Back to Journals » International Journal of Nanomedicine » Volume 15
Etoposide Amorphous Nanopowder for Improved Oral Bioavailability: Formulation Development, Optimization, in vitro and in vivo Evaluation
Authors Wang Y, Wang S, Xu Y, Wang P, Li S, Liu L, Liu M, Jin X
Received 10 June 2020
Accepted for publication 3 September 2020
Published 8 October 2020 Volume 2020:15 Pages 7601—7613
DOI https://doi.org/10.2147/IJN.S265817
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Yue Wang, Shuhang Wang, Yingju Xu, Ping Wang, Sukai Li, Lu Liu, Mengyan Liu, Xiangqun Jin
School of Pharmacy, Jilin University, Changchun, People’s Republic of China
Correspondence: Xiangqun Jin Tel +86-138-4409-0377
Fax +86-431-8561-9662
Email [email protected]
Introduction: Etoposide refers to a derivative of podophyllotoxin, which plays an important role in the treatment of cancer due to its prominent anti-tumor effect. As a BCS IV drug, etoposide exhibits insufficient aqueous solubility and permeability, thereby limiting its oral absorption. To enhance the oral bioavailability of etoposide, this study developed an amorphous nanopowder.
Methods: Based on preliminary screening and experimental design, the stabilizer and preparation process of etoposide nanosuspension were explored. Subsequently, using a Box–Behnken design, the effects of independent factors (ultrasonication time, ratio of two phases and stabilizer concentration) on response variables (particle size and polydispersity index) were studied, and then the formulation was optimized. Finally, nanosuspension was further freeze dried with 1% of mannitol resulting in the formation of etoposide amorphous nanopowder.
Results: The optimized etoposide nanopowder showed as spherical particles with an average particle size and polydispersity index of 211.7 ± 10.4 nm and 0.125 ± 0.028. X-ray powder diffraction and differential scanning calorimetry confirmed the ETO in the nanopowder was amorphous. Compared with coarse powder and physical mixture, etoposide nanopowder achieved significantly enhanced saturated solubility and dissolution in various pH environments. The Cmax and AUC0–t of etoposide nanopowder after oral administration in rats were respectively 2.21 and 2.13 times higher than the crude etoposide suspension. Additionally, the Tmax value of nanopowder was 0.25 h, compared with 0.5 h of reference group.
Discussion: In the present study, the optimized amorphous nanopowder could significantly facilitate the dissolution and oral absorption of etoposide and might act as an effective delivery method to enhance its oral bioavailability.
Keywords: etoposide, amorphous nanopowder, oral absorption, poor aqueous solubility, bioavailability enhancement
Introduction
Etoposide (ETO) is a semisynthetic podophyllotoxin derivative, with the molecular formula of C29H32O13 (Figure 1). It is capable of interfering with mammalian DNA topoisomerase II in the late S or G2 phase of the cell cycle, and combines with enzymes and DNA to fabricate a drug-enzyme-DNA stable reversible complex, which can irreparably impair DNA, eventually causing apoptosis.1,2 For its prominent anti-tumor effect, it has been applied for treating various tumors, such as leukemia, lymphoma, small cell lung cancer and oropharyngeal cancer.3,4 However, as a BCS IV drug, etoposide exhibits insufficient aqueous solubility and permeability,5,6 resulting in many formulations of etoposide being prepared as injections.7–9 However, injections have many disadvantages, such as the need to add a large amount of surfactants and organic solvents,10–12 which may cause allergic reactions in patients.13 Therefore, the development of oral preparations of etoposide has become an attractive research field. To facilitate the oral absorption of ETO, several strategies have been adopted, including nanostructured lipid carriers and self-emulsifying delivery systems. Some of them exhibit high in vivo performance.14,15
 |
Figure 1 The chemical structure of ETO. |
Drug nanoparticles composed of drugs and stabilizers are considered one of the most promising nanotechnologies to promote insoluble drugs to exhibit enhanced solubility and oral bioavailability.16–18 With the decrease in particle size (PS) and the increase in specific surface area, drug nanoparticles can significantly elevate the saturation solubility and dissolution rate of drugs.19 Moreover, under the high drug loading capacity of drug nanoparticles, patients can have lower dosage and better compliance in oral administration.20 The role of drug nanoparticles in improving solubility and oral bioavailability has been confirmed in various literatures.21–23 Drug nanoparticles may act as a preferable nanotechnology to facilitate the solubility and oral absorption of ETO, but this has rarely been studied. In this work, an ETO nanosuspension was prepared using a sonoprecipitation–high pressure homogenization method; subsequently, the formulation was optimized using a Box–Behnken design (BBD) of response surface method; lastly, it was transformed into ETO nanopowder by freeze-drying. The optimized ETO nanopowder was systematically analyzed in terms of morphology, particle size and physicochemical properties. Finally, in vitro dissolution and in vivo pharmacokinetics of optimized ETO nanopowder were assessed.
Materials and Methods
Materials
The following materials were provided by or purchased from the sources in parentheses: etoposide (Shanghai Xiandai Pharma Ltd, Shanghai, China); sodium dodecyl sulfate (SDS) and sodium deoxycholate (Tianjin Yong da Chemical Reagent Co. Ltd, Tianjin, China); D-alpha-Tocopheryl polyethylene glycol 1000 succinate (TPGS), poloxamer 188 (Pluronic F68), and poloxamer 407 (Pluronic F127) (BASF, Ludwigshafen, Germany); polyvinyl alcohol 217 (PVA 217) (Kuraray Co. Ltd, Japan, Osaka); polyethylene glycol 2000 (PEG-2000) and polyvinylpyrrolidone K30 (PVP K30) (Tianjin Bodi Chemical Co. Ltd, Tianjin, China); sodium carboxyl methyl cellulose (CMC-Na) (Nanjing Zhongnuo Bioengineering Co. Ltd, Nanjing, China); dimethyl sulfoxide (DMSO), methanol, acetone and acetonitrile of chromatographic grade (Concord Technology Co. Ltd, Tianjin, China). All reagents and chemicals used were of analytical or chromatographic grade.
