Back to Journals » Clinical Ophthalmology » Volume 14
Etiologies of Acute Optic Neuritis in Thailand: An Observational Study of 171 Patients
Authors Vanikieti K, Janyaprasert P, Lueangram S, Nimworaphan J ![]() , Rattanathamsakul N, Tiraset N, Chokthaweesak W, Samipak N
, Rattanathamsakul N, Tiraset N, Chokthaweesak W, Samipak N ![]() , Padungkiatsagul T
, Padungkiatsagul T ![]() , Preechawat P, Poonyathalang A
, Preechawat P, Poonyathalang A ![]() , Pulkes T
, Pulkes T ![]() , Tunlayadechanont S, Siriyotha S, Jindahra P
, Tunlayadechanont S, Siriyotha S, Jindahra P ![]()
Received 11 July 2020
Accepted for publication 31 August 2020
Published 30 September 2020 Volume 2020:14 Pages 2935—2942
DOI https://doi.org/10.2147/OPTH.S271820
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Kavin Vanikieti,1 Pavarut Janyaprasert,1 Sirin Lueangram,1 Jirat Nimworaphan,1 Natthapon Rattanathamsakul,1 Nanida Tiraset,1 Wimonwan Chokthaweesak,1 Narong Samipak,1 Tanyatuth Padungkiatsagul,1 Pisit Preechawat,1 Anuchit Poonyathalang,1 Teeratorn Pulkes,2 Supoch Tunlayadechanont,2 Sukanya Siriyotha,3 Panitha Jindahra2
1Department of Ophthalmology, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok 10400, Thailand; 2Division of Neurology, Department of Medicine, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok 10400, Thailand; 3Department of Clinical Epidemiology and Biostatistics, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok 10400, Thailand
Correspondence: Panitha Jindahra
Division of Neurology, Department of Medicine, Faculty of Medicine Ramathibodi Hospital, Mahidol University, 270 Rama VI Road, Thung Phaya Thai, Ratchathewi, Bangkok 10400, Thailand
Tel +6622011386
Email [email protected]
Purpose: To analyze the demographic patterns, clinical characteristics and etiologies of acute optic neuritis (ON).
Methods: This retrospective observational study included patients with acute ON who presented to a university hospital in Bangkok, Thailand, between January 2010 and March 2020. The demographic details, clinical characteristics and etiologies of acute ON were evaluated.
Results: A total of 171 patients were included in the study (78.4% [n=134] female; mean age 45 years [standard deviation 15.4 years]; 32.2% [n=55] bilateral involvement). The most common type of acute ON was idiopathic (51.5%), followed by neuromyelitis optica spectrum disorder (NMOSD, 30.9%), other autoimmune disorders (9.9%), myelin oligodendrocyte glycoprotein antibody-associated disorder (MOGAD, 5.3%), multiple sclerosis (MS, 1.8%), and postinfection (0.6%). In the other autoimmune disorders group, 2 patients developed systemic lupus erythematosus (1.2%), 2 Sjogren’s syndrome (1.2%), 1 RA (0.6%), 1 anti-NMDAR (0.6%), 3 anti-Jo1 (1.8%), 2 c-ANCA (1.2%), 1 anti-centromere (0.6%), and 5 nonspecific autoimmune disorders (2.9%). In the idiopathic group, 38.6% developed single isolated ON, 1.8% relapsing isolated ON and 11.1% chronic relapsing inflammatory optic neuropathy.
Conclusion: The most common form of acute ON in this study, similar to other Asian countries, was idiopathic. Idiopathic-ON shared some phenotypes with NMOSD and MOGAD. We also reported patients with anti-NMDAR, anti-Jo1, c-ANCA and anti-centromere disorders. Improvements in antibody detection have widened the range of possible etiologies of acute ON. The study highlighted the important role of antibodies in creating effective treatments in the future.
Keywords: optic neuritis, multiple sclerosis, neuromyelitis optica spectrum disorder, myelin oligodendrocyte glycoprotein antibody-associated disorder, autoimmune disorders, postinfection
Introduction
Optic neuritis (ON) is an acute inflammatory disorder of the optic nerve. The annual incidence of ON worldwide is 1–6.4 per 100,000 adults.1–3 It is a common disease with various etiologies, including infectious and immune-mediated processes.1 The present study focused mainly on immune-mediated ON. Multiple sclerosis-associated ON (MS-ON), or so-called typical ON, has strongly affected white people of European background.4 Approximately 50% of patients with typical ON will develop MS within 15 years, according to the Optic Neuritis Treatment Trial.5 Patients with MS-ON carry a good visual prognosis. The MS conversion rate following initial ON manifestation varies substantially among different countries. It was estimated to be 13–87% in Europe and North America, 8.3% in Japan, 12% in Mexico and 14.3% in Taiwan.6–13 Additionally, a study in Taiwan revealed that the cumulative incidence of MS after a new diagnosis of ON was only 0.78%.14 Different etiologies of ON could be accountable for this variety across studies. Specific biomarkers of ON were recently discovered, including aquaporin-4 immunoglobulin (AQP4-IgG) and myelin oligodendrocyte glycoprotein immunoglobulin (MOG-IgG).15 These antibodies establish the distinct entities of neuromyelitis optica spectrum disorder (NMOSD) and MOG-IgG-associated disorder (MOGAD), respectively.15 Their clinical manifestations, prognoses and treatments differ from those for MS. Therefore, it is essential to reinvestigate the causes of ON in the community. The goal of this study was to describe the etiologies and clinical characteristics of ON among patients in a university hospital in Bangkok.
