Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 9
Etanercept (Enbrel®) alternative storage at ambient temperature
Authors Shannon E, Daffy J, Jones H, Paulson A, Vicik SM ![]()
Received 7 January 2017
Accepted for publication 8 June 2017
Published 21 July 2017 Volume 2017:9 Pages 87—99
DOI https://doi.org/10.2147/CPAA.S131832
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Arthur E. Frankel
Edel Shannon,1 Joanne Daffy,2 Heather Jones,3 Andrea Paulson,4 Steven M Vicik5
1Global Chemistry, Manufacturing, and Controls Regulatory, 2Contract Operations Quality Assurance, Pfizer Ireland Pharmaceuticals, Clondalkin, Dublin, Ireland; 3Medical Affairs, Pfizer, Collegeville, PA, USA; 4Pharmaceutical Research and Development, 5Global Supply Product Portfolio Management, Pfizer Biotech, Andover, MA, USA
Background: Biologic disease-modifying antirheumatic drugs, including tumor necrosis factor inhibitors such as etanercept (Enbrel®), have improved outcomes for patients with rheumatic and other inflammatory diseases, with sustained remission being the optimal goal for patients with rheumatoid arthritis. Flexible and convenient treatment options, compatible with modern lifestyle, are important in helping patients maintain treatment and manage their disease. Etanercept drug product (DP) is available in lyophilized powder (Lyo) for solution injection, prefilled syringe, and prefilled pen presentations and is typically stored under refrigerated conditions. We aimed to generate a comprehensive analytical data package from stability testing of key quality attributes, consistent with regulatory requirements, to determine whether the product profile of etanercept is maintained at ambient temperature.
Methods: Test methods assessing key attributes of purity, quality, potency, and safety were performed over time, following storage of etanercept DP presentations under a range of conditions.
Results: Results and statistical analysis from stability testing (based on size exclusion high-performance liquid chromatography, hydrophobic interaction chromatography, and sodium dodecyl sulfate-polyacrylamide gel electrophoresis Coomassie) across all etanercept presentations (10 and 25 mg/vial Lyo DP; 25 and 50 mg prefilled syringe DP; 50 mg prefilled pen DP) showed key stability-indicating parameters were within acceptable limits through the alternative storage condition of 25°C±2°C for 1 month.
Conclusion: Stability testing performed in line with regulatory requirements supports a single period of storage for etanercept DP at an alternative storage condition of 25°C±2°C for up to 1 month within the approved expiry of the product. This alternative storage condition represents further innovation in the etanercept product lifecycle, providing greater flexibility and enhanced overall convenience for patients.
Keywords: etanercept, ambient, storage, temperature, stability
Plain language summary
Why was the study conducted?
Use of biologic agents such as etanercept in the treatment of rheumatic diseases has brought considerable benefits in terms of symptom control as well as in other patient-relevant outcomes. However, due to their complexity, these drugs are normally stored under refrigerated conditions to maintain stability. Establishing the stability of etanercept DP at an alternative storage condition of ambient temperature would offer improved flexibility for patients.
What did the researchers do and find?
The researchers performed a comprehensive range of stability tests across all etanercept DP presentations of key product quality indicators, in line with regulatory requirements, which showed the product was stable following a single period of storage at ambient temperature (25°C±2°C) for up to 1 month.
What do these results mean?
These results mean that etanercept DP can be stored under conditions beyond the originally marketed conditions requiring refrigeration (5°C±3°C), thereby providing greater flexibility for patients in managing their disease and in fulfilling their daily lives in terms of work, leisure, and travel.
Introduction
Introduction of biologic disease-modifying antirheumatic drugs (DMARDs) for the treatment of autoimmune rheumatic diseases and spondyloarthropathies began with the approval of the tumor necrosis factor (TNF) inhibitor etanercept and has since revolutionized the management of patients with rheumatic diseases.1 Following its approval in 1998 for the treatment of patients with moderate to severe refractory rheumatoid arthritis (RA), etanercept has been at the forefront of the expansion of therapeutic options available for patients with other inflammatory diseases (psoriatic arthritis, ankylosing spondylitis, plaque psoriasis,2 nonradiographic axial spondyloarthritis,3 as well as polyarticular-course juvenile idiopathic arthritis, and the juvenile idiopathic arthritis categories extended oligoarthritis, enthesitis-related arthritis, and psoriatic arthritis).4
Use of biologic therapy, including TNF inhibitors, has improved outcomes for patients with RA, with sustained remission now considered the optimal treatment goal to prevent joint damage.1 Therefore, flexible and convenient treatment options that are compatible with modern lifestyle are important aspects in helping patients maintain treatment and manage their disease.