Methods
Preparation of Amorphous Nanopowder
ETO amorphous nanopowder was prepared using a sonoprecipitation–high pressure homogenization method. Briefly, the crude etoposide was completely dissolved in DMSO. In the presence of sonication (SCIENTZ-IID ultrasonic processor, Scientz Biotechnology Co., Ltd, Ningbo, China), the ETO solution was rapidly added to the aqueous solution containing the stabilizer (Pluronic 127, PVP K30, CMC-Na, sodium deoxycholate, SDS, PVA, TPGS, Pluronic F68 or PEG-2000) to obtain a starting suspension. The starting suspension was then placed in a high-pressure homogenizer (HPH, Pharmaceutical ultra-high-pressure homogenizer of AH100D, ATS Engineering Inc., Shanghai, China) at 4°C. After that, the nanosuspension was concentrated by centrifugation of 15,000 rpm/min for 15 minusing a high-speed refrigerated centrifuge (Anhui Zhongke Zhongjia Scientific Instrument Co. Ltd, Anhui, China). Finally, the supernatant was removed and the precipitate was redistributed in an aqueous solution containing the stabilizer.
Further, the nanosuspension and cryoprotectant were lyophilized together (Labconco, USA) for 24 hours. The lyophilized ETO nanopowder was evaluated for appearance, re-dispersibility and Sf (particle size after lyophilization)/Si (initial particle size) ratio.
Formulation Optimization of ETO Nanopowder
Through the preliminary experiments of different process parameters, X1 (ultrasonication time), X2 (ratio of two phases) and X3 (stabilizer concentration) were selected as the critical process parameters affecting PS (Y1) and polydispersity index (PDI, Y2) of ETO nanopowder. Then the prescription was further optimized by BBD. This study was a three-factor central composite design (CCD) using 17 batches. The ultimate goal of optimization was to obtain ETO nanopowder with minimum PS and PDI. The experiment was designed and analyzed by Design-Expert software (Version 8.0.6.1, Stat-Ease Inc., Minneapolis, MN). The variables and their levels are listed in Table 1.
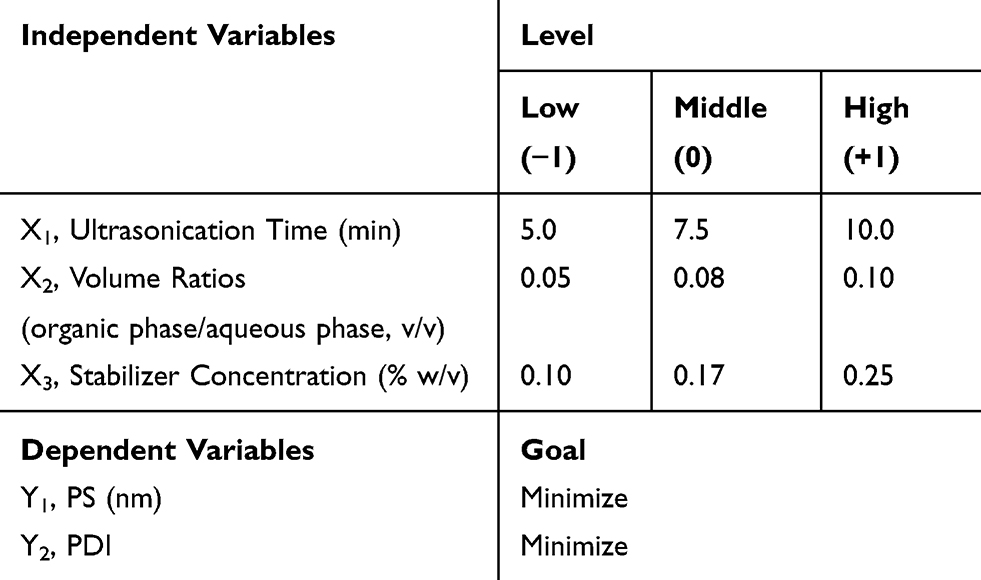 |
Table 1 Independent Factors and Responses in the Box–Behnken Design |
Particle Size and Size Distribution Analysis
ETO nanopowder was re-dispersed in water and evaluated for PS and PDI using dynamic light scattering (ZEN 3600, Zetasizer nano ZS 90, Malvern, UK).24 The re-dispersed ETO nanopowder was diluted 5-fold with distilled water before measurement. Each sample was measured in triplicate at room temperature.
Morphology Observation
The morphology of the ETO nanopowder was analyzed with a transmission electronic microscope (TEM, JEM 2010; JEOL, Japan) operated at 120 kV.25 The ETO nanopowder was diluted with distilled water before measurement. One drop of the re-dispersed ETO nanopowder was placed on the surface of a copper grid, and the excess liquid was drained on filter paper. The copper grid was strained in 2% phosphotungstic acid for 120 s.
Differential Scanning Calorimetry (DSC)
Differential scanning calorimetry analysis was carried out (DSC-3; METTLER TOLEDO, Switzerland). The samples were accurately weighed (2–5 mg) and placed in a standard aluminum pan, sealed with a lid and perforated. The samples were scanned at a rate of 10°C/min over a range of 40–300°C under 50 mL/min nitrogen flow rate.26
Powder X-Ray Diffraction (PXRD)
The powder X-ray diffraction patterns of the ETO nanopowder were analyzed by D/Max-2400 X-ray fluorescence spectrometer (Rigaku, Osaka, Japan).27 Data were collected with Cu-Ka radiation and a voltage of 40 kV, and the scanning speed was 4°/min from 3° to 50°.