Patients and Methods
This retrospective observational study included patients with acute ON, who presented to Ramathibodi Hospital, a university hospital in Bangkok, Thailand, between January 1, 2010 and March 31, 2020. The diagnosis of ON was made clinically based on typical signs of optic neuropathy, including acute loss of vision, dyschromatopsia, positive relative afferent pupillary defect, and visual field defect, with or without optic disc swelling. All patients underwent thorough neuro-ophthalmological, neurological and systemic examinations. Inclusion criteria were age ≥ 16 years and first presentation of acute immune-mediated ON. Exclusion criteria were age < 16 years; anterior segment, vitreous or retinal involvement; and other causes of optic neuropathy, such as infection, compression, toxin, ischemia, hereditary or trauma. The patients’ medical records were retrospectively reviewed for age, sex, presence of pain on ocular movement, bilaterality, initial visual acuity (VA), presence of swollen discs and blood tests for autoimmune disease biomarkers. The Snellen VA chart was converted to the logarithm of the minimal angle of resolution (logMAR) equivalent. Autoimmune disease biomarkers were available in a routine panel, including AQP4-IgG (cell-based assay), MOG-IgG, antinuclear antibody, anti-double-stranded DNA antibody, anti-centromere, anti-Ro/SSA, anti-La/SSB, rheumatoid factors, anti-neutrophil cytoplasmic antibody and anti-Jo1 antibody. Note that MOG-IgG tests only became available recently (in the last few years). Anti-N-methyl D-aspartate receptor (anti-NMDAR) antibody tests were performed only in suspected cases. Magnetic resonance imagings (MRIs) of the orbit, brain, and spinal cord were reviewed retrospectively. The orbit and brain MRIs were carried out in all patients, while the spinal cord MRIs were performed only in patients with symptoms and signs of transverse myelitis. Spinal cord MRIs confirmed the presence of transverse myelitis. The MRI examinations were performed on two different scanners, a 3.0T scanner (Ingenia; Philips Healthcare, Best, the Netherlands) and a 1.5T scanner (Signa TwinSpeed; GE Healthcare), using our standard brain and orbit MRI protocols that included axial and coronal contrast-enhanced T1-weighted images with fat suppression (CE-T1W/FS), T2-weighted images with fat suppression and axial fluid-attenuation inversion recovery images (FLAIR). Etiologies of acute ON were categorized into six types: MS, NMOSD, MOGAD, other autoimmune disorders, postinfection (possible autoimmune reaction) and idiopathic. NMOSD was further classified into two subtypes according to seropositivity to AQP4-IgG. Other autoimmune disorders included systemic lupus erythematosus (SLE), Sjogren’s syndrome, rheumatoid arthritis (RA), anti-NMDAR, anti-Jo1, c-ANCA, anti-centromere, and nonspecific autoimmune disorders. Patients with nonspecific autoimmune disorders had positive antinuclear antibodies, but there was not enough evidence to fulfill any criteria of an autoimmune disorder. Idiopathic-ON was further classified into single isolated ON (SION), relapsing isolated ON (RION), and chronic relapsing inflammatory optic neuropathy (CRION) (see below).