Since the first clinical trials of etanercept in 1993, over 4.5 million patient-years of post-marketing experience has been accumulated, with the safety and efficacy of etanercept being consistent in the treatment of patients across a spectrum of inflammatory diseases.5 In all of these populations, etanercept (with or without methotrexate) effectively reduced signs and symptoms, disease activity, and disability, and improved health-related quality of life, with these benefits being sustained during long-term treatment.2
The biochemical and physical structures of recombinant protein biologics are dictated by their primary amino acid sequence and, where present, by posttranslational modifications such as glycosylation. These can have a role in structure, activity, signaling, clearance, and immunogenicity. Etanercept is a technically complex 934-amino acid protein (MW ~150 kDa), which is heavily glycosylated, containing both N- and O-linked oligosaccharides.6 Extensive characterization of the drug product (DP) is performed using a range of sensitive physical, chemical, and biologic analytical methodologies.7 Regulation of the manufacture of etanercept and other biologic drugs is governed by a requirement to perform robust analytical and stability testing to ensure quality is maintained.8
Protein instability can stem from degradation, due to fragmentation of the peptide backbone or hydrolysis of amino acid side chain amides (Asn and Gln residues), oxidation (of Met, His, Cys, Tyr, and Trp residues), denaturation (loss of 3D structure), or aggregation (covalent or noncovalent association of monomers or native multimers).9 Moreover, increases in physical and chemical degradation can occur during long-term storage.10
Specific guidance is provided on the stability testing of biologic products,11 particularly since these are sensitive to environmental factors such as temperature, oxidation, light, ionic content, or shear, and stringent conditions for storage are usually required. The International Conference on Harmonization Q5C provides guidance to applicants regarding the type of stability studies to be provided in support of marketing authorization applications and amendments for biologic medicinal products.11
As originally manufactured, etanercept DP was provided as a 25 mg/vial lyophilized powder (Lyo), requiring reconstitution prior to administration by subcutaneous injection. Etanercept 25 and 50 mg prefilled syringe (PFS) DP and 50 mg prefilled pen (PFP) presentations were subsequently added to enhance patient convenience.6 An etanercept 10 mg/vial Lyo DP presentation was introduced for use in pediatric populations.6 The long-term recommended storage conditions for all etanercept DP configurations marketed in the European Union is 5°C±3°C. Here we describe the comprehensive analytical data package obtained from stability testing of key product quality attributes, performed in line with regulatory requirements, to support an alternative short-term storage condition for etanercept at 25°C±2°C, to supplement the long-term storage.
Methods
Stability studies were conducted on multiple batches manufactured at three commercial manufacturing sites.
Assays
The stability assays used to assess the key attributes of purity, quality, potency, and safety are summarized in Table 1. Size exclusion high-performance liquid chromatography (SE-HPLC), hydrophobic interaction chromatography (HIC), and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) Coomassie were used as the key quantitative stability-indicating assays.
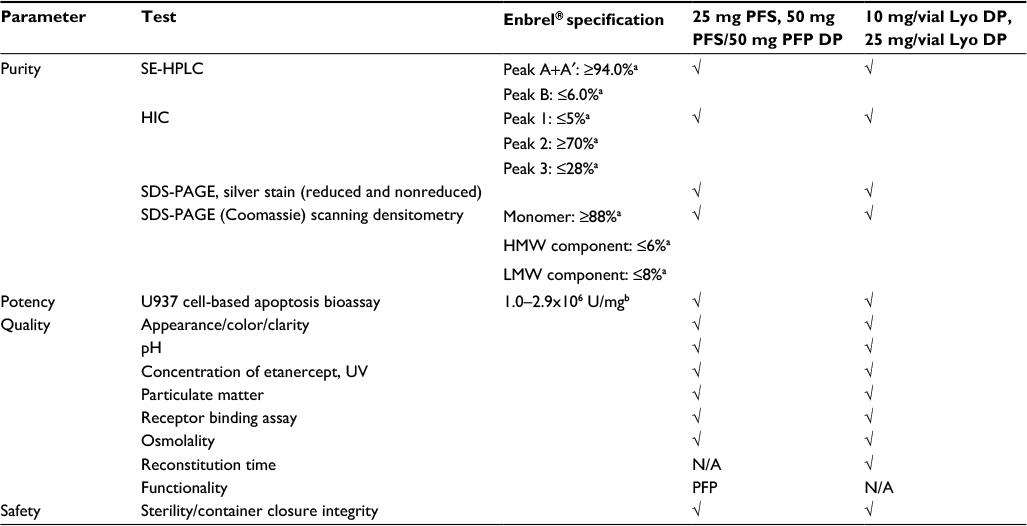 | Table 1 Stability tests performed for etanercept DP presentations Notes: aRelative % peak area. bAt the time of this analysis, the A375 cell-based assay (specification limit 0.8–2.9×106 U/mg) was used for assessment of potency of etanercept 10 mg/vial (n=3 batches) and 25 mg/vial Lyo DP (n=5 batches), at the alternative storage condition of 25°C±2°C through to 24 months. The A375 assay has since been replaced by the U937 cell-based apoptosis bioassay. Abbreviations: DP, drug product; PFS, prefilled syringe; PFP, prefilled pen; Lyo, lyophilized powder; SE-HPLC, size exclusion high-performance liquid chromatography; HIC, hydrophobic interaction chromatography; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; HMW, high molecular weight; LMW, low molecular weight; UV, ultraviolet; N/A, not applicable. |
Size exclusion high-performance liquid chromatography
SE-HPLC is performed to assess the purity of etanercept by separation of the high-molecular-weight component (HMWC) from homogenous dimeric etanercept and low-molecular-weight component (LMWC; clipped species) based on molecular size, and resolves etanercept into two principal peaks: Peak B=HMWC (aggregation); Peak A+A′=etanercept dimer (desired active product)+LMWC. Results are expressed as relative percent peak area.