Determination of Saturation Solubility
The saturation solubility of coarse powder (PS = 13.33 ± 3.44 μm), physical mixture and nanopowder of ETO in different media (distilled water, pH 1.2 solution or pH 6.8 PBS) were analysed by a shaking-flask method.28 Briefly, an excess amount of sample powder was dispersed into 0.5 mL different media, and then shaken for 72 h at 100 rpm and 37°C in the HCR-D210 shaking-air bath (Henan Instruments Co., Ltd, Henan, China). These samples were centrifuged at 15,000 rpm for 15 min, and the supernatant was immediately filtered through a 0.22 μm syringe filter. Finally, the content of etoposide in samples was detected by high performance liquid chromatography (HPLC) analysis in triplicate. Drug analysis was conducted on a Kromasil 100–5 Phenyl column (250 × 4.6 mm i.d., 5 μm, Zhonghuida Technologies, China). The HPLC mobile phase consisted of acetonitrile and 0.02 M sodium acetate (pH 4.0, 30:70, v/v) at a flow rate 1.0 mL/min, column temperature was maintained at 40°C, and injection volume was 10 μL. The detection wavelength was set at 245 nm.29
In vitro Dissolution Test
The dissolution profiles of coarse powder (PS = 13.33 ± 3.44 μm), physical mixture and nanopowder of ETO in different media (pH 1.2 solution, pH 6.8 PBS or pH 7.4 PBS) were tested with a dissolution apparatus (RC806D). The medium was stirred at 100 rpm and the temperature was set at 37°C. The accurately weighed powder (containing 5 mg etoposide) was added into 200 mL different media. Aliquots of 5 mL were withdrawn at 1, 2, 3, 5, 10, 15, 30, 45 and 60 min and replaced with an equal volume of fresh media. The samples were immediately filtered through a 0.22 μm syringe filter and quantified by HPLC in triplicate.29
Pharmacokinetics Study
Twelve SD male rats weighting 180–220 g were purchased from the Institute of Laboratory Animal Science, Shenyang Pharmaceutical University (Shenyang, China). This study was reviewed and approved by the Laboratory Animal Welfare Ethics Committee of Shenyang Pharmaceutical University (Animal ethical numberSYPU - IACUC - C2019 - 7 - 10 - 203) and all the operations were conducted in accordance with the “Guidelines of Shenyang Pharmaceutical University for the Care and Use of Experimental Animals.” Rats were fasted in a specific pathogen-free (SPF) animal house for 12–16 h with free access to water. The rats were randomly divided into two groups (n=6) and orally administered with the suspension of coarse etoposide powder (PS = 13.33 ± 3.44 μm) and ETO nanopowder respectively, each equivalent to 180 mg/kg ETO. Equivalent volume of blood samples (0.5 mL) were obtained from orbital veins and transferred into heparinized tubes at 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12 and 24 h after oral administration. Then, the blood sample was centrifuged at 6000 rpm/min for 10 min.
The plasma was separated and stored at −80°C before analysis. Etoposide concentration in rat plasma was determined by UPLC/MS/MS. The UPLC/MS/MS method is as follows: Chromatographic separation was carried out with an ACQUITY™ UPLC system (Waters Corp., Milford, MA, USA). The separation was conducted on an ACQUITY UPLC™ BEH C18 column (50 mm × 2.1 mm i.d., 1.7 μm, Waters Corp., Milford, MA, USA). The mobile phase consisted of acetonitrile (A) and 0.1% formic acid in water (B), and was delivered at a flow rate of 0.2 mL/min. The column was kept at 35°C, and the autosampler was maintained at 4°C. The linear gradient elution program was (1) A decreased from 70% to 30% during the first 1.0 min; (2) A was held at 30% for 1.0 min; (3) A was reset to the initial composition in 0.2 min; (4) A was held at 70% for 0.8 min. The injection volume was 5 μL using the partial loop mode. Mass spectrometric detection was carried out with a Waters ACQUITY™ TQD triple quadrupole tandem mass spectrometer (Waters Corp., Manchester, UK) in positive ESI mode. The optimized source/gas parameter was capillary 3.1 kV; cone voltage 3.0 V; radio frequency 0.3 V; source temperature 100°C; desolvation temperature 400°C. Nitrogen was used as the desolvation gas (450 L/h) and cone gas (50 L/h). For collision-induced dissociation, argon was used as the collision gas at a flow rate of 0.2 mL/min. Quantitation was performed using multiple reaction monitoring of the fragmentation transitions of m/z 589.05→228.90 amu for ETO, and m/z 656.89→382.91 amu for the internal standard, with a scan time of 0.02 s per transition.30,31 Pharmacokinetic data were collected by Masslynx™ NT4.1 software (Waters Corp., Milford, MA, USA) and processed with the QuanLynx™ program (Waters Corp, Milford, MA, USA).31 Pharmacokinetic parameters were analyzed with DAS 2.0 software (Mathematical Pharmacology Professional Committee of China, Shanghai, China).4 All descriptive parameters are expressed as mean ± SD.
Results and Discussion
Formulation Development and Optimization
Selection of Organic Solvent, Stabilizers and Process Parameters
In this section, the results and scientific explanations of the formulation selection of ETO nanopowder are presented. The content of the preliminary experiment aimed to delve into the influence of various experimental parameters on PS and PDI, such as the type of organic phase, ratio of two phases, ultrasonication time, the type of stabilizer, stabilizer concentration and HPH operation parameters. Then, the critical process parameters were selected to optimize the formulation of ETO nanopowder by BBD.
The sonoprecipitation–high pressure homogenization method is a combination of top-down and bottom-up methods.32,33 In the bottom-up method, the nucleation is critical to harvest uniform drug nanoparticles in a narrow size distribution. The supersaturation induced by the antisolvent is vital for the control of the nucleation process. By selecting a suitable antisolvent to achieve high supersaturation, more rapid nucleation can be induced, thereby resulting in crystals of similar size.15 In this study, the effects of different organic solvents on PS and PDI of ETO nanosuspension were compared (Figure 2A). As revealed from the results, the precipitation system with acetone and acetonitrile led to the formation of large particulate aggregates, which was not conducive to subsequent processing with a high-pressure homogenizer. However, the suspension with DMSO as antisolvent did not produce this phenomenon. Moreover, the solubility of ETO in DMSO was significantly higher than that in other solvents. Therefore, choosing DMSO as antisolvent can reduce the volume of the organic solvent in the suspension, which is conducive to reduce the PS and PDI of the nanosuspension (Figure 2B). In existing studies, ultrasonication was able to enhance nucleation by producing acoustic cavitation in solution and subsequently shortening the induction period, so as to obtain a suspension with uniform particle size distribution.34,35 Accordingly, ultrasonication time is also an important factor affecting PS and PDI of a nanosuspension. As shown in Figure 2C, with the increase in ultrasonication time, PS and PDI of the nanosuspension decreased first and then increased. The reason might be that with the increase in ultrasonication time, the input energy increases, generating heat and thus leading to the aggregation of particles.
 |
Figure 2 The effect of various parameters, (A) type of organic solvent, (B) ratio of two phases, (C) ultrasonication time, (D) homogenization pressure, (E) type of stabilizer, (F) stabilizer concentration, (G) homogenization cycles in each prescription, on mean PS and PDI of ETO nanopowder. |
High-pressure homogenization refers to a top-down method capable of effectively reducing PS and PDI of nanoparticles.36,37 The effect of different homogenization pressure and cycle times on the PS and PDI of the nanosuspension was explored in this study. As demonstrated from the results, with the increase in homogenization pressure and cycle times of high-pressure homogenization, the energy input increased, and the PS and PDI of ETO nanosuspension declined (Figure 2D and G). Moreover, it is noteworthy that with the increase in homogenization cycle times, the decrease in d90 was greater than that of the average particle size, indicating that high-pressure homogenization could effectively reduce the large particles in the system and promote the system to be more uniform.