Diagnoses
MS was diagnosed based on the 2010 McDonald criteria.16 NMOSD was diagnosed based on the 2015 Wingerchuk diagnostic criteria.17 MOGAD was diagnosed based on the 2018 MOG-IgG-associated disorder diagnostic criteria.18 SLE was diagnosed based on the 2019 American College of Rheumatology/European League Against Rheumatism Collaborative (ACR/EULAR) initiative classification criteria for SLE.19 Sjögren’s syndrome was diagnosed based on the 2012 American College of Rheumatology classification criteria for Sjögren’s syndrome.20 Rheumatoid arthritis (RA) was diagnosed based on the 2010 ACR/EULAR classification criteria for RA.21 Postinfectious ON (possible autoimmune reaction) was diagnosed when ON developed following resolved infections and was responsive to steroid treatments. Idiopathic-ON was diagnosed when no cause was found. SION was defined as a single and isolated episode of ON. RION was defined as spontaneously relapsing isolated episodes of ON. CRION referred to isolated episodes of relapses of ON on steroid withdrawal.22
Statistical Analysis
Descriptive analyses (frequency and percentage) were performed for clinical and radiological features, including sex, age, initial VA, race, pain, bilaterality of ON, optic disc appearances, and enhancement of the anterior visual pathway. The mean (SD) and median (range) were presented for continuous variables. One-way analysis of variance was analyzed to compare age and initial VA between groups. Chi-square tests were analyzed to compare other clinical and radiological features between groups. A p-value of less than 0.05 was considered statistically significant. Stata version 16 (College Station, TX: Statacorp LLC.) was used in the analysis.
Ethical Issue
The study was approved by the Ethical Clearance Committee on Human Rights Related to Research Involving Human Subjects, Faculty of Medicine Ramathibodi Hospital, Mahidol University (Trial number MURA 2019/156). All participants and carers gave written informed consent to participate in the study.
Results
A total of 171 patients were included in the study (78.4% [n=134] female; mean age 45 years [standard deviation (SD) 15.4 years]; 32.2% [n=55] bilateral involvement; and 51.5% [n=88] pain on ocular movement). The mean initial logMAR VA was 1.7 (SD 1.1). Of all affected eyes, 73 (33.3%) had swollen optic discs. Of all patients with ON, there were 3 patients with MS (1.8%), 53 NMOSD (30.9%), 9 MOGAD (5.3%), 17 other autoimmune disorders (9.9%), 1 postinfection (0.6%) and 88 idiopathic (51.5%) (Figure 1). Among those with NMOSD, 47 patients were seropositive for AQP4-IgG (27.5%) and 6 were seronegative (3.5%). These six patients with seronegative NMOSD developed transverse myelitis in the course of their illnesses. In the other autoimmune disorders group, 2 patients developed SLE (1.2%), 2 Sjogren’s syndrome (1.2%), 1 RA (0.6%), 1 anti-NMDAR (0.6%), 3 anti-Jo1 (1.8%), 2 c-ANCA (1.2%), 1 anti-centromere (0.6%), and 5 nonspecific autoimmune disorders (2.9%). In the idiopathic group, 66 