Each sample (14 µL; 2.5 mg/mL) was injected onto an analytical HPLC column (8×300 mm) containing hydrophilic silica gel (5 μm with a pore size of 30 nm and of a grade suitable for fractionation of globular proteins in the relative molecular mass range of 10,000–1,000,000) at 25°C. Monitoring was performed by ultraviolet detection (λ=220 nm). A mobile phase consisting of 220 mL of solution A (sodium dihydrogen phosphate [15.6 g] and sodium chloride [8.8 g] in 1000 mL water for chromatography) and 780 mL of solution B (anhydrous disodium hydrogen phosphate [14.2 g] and sodium chloride [8.75 g] in 1000 mL water for chromatography), adjusted to pH 7.2, was used. The flow rate was 1.0 mL/min. Etanercept was resolved into two principal peaks based on molecular size: Peak B=HMWC; Peak A+A′=etanercept dimer+LMWC.
Hydrophobic interaction chromatography
HIC is performed to assess the purity of etanercept and the related product species present. The method is used to resolve etanercept into three variants that differ in biologic activity: Peak 1 is predominantly clipped species (fragmentation); Peak 2 is homogenous etanercept (desired active product); and Peak 3 consists of misfolded species, aggregates, etanercept fragments, and other process-related impurities. Results are expressed as relative percent peak area.
Each sample (5 µL; 2 mg/mL) was injected onto a TSK-GEL Butyl-NPR (2.5 µm; Tosoh) analytical column (4.6×350 mm) at 35°C, connected to an HPLC system. Product-related impurities were separated by gradient elution using mobile phase A (ammonium sulfate [475.9 g] and anhydrous disodium hydrogen phosphate [28.4 g] in 1950 mL water for chromatography, adjusted to pH 7.0 with phosphoric acid and diluted to 2000 mL with water for chromatography) and mobile phase B (anhydrous disodium hydrogen phosphate [28.4 g] in water for chromatography and diluted to 1950 mL with water, adjusted to pH 7.0 with phosphoric acid and diluted to 2000 mL with water). The flow rate was 1.0 mL/min, and chromatography was monitored by fluorescence detection (278 nm for excitation; 350 nm for emission). Elution of the protein molecules occurred in order of increasing hydrophobicity, as the salt concentration decreased throughout the run, separating the protein into HIC Peak 1 (clipped species of etanercept), HIC Peak 2 (etanercept), and HIC Peak 3 (misfolded and aggregated etanercept). The peaks were integrated and reported as relative % Peak 1, % Peak 2, and % Peak 3.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Coomassie)
Etanercept was analyzed for impurities by SDS-PAGE under reducing conditions and stained with a Coomassie stain. This procedure was used to separate proteins based primarily on their molecular weights (MWs). SDS binds along the polypeptide chain and the charge of the reduced SDS protein complex is proportional to its MW. The distance migrated on the gel is inversely proportional to the sample MWs. Samples were initially diluted in water or formulation buffer, following which they were diluted in reducing sample buffer containing 1,4-dithiothreitol and heated at 95°C±5°C for 5 min. Twenty micrograms of each sample was loaded onto a precast 8%–16% Tris-glycine gel. A MW ladder was also loaded onto the gel to determine the apparent MW of each sample fragment. The proteins were separated by electrophoresis and the gel was stained with 0.25% w/v Coomassie R-250, destained to clarity, and scanned.
The results are reported as % HMW (MW >76 kDa; aggregation; denaturation), % main band (MW ~76 kDa; indicator of desired active product), and % LMW (MW <76 kDa; fragmentation). Sample banding profiles are compared with the reference standard banding profile.
Cell-based bioassay
Neutralization of TNFα-mediated apoptosis in U937 cells
The potency of etanercept samples was quantified by measurement of their ability to neutralize TNFα-mediated apoptosis in histiocytic lymphoma cell-line U937 (ATCC No. CRL-1593.2) via caspase activation. The U937 cells were incubated at 36.0°C–38.0°C for 30–60 min in a humidified incubator using 5%±2% CO2 with varying dilutions of test and reference preparations of etanercept in the presence of TNFα. They were then incubated with Caspase-Glo 3/7 reagent at room temperature for 30–60 min, which resulted in cleavage of a luminogenic substrate by caspase, subsequent release of a luciferase substrate, and generation of a luminescent signal. The luminescence produced was proportional to the amount of caspase activity present. Biologic activity of the sample was calculated based on the ratio of IC50 values of calibration curve, relative to each control and test sample curve. Sample-specific activity results are reported as U/mg.
Neutralization of TNFα-mediated growth inhibition of A375 cells
The potency of etanercept was quantified by measurement of its ability to neutralize TNFα-mediated growth inhibition of cells from the human melanoma cell line A375.S2 by co-incubation with TNFα and varying concentrations of etanercept. Samples and control were diluted to 75 ng/mL and added to 96-well cell culture plates, where a further nine 1/1.5 serial dilutions were prepared. Reference standard diluted to 75 ng/mL was applied to the plate as a calibrator. A375.S2 cells were diluted to 4×105 cells/mL and added to the plate at 25 mL/well, followed by TNFα at ~5 ng/mL that is added to the plate at 25 mL/well. TNFα was not added to the positive control wells and etanercept was not added to the negative control wells. Following a 72-hour co-incubation at 37°C, nonviable cells were removed by washing with phosphate-buffered saline and the remaining cells were methanol-fixed, stained with 0.1% (w/v) crystal violet, washed again with water, and then solubilized with 2% (w/v) deoxycholate to release the stain into the solution. The intensity of the color produced was directly proportional to the activity of Enbrel, allowing sample activities to be calculated relative to the standard calibration curve.