The selection of the stabilizer is the most important step in the preparation of a nanosuspension. The PS and PDI of a nanosuspension can be altered by adding different stabilizers in the crystallization milieu.38,39 As suggested in Figure 2E, under identical preparation conditions, the use of charged stabilizers (SDS or sodium deoxycholate) can effectively reduce PS and PDI of ETO nanosuspension. However, the addition of polymers such as Pluronic 127, PVP K30, CMC-Na, PVA 217, TPGS, Pluronic F68 or PEG-2000 could not reduce PS and PDI of ETO nanosuspension. It was therefore demonstrated that electrical repulsion is dominant in the formation of the ETO nanosuspension system. However, though sodium deoxycholate could give excellent PS and PDI to a nanosuspension, it is unstable in gastric acid. Accordingly, SDS was chosen as the stabilizer of ETO nanosuspension in this study. Subsequently, the effect of stabilizer concentration on PS and PDI of the nanosuspension was determined. It was demonstrated that with an increase in SDS concentration, PS of ETO nanosuspension decreased. However, when a higher stabilizer concentration was added to the formula, the nanosuspension exhibited smaller PS as expected, whereas PDI increased (Figure 2F). Similar results were identified in previous studies.40 An explanation for these results is that when the concentration of stabilizer is low, the stabilizer should cover the whole hydrophobic surface of nanoparticles to mask their hydrophobic region, minimizing the free stabilizer in the system. However, when a higher concentration of stabilizer is added, the free stabilizer in the system increases. Under the concentration of free stabilizer higher than the critical micelle concentration, the mentioned free stabilizers tend to form micelles, which may be the main reason for the uneven particle size distribution of the system.40
Formulation Optimization
The design matrix of the 17 experiments was designed by BBD with three factors and three levels, as shown in Table 2. These experiments were carried out randomly to reduce systematic error caused by extraneous factors. Then, the responses and relevant data of the 17 experiments were simultaneously fitted to linear, two-factor (2F) and quadratic models by Design Expert software. As shown in Table 3, quadratic models were the best fitted model for PS and PDI, with coefficient of multiple determinations (r2) of 0.9887 and 0.9605, respectively. Accordingly, the best formulation of ETO nanopowder was calculated and predicted by the quadratic equations. The polynomial regression equations of the quadratic model are as follows:
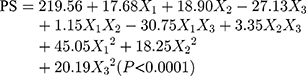

 |
Table 2 Arrangement and Response Variables of Box–Behnken Design |
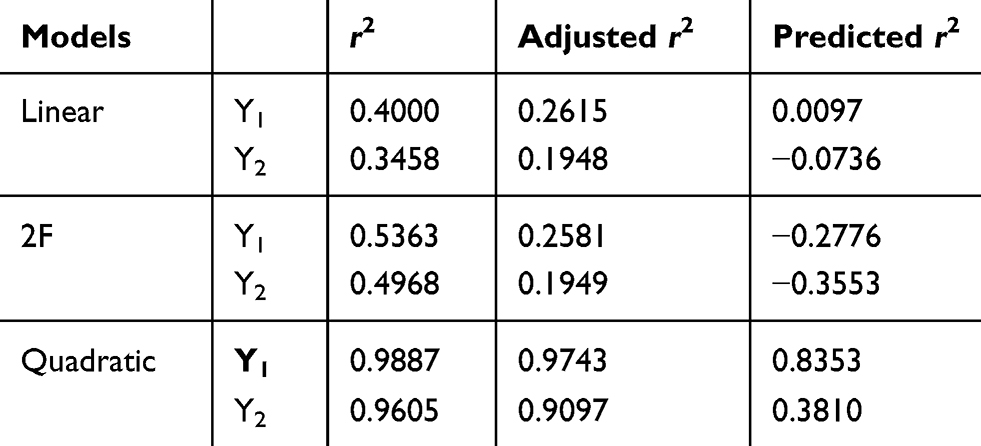 |
Table 3 Results of Statistical Models for Responses |
In an analysis of variance (ANOVA, Table 4), it was found that ultrasonication time (X1), ratio of two phases (X2) and stabilizer concentration (X3) were the main factors affecting the particle size of ETO nanopowder. And the influence of the ratio of two phases (X2) and stabilizer concentration (X3) was the most significant. The second-order interaction of ultrasonication time and stabilizer concentration (X1X3) also had a significant effect on the particle size of ETO nanopowder. For the PDI of ETO nanopowder, in addition to X1, X2 and X3, the second-order interaction of ultrasonication time and stabilizer concentration (X1X3) was also statistically significant on the PDI of the developed formulations. For a more intuitive assessment of the factor effects on the CQAs, the three-dimensional response surface plots are shown in Figure 2. With the increase in ultrasonication time, PS and PDI of ETO suspension first ascended and then increased (Figure 3A). In Figure 3B and C, the higher the concentration of SDS, the smaller the particle size of ETO nanosuspension, but the PDI first decreased and then increased. These results were consistent with the previous preliminary experimental results. In Figure 3A and C, both PS and PDI decreased with the decrease of the proportion of organic phase to aqueous phase. Therefore, reducing the volume of organic phase not only helps obtain excellent PS and PDI nanosuspension, but also solves the problem of drug precipitation being difficult to recover after centrifugation of nanosuspension due to more organic solvents.