developed SION (38.6%), 3 RION (1.8%) and 19 CRION (11.1%). Clinical manifestations of each etiology of ON are shown in Table 1. Radiological phenotypes are shown in Table 2 and Figure 2. Sex and optic disc appearances were significantly different among the groups. The MOGAD group had the lowest female to male ratio. Most patients with MOGAD developed optic disc swelling, whereas most patients with NMOSD, other autoimmune disorders, and idiopathic had normal optic discs. There was no statistically significant difference regarding age, pain, bilaterality, initial VA, and location of optic nerve enhancement among all groups. Subgroup analysis was performed for the MS, NMOSD, and MOGAD groups. Sex and optic disc appearances were significantly different among the three groups. There was no significant difference regarding age, pain, bilaterality, initial VA, and location of optic nerve enhancement in the subgroup analysis.
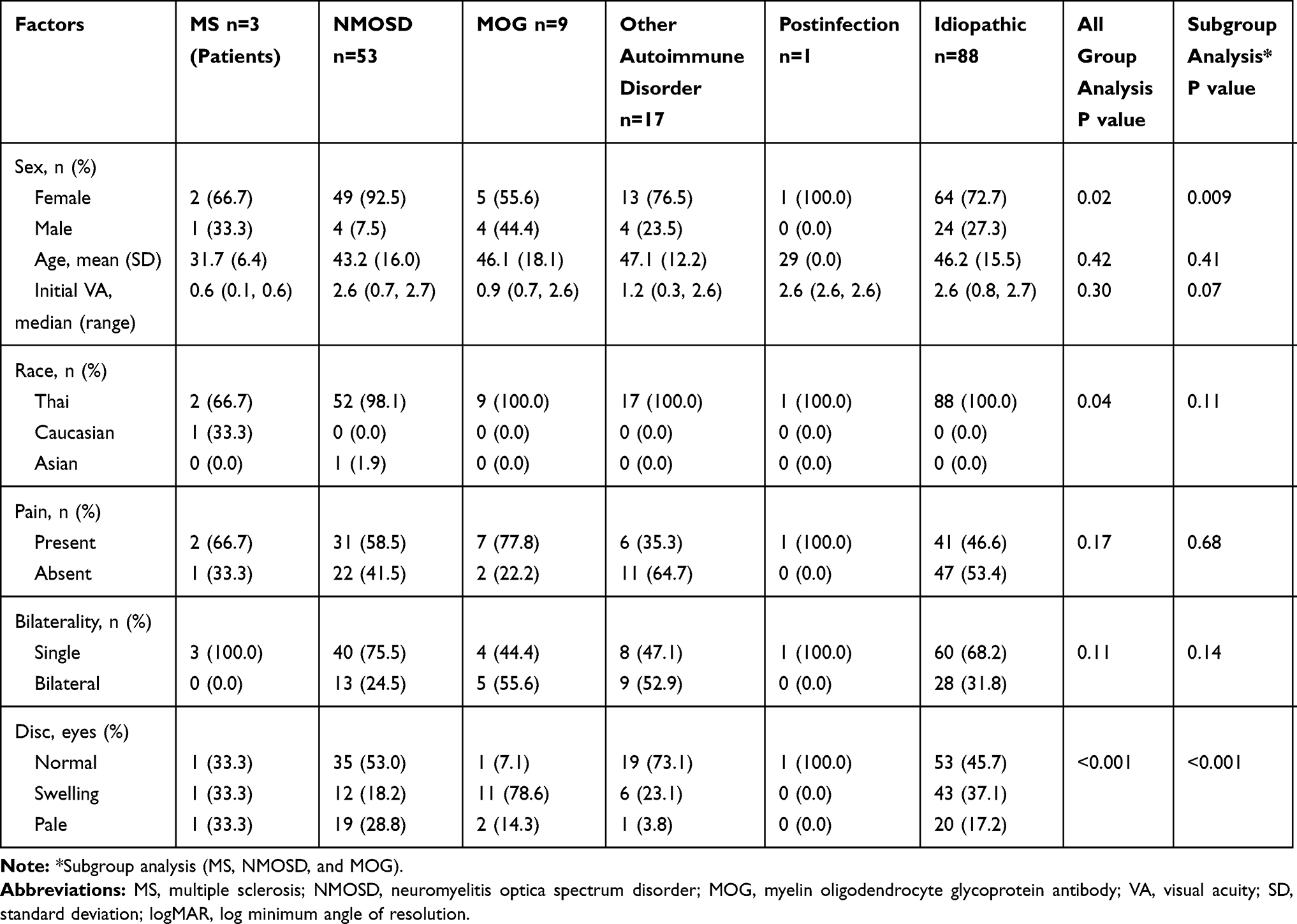 |
Table 1 Patients’ Demographics, Grouped by Diagnosis |
 |
Table 2 Frequency of Enhanced Optic Nerves on Contrast-Enhanced T1-Weighted Orbital Magnetic Resonance Imaging with Fat Suppression in Different Segments of the Optic Nerve |
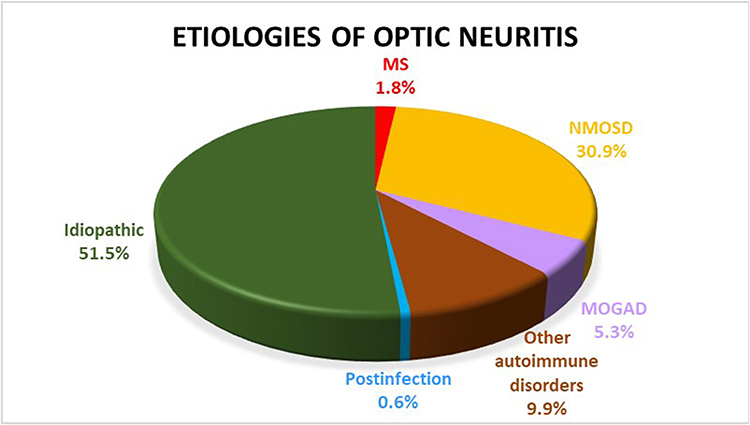 |
Figure 1 Pie-chart showing the frequencies of etiologies of acute optic neuritis. |
 |
Figure 2 Fat-suppressed T1-weighted magnetic resonance imaging of the orbits with contrast shows (A) right optic nerve enhancement at the intraorbital segment in a patient with MS; (B) right optic nerve enhancement from the intraorbital segment to the intracranial segment in a patient with NMOSD; (C) bilateral optic nerve enhancement at the intraorbital and intracanal segments with optic nerve sheath and peribulbar fat involvement in a patient with MOGAD; (D) bilateral optic nerve involvement in a patient with CRION and anti-Jo1; and (E) mild enhancement of the perioptic nerve sheath complex at the intracanalicular segment of the left optic nerve in a patient with RION and idiopathic ON. |
A patient with anti-NMDAR antibody was a 22-year-old man with a history of aseptic meningitis. He initially presented with two episodes of bilateral ON, followed by transverse myelitis, double vision, seizure, hallucination and confusion within a few months. His logMAR VA at the start of the second episode was 1.1 bilaterally, which was restored to 0.18 and 0.0 following steroid treatment. Both optic discs and other ophthalmologic findings were unremarkable. Contrast-enhanced T1-weighted (CE-T1W) orbital MRI demonstrated enhancement of both optic nerves in the intracanal and intracranial segments. CE-T1W brain MRI revealed enhanced lesions at the bilateral temporal lobes, basal ganglia, periventricular area and right cerebellar peduncle. Spinal cord MRIs revealed extensive longitudinal cord involvement. AQP-IgG and other antibodies were negative. He was later tested for anti-NMDAR antibody, which turned out to be positive. He was subsequently diagnosed with anti-NMDAR limbic encephalitis.