At the time of this analysis, the A375 cell-based assay was used for assessment of potency of etanercept 10 and 25 mg/vial Lyo DP, at the alternative storage condition of 25°C±2°C through to 24 months. The A375 assay has since been replaced by the U937 cell-based apoptosis bioassay.
Storage conditions
The storage conditions at which stability tests were performed on the various etanercept DP presentations are summarized in Table 2. While the aim of the analysis was to establish whether the product profile of etanercept DP was maintained at an ambient temperature of 25°C, the samples were subjected to a higher temperature of 30°C (following storage at 5°C) to allow for some variability in local ambient temperature conditions.
 | Table 2 Storage conditions for stability studies and approved shelf life in EU for etanercept DP presentations Notes: aSince the 50 mg PFP encloses a single etanercept 50 mg DP solution for injection in PFS, and the PFP components do not have any contact with the DP contained in the PFS or with the needle that is preattached to the syringe, the analysis performed on etanercept 50 mg DP PFS is considered applicable to the 50 mg PFP presentation. bSamples were stored for 39 months at 5°C±3°C prior to storage at 30°C±2°C. cSamples were stored for 36 months at 5°C±3°C prior to storage at 30°C±2°C. dSamples were stored for 30 or 36 months at 5°C±3°C prior to storage at 30°C±2°C. Abbreviations: EU, European Union; DP, drug product; Lyo, lyophilized powder; PFS, prefilled syringe; PFP prefilled pen. |
Analysis
Statistical analysis was performed on the following quantitative stability-indicating parameters: HIC, SE-HPLC, SDS-PAGE (Coomassie). The stability data were statistically analyzed using linear regression to determine whether individual batches had the same nominal rate of change, as indicated by common slopes using analysis of covariance. A common slope model was fitted if the P value for the batch by timepoint interaction was >0.05 (5%). If the P value was <0.05 (5%), the batch with the greatest rate of change was used to describe the data (i.e., a separate slopes model was used) to assess compatibility with the proposed storage time. Residual analysis was used to check the validity of the linear models. If curvature was observed in the residuals plotted against the predicted values, then nonlinear models were sought. Data analysis was performed using SAS JMP v.8.0.1. (SAS Institute, Cary, NC, USA).
Results
Results and statistical analysis from stability testing across the etanercept DP presentations are described below.
Etanercept 10 and 25 mg/vial Lyo DP
Stability studies were initially performed on etanercept 10 mg/vial (n=3 batches) and 25 mg/vial Lyo DP (n=5 batches) at the alternative storage condition of 25°C±2°C through to 24 months and data for the quantitative stability-indicating assays (HIC, SE-HPLC, and SDS-PAGE Coomassie; Figure 1) were assessed by statistical methods. The rates of change of the etanercept 10 and 25 mg/vial Lyo DP based on these key quantitative stability-indicating assays over 1-month storage at 25°C±2°C are listed in Table 3. All results are well within specification for up to 1 month at 25°C±2°C.
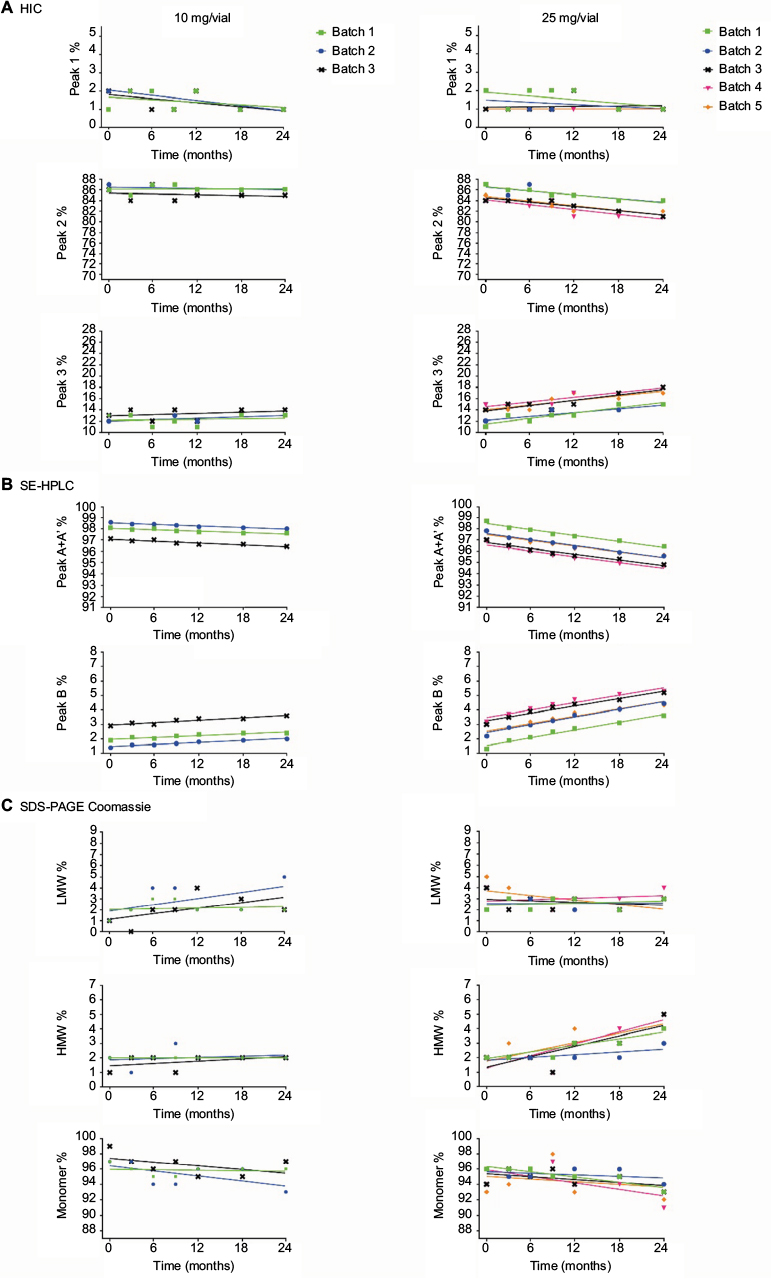