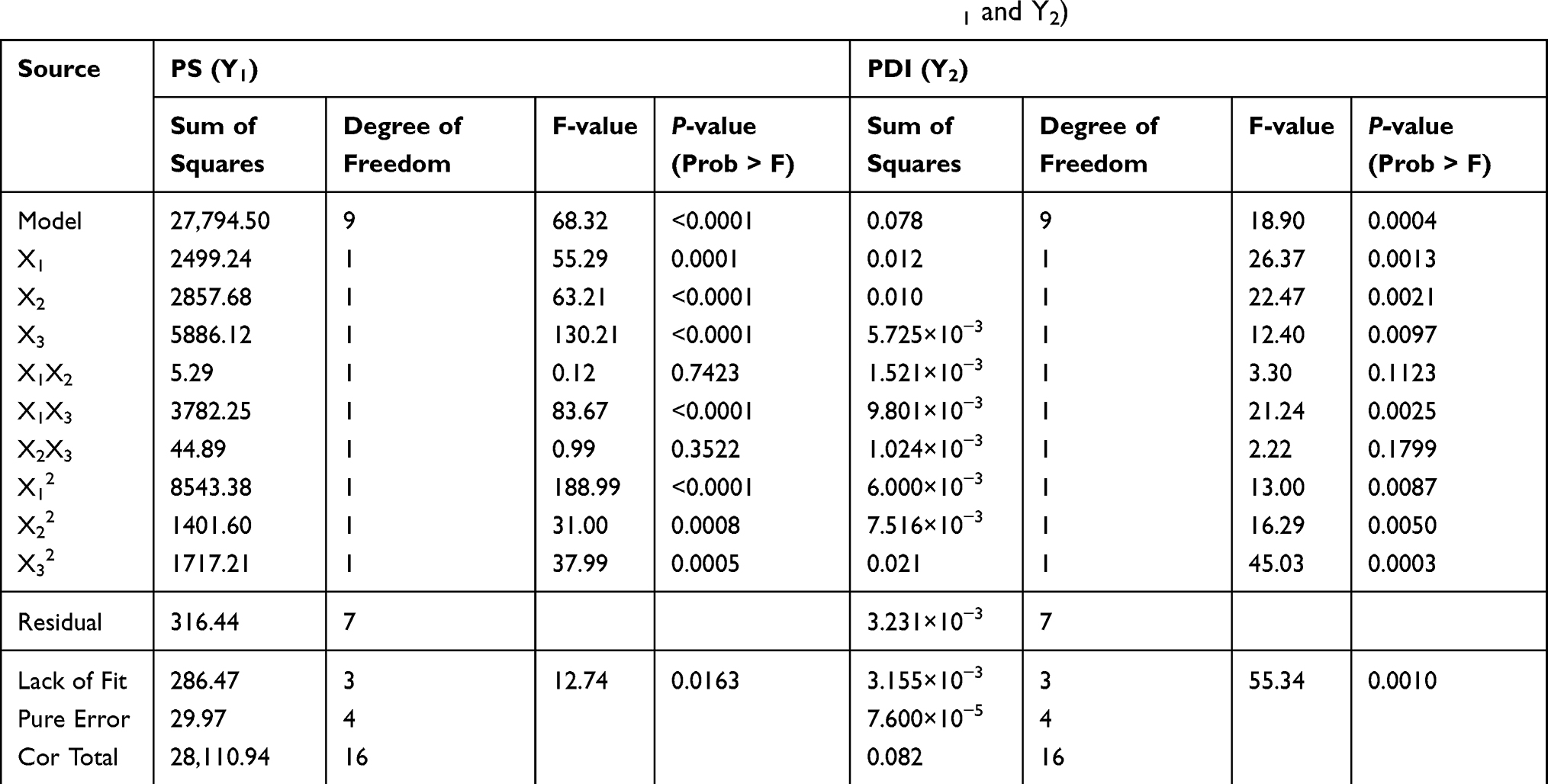 |
Table 4 Analysis of Variance (ANOVA) in the Quadratic Model for Responses (Y1 and Y2) |
 |
Figure 3 Response surface profiles showed the effects of ultrasonication time (X1), ratio of two phases (X2) and stabilizer concentration (X3) on PS (Y1) and PDI (Y2) of ETO nanopowder. (A) The effect of second-order interaction of ultrasonication time and stabilizer concentration (X1X2) on the PS and PDI of ETO suspension. (B) The effect of second-order interaction of ultrasonication time and stabilizer concentration (X1X3) on the PS and PDI of ETO suspension. (C) The effect of second-order interaction of ultrasonication time and stabilizer concentration (X2X3) on the PS and PDI of ETO suspension. |
According to the results of the BBD experiment, the optimal formulation is as follows: the ultrasonication time was 7.22 min, the content of stabilizer was 0.2% w/v and the volume ratio of water phase to organic phase was 0.06. The best prediction value was PS of 207.2 nm, PDI of 0.112. The actual particle size and PDI of the best prescription were 211.7 ± 10.4 nm and 0.125 ± 0.028, which were consistent with the predicted values.
Lyophilization Study
Solidification is a very important step for the preparation of nanopowder. In this paper, we evaluated the type and concentration of cryoprotectant on the appearance, re-dispersibility, PS, PDI and Sf/Si ratio of ETO nanopowder. The results are shown in Table 5. The ETO nanopowder using 1% (w/v) and 2% (w/v) mannitol as the cryoprotectant had excellent performance in all aspects. In order to increase the drug loading, we selected 1% (w/v) mannitol as the cryoprotectant of ETO nanopowder.
 |
Table 5 Effect of Different Cryoprotectants on Physical Characterization |
Characterization of ETO Nanosuspension
Morphology and Particle Size
The PS of the fresh ETO nanosuspension was 211.7 ± 10.4 nm, PDI was 0.125 ± 0.028 and zeta potential (ZP) was −46.2 mv. The PS of the lyophilized ETO nanopowder was 217.6 ± 14.3 nm and PDI was 0.147 ± 0.036. After freeze-drying, the particle size of the re-dispersed nanopowder was slightly larger than that of the fresh nanosuspension, which indicated that a certain degree of particle aggregation occurred in the process of freeze-drying. The TEM image of re-dispersed nanopowder was spherical particles with a diameter range of about 200 nm (Figure 4).