There were three patients with anti-Jo1. The first patient was a 61-year-old man with 3+ anti-Jo1 and a history of chronic obstructive pulmonary disease. He had painless bilateral CRION, logMAR VA 1.3 in both eyes, and normal optic discs. Other neurological examinations were normal. Both optic nerves were enhanced from the intraorbital segment to the chiasm on CE-T1W orbital MRI. His brain MRI was normal. He responded well to steroid treatment and experienced no further relapses of ON following initiation of mycophenolate mofetil. The second case was a 39-year-old woman with 1+ anti-Jo1. She had painless bilateral SION, logMAR VA 0.7 in both eyes, normal optic discs, normal neurological examinations, and no optic nerve enhancement. Brain MRI (FLAIR) revealed nonspecific, hyperintense foci in the subcortical white matter of both fronto-parieto-temporal lobes. Her vision recovered well after steroid treatment. The third case was a 45-year-old woman with 1+ anti-Jo1. She had painless unilateral SION, logMAR VA 2.6, normal optic discs, and normal neurological examinations. There was enhancement of the optic nerve in the intracanal and intracranial segments. Her brain MRI revealed pachymeningitis at the bilateral anterior cranial fossa and bilateral olfactory bulbs. Her vision recovered well following steroid treatment.
Two patients were seropositive for c-ANCA but did not report any systemic symptoms. The first case was a 45-year-old woman with painless bilateral SION, logMAR VA 2.6 on the right and 2.7 on the left, normal optic discs, and no optic nerve enhancement. There were bilateral periventricular lesions on FLAIR. The second patient was a 58-year-old woman with painful bilateral SION, logMAR VA 2.9 in both eyes, mild disc swelling, enhancement of optic nerves at the intraorbital and intracanal segments, and normal brain MRIs. Both patients responded well to steroids.
A 54-year-old woman with anti-centromere antibody had painful unilateral CRION, logMAR VA 0.9, normal optic disc appearance, an enhanced optic nerve at the intracranial segment, and pachymeningitis on brain MRIs.
Discussion
This study demonstrated that the most common etiology of acute ON at a university hospital in Bangkok, Thailand, was idiopathic, followed by NMOSD, other autoimmune disorders, MOGAD, MS, and postinfection. Severe visual loss and enhancement of the long segment of the optic nerve were frequent findings. Sex and optic disc appearances were important clinical features that are helpful in clinical consideration. The etiologies were generally similar to those observed in other Asian countries.23–27 The results were also comparable to those reported in Chiangmai, which lies in northern Thailand.28 A slight difference is that the number of MS cases was relatively higher in Chiangmai Province. In China, the most frequent etiology was idiopathic according to Zhang et al.23 Other autoimmune disorders and NMOSD were common etiologies in another Chinese series.24 Chinese patients ≥ 65-year-old were less likely to develop NMOSD or MS. They experienced severe visual loss and a poor visual prognosis.24 Japanese and Korean studies revealed that the most common etiology was idiopathic, followed by NMOSD, MOGAD and MS.25,26 The proportion of MS in these studies was relatively low. Meanwhile, the majority of South African patients with immune-mediated ON developed idiopathic-ON with atypical features.29 Idiopathic-ON clinical profiles in South African patients were different from those in Caucasian patients. South African patients tended to have bilateral involvement, severe visual loss, disc swelling and a weak association with MS.30 Patients with ON from African or African-Caribbean backgrounds were > 3 times more likely to develop NMOSD, compared with patients of Caucasian backgrounds.31 Conversely, MS was the most common etiology of ON in Caucasian, Saudi Arabian and Indian patients.4,32–34 A study in Turkey revealed that idiopathic-ON and MS were the most frequent etiologies, whereas NMOSD was the least frequent etiology.35 These studies suggest that the frequency of ON etiologies varies among patients of different ethnic backgrounds. Early recognition of the specific cause of ON is important to prevent severe visual loss and avoid inappropriate treatments. It is also necessary to make thorough examinations in Asian and African patients before diagnosing MS.
