:10.1182/blood-2009-05-223727
35. Asshoff M, Petzer V, Warr MR, et al. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood. 2017;129(13):1823–1830. doi:10.1182/blood-2016-09-740092
36. Xin Y, Kawashima J, Weng W, Kwan E, Tarnowski T, Silverman JA. Pharmacokinetics and Safety of Momelotinib in Subjects With Hepatic or Renal Impairment. J Clin Pharm. 2018;58(4):522–532. doi:10.1002/jcph.1050
37. Pardanani A, Laborde RR, Lasho TL, et al. Safety and efficacy of CYT387, a JAK1 and JAK2 inhibitor, in myelofibrosis. Leukemia. 2013;27(6):1322–1327. doi:10.1038/leu.2013.71
38. Pardanani A, Gotlib J, Roberts AW, et al. Long-term efficacy and safety of momelotinib, a JAK1 and JAK2 inhibitor, for the treatment of myelofibrosis. Leukemia. 2018;32(4):1034–1037. doi:10.1038/leu.2017.330
39. Tefferi A, Barraco D, Lasho TL, et al. Momelotinib therapy for myelofibrosis: a 7-year follow-up. Blood Cancer J. 2018;8(3):29. doi:10.1038/s41408-018-0067-6
40. Mesa RA, Kiladjian -J-J, Catalano JV, et al. SIMPLIFY-1: A Phase III Randomized Trial of Momelotinib Versus Ruxolitinib in Janus Kinase Inhibitor–Naïve Patients With Myelofibrosis. J Clin Oncol. 2017;35(34):3844–3850. doi:10.1200/JCO.2017.73.4418
41. Mesa RA, Catalano J, Cervantes F, et al. Dynamic and time-to-event analyses demonstrate marked reduction in transfusion requirements for janus kinase inhibitor-naïve myelofibrosis patients treated with momelotinib compared head to head with ruxolitinib. 2019 ASH Annual Meeting & Exposition. 2019.
42. Harrison CN, Vannucchi AM, Platzbecker U, et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): a randomised, open-label, phase 3 trial. Lancet Haematol. 2018;5(2):e73–81. doi:10.1016/S2352-3026(17)30237-5
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.