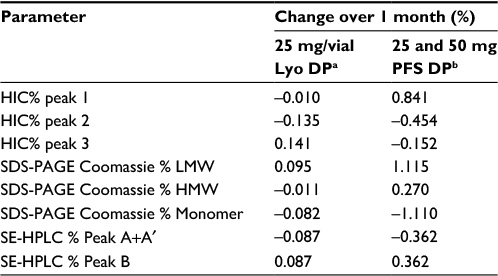 | Table 3 Statistical analysis of key stability-indicating assays for etanercept DP presentations over 1 month storage at 25°C±2°C Notes: a10 mg/vial Lyo presentation was considered to have greater stability than the 25 mg/vial Lyo presentation; therefore, statistical analysis was performed only on the 25 mg/vial Lyo presentation. bSince the 50 mg PFP encloses a single etanercept 50 mg DP solution for injection in PFS, and the PFP components do not have any contact with the DP contained in the PFS or with the needle that is preattached to the syringe, the analysis performed on the etanercept 50 mg DP PFS is considered applicable to the 50 mg PFP presentation. Abbreviations: DP, drug product; Lyo, lyophilized powder; PFS, prefilled syringe; HIC, hydrophobic interaction chromatography; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; LMW, low molecular weight; HMW, high molecular weight; SE-HPLC, size exclusion high-performance liquid chromatography; PFP, prefilled pen. |
Based on the supportive stability results obtained for the etanercept Lyo DP at 25°C, a further phase of studies was initiated to support long-term storage of etanercept DP at refrigerated temperature, including ambient storage at the end of shelf life for up to 4 weeks out of the refrigerator.
Etanercept 10 mg/vial (n=3 batches) and 25 mg/vial Lyo DP material (n=2 batches) were stored at the long-term recommended storage condition of 5°C±3°C for 39 and 36 months, respectively, and were then transferred to 30°C±2°C for 4 weeks’ study duration. There was no significant degradation observed, reflecting a satisfactory stability profile, and all data remained within specification for 4 weeks (Figure 2).
 | Figure 2 Stability data for the quantitative stability-indicating assays (A) HIC, (B) SE-HPLC, and (C) SDS-PAGE Coomassie for etanercept 10 mg/vial (n=3 batches) and 25 mg/vial Lyo DP (n=2 batches) stored at 5°C±3°C for 30 and 36 months, respectively, followed by 4 weeks at 30°C±2°C (only the 4 weeks storage period at 30°C±2°C is shown). Abbreviations: HIC, hydrophobic interaction chromatography; SE-HPLC, size exclusion high-performance liquid chromatography; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; LMW, low molecular weight; HMW, high molecular weight; Lyo, lyophilized powder; DP, drug product. |
In conclusion, the data support a single period of storage for up to 1 month at the alternative storage condition, 25°C±2°C, within an expiry period of 36 months for etanercept 10 and 25 mg/vial Lyo DP. (The period for expiry of the 25 mg/vial Lyo DP was recently extended to 48 months, including the 1-month alternative storage, in some jurisdictions.)12
Etanercept 25 and 50 mg PFS DP
Assays were performed to monitor the stability of the etanercept 25 and 50 mg PFS DP presentations stored at the alternative storage condition of 25°C±2°C and at the accelerated storage condition of 30°C±2°C (Table 2).
The stability data for the quantitative stability-indicating assays (HIC, SE-HPLC, and SDS-PAGE Coomassie) for etanercept 25 mg (n=7) and 50 mg (n=7) PFS DP at 25°C±2°C through to 12 months were assessed by statistical methods. All results were within specification for 1 month at the alternative storage condition (25°C±2°C), as shown in Table 3.
Based on the supportive stability results for the etanercept 25 and 50 mg PFS DP, studies were initiated to support long-term storage at refrigerated temperatures that included ambient storage at the end of shelf life for up to 4 weeks out of the refrigerator.