 |
Figure 4 TEM images of ETO nanopowder. (Scale bar, 200 nm). |
Crystalline Characterization
The X-ray powder diffraction patterns of the ETO samples are illustrated in Figure 5A. The major characteristic peaks of ETO (at 2θ of 13.486°, 17.987° and 22.489°) were detected in diffractograms of the coarse powder and physical mixture of ETO (Figure 5A (B and C)), indicating that the crystalline state was maintained in the physical mixture. However, compared with the coarse powder and physical mixture, the ETO nanopowder showed a relatively flat curve without a strong peak (Figure 5A (A)), which confirmed that the ETO in the nanopowder was amorphous.
 |
Figure 5 PXRD patterns (A) and DSC thermograms (B) of ETO nanopowder (a); physical mixture of ETO (b); coarse ETO (c); SDS (d); mannitol (e). |
DSC patterns of coarse ETO powder, SDS, mannitol, the physical mixture and nanopowder are shown in Figure 5B. The thermogram of coarse ETO powder showed a characteristic peak at 286.5°C, which revealed the crystalline nature of the drug (Figure 5B (C)). But a broad peak at around 280°C was observed in the physical mixture (Figure 5B (B)), which may be caused by the overlapping of the melting endothermic peak of ETO and SDS. Mannitol showed a sharp endothermic peak at 168°C, which could be observed in the thermogram of physical mixture and nanopowder of ETO (Figure 5B (E)). However, the DSC curves of ETO nanopowder showed the ETO melting peak was absent (Figure 5B (A)), which indicates that ETO in the nanopowder was mainly amorphous. This result was consistent with that of XRPD. The melting peaks at 142°C and 221°C indicated that the ETO nanopowder had changed, which may be a result of the interaction between the drug and the excipients.
Determination of Saturation Solubility
The saturated solubility of coarse ETO powder, physical mixture and optimized ETO nanopowder in different media are listed in Table 6. In comparison to the coarse powder, the solubility of ETO were significantly increased by 2.31-fold in distilled water, 2.74-fold in pH 1.2 solution and 2.41-fold in pH 6.8 PBS for the nanopowder. The solubility of ETO in physical mixture was also improved, but the solubility of nanopowder in distilled water, pH 1.2 solution and pH 6.8 PBS were still 1.90-, 2.36- and 1.92-fold higher than that of physical mixture. The Kelvin and Ostwald–Freundlich equations are applicable to illustrate the reason for this phenomenon. According to the Kelvin equation, the decrease of the particle size of nanopowder (from 13.33 ± 3.44 μm to 217.6 ± 14.32 nm) is helpful to the increase of the surface curvature, which leads to the increase of the dissolution pressure. This causes the equilibrium to move in the direction of dissolution, resulting in an increase in saturated solubility.41 The Ostwald–Freundlich equation more directly explains that the saturation solubility of drug increases with the decrease of the particle size.42 Moreover, the solubilizing effect of SDS is also one of the reasons for the increase of solubility of nanopowder and physical mixture.43
 |
Table 6 Saturation Solubility of the Coarse Powder, Physical Mixture and Nanopowder of ETO in Different Media |
In vitro Dissolution Test
The dissolution profile of coarse ETO powder, physical mixture and optimized ETO nanopowder in different media are shown in Figure 6. ETO nanopowder showed significantly improved dissolution performance in all media compared with coarse powder and physical mixture of ETO. The dissolution rate of ETO nanopowder in pH 1.2 solution improved by 1.70-fold and 1.55-fold, with 98.01 ± 0.82% release in 15 min when compared with coarse powder (57.60 ± 0.29%) and physical mixture (63.03 ± 0.86%) of ETO, respectively. Whereas, the concentration of ETO of all samples in pH 1.2 solution began to decrease gradually after 15 minutes, which possibly was due to the stability of etoposide in strong acid solvent. Moreover, the dissolution rate of optimized ETO nanopowder was improved in pH 6.8 PBS (98.97 ± 0.78% vs 66.73 ± 1.68%, 73.82 ± 0.86%) and pH 7.4 PBS (98.53 ± 1.11% vs 67.95 ± 2.79%, 74.44 ± 1.23%) in 60 min, respectively. In addition, the dissolution velocities of ETO nanopowder in all different media increased significantly. Compared with coarse powder and physical mixture of ETO (<45.3% and <55%, respectively), the dissolution of ETO nanopowder in 3 min was more than 80%. These results suggested that the formation of nanopowder strongly improves the dissolution rate and degree of ETO. The increase of dissolution rate and degree of physical mixture was due to the presence of SDS.43 However, the further dramatic increase of the dissolution rate and degree mainly depends on the decrease of the particle size (from 13.33 ± 3.44 μm to 217.6 ± 14.32 nm). According to the Noyes–Whitney equation, the decrease of the drug particle size will lead to an increase of the surface area and a decrease of the diffusion layer thickness, thus increasing the dissolution rate.41
 |
Figure 6 Dissolution profile of coarse powder, physical mixture and nanopowder of ETO in pH 1.2 solution (A); pH 6.8 PBS (B) and pH 7.4 PBS (C). |
Pharmacokinetics Study
In order to further verify the advantages of nanopowder in improving the oral absorption of etoposide, the differences between the pharmacokinetic parameters of nanopowder and coarse powder of ETO in rats were compared. The key pharmacokinetic parameters are shown in Table 7, and the plasma drug concentration-time curve is shown in Figure 7. After oral administration, the Cmax and AUC0-t values of nanopowder were 2.21- and 2.13-fold higher than those of coarse ETO. This improvement of oral bioavailability of ETO could be related to the increase of saturation solubility and dissolution of drug via nanopowder formulation. Additionally, the Tmax values were 0.250.5 h for nanopowder and coarse ETO, respectively. The shortened Tmax of nanopowder could be related to its ability to accelerate the dissolution of drugs, leading to the rapid drug absorption.44 These results suggested that the nanopowder has a good contribution in improving the oral absorption of etoposide.
 |
Table 7 Pharmacokinetic Parameters of Oral ETO Coarse Powder and Nanopowder |
 |
Figure 7 Plasma concentration-time curves of oral ETO coarse powder and the optimized nanopowder. |
Conclusion
In this study, etoposide amorphous nanopowder was successfully prepared using a sonoprecipitation–high pressure homogenization method and then optimized with BBD. The physicochemical properties of the optimized formulation were evaluated by TEM, DSC and PXRD. Compared with the coarse powder and physical mixture, the aqueous solubility and in vitro dissolution rate of ETO in nanopowder were significantly improved. Pharmacokinetic study demonstrated that the nanopowder formulation effectively facilitated drug absorption of ETO in rats after oral administration. All the findings revealed that amorphous nanopowder is a promising strategy for enhanced oral bioavailability of hydrophobic compounds, such as ETO.