The number of patients with MS patients in this study was small. Like Western studies, their clinical and radiological presentations were classical for MS-ON.1,36 They developed unilateral visual loss with variable severity. Ocular pain was a common finding. Optic nerve enhancement was localized anteriorly at the intraorbital and intracanal segments.
Clinical features in patients with NMOSD in this study were in good agreement with those in Western literature.17,37 They presented with severe visual loss, simultaneous bilateral involvement, enhancement of the long segment of the optic nerve, and chiasmal involvement.
Several features of MOGAD in this study were consistent with those in other Western and Asian studies.38 Unlike MS and NMOSD, many studies have revealed that MOGAD equally affects males and females. The prominent clinical feature is optic disc swelling. The proportion of the age of onset, VA, and optic nerve neuroimaging is similar between patients of different races. Caucasian patients are more likely to experience ocular pain, recurrent attacks, and other central nervous system involvement. Asian patients tend to have disease isolated to the optic nerve.38
ON is relatively uncommon in the course of SLE, Sjogren’s syndrome and RA.39,40 It can be either the first presentation of the autoimmune disorders or present after the patient receives medications. The disorders have specific treatments and are steroid-dependent. In this study, we reported anti-NMDAR-associated ON. The observations suggest that ON or transverse myelitis can be an early manifestation of anti-NMDAR limbic encephalitis.41 The clinical phenotypes mimic seronegative NMOSD. Moreover, we have also reported three cases of ON with anti-Jo1 antibody. The severity of ON was related to the level of the antibody. The patient with 3+ anti-Jo1 developed severe bilateral visual loss, long optic nerve involvement, and steroid-dependence. The other two patients, with 1+ anti-Jo1, had a relatively mild course of the disease. As far as we are aware, there has been no previous report of anti-Jo1-associated ON. The antibody usually causes anti-synthetase syndrome, interstitial lung disease and myositis.39,40,42,43 Anti-centromere and c-ANCA in ON have been occasionally reported in systemic sclerosis and granulomatosis with polyangiitis, respectively.44,45
Idiopathic-ON had some special characteristics, such as severe visual loss, abnormal enhancement of the chiasm and tract, and involvement of the long segment of the optic nerve. These cases shared the same phenotypes as NMOSD and MOGAD. It is possible that these idiopathic cases were caused by undetected AQP4-IgG, MOG-IgG or unknown antibodies. In a cohort study, patients with SION, RION, CRION and MS-ON were reinvestigated for AQP4-IgG and MOG-IgG. More cases were reclassified to the NMOSD and MOGAD groups according to the antibody status.46 The number of idiopathic cases may decline with improved detection of antibodies. A limitation of the study was that visual evoked potential and optical coherence tomography were not evaluated.
Conclusion
The most common cause of acute ON in this study was idiopathic. The finding was similar to those from other Asian countries. Idiopathic-ON shared some phenotypes with NMOSD and MOGAD. We have also reported cases of patients with ON with anti-NMDAR, anti-Jo1, c-ANCA and anti-centromere antibodies. The improvement in antibody detection has widened the range of possible etiologies of acute ON. The study highlighted the important role of antibodies in creating effective treatments in the future.
Acknowledgments
The study was supported by Division of Neurology, Department of Medicine, Faculty of Medicine Ramathibodi Hospital, Mahidol University.
Disclosure
Natthapon Rattanathamsakul reports personal fees from Novartis, Thailand, outside the submitted work. The authors report no other potential conflicts of interest in this work.
References
1. Pau D, Zubidi NAI, Yalamanchili S, Plant GT, Lee AG. Optic neuritis. Eye (Lond). 2011;25(7):833–842. doi:10.1038/eye.2011.81
2. Lee JY, Han J, Yang M, Oh SY. Population-based incidence of pediatric and adult optic neuritis and the risk of multiple sclerosis. Ophthalmology. 2020;127(3):417–425. doi:10.1016/j.ophtha.2019.09.032
3. Wakakura M, Ishikawa S, Oono S, et al. Incidence of acute idiopathic optic neuritis and its therapy in Japan. Optic neuritis treatment trial multicenter cooperative research group (ONMRG). Nippon Ganka Gakkai Zasshi. 1995;99(1):93–97.
4. Amezcua L, McCauley JL. Race and ethnicity on MS presentation and disease course. ACTRIMS forum 2019. Mult Scler. 2020;26(5):561–567. doi:10.1177/1352458519887328
5. The Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final optic neuritis treatment trial follow-up. Arch Neurol. 2008;65(6):727–732.