Etanercept 25 mg (n=6) and 50 mg (n=7) PFS DP were stored at the long-term recommended storage condition of 5°C±3°C for 30 or 36 months and then transferred to 30°C±2°C for the 4-week study duration. All data reflected a satisfactory stability profile and remained in specification throughout the 4-week study for all batches (Figure 3).
 | Figure 3 Stability data for the quantitative stability-indicating assays (A) HIC, (B) SE-HPLC, and (C) SDS-PAGE Coomassie for etanercept 25 mg (n=6 batches) and 50 mg PFS DP (n=7 batches) at the alternative storage condition of 4 weeks at 30°C, following storage at the recommended condition of 5°C±3°C for 30 or 36 months. Abbreviations: HIC, hydrophobic interaction chromatography; PFS, prefilled syringe; SE-HPLC, size exclusion high-performance liquid chromatography; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; LMW, low molecular weight; HMW, high molecular weight; DP, drug product. |
In conclusion, the data support a single period of storage for up to 1 month at the alternative storage condition, 25°C±2°C, within the approved expiry period of 30 months for etanercept 25 and 50 mg PFS DP.
Etanercept 50 mg PFP DP
The etanercept 50 mg PFP DP presentation is a spring-powered injection system consisting of two subassemblies, front and rear, enclosing a single etanercept 50 mg solution for injection in PFS, the contents of which are delivered to the patient subcutaneously. The PFP subassemblies do not have any contact with the DP contained in the PFS or with the needle that is preattached to the syringe. The analysis performed on the etanercept 50 mg PFS DP is, therefore, considered applicable to the 50 mg PFP DP preparation.
The results of the stability study testing, indicating the period at which the various etanercept DP presentations remain within specification, are summarized in Table 2. Additional tests were performed (Table 1; Figures S1–S3), and the stability data and statistical analysis collectively support the alternative storage condition 25°C±2°C for up to 1 month.
Discussion
In recent years, there has been a considerable change in the management of patients with RA. Early and aggressive treatment, including the use of novel biologic agents, has been shown to lead to favorable patient outcomes in reducing synovial inflammation, delaying joint damage, and maintaining functional status.13 Prompt treatment initiation and a tight control follow-up aimed at achieving low disease activity or remission are now regarded as the standard of care.14 Such treatment strategies have been shown in clinical trials to lower the levels of disease activity and to improve psychologic well-being and physical functionality, and are now reflected in the classification criteria and recommendations for the utilization of conventional DMARDs and biologic agents in the treatment of RA.1
There is evidence to suggest that initiation of treatments early in the disease course not only offers substantial improvements in functional ability and quality of life,15 but also brings significant benefits in terms of productivity, due to reduced sick leave and lost employment,16 and a decline in work disability.17 Much of this progress in disease management and patient outcomes has been due to the availability of biologic DMARDs, such as etanercept. However, there has been a need for treatment options to evolve to meet the changing habits and expectations of patients. For instance, previously, many patients with RA may have experienced impaired ability to travel, but with improvements in functioning and quality of life, these restrictions are diminished;18 therefore, flexible and convenient treatment options that are compatible with modern lifestyle, including working and traveling, are important aspects in helping patients maintain treatment and manage their disease.
Cold storage conditions are not fully compatible with many of the activities of modern lifestyle and many of these drawbacks could potentially be mitigated if stability of the product could be demonstrated at temperatures encountered under ambient conditions. Moreover, short storage limits contribute to product wastage due to routine discarding of unused diluted drug19,20 and inconvenience for patients; this too may be reduced if product stability were extended to out-of-refrigeration storage.21 The development and availability of PFS and PFP presentations, to complement the vial presentation of etanercept DP, provided patients with greater self-administration options to manage their disease. However, offering an option of short-term storage at ambient conditions as an alternative to the originally marketed conditions of 5°C±3°C enhances overall patient convenience and provides greater flexibility, particularly while traveling, with etanercept.
The International Conference on Harmonization Q5C specifies the type of stability studies to be provided in support of marketing authorization for biologic products.11 These specific guidelines for biologics are necessary on account of both their higher level of structural complexity compared with small molecules, with maintenance of biologic activity often dependent on noncovalent interactions, and also the distinct pathways leading to their instability. To comply with these guidelines, a sequential approach was followed in line with regulatory requirements, to support an alternative short-term storage condition for etanercept at 25°C±2°C, to supplement the long-term storage.
Using SE-HPLC, HIC, and SDS-PAGE (Coomassie) as the key stability-indicating assays demonstrated a sufficiently broad and robust package of data to comply with the regulatory requirements,22 to support a single period of storage for etanercept at an alternative storage condition of 25°C±2°C for up to 1 month within the approved expiry of the product. In particular, initial studies that showed only a minimal rate of change for the 25 mg/vial Lyo DP suggested that etanercept DP had the potential to be stored under conditions beyond the originally European Union-marketed conditions of 5°C±3°C. More importantly, subsequent demonstration of the continued stability of etanercept 25 mg Lyo DP under the long-term recommended storage condition of 5°C±3°C for 36 months, including a period of 4 weeks under the accelerated condition of 30°C±2°C, reflected a satisfactory stability profile. This has the potential to provide greater flexibility for patients in managing their disease and in fulfilling their daily lives in terms of work, leisure, and travel. This alternative storage condition for etanercept thus represents further innovation in the product lifecycle and offers enhanced overall patient convenience.