Abbreviations
AUC, area under the curve; BBD, Box–Behnken design; CCD, central composite design; CMC-Na, sodium carboxyl methyl cellulose; DMSO, dimethyl sulfoxide; DSC, differential scanning calorimetry; ETO, etoposide; HPH, high-pressure homogenizer; PBS, phosphate buffered saline; PDI, polydispersity index; PEG-2000, polyethylene glycol 2000; PS, particle size; PVA 217, polyvinyl alcohol 217; PVP K30, polyvinylpyrrolidone K30; SDS, sodium dodecyl sulfate; SPF, specific pathogen-free; TPGS, D-alpha-Tocopheryl polyethylene glycol 1000 succinate; TEM, transmission electronic microscopy; ZP, zeta potential.
Acknowledgments
The authors wish to thank the School of Pharmacy, Shenyang Pharmaceutical University, Shenyang, for their technical support.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Hande KR. Etoposide: four decades of development of a topoisomerase II inhibitor. Eur J Cancer. 1998;34(10):1514–1521. doi:10.1016/S0959-8049(98)00228-7
2. Satoshi I, Masayuki S, Takahiro O, et al. Comparison of carboplatin plus etoposide with amrubicin monotherapy for extensive-disease small cell lung cancer in the elderly and patients with poor performance status. Thoracic Cancer. 2018;9(8):967–973. doi:10.1111/1759-7714.12772
3. Rang KY, Bada L, Ran BM, Kil LJ, Woo CJ, Muy-Teck T. Evaluation of pemetrexed and etoposide as therapeutic regimens for human papillomavirus-positive oral and oropharyngeal cancer. PLoS One. 2018;13(7):e0200509. doi:10.1371/journal.pone.0200509
4. Liu X, Luo L, Qi P, et al. A comprehensive preclinical evaluation of intravenous etoposide lipid emulsion. Pharm Res. 2019;36(7):1–11. doi:10.1007/s11095-019-2637-0
5. Smyth RD, Pfeffer M, Scalzo AJ, Comis RL. Bioavailability and pharmacokinetics of etoposide (VP-16). Semin Oncol. 1985;12:48–51.
6. Li X, Choi J. Effects of quercetin on the pharmacokinetics of etoposide after oral or intravenous administration of etoposide in rats. Anticancer Res. 2009;29(4):1411–1415.
7. Reddy LH, Murthy RSR. Etoposide-loaded nanoparticles made from glyceride lipids: formulation, characterization, in vitro drug release, and stability evaluation. AAPS Pharmscitech. 2005;6(2):E158E166. doi:10.1208/pt060224
8. Yadav KS, Sawant KK. Formulation optimization of etoposide loaded PLGA nanoparticles by double factorial design and their evaluation. Curr Drug Deliv. 2010;7(1):51–64. doi:10.2174/156720110790396517
9. Reddy LH, Sharma RK, Chuttani K, Mishra AK, Murthy RSR. Etoposide-incorporated tripalmitin nanoparticles with different surface charge: formulation, characterization, radiolabeling, and biodistribution studies. Aaps J. 2004;6(3):55–64. doi:10.1208/aapsj060323
10. Peltier S, Oger J, Lagarce F, Couet W, Benoit J. Enhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded lipid nanocapsules. Pharm Res. 2006;23(6):1243–1250. doi:10.1007/s11095-006-0022-2
11. Jahangir MA, Khan R, Imam SS. Formulation of sitagliptin-loaded oral polymeric nano scaffold: process parameters evaluation and enhanced anti-diabetic performance. Artificial Cells Nanomed Biotechnol. 2018;46:66–78. doi:10.1080/21691401.2017.1411933
12. Imam SS, Aqil M, Akhtar M, Sultana Y, Ali A. Formulation by design-based proniosome for accentuated transdermal delivery of risperidone: in vitro characterization and in vivo pharmacokinetic study. Drug Deliv. 2015;22(8):1059–1070. doi:10.3109/10717544.2013.870260
13. Van Zuylen L, Verweij J, Sparreboom A. Role of formulation vehicles in taxane pharmacology. Invest New Drugs. 2001;19(2):125–141. doi:10.1023/A:1010618632738
14. Zhang T, Chen J, Zhang Y, Shen Q, Pan W. Characterization and evaluation of nanostructured lipid carrier as a vehicle for oral delivery of etoposide. European J Pharmaceutical Sci. 2011;43(3):174–179. doi:10.1016/j.ejps.2011.04.005
15. Zhang H, Hollis CP, Zhang Q, Li T. Preparation and antitumor study of camptothecin nanocrystals. Int J Pharm. 2011;415(1):293–300. doi:10.1016/j.ijpharm.2011.05.075
16. Borhade V, Pathak S, Sharma S, Patravale VB. Formulation and characterization of atovaquone nanosuspension for improved oral delivery in the treatment of malaria. Nanomedicine. 2014;9(5):649–666. doi:10.2217/nnm.13.61
17. Alshehri S, Imam S, Altamimi M, et al. Enhanced dissolution of luteolin by solid dispersion prepared by different methods: physicochemical characterization and antioxidant activity. ACS Omega. 2020;5(12):6461–6471. doi:10.1021/acsomega.9b04075
18. Gurunath S, Kumar SP, Basavaraj NK, Patil PA. Amorphous solid dispersion method for improving oral bioavailability of poorly water-soluble drugs. J Pharmacy Res. 2013;6(4):476–480. doi:10.1016/j.jopr.2013.04.008
19. Lei G, Liu G, Ma J, et al. Application of drug nanocrystal technologies on oral drug delivery of poorly soluble drugs. Pharm Res. 2013;30(2):307–324. doi:10.1007/s11095-012-0889-z
20. Pawar VK, Singh Y, Meher JG, Gupta S, Chourasia MK. Engineered nanocrystal technology: in-vivo fate, targeting and applications in drug delivery. J Controlled Release. 2014;183:51–66. doi:10.1016/j.jconrel.2014.03.030