6. Söderström M. Optic neuritis and multiple sclerosis. Acta Ophthalmol Scand. 2001;79(3):223–227. doi:10.1034/j.1600-0420.2001.790302.x
7. Sandberg-Wollheim M, Bynke H, Cronqvist S, et al. A long-term prospective study of optic neuritis: evaluation of risk factors. Ann Neurol. 1990;27(4):386–393. doi:10.1002/ana.410270406
8. Rodriguez M, Siva A, Cross SA, et al. Optic neuritis: a population-based study in Olrnsted County, Minnesota. Neurology. 1995;45(2):244–245. doi:10.1212/WNL.45.2.244
9. O’Riordan JI, Thompson AJ, Kingsley DP, et al. The prognostic value of brain MRI in clinically isolated syndromes of the CNS: a 10-year follow-up. Brain. 1998;121(pt3):495–503. doi:10.1093/brain/121.3.495
10. Söderström M, Ya-Ping J, Hillert J, et al. Optic neuritis: prognosis for multiple sclerosis from MRI, CSF, and HLA findings. Neurology. 1998;50(3):708–714. doi:10.1212/WNL.50.3.708
11. Isayama Y, Takahashi T, Shimoyoma T, et al. Acute optic neuritis and multiple sclerosis. Neurology. 1982;32(1):73–76. doi:10.1212/WNL.32.1.73
12. Corona-Vazquez T, Ruiz-Scadoval J, Arriada-Mendicoa N. Optic neuritis progression to multiple sclerosis. Acta Neurol Scand. 1997;95(2):85–89. doi:10.1111/j.1600-0404.1997.tb00074.x
13. Lin YC, Yen MY, Hsu WM, Lee HC, Wang AG. Low conversion rate to multiple sclerosis in idiopathic optic neuritis patients in Taiwan. Jpn J Ophthalmol. 2006;50(2):170–175. doi:10.1007/s10384-005-0281-1
14. Woung L-C, Lin C-H, Tsai C-Y, et al. Optic neuritis among national health insurance enrollees in Taiwan, 2000–2004. Neuroepidemiology. 2007;29(3–4):250–254. doi:10.1159/000112858
15. Chen JJ, Pittock SJ, Flanagan EP, et al. Optic neuritis in the era of biomarkers. Surv Ophthalmol. 2020;65(1):12–17. doi:10.1016/j.survophthal.2019.08.001
16. Polman CH, Reingold SC, Banwaell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292–302. doi:10.1002/ana.22366
17. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177–189. doi:10.1212/WNL.0000000000001729
18. López-Chiriboga AS, Majed M, Fryer J, et al. Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG-associated disorders. JAMA Neurol. 2018;75(11):1355–1363. doi:10.1001/jamaneurol.2018.1814
19. Aringer M, Costenbader K, Daikh D, et al. 2019 European league against rheumatism/American college of rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 2019;71(9):1400–1412. doi:10.1002/art.40930
20. Shiboski SC, Shiboski CH, Criswell LA, et al. American college of rheumatology classification criteria for Sjögren’s syndrome: a data-driven, expert consensus approach in the SICCA cohort. Arthritis Care Res (Hoboken). 2012;64(4):475–487. doi:10.1002/acr.21591
21. Aletaha D, Neogi T, Silman AJ, et al. 2010 rheumatoid arthritis classification criteria. An American college of rheumatology/European league against rheumatism collaborative initiative. Arthritis Rheumatol. 2010;62(9):2569–2581. doi:10.1002/art.27584
22. Petzold A, Wattjes MP, Costello F, et al. The investigation of acute optic neuritis: a review and proposed protocol. Nat Rev Neurol. 2014;10(8):447–458. doi:10.1038/nrneurol.2014.108
23. Zhang X, Wang W, Wei W, et al. Etiological profile of presumptive optic neuritis in China. J Clin Neurosci. 2008;15(12):1346–1349. doi:10.1016/j.jocn.2008.01.012
24. Wang J, Zhou H, Qin L, et al. Optic neuritis in the older Chinese population: a 5-year follow-up study. J Ophthalmol. 2017;2017:3458356. doi:10.1155/2017/3458356
25. Kim HJ, Park K-A, S Y. O, et al. Association of optic neuritis with neuromyelitis optica spectrum disorder and multiple sclerosis in Korea. Korean J Ophthalmol. 2019;33(1):82–90. doi:10.3341/kjo.2018.0050
26. Ishikawa H, Kezuka T, Shikishima K, et al. Epidemiologic and clinical characteristics of optic neuritis in Japan. Ophthalmology. 2019;126(10):1385–1398. doi:10.1016/j.ophtha.2019.04.042