Acknowledgments
The study was funded by Pfizer. Editorial/medical writing support was provided by Iain McDonald of Engage Scientific Solutions and was funded by Pfizer.
The abstract of this paper was accepted for publication at the Annual Congress of the European League Against Rheumatism (EULAR); June 8–11, 2016; London, UK and published in Ann Rheum Dis. 2016;75(Suppl 2):1014 http://ard.bmj.com/content/75/Suppl_2/1014.1.abstract?sid =13,47,582c-a382–443c-b923–3,34,64,34,006f7 doi:10.1136/annrheumdis- 2016-eular.1855.
Author contributions
All the authors made substantial contributions to the conception and design, acquisition of data, and analysis and interpretation of data; were involved in drafting the manuscript and revising the manuscript critically for important intellectual content; and have given final approval of the version to be published.
Disclosure
All the authors are full-time employees of Pfizer and hold stock or stock options. The authors report no other conflicts of interest in this work.
References
Smolen JS, Landewé R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. 2014;73(3):492–509. | ||
Scott LJ. Etanercept: a review of its use in autoimmune inflammatory diseases. Drugs. 2014;74(12):1379–1410. | ||
Maksymowych WP, Dougados M, van der Heijde D, et al. Clinical and MRI responses to etanercept in early non-radiographic axial spondyloarthritis: 48-week results from the EMBARK study. Ann Rheum Dis. 2015;75(7):1328–1335. | ||
Windschall D, Müller T, Becker I, Horneff G. Safety and efficacy of etanercept in children with the JIA categories extended oligoarthritis, enthesitis-related arthritis and psoriasis arthritis. Clin Rheumatol. 2015;34(1):61–69. | ||
Hassett B, Singh E, Mahgoub E, O’Brien J, Vicik SM, Fitzpatrick B. Manufacturing History of Enbrel: Description of Changes to Impact Overall Safety Profile and Process Performance. J Clin Rheumatol. 2016;22:158. | ||
Pfizer. Enbrel (etanercept) SmPC. [homepage on the Internet] 2010. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000262/WC500027361.pdf. Accessed June 23, 2016. | ||
Graumann K, Premstaller A. Manufacturing of recombinant therapeutic proteins in microbial systems. Biotechnol J. 2006;1(2):164–186. | ||
Committee for proprietary medicinal products (CPMP). Guideline on stability testing: stability testing of existing active substances and related finished products (2003) CPMP/QWP/122/02, rev 1 corr. [homepage on the Internet]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003466.pdf. Accessed June 10, 2016. | ||
Manning MC, Chou DK, Murphy BM, Payne RW, Katayama DS. Stability of protein pharmaceuticals: an update. Pharm Res. 2010;27(4):544–575. | ||
Wang W. Advanced protein formulations. Protein Sci. 2015;24(7):1031–1039. | ||
International Conference on Harmonization (ICH). Quality of biotechnological products: stability testing of biotechnological/biological products. Q5C. [homepage on the Internet] 1995. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5C/Step4/Q5C_Guideline.pdf. Accessed June 10, 2016. | ||
Pfizer. Enbrel 25 mg powder and solvent for solution for injection. SPC. Last Updated 6 May 2016. Available from: http://www.medicines.org.uk/emc/medicine/3343. Accessed November 22, 2016. | ||
Ruderman EM, Nola KM, Ferrell S, Sapir T, Cameron DR. Incorporating the treat-to-target concept in rheumatoid arthritis. J Manag Care Pharm. 2012;18(9):1–18. | ||
Monti S, Montecucco C, Bugatti S, Caporali R. Rheumatoid arthritis treatment: the earlier the better to prevent joint damage. RMD Open. 2015;1(Suppl 1):e000057. | ||
van der Kooij SM, de Vries-Bouwstra JK, Goekoop-Ruiterman YP, et al. Patient-reported outcomes in a randomized trial comparing four different treatment strategies in recent-onset rheumatoid arthritis. Arthritis Rheum. 2009;61(1):4–12. | ||
Birnbaum H, Pike C, Kaufman R, Marynchenko M, Kidolezi Y, Cifaldi M. Societal cost of rheumatoid arthritis patients in the US. Curr Med Res Opin. 2010;26(1):77–90. | ||