21. Chang D, Ma Y, Cao G, et al. Improved oral bioavailability for lutein by nanocrystal technology: formulation development, in vitro and in vivo evaluation. Artificial Cells, Nanomed Biotechnol. 2017;46(5):1018–1024. doi:10.1080/21691401.2017.1358732
22. Prachi S, Milind B, Diptee E, Varsha P. Enhanced dissolution/caco-2 permeability, pharmacokinetic and pharmacodynamic performance of re-dispersible eprosartan mesylate nanopowder. European J Pharmaceutical Sci. 2019;132:72–85. doi:10.1016/j.ejps.2019.02.021
23. Pu X, Sun J, Han J, et al. Nanosuspensions of 10-hydroxycamptothecin that can maintain high and extended supersaturation to enhance oral absorption: preparation, characterization and in vitro/in vivo evaluation. J Nanoparticle Res. 2013;15(11):1–13. doi:10.1007/s11051-013-2043-1
24. Hoo CM, Starostin N, West P, Mecartney ML. A comparison of atomic force microscopy (AFM) and dynamic light scattering (DLS) methods to characterize nanoparticle size distributions. J Nanoparticle Res. 2008;10(1):89–96. doi:10.1007/s11051-008-9435-7
25. Panthani MG, Hessel CM, Reid D, Casillas G, José-Yacamán M, Korgel BA. Graphene-supported high-resolution TEM and STEM imaging of silicon nanocrystals and their capping ligands. J Physical Chemistry C. 2012;116(42):22463–22468. doi:10.1021/jp308545q
26. Ali HSM, York P, Blagden N. Preparation of hydrocortisone nanosuspension through a bottom-up nanoprecipitation technique using microfluidic reactors. Int J Pharm. 2009;375(1):107–113. doi:10.1016/j.ijpharm.2009.03.029
27. Leng D, Chen H, Li G, et al. Development and comparison of intramuscularly long-acting paliperidone palmitate nanosuspensions with different particle size. Int J Pharm. 2014;472(12):380–385. doi:10.1016/j.ijpharm.2014.05.052
28. Mitri K, Shegokar R, Gohla S, Anselmi C, Müller RH. Lutein nanocrystals as antioxidant formulation for oral and dermal delivery. Int J Pharm. 2011;420(1):141–146. doi:10.1016/j.ijpharm.2011.08.026
29. Duncan GF, Farmen RH, Movahhed HS, Pittman KA. High-performance liquid chromatographic method for the determination of etoposide in plasma using electrochemical detection. J Chromatogr B Biomed Sci Appl. 1986;380(2):357–365. doi:10.1016/S0378-4347(00)83664-6
30. Zhuo X, Lei T, Miao L, et al. Disulfiram-loaded mixed nanoparticles with high drug-loading and plasma stability by reducing the core crystallinity for intravenous delivery. J Colloid Interface Sci. 2018;529:34–43. doi:10.1016/j.jcis.2018.05.057
31. Chen H, Shi S, Zhao M, Zhang L, He H, Tang X. A lyophilized etoposide submicron emulsion with a high drug loading for intravenous injection: preparation, evaluation, and pharmacokinetics in rats. Drug Dev Ind Pharm. 2010;36(12):1444–1453. doi:10.3109/03639045.2010.487267
32. Shegokar R, Müller RH. Nanocrystals: industrially feasible multifunctional formulation technology for poorly soluble actives. Int J Pharm. 2010;399(12):129–139. doi:10.1016/j.ijpharm.2010.07.044
33. Rabinow BE. Nanosuspensions in drug delivery. Nat Rev Drug Discov. 2004;3(9):785. doi:10.1038/nrd1494
34. Zhou T, Qian G, Zhou XG, Yuan WK. Effect of ultrasound on anti-solvent crystallization process of glycin. Chinese J Process Engineering. 2007;10(7):413–416.
35. Hem SL. The effect of ultrasonic vibrations on crystallization processes. Ultrasonics. 1967;5(4):202–207. doi:10.1016/0041-624X(67)90061-3
36. Moschwitzer JP. Drug nanocrystals in the commercial pharmaceutical development process. Int J Pharm. 2013;453(1):142–156. doi:10.1016/j.ijpharm.2012.09.034
37. Moschwitzer JP, Muller RH. New method for the effective production of ultrafine drug nanocrystals. J Nanosci Nanotechnol. 2006;6(9):3145–3153. doi:10.1166/jnn.2006.480
38. Alshehri S, Imam SS, Altamimi MA, et al. Host-guest complex of β-cyclodextrin and pluronic F127 with Luteolin: physicochemical characterization, anti-oxidant activity and molecular modeling studies. J Drug Deliv Sci Technol. 2020;55:101356. doi:10.1016/j.jddst.2019.101356
39. Ma M, Zhang G, Li W, Li M, Fu Q, He Z. A carbohydrate polymer is a critical variable in the formulation of drug nanocrystals: a case study of idebenone. Expert Opin Drug Deliv. 2019;16(12):1–9. doi:10.1080/17425247.2019.1682546
40. Patel K, Patil A, Mehta M, Gota V, Vavia P. Oral delivery of paclitaxel nanocrystal (PNC) with a dual Pgp-CYP3A4 inhibitor: preparation, characterization and antitumor activity. Int J Pharm. 2014;472(1):214–223. doi:10.1016/j.ijpharm.2014.06.031
41. Junyaprasert VB, Morakul B. Nanocrystals for enhancement of oral bioavailability of poorly water-soluble drugs. Asian J Pharmaceutical Sci. 2015;10(1):13–23. doi:10.1016/j.ajps.2014.08.005
42. Buckton G, Beezer AE. The relationship between particle size and solubility. Int J Pharm. 1992;82(3):R7–R10. doi:10.1016/0378-5173(92)90184-4
43. Roig MG, Rashid DH, Kennedy JF. Rheological studies on the solubilization by the surfactant SDS of complexes between three acidic polysaccharides and an organic base chloride. J Biomaterials Science-Polymer Edition. 1997;8(7):493–516. doi:10.1163/156856297X00416
44. Sahu BP, Das MK. Preparation and in vitro/in vivo evaluation of felodipine nanosuspension. Eur J Drug Metab Pharmacokinet. 2014;39(3):183–193. doi:10.1007/s13318-013-0158-5
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.