27. Lim SA, Goh KY, Tow S, et al. Optic neuritis in Singapore. Singapore Med J. 2008;49(9):667–671.
28. Hansapinyo L, Vivattanaseth C. Clinical characteristics, treatment outcomes and predictive factors in optic neuritis. Open Ophthalmol J. 2018;12:247–255. doi:10.2174/1874364101812010247
29. Mustak H, Cook C. Clinical profile and outcomes of optic neuritis in an HIV prevalent urban community in South Africa. Middle East Afr J Ophthalmol. 2017;24(3):131–135. doi:10.4103/meajo.MEAJO_133_17
30. Pokroy R, Modi G, Saffer D. Optic neuritis in an urban black African community. Eye (Lond). 2001;15(Pt 4):469–473. doi:10.1038/eye.2001.157
31. Storoni M, Pittock SJ, Weinshenker BG, Plant GT. Optic neuritis in an ethnically diverse population: higher risk of atypical cases in patients of African or African-Caribbean heritage. J Neurol Sci. 2012;312(1–2):21–25. doi:10.1016/j.jns.2011.08.030
32. Deschamps R, Lecler A, Lamirel C, et al. Etiologies of acute demyelinating optic neuritis: an observational study of 110 patients. Eur J Neurol. 2017;24(6):874–879. doi:10.1111/ene.13315
33. Alamgir MJ, Ali SA, Hamdy NA, Khan MZ, Mohammad EE. Optic neuritis: observation and experience at a tertiary care hospital in Qassim region, Saudi Arabia. Int J Health Sci (Qassim). 2017;11(5):30–34.
34. Pandit L, Shetty R, Misri Z, et al. Optic neuritis: experience from a south Indian demyelinating disease registry. Neurol India. 2012;60(5):470–475. doi:10.4103/0028-3886.103186
35. Karti O, Karti DT, Kilic İH, Gokcay F, Celebisoy N. Baseline demographics, clinical features, and treatment protocols of 240 patients with optic neuropathy: experiences from a neuro-ophthalmological clinic in the Aegean region of Turkey. Int Ophthalmol. 2019;39(1):155–166. doi:10.1007/s10792-017-0799-5
36. Mealy MA, Whetstone A, Orman G, Izbudak I, Calabresi PA, Levy M. Longitudinally extensive optic neuritis as an MRI biomarker distinguishes neuromyelitis optica from multiple sclerosis. J Neurol Sci. 2015;355(1–2):59–63. doi:10.1016/j.jns.2015.05.013
37. Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology. 2015;84(11):1165–1173. doi:10.1212/WNL.0000000000001367
38. Padungkiatsagul T, Chen JJ, Jindahra P, et al. Differences in clinical features of myelin oligodendrocyte glycoprotein antibody-associated optic neuritis in white and Asian race [published online ahead of print, 2020 Jul 15]. Am J Ophthalmol. 2020:
39. Lin YC, Wang AG, Yen MY. Systemic lupus erythematosus-associated optic neuritis: clinical experience and literature review. Acta Ophthalmol. 2009;87(2):204–210. doi:10.1111/j.1755-3768.2008.01193.x
40. Akpek EK, Bunya VY, Saldanha IJ. Sjögren’s syndrome: more than just dry eye. Cornea. 2019;38(5):658–661. doi:10.1097/ICO.0000000000001865
41. Kruer MC, Koch TK, Bourdette DN, et al. NMDA receptor encephalitis mimicking seronegative neuromyelitis optica. Neurology. 2010;74(18):1473–1475. doi:10.1212/WNL.0b013e3181dc1a7f
42. Jensen ML, Løkke A, Hilberg O, Hyldgaard C, Bendstrup E, Tran D. Clinical characteristics and outcome in patients with antisynthetase syndrome associated interstitial lung disease: a retrospective cohort study. Eur Clin Respir J. 2019;6(1):1583516. doi:10.1080/20018525.2019.1583516
43. Damoiseaux J, Vulsteke JB, Tseng CW, et al. Autoantibodies in idiopathic inflammatory myopathies: clinical associations and laboratory evaluation by mono- and multispecific immunoassays. Autoimmun Rev. 2019;18(3):293–305. doi:10.1016/j.autrev.2018.10.004
44. Baharani A, Raja Rami Reddy P. Bilateral retinopathy and optic neuropathy associated with systemic sclerosis. Ocul Immunol Inflamm. 2019. doi:10.1080/09273948.2019.1629604
45. Rothschild PR, Pagnoux C, Seror R, Brézin AP, Delair E, Guillevin L. Ophthalmologic manifestations of systemic necrotizing vasculitides at diagnosis: a retrospective study of 1286 patients and review of the literature. Semin Arthritis Rheum. 2013;42(5):507–514. doi:10.1016/j.semarthrit.2012.08.003
46. Petzold A, Woodhall M, Khaleeli Z, et al. Aquaporin-4 and myelin oligodendrocyte glycoprotein antibodies in immune-mediated optic neuritis at long-term follow-up. J Neurol Neurosurg Psychiatry. 2019;90(9):1021–1026. doi:10.1136/jnnp-2019-320493
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.