Nikiphorou E, Guh D, Bansback N, et al. Work disability rates in RA. Results from an inception cohort with 24 years follow-up. Rheumatology (Oxford). 2012;51(2):385–392. | ||
Orenstein R. Travel in patients receiving TNF-alpha inhibitors. Travel Med Infect Dis. 2005;3(2):105–109. | ||
Román Ivorra JA, Ivorra J, Monte-Boquet E, Canal C, Oyagüez I, Gómez-Barrera M. Cost analysis of biologic drugs in rheumatoid arthritis first line treatment after methotrexate failure according to patients’ body weight. Reumatol Clin. 2016;12(3):123–129. | ||
Benucci M, Stam WB, Gilloteau I, et al. Abatacept or infliximab for patients with rheumatoid arthritis and inadequate response to methotrexate: an Italian trial-based and real-life cost-consequence analysis. Clin Exp Rheumatol. 2013;31(4):575–583. | ||
Ikeda R, Vermeulen LC, Lau E, et al. Stability of infliximab in polyvinyl chloride bags. Am J Health Syst Pharm. 2012;69(17):1509–1512. | ||
Committee for Medicinal Products for Human Use (CHMP). Guideline on Stability Testing for Applications for Variations to a Marketing Authorization (EMA/CHMP/CVMP/QWP/4,41,071/2011-Rev.2). [homepage on the Internet] 2014. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/04/WC500164972.pdf. Accessed June 10, 2016. | ||
Pfizer. Enbrel 10 mg powder and solvent for solution for injection for pediatric use. SPC. [homepage on the Internet]. Last Updated 6 May 2016. Available from: http://www.medicines.org.uk/emc/medicine/24761. Accessed November 22, 2016. | ||
Pfizer. Enbrel 25mg solution for injection in pre-filled syringe. SPC. [homepage on the Internet]. Last Updated 6 May 2016. Available from: http://www.medicines.org.uk/emc/medicine/19161. Accessed November 22, 2016. | ||
Pfizer. Enbrel 50mg solution for injection in pre-filled syringe. SPC. [homepage on the Internet]. Last Updated 6 May 2016. Available from: http://www.medicines.org.uk/emc/medicine/19162. Accessed November 22, 2016. | ||
Pfizer. Enbrel 50 mg solution for injection in pre-filled pen. SPC. [homepage on the Internet]. Last Updated 6 May 2016. Available from: http://www.medicines.org.uk/emc/medicine/22143. Accessed November 22, 2016. |
Supplementary materials
 | Figure S1 Stability data for the neutralization of TNFα-mediated growth inhibition in A375 cells for (A) etanercept 10 mg/vial (n=3 batches) and (B) 25 mg/vial Lyo DP (n=5 batches), and for the neutralization of TNFα-mediated apoptosis in U937 cells for (C) etanercept 25 mg/vial Lyo DP (n=3 batches), at the alternative storage condition of 25°C±2°C through to 24 months. At the time of analysis of the 18-month timepoint for the etanercept 25 mg/vial Lyo DP, the assay was transitioning from the A375 assay to the U937 apoptosis assay. Both assays were run on the same batches in an analytical method crossover exercise at the 18- and 24-month timepoints. Abbreviations: TNF, tumor necrosis factor; Lyo, lyophilized powder; DP, drug product. |
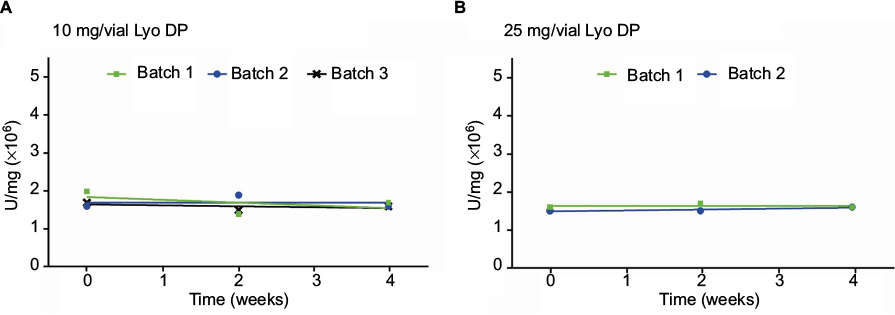 | Figure S2 Stability data for the neutralization of TNFα-mediated apoptosis in U937 cells for (A) etanercept 10 mg/vial (n=3 batches) and (B) 25 mg/vial Lyo DP (n=2 batches), stored at 5°C±3°C for 30 and 36 months, respectively, followed by 4 weeks at 30°C±2°C (only the 4 weeks storage period at 30°C±2°C is shown). Abbreviations: TNF, tumor necrosis factor; Lyo, lyophilized powder; DP, drug product. |
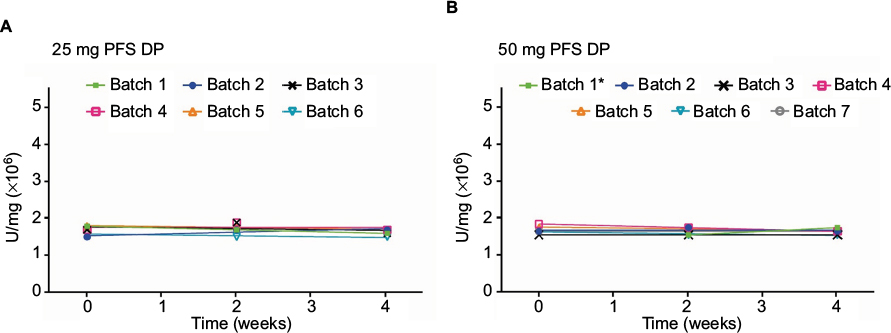 | Figure S3 Stability data for the neutralization of TNFα-mediated apoptosis in U937 cells for (A) etanercept 25 mg (n=6 batches) and (B) 50 mg PFS DP (n=7 batches) at the alternative storage condition of 4 weeks at 30°C ±2°C, following storage at the recommended condition of 5°C±3°C for 30 or 36 months (only the 4 weeks storage period at 30°C ±2°C is shown). *t=0 timepoint not scheduled. Abbreviations: TNF, tumor necrosis factor; PFS, prefilled syringe; DP, drug